
Abstract
Significance:
Skin protects the body from dehydration, pathogens, and external mutagens. NADPH oxidases are central components for regulating the cellular redox balance. There is increasing evidence indicating that reactive oxygen species (ROS) generated by members of this enzyme family play important roles in the physiology and pathophysiology of the skin.
Recent Advances:
NADPH oxidases are active producers of ROS such as superoxide and hydrogen peroxide. Different isoforms are found in virtually all tissues. They play pivotal roles in normal cell homeostasis and in the cellular responses to various stressors. In particular, these enzymes are integral parts of redox-sensitive prosurvival and proapoptotic signaling pathways, in which they act both as effectors and as modulators. However, continuous (re)activation of NADPH oxidases can disturb the redox balance of cells, in the worst-case scenario in a permanent manner. Abnormal NADPH oxidase activity has been associated with a wide spectrum of diseases, as well as with aging and carcinogenesis.
Critical Issues:
Sunlight with its beneficial and deleterious effects induces the activation of NADPH oxidases in the skin. Evidence for the important roles of this enzyme family in skin cancer and skin aging, as well as in many chronic skin diseases, is now emerging.
Future Directions:
Understanding the precise roles of NADPH oxidases in normal skin homeostasis, in the cellular responses to solar radiation, and during carcinogenesis will pave the way for their validation as therapeutic targets not only for the prevention and treatment of skin cancers but also for many other skin-related disorders. Antioxid. Redox Signal. 28, 1238–1261.
Introduction
R

Tissues of highest expression are indicated in bold (37, 47, 54, 100, 114, 134, 155). Specific skin-related publications are indicated in the table. There are more skin-related disorders, which are characterized by increased levels of reactive oxygen species and NOX activity. However, there is no indication of which specific NOX isoform is involved [see Babalola et al. (13)].
H, human; M, mouse; N, not expressed; n.a., not applicable; n.d., not defined; NOX, NADPH oxidase; UV, ultraviolet; Y, expressed.
The skin protects the body from dehydration, pathogens, and external mutagens. NADPH oxidases play important roles in skin homeostasis and are central mediators in the cellular stress response (Table 1). However, they are also key to the establishment of chronic oxidative stress and metabolic alterations (13, 17). Hence, particular focus is on their suitability as therapeutic targets for both the prevention and treatment of many chronic and inherited diseases, as well as cancer (131). Here we present a review of the current literature regarding the NADPH oxidases family and its functions in skin in health and disease, with a focus on UV radiation-induced skin cancers.
Roles of NADPH oxidases: The Good, the Bad, and the Ugly
Despite their bad reputation, ROS are essential for the cell. The level of ROS that is required to maintain normal physiological processes is highly condition and cell-type specific (Fig. 2 and Table 1). Many of the components of signaling pathways are redox sensitive. For instance, the activation or deactivation of many kinases and phosphatases is mediated by the oxidation of thiol residues (90). Similarly, the activity of several transcription factors is profoundly modified by ROS. Furthermore, ROS produced by NADPH oxidases are fundamental for processes such as the host immune defense, angiogenesis, wound healing, cell differentiation, and thyroid hormone production. In addition, low levels of ROS are required for adult stem cell maintenance and genetic reprogramming during developmental processes (200). High amounts of ROS are produced in some physiological circumstances. For instance, NOX2 produces large amounts of ROS in phagocytes, which are indispensable for the destruction of pathogens (19).
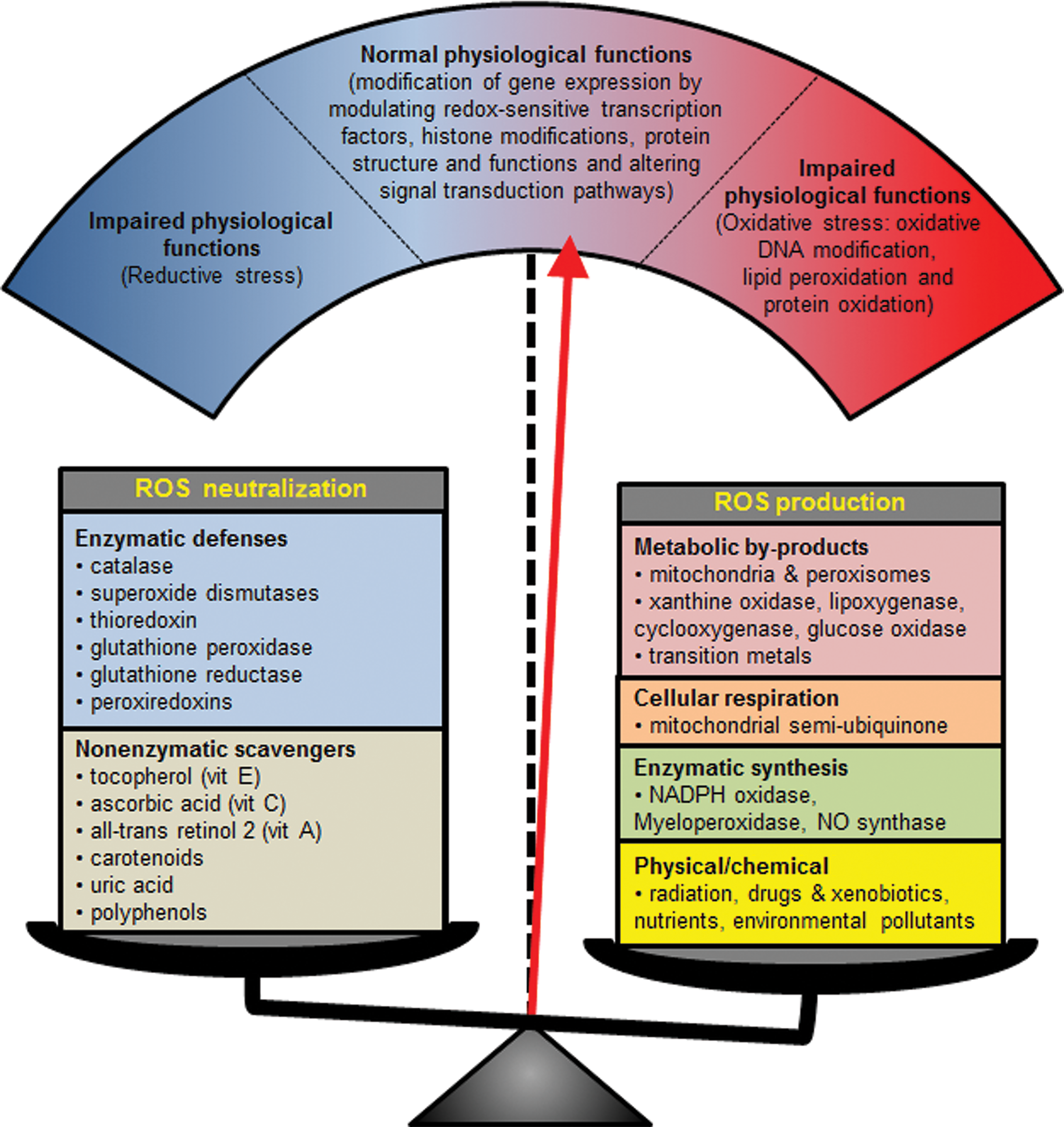
The transient production of pathologically high levels of ROS by NADPH oxidases presents a serious challenge to the redox balance of the cell (Fig. 2). It can cause genomic instability, damage to other biomolecules, alterations in posttranslational modifications, and both metabolic and epigenetic reprogramming (38, 200). These changes increase the risk of developing chronic wounds, fibrosis of various epithelia, diabetes, high blood pressure, asthma, or hypothyroidism (13, 100).
In the worst-case scenarios, chronically decreased or increased NADPH oxidase activity causes life-threatening diseases, such as cancer in almost any tissue, chronic renal failure, granulomatous disease (immune defect), or cardiovascular disease, including arteriosclerosis and myocardial infarction (19, 100). Furthermore, NADPH oxidases have been shown to contribute to the pathogenesis of many neurodegenerative diseases such as Parkinson's and Alzheimer's (114).
NADPH Oxidases in Skin Cells
The three main cell types of the skin—epidermal keratinocytes, dermal fibroblasts, and melanocytes—exhibit different expression patterns of NADPH oxidases.
NADPH oxidases in keratinocytes
In untreated normal human keratinocytes, the expression of NOX1, NOX2, and NOX4 is detectable by Western blot (145, 151). Immortalized keratinocyte cell lines such as HaCaT produce NOX1, 2, 4, and 5 at the messenger RNA (mRNA) level (21, 34). NOX1, 2, and 4 proteins are strongly expressed and appear to be the main producers of ROS in HaCaT (34, 102), while calcium-modulated NADPH oxidase activity was attributed to NOX5 (21). In normal untreated mouse keratinocytes, protein levels of NOX1, NOX2, and NOX4 are detectable (77, 145). There is no homologue of human NOX5 in rodents, including mice and rats (86).
Aging keratinocytes of mice exhibit elevated levels of ROS, which were attributed to the activity of NOX1 (77). Other studies in xeroderma pigmentosum factor C (XPC) patients and respective mouse models confirmed the link between pathologically high levels of NOX1 and 2, and premature skin aging features increased UV light sensitivity and carcinogenesis (109, 145, 151). Furthermore, the expression of DUOX1 is specifically upregulated in response to a testosterone-triggered increase in intracellular calcium levels in mouse epidermal keratinocytes (MPEK-BL6) and human skin equivalents (96). In particular, H2O2 produced by DUOX1 was shown to trigger apoptosis in skin keratinocytes during stratification, which facilitates cornified envelope development and hence ensures proper barrier formation.
NADPH oxidases in fibroblasts
Untreated primary human fibroblasts exhibit high mRNA levels of NOX and NOX5, as well as lower levels of DUOX1 and 2 (190). Interestingly, in contrast to untreated keratinocytes where NOX1 and 2 are the dominant isoforms, they are not expressed in untreated fibroblasts. This is a nice example of cell type-specific gene expression. Aberrant or pathologically high activity of NADPH oxidase isoforms in fibroblasts is associated with a wide range of skin disorders. NOX4 plays a role in cutaneous aging as shown by Noh et al. (132). In their model, senescent fibroblasts exhibit increased levels of NOX4-dependent ROS as well as matrix metalloproteinase (MMP)-1, the latter leading to the degradation of collagen.
One of the most widely studied relationships is probably the link between oxidative stress and skin fibrosis (13, 164). Stimulation with growth factors, in particular transforming growth factor beta (TGFβ) or cytokines such as tumor necrosis factor alpha (TNFα), provokes the expression and activation of NOX2 or NOX4, which facilitates fibroblast proliferation, differentiation, and increased extracellular matrix (ECM) deposition (13, 140). These actions are indispensable for the inflammatory response, in particular for wound healing (see NOXs and Their Role During Wound Healing). However, sustained activation can lead to hypertrophic scarring, scleroderma, and cancer development (13, 187). Hence, modulating the activity of NADPH oxidases could be a way to reinstate homeostasis. There is also a potential link between transplant rejection by fibroblast activation, graft damage by ROS, and the activation of NADPH oxidases by immunosuppressive drugs (83).
NADPH oxidases in melanocytes
The reported presence of specific NADPH oxidase isoforms in normal human epidermal melanocytes (NHEM) and melanoma cell lines lacks consistency. For example, in human melanocytes, Brar et al. (26) detected NOX4 and p22phox, while Liu et al. (107) identified only NOX1, NOXO1, and p22phox. Prasad et al. (142) also showed weak expression of NOX1 in NHEM. There are as yet no data attesting to NADPH oxidase expression in mouse melanocytes. The results for various melanoma cell lines are similarly perplexing (Table 2). Brar et al. showed protein expression for only NOX2, p47phox, and p67phox in A375 cells. Liu et al. tested different melanoma cell lines and identified NOX4 in some of them, while all tested cell lines expressed NOX1, NOXO1, and p22phox. Prasad et al. (142) also identified NOX1 in all tested melanoma cell lines at various expression levels.
Listed is a selection of melanoma cell lines described by publications mentioned in the main text. In addition to the indicated references, data on cell lines were collected from the Wistar Institute (
, stop codon; (*), see Cellosaurus entry for a more comprehensive list of mutations; DEL, protein not expressed; EGFR, epidermal growth factor receptor; F, female donor; M, male donor; RGP, radial growth phase; VGP, vertical growth phase; WT, wild type.
However, in another study by Yamaura et al. (195), the only isoform detected in the metastatic MM-BP cell line was NOX4. Recently, NOX5 was described as being expressed in the nonepithelial metastatic human melanoma cell line UACC-257 (9). In mouse melanoma B16 cells though, NOX4 was found to be the major isoform consistent with the majority of publications on human melanoma (108, 195). The expression of NOX4 is regulated by microphthalmia-associated transcription factor (MiTF), the master regulator of melanogenesis, which is activated on stimulation of the melanocortin-1 receptor (MC1R). NOX4 activity is associated with the inhibition of melanin synthesis and thus provides natural negative feedback regulation.
However, increased NADPH oxidase-dependent ROS production by both melanocytes and perilesional keratinocytes is thought to contribute to the destruction of pigment-producing cells in vitiligo skin (16, 59). Moreover, the direct association of NADPH oxidases with melanogenesis could explain at least some of the contradictory results obtained for NHEM and different melanoma cell lines. In another study, Henri et al. (73) uncovered a direct link between MC1R activity and ROS production in HaCaT, while Chou et al. (40) showed that the migration of melanocyte stem cells during the repopulation of a skin section after injury is dependent on MC1R activity. Hence, there is a good chance that NOX4 facilitates the migration and proliferation of melanocyte stem cells.
In conclusion, melanocytes express p22phox , NOX1, and perhaps NOX4, while melanomas express NOX4 and perhaps NOX1, NOX2, and/or NOX5. Distinct NADPH oxidase isoforms may be expressed in melanocytes, during different stages of melanomagenesis and metastasis. For a complete understanding and to avoid contradictory results, it is essential to make a clear distinction when it comes to the origin of tissues, cells, and cell lines. In addition, a more careful description of culture conditions, cellular appearances, and genotypic features when comparing experimental outcomes is necessary.
This is of importance when dealing with an enzyme involved in various stress response pathways and cellular malignant transformation. In this instance, information about both the metastatic stage and the surrounding tissue from which a tumor is recovered is essential for understanding experimental results. Therefore, a more systematic approach is necessary to reach proper conclusions about the functions of NADPH oxidases in the melanocytic lineage.
NADPH Oxidases and the Cellular Responses to UV Irradiation
UV irradiation triggers oxidative stress, immunosuppression, inflammation, and DNA mutations. It is the major cause of skin carcinogenesis and dramatically accelerates cutaneous aging (66). The UV radiation, through the ozone layer to the surface of our planet, consists of ∼5% UVB and 95% UVA (Fig. 3). For a long time, UVA was considered “harmless” until in 2009 the World Health Organization upgraded it to a class 1 carcinogen (58). While most materials absorb UVB, UVA is able to pass through glass (139). Furthermore, while UVB does not penetrate deeply into the skin, UVA can reach and damage the dermal layer and is assumed to be the main cause of cutaneous aging (Fig. 3).

NADPH oxidases in cellular responses to UVA
UVA signature mutations are typically of oxidative nature (64, 137, 192). They include 8-oxo-7,8-dihydroguanine (8-oxoG), abasic sites, protein/protein and protein/DNA crosslinks, DNA single-strand and double-strand breaks (SSBs/DSBs), as well as oxidative damage to other biomolecular structures of the cell. The formation of DSBs by UVA is, however, a subject of debate (63, 153). Furthermore, UVA has been shown to induce an antioxidant response by increasing the activity and expression of several antioxidant enzymes (121).
NADPH oxidases have been found to play a major role in generating ROS post-UVA irradiation (73, 182) (Fig. 4). UVA has been shown to provoke biphasic activation of NOX1 (182). The first peak at around 30 min diminishes to base level within an hour and a second peak appears at around 120 min. However, there is the possibility of further activation at later stages. The production of ceramide and increased intracellular calcium levels following UVA irradiation are two possibilities leading to NOX1 activation. In the first scenario, UVA-induced ceramide release activates the small GTPase Rac1 (120, 198) (Fig. 1). In the second scenario, an increase in intracellular calcium levels activates protein kinase C (PKC), which phosphorylates the inhibitor of Rac1. Rac1 is then released and translocates to the cell membrane to bind NOX1 (88, 122).

Furthermore, calcium activates phospholipase A2 (PLA2). The role of PLA2 is to release arachidonic acid (ARA) from membranes to activate cyclooxygenase 2 (COX2) facilitated synthesis of prostaglandin E2 (PGE2), which in turn modulates inflammation (157). ARA can also bind the redox-sensitive S100A8/A9 proteins in a calcium-dependent manner. These can activate NADPH oxidases through subunit interactions (21, 87). The activation of NOX1 in this cascade is part of a composite positive feedback loop as evidenced by NOX1 knockdown experiments (182). Indeed, NOX1 silencing resulted not only in decreased levels of ROS but also in reduced PGE2 synthesis and inflammation. In mouse primary keratinocytes, UVA additionally induces the production of NOX2, although NOX2 knockout reduced ROS levels by only 30% (69). NOX2 can also be activated by a UVA-associated calcium increase in macrophages, which reside in the skin predominantly in the dermis (105). UVA-induced NOX2 is thought to be involved in mediating calcium oscillation and exocytosis in macrophages (105).
UVA is capable of introducing small amounts of both cyclobutane pyrimidine dimers (CPDs) and pyrimidine (6-4) pyrimidone photoproducts (6-4PPs) into the DNA (125, 178). Interestingly, most studies have found that UVA-induced CPDs are repaired more slowly than UVB-induced ones. The mechanisms by which photoproducts are introduced include mediators in the form of photosensitizers since DNA does not absorb UVA directly. Endogenous photosensitizers include a series of proteinogenic chromophores such as porphyrins, flavins, bilirubin, vitamins, and amino acids (192).
Melanin can also act as a substrate for photosensitization. This is rather unexpected and counterintuitive, as one thinks of melanin as the body's natural sunscreen, which is supposed to protect the skin from solar damage (27). However, reality paints a very different picture. For example, pigmented mice exhibit two to three times more DSB after UV exposure compared to albino mice (174).
Another observation is that about half of all CPDs introduced into melanocyte DNA post-UV are generated hours later in a radiation-independent mechanism (143). Both NADPH oxidases and the nitric oxide synthase (inducible nitric oxide synthase) are UV inducible and inhibition of either of them completely abolishes the synthesis of these “dark CPDs” (143). In the proposed model, superoxide and nitric oxide generate peroxynitrite, which not only fragments melanin polymers but has also the potential to generate excited triplet states in redox-transformed tyrosines. This energy is subsequently released onto DNA to create photodamage-like alterations.
NADPH oxidases in cellular responses to UVB
Although the proportion of UVB within the total UV seems relatively small, its effect is enormous (Fig. 3). Because of its wavelength (315–280 nm), UVB is easily absorbed by DNA and is therefore highly mutagenic (139). The most prominent alterations to DNA include CPDs and 6-4PP. It is estimated that UVB creates DNA damage in the form of pyrimidine dimers and oxidation at a ratio of about 200:1 (89). Photoproducts appear within nanoseconds on UVB exposure and they are repaired by the nucleotide excision DNA repair (NER) pathway (116).
The basic steps of detection, excision, and gap-filling encompass a set of core proteins commonly known as xeroderma pigmentosum (XP)-factors, named after the associated disorder XP. Particular mutational hotspots are CpG islands of mammalian promoters, where UV-induced C-T transitions can lead to aberrant expression of oncogenes. This phenomenon is more prominent in genes that are transcriptionally active at the time of UV exposure (138). A major factor is that these DNA sections are less likely to benefit from the protective effects of nucleosomal DNA packaging if active. In addition to its ability to inflict direct damage to DNA, UVB irradiation also increases the production of intracellular ROS (118). This causes DNA alterations characteristic of oxidative damage as discussed in the section NOXs in cellular responses to UVA.
Numerous studies point to the production of ROS through NADPH oxidase activation following UVB irradiation (146, 148, 150, 159) (Fig. 4). Rahman et al. (146) found that UVB specifically induced the expression of COX2 and NOX4 in HR-1 hairless mice. This expression is mediated by the transcription factor, nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), which is stabilized and activated in response to UVB. Furthermore, specific inhibition of the kinase MSK1, which is downstream of the p38 MAP kinase, decreased the phosphorylation of NF-κB and thereafter the expression of both COX2 and NOX4. On the contrary, Ryu et al. (159) showed that UVB-induced NOX1 activation in keratinocytes is mediated by the leukotriene B4 receptor 2 (BLT2). BLT2 binds a variety of oxidized ARA products and is upregulated following UVB.
Highlighting the role of NOX1 on UVB-induced ROS production, we recently reported that UVB irradiation results in the biphasic activation of NOX1 (145). This feature plays a critical role in defining keratinocyte fate by modulating the DNA damage response network. The first immediate peak of NOX1 activation following UVB diminished to base level within an hour, similarly to what has been described for UVA-initiated NOX1 activation (182). However, the second peak appeared much later and lasted from about 9–13 h postirradiation (145).
Interestingly, the inhibition of NOX1 after UVB irradiation by using an NOX1-specific peptide inhibitor (InhNOX1) was associated with increased NER efficiency and a reduction in apoptosis. This was eventually translated into decreased overall photocarcinogenesis. These results suggest a redox-sensitive balance between DNA repair and apoptotic pathways, which may reflect a critical regulatory mechanism through which NOX1 activation is directly proportional to the amount of DNA damage. In line with this hypothesis, the expression and activity of NOX1 respond directly to elevated levels of damaged DNA in XPC KO mice, which exhibit high UV sensitivity and a predisposition to developing skin cancers, as well as symptoms of premature aging (77, 145, 149, 151). XPC silencing in normal human keratinocytes correlates with the accumulation of mutations that drive metabolic alterations and tumorigenesis facilitated by overactivation of NOX1 (149, 151). These features were prevented either by overexpressing XPC or by using InhNOX1.
The activities of NADPH oxidases contribute to the determination of cell fate following UVB irradiation. Of particular interest are the activities of the redox-sensitive transcription factors activator protein 1 (AP-1) and signal transducer and activator of transcription (STAT1)/3. They are modulated by p38 and ERK1/2 mitogen-activated protein kinase (MAPK) signaling pathways and regulate the expression of several NADPH oxidase family members (115). The p38 and ERK1/2 MAPK signaling pathways are activated in response to UV irradiation and are considered pro- and antiapoptotic, respectively (80, 163). The underlying regulatory mechanism between these is thought to be mutually inhibitory, a common network motif that has the function of a switch (4).
For instance, in the case of UVB-induced apoptosis, the activation of p38 occurs more quickly and suppresses the full activation of ERK, thereby initiating cell cycle arrest, DNA repair, and apoptosis. The phosphorylation level of ERK remains low unless the concentration of activated p38 declines. In which case, the inhibitory effect on ERK diminishes, stimulating ERK phosphorylation and prosurvival cell signaling, while further inhibiting p38. Therefore, the decision as to which pathway is switched on depends on the signal strength of each component at any particular moment in time and the strength of the individual antagonism (199). This concept is supported by the observation that inhibition of p38α induced the expression of NOX2 and cell proliferation, indicating the activation of the ERK1/2 prosurvival cascade (109).
The activation of NADPH oxidases on UV exposure can have serious long-term consequences for the skin. An increase in the concentration of ROS causes prolonged inflammation, alterations in metabolic pathways, and the remodeling of epigenetic modifications leading to altered nuclear dynamics and gene expression profiles. Consequently, chronic NADPH oxidase activation and a shift in the intracellular redox equilibrium could be the key events leading to the tumoral transformation of cells and may offer a target for early intervention.
NADPH Oxidases and Skin Cancers
Melanoma
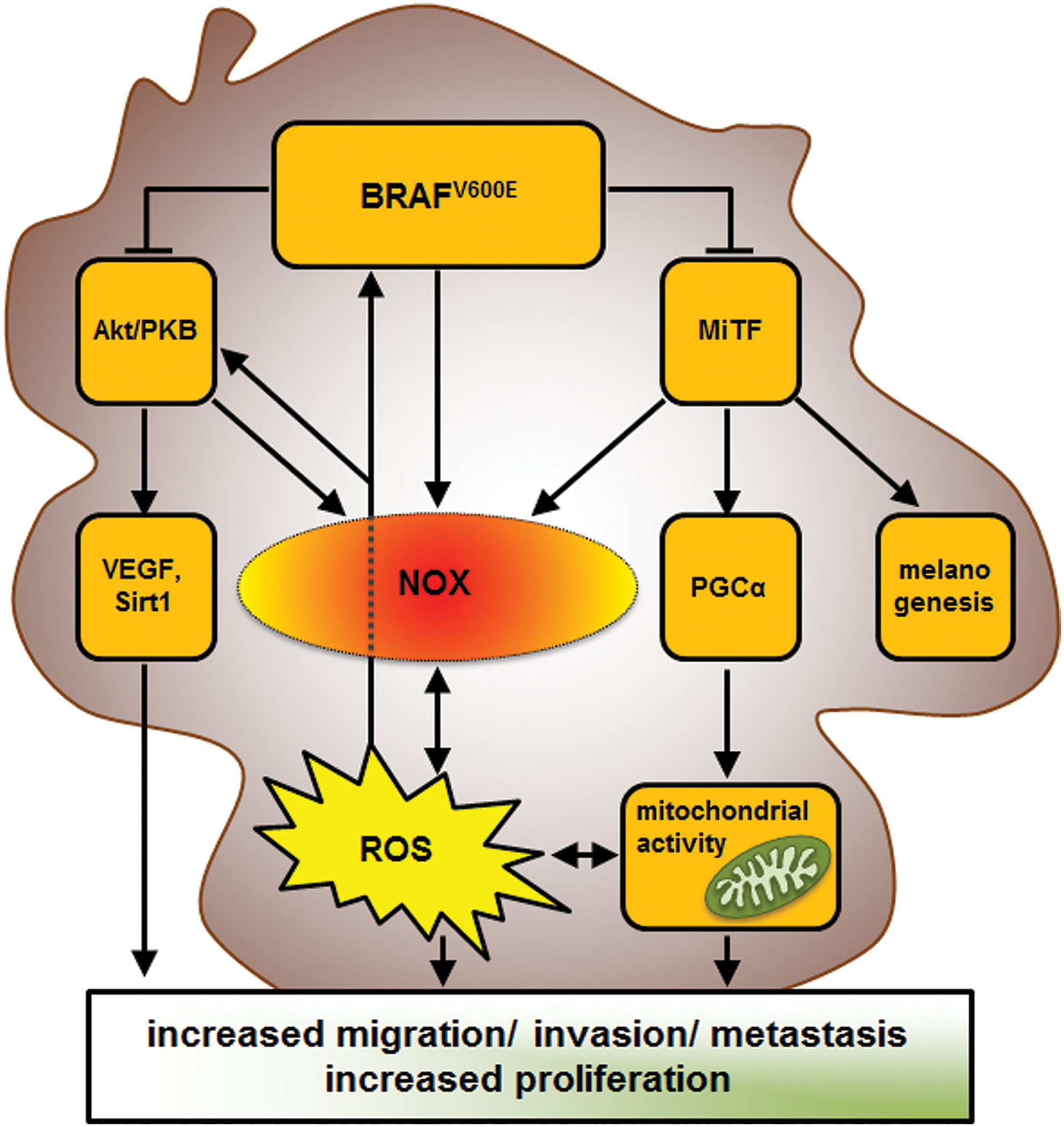
Melanoma accounts for only 4% of all skin cancers, yet it causes the majority of skin cancer-related deaths owing to its rapid growth, its tendency to metastasize, and its often-aggressive colonization of other tissues (7). There is accumulating evidence for a crucial role of NADPH oxidases in the malignant transformation of melanocytes: (i) Melanoma mainly develops on sun-exposed areas of the skin. As mentioned in the section NOXs and the Cellular Responses to UV Irradiation, photodamage to DNA can occur directly (UVB) or indirectly in melanocytes. Indirect damage in the form of “dark CPDs” can arise from subsequent transfer of solar energy and the creation of excited triplet state electrons in degrading melanin molecules (143). The treatment of the skin with antioxidants such as vitamin E or the NADPH oxidases inhibitor VAS2870 after irradiation successfully prevents this kind of DNA damage in melanocytes. (ii) Cancer cells, including melanoma, exhibit increased ROS levels (123, 193). Traditionally, this process has been attributed to mitochondria and metabolic activity (186). In addition, several publications have highlighted the impact of metabolic reprogramming on steady-state levels of ROS in melanoma (184). However, it is still unclear whether and how ROS generated by mitochondria contribute to the initial steps of transformation and melanoma development. On the contrary, NADPH oxidases are activated immediately after UV irradiation and it has been shown in keratinocytes that NADPH oxidase activation precedes metabolic alterations (145, 149, 182). Furthermore, the expression of different isoforms by melanoma cell lines and tumors contributes to the neoplastic transformation of cells and melanomagenesis (9, 26, 107, 195). In line with this finding, the inhibition of NADPH oxidases by using an NADPH analog has been shown to reduce ROS and to induce apoptosis in melanoma cells, while melanocytes remained unaffected (154). (iii) One of the most commonly mutated genes in melanoma cells is BRAF, the main variant being V600E (46). BRAF is downstream of Ras and activates the MEK/ERK1/2 MAPK prosurvival signaling cascade. BRAFV600E is not dependent on Ras and has constitutive kinase activity (110). However, many benign nevi also contain mutated forms of BRAF, which means that other factors contribute to the initial stages of neoplastic transformation (48). Importantly, elevated gene expression for NOX1 and NOX4 has already been reported in several cell lines harboring activation of the Ras/RAF/MEK/ERK signaling pathway (1, 61, 124, 190, 195) (Table 2). Therefore, one can safely assume that NOX1/4-mediated ROS production plays an important role during various stages of malignant transformation of BRAFV600E-positive melanoma cells (Fig. 5).
While NOX1 and/or NOX4 play a crucial role in melanoma initiation, they are also important during cancer progression and metastasis. The overexpression of NOX1 in melanoma cells increases their invasiveness through upregulation of MMP-2 and the induction of the epithelial/mesenchymal transition (EMT) (95, 107). In particular, small interfering RNA (siRNA)-mediated downregulation of NOX1 in cultured melanoma cells inhibits NF-κB-induced activation of vascular endothelial growth factor (VEGF) and MMPs.
Furthermore, the subcutaneous injection of melanoma cells in NOX1-deficient mice or in wild-type mice treated with the NOX1/4-specific inhibitor GKT136901 reduces tumor angiogenesis (56). This effect is mediated by the AKT-dependent suppression of the anti-inflammatory nuclear hormone receptor peroxisome-proliferator activated receptor-α (PPARα), a negative regulator of NF-κB signaling. NOX4-derived ROS have also been shown to promote melanoma cell survival through either NF-κB (26) or AKT/ERK (128). For instance, when noninvasive WM35 melanoma cells were transfected with AKT, they switched to the aggressive invasive state (62). This switch was accompanied by stimulated expression of NOX4, VEGF, and SIRT1 leading to increased ROS signaling, angiogenesis, and proliferation. Yamaura et al. (195) further showed that knockdown of NOX4 in melanoma cell lines resulted in an accumulation of cells at the G2/M phase of the cell cycle. They suggested that post-transformation, NOX4 promotes the invasion and proliferation of melanoma cells by responding to increased AKT signaling.
NADPH oxidases should hence be examined in more detail for their suitability for the prevention of melanoma and for their use as targets in combination with current melanoma treatments, particularly for their role during the emergence of resistance to targeted therapies (Fig. 5). Indeed, a sudden inhibition of BRAFV600E can cause enormous oxidative stress in cells by several different mechanisms:
(i) BRAFV600E represses MiTF by upregulation of its repressor BRN2 (60, 188). Furthermore, as discussed in the section NOXs in melanocytes, MiTF stimulates the expression of NOX4 (108). As a result, BRAFV600E-targeted therapy may trigger increased ROS production by NOX4.
(ii) The activity of BRAFV600E prompts the repression of peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC1α), a potent activator of mitochondrial biogenesis, via MiTF (67). Consequently, the inhibition of BRAFV600E fully activates MiTF and subsequently increases PGC1α activity. This leads to a metabolic switch from glycolytic to high oxidative phosphorylation in the mitochondria, which has been reported to cause a boost in tumor growth (67, 184).
(iii) BRAFV600E was shown to inhibit the Akt/PKB signaling pathway by interaction with the mTORC2 complex (36). Therefore, the inhibition of BRAFV600E would activate Akt, which could potentially activate both NOX1 and NOX4 (62, 149). These examples show that elevated levels of ROS could be one of the underlying reasons for the frequently occurring resistance to treatment with BRAFV600E-specific inhibitors in patients with metastatic melanoma. Therefore, synergistic therapeutic approaches that use NADPH oxidase inhibitors could minimize the likelihood of emerging resistance and improve the long-term prognosis for cancer patients (67) (Fig. 5).
Nonmelanoma skin cancer
The most common nonmelanoma skin cancer (NMSC) is basal cell carcinoma (BCC) and squamous cell carcinoma (SCC). Both cancers originate from keratinocytes, grow slowly, and exhibit a very low mortality rate. However, they are the most common cancers in human and therefore present an increasing economic burden to society in general and to patients in particular (111). In addition, they often occur in highly visible areas of the skin, causing immense psychological distress for affected patients. As for melanoma, they also mainly arise on sun-exposed areas of the skin. Furthermore, there are a number of inherited disorders that can also cause NMSC (130).
The development of BCC is linked to the constitutive activation of the sonic hedgehog/patched receptor for sonic hedgehog (PTCH)/Gli signaling pathway and is thought to originate from hair follicle stem cells (93, 112) (Fig. 6). The clonal expansion of dysregulated stem cells, which have escaped the stem cell niche, may be the main cause of BCC (51). While BCC does not tend to metastasize, it can leave severe disfigurement and damage to neighboring tissue when increasing in size. There are not many publications on the role of ROS in BCC, and no publications yet about NADPH oxidases in relation to the pathology of BCC (33, 179).

Unlike BCC, the relationship between NADPH oxidases and the development of SCC is far better understood. SCC shows on average more UVA (A:T—>C:G transversions) than UVB (G:C—>A:T transitions) signature mutations, suggesting oxygen radicals to be main culprits (2). Furthermore, SCC tumors display increased NADPH oxidase activity compared to normal skin (151). Recent studies suggest various mechanisms for the upregulation of NADPH oxidases.
(i) Calcium signaling
Increased calcium levels in keratinocytes are usually associated with differentiation (24, 72). However, SCC cells show only a low degree of differentiation despite having high levels of calcium (141). Malignant cells are characterized by increased expression of the PGE2 receptors EP1 and EP2, as well as epidermal growth factor receptor (EGFR) and COX2 (157) (Fig. 4). The EP1 receptor can facilitate the mobilization of internal calcium stores by activating phospholipase C (PLC) and PKC. This can potentially activate the expression of NOX1 and increase ROS production in a UVA-independent manner (182). The link between ROS, elevated calcium levels, and NADPH oxidases has also been shown in macrophages, whose treatment with low concentrations of H2O2 stimulated PLC followed by increased intracellular calcium, which triggered the full respiratory burst by NOX2 (53).
(ii) Accumulation of damaged DNA
XPC is essential for damage detection and the initiation of the NER process (116). In XPC-deficient cells, the accumulation of DNA mutations in keratinocytes results in upregulated expression of NOX1 (149, 151). This is accompanied by an elevation in ROS levels and a rise in the number of neoplastic transformations leading to tumor development. Moreover, XPC knockout mice present premature skin aging owing principally to a rise in NOX1-mediated ROS levels (77). Consistent with these results, the most common mutations in aged XPC−/− mice are representative of oxidative damage to nucleotides, which are not repaired by NER (191). DNA damage accumulation on repeated UVB exposure has been shown to be associated with chronic activation of NOX1 (145). This was circumvented by administration of InhNOX1 and promising results have been obtained in the prevention and treatment of UVB-induced cancers in both NER-proficient and NER-deficient mice.
(iii) MAPK kinase signaling
About 90% of all SCC tumors have UV signature mutations in the p53 gene (22). p53 mutations appear in particular hotspots and are usually indicative of UVB damage (139). A reduction in p53 activity is often accompanied by a reduction in the levels of p38 isoforms, in particular p38α (109, 144). p38 MAPKs are strongly activated in response to various stresses, including UV irradiation (44). Like p53, p38 MAPKs play a role in cell cycle arrest and apoptosis. However, the simultaneous loss of p38α and p53 dramatically increases the aggressiveness of SCC cells, which seems to be mediated by an upregulation of NOX2 (86).
Another interesting fact is that melanoma patients who are treated with the BRAFV600E inhibitor vemurafenib can develop SCC in cells that are wild type for BRAF (170). Sixty percent of these tumors exhibited mutations in the Ras oncogene, the majority of which were H-Ras Q61L. In these probands, inhibition with targeted inhibitors induced BRAF to bind to CRAF (71). This resulted in the reactivation of the ERK1/2 pathway and prompted the formation of SCC. In this context, the phosphorylation (activation) of the transcription factor GATA-6 by ERK1/2 upregulates the expression of NOX1, which is one of the fundamental events leading to cancer development (1, 124). Furthermore, ROS production by NADPH oxidases is required for the phosphorylation of ERK1/2, further enhancing oncogenic cellular remodeling (135, 158). In fact, mice that lack the Rac activator Tiam1 are resistant to Ras-induced skin cancers. Hence, targeting NOX1 is particularly attractive in patients who are treated with RAF inhibitors.
(iv) NAD+/NADH and NADP+/NADPH ratios
NAD+/NADH and NADP+/NADPH are fundamental mediators in various biological processes, including energy metabolism, redox regulation, and calcium homeostasis (104). Several in vitro and in vivo studies have demonstrated that administration of low doses of precursors such as nicotinamide and niacin (vitamin B3) protects the skin against chemical- and UV-induced carcinogenesis (20, 172). The protective functions of nicotinamide have been ascribed to its ability to affect the energy metabolism and DNA repair capacity (20, 172).
Recent studies have shown that changes in the energy metabolism and overactivation of NADPH oxidases are early key events during photocarcinogenesis (145). Benavente and Jacobson (20) suggested that under conditions of nicotinamide starvation, HaCaT utilizes glutamate as an energy source to produce NADPH. The increased availability of the latter would not only ensure mitochondrial functionality but also enhance the activity of NADPH oxidases and ROS production. In line with these findings, a clinical trial obtained promising results in the prevention of NMSC by giving oral doses of vitamin B3 (35).
However, this trial was conducted on older patients with a history of NMSCs. Older individuals have among others significantly lower NAD+ levels and a decreased DNA repair capacity (119). Oral doses of vitamin B3 may just counteract these deficiencies and stimulate DNA repair. Therefore, the question would still be whether vitamin B3 is beneficial to all age groups or just to those with lower NAD+ levels. Further studies are also needed to clarify the precise interconnections between reprogramming of the energy metabolism, NADPH oxidase activation, and ratios of NAD+/NADH-NADP+/NADPH.
It is becoming clear that NADPH oxidases, especially NOX1, play a central role during the initiation of the malignant transformation of keratinocytes and SCC development. A reduction in NOX1 activity and ROS can prevent or delay cancer development significantly (145). However, like the observation on BRAF inhibition in melanoma, adaptation and/or compensation for the loss of NOX1 have been observed. Hence, more work is necessary for a more comprehensive understanding of the respective signal transduction and gene regulatory networks.
NADPH Oxidases and Their Role During Wound Healing
Wound healing is one of the most important processes of tissue reconstitution. It has long been recognized that the principal mechanisms that are paramount to restoring tissue integrity are the same as those that trigger neoplastic transformation and the survival of cancer cells (10, 50). The three main stages of wound healing are (i) the inflammatory, (ii) the proliferative, and (iii) the tissue remodeling phase [reviewed in Barrientos et al. (15) and Singer and Clark (165)].
On wounding, DUOX enzymes are involved in the rapid production of a hydrogen peroxide gradient that repels bacteria from the epithelial surface and minimizes the risk of infection (129). Furthermore, the generated reactive oxygen moieties also modify components of autocrine and paracrine cellular signaling pathways that control the expression of various genes necessary for the initial inflammatory response phase (183). In particular, DUOX-derived ROS activate ERK1/2, MMP-9, NF-κB, IL-8, Src family kinases, and mucin family glycoproteins (98, 161, 189, 197). Leukocytes are responsible for cleansing wounds of invading pathogens and necrotic tissue.
They are recruited to a wounded site by two possible mechanisms: (i) by using hydrogen peroxide directly as a chemoattractant and (ii) by the H2O2-mediated production of chemokines and growth factors by epithelial cells (52, 129). A number of publications show that the rapid recruitment of immune cells during the inflammatory phase is also dependent on the activity of DUOX enzymes (129, 161, 189). Phagocytotic NOX2 is also indispensable for proper wound healing, and reduced NOX2 activity leads to a prolonged and more intense inflammatory response in patients with chronic granulomatous disease (CGD) (30).
During the proliferative phase, chemokines and growth factors produced during the previous stage stimulate the formation of new blood vessels, the production of granulation tissue from fibroblasts at the bottom of the wound, the secretion of ECM proteins, the induction of the EMT, and both the proliferation and migration of keratinocytes from the edge of the wound (15, 196). It has been shown that NOX1/4-derived ROS are required to enhance migration of HaCaT cells following treatment with growth factors such as TGFβ and hepatocyte growth factor (HGF) (127).
In another study, neuregulin, a member of the epidermal growth factor (EGF) family, was reported to induce NOX1- and NOX2-derived ROS generation, increasing cofilin dephosphorylation and promoting cell migration in HaCaT keratinocytes (92). Granulation tissue is formed in the final step of the proliferation phase from fibroblasts entering senescence. Cell cycle arrest is necessary to reduce wound fibrosis and tumor growth.
Jun and Lau (84) demonstrated that fibroblast senescence is mediated by ROS produced by NOX1 and by subsequent activation of the p16INK4a/pRb pathway. NOX1 is regulated by cysteine-rich angiogenic inducer 61 (CYR61/CNN1), which is highly expressed during wound healing in the granulation tissue, where it orchestrates not only fibroblast senescence but also angiogenesis, cell adhesion, and migration. Collagen is modified during the remodeling phase (84). NOX4 knockdown is associated with a decrease in collagen deposition and dityrosine crosslinking of the ECM in the granulation tissue as well as the delayed recruitment of phagocytes (103). In line with this, wounds inflicted to NOX4 knockout mice needed longer to heal than those in wild-type controls.
While NADPH oxidases are essential for the healing process of injuries, it has also been demonstrated that downregulation can be beneficial for the treatment of chronic wounds (167, 194). Sotomayor et al. (167) showed that the NADPH oxidases inhibitor S42909 led to a decrease in vascular inflammation, reduced wound contraction, and increased collagen deposition. The latter may seem contradictory in view of the results obtained by Lévigne et al. (103). However, it could be seen as a confirmation of the ROS thresholding hypothesis (80) (Fig. 2). In the case of chronic wounds where ROS levels are expected to be high, NADPH oxidase activity could also be high, which would prolong the inflammatory process and enhance collagen degradation. Interestingly, inhibition of NADPH oxidase in chronic wounds increases the expression of TGFβ, which has been shown to activate NADPH oxidases in HaCaT cells (127). This indicates a regulatory feedback inhibition. Furthermore, in the context of chronic diabetic wounds in patients with microvascular complications, an accumulation of oxidized protein derivatives was detected (173). These were linked to the disruption of the mitochondrial membrane potential and excessive ROS production by NOX4 leading to an elevation in apoptosis biomarkers and increased apoptosis (171).
In conclusion, NADPH oxidases and their actions are indispensable for proper wound closure. However, if the equilibrium is upset, increased ROS production and its downstream effects can significantly impair the healing process by augmenting and prolonging the inflammatory response. Therefore, NADPH oxidases could present promising therapeutic targets in the context of chronic wound treatment.
NADPH Oxidases in Noncancer Skin Disorders
In addition to carcinogenesis, NADPH oxidases have been associated with various skin diseases and chronic inflammation, including systemic sclerosis, psoriasis, atopic dermatitis (AD), and a number of infectious diseases (13).
Skin fibrosis, a frequent and progressive reaction to chronic injury or inflammation, is characteristic of many skin disorders such as systemic sclerosis, hypertrophic and keloid scarring, and dermatofibrosis (13, 164). Persistent injury, infection, and inflammation are the best-known triggers of fibrosis. Subsequent damage to endothelial cells initiates the secretion of cytokines and the recruitment of immune cells.
These cells then release key fibrotic growth factors, such as TGFβ, connective tissue growth factor (CTGF), and platelet-derived growth factor (PDGF), resulting in the activation and proliferation of fibroblasts in addition to their differentiation into myofibroblasts. An accumulation of these cells in the dermis is associated with excessive synthesis of the ECM that characterizes fibrosis. Furthermore, myofibroblasts express alpha-smooth muscle actin (α-SMA) leading to an enhanced contraction of the ECM (13). Numerous studies have shown that NOX4 is involved in the initiation, establishment, and development of tissue fibrosis in various organs, including the lung, kidney, liver, and skin (6, 13, 14, 25).
Profibrotic polypeptides such as TGFβ, PDGF, angiotensin II, and endothelin-1 have been shown to modulate the expression of NADPH oxidases (8, 45, 70, 169). For instance, TGFβ1-mediated expression of the collagen type I gene, α-SMA, and fibronectin 1 was shown to be NOX4 dependent (49). Recent studies identified AP-1 and Smad3 transcription factor binding sites upstream of the human NOX4 promoter (49, 81). These transcription factors are involved in the TGFβ-dependent regulation of NOX4 gene expression. Furthermore, NOX4 is regulated by polymerase delta-interacting protein 2 (PDIP2) (113) (Fig. 1). PDIP2 is highly expressed in fibrotic tissue and could potentially activate NOX4 under pathophysiological conditions (49).
Psoriasis is characterized by itchy, raised patches, epidermal hyperplasia, leukocyte infiltration, and chronic inflammation of the skin (31). However, the precise underlying mechanisms remain unclear. Recent studies have suggested failure in skin barrier functions, IL-17 and IL-23 signaling, as well as TNFα and NF-κB signaling (31, 117). In an IL-23-induced psoriasis mouse model, NOX2-mediated ROS production in keratinocytes was TNFα dependent and led to the continuous activation of NF-κB (68).
In this model, NOX2-derived hydrogen peroxide was imported by aquaporin-3 and was shown to be a crucial mediator in NF-κB activation. Similar results were obtained for the upregulation of NOX2 and DUOX2 in an imiquimod-induced psoriasis-like murine skin model (126).
Another chronic inflammatory skin disorder—AD—results from excessive stimulation of immune cells and leads to an allergic reaction (75, 97). The release of specific proinflammatory cytokines in epidermal keratinocytes, such as IL-4/IL-13 (DUOX1) and IL-8/CCL20 (DUOX2), results not only in an upregulation of DUOX enzymes but also creates a positive feedback loop to sustain cytokine production. In addition, several studies have shown the activation of COX2/PGE2/NF-κB signaling and downregulation of filaggrin in the skin of patients with AD (85, 177). Recently, the topical application of NADPH oxidase-specific inhibitors was used successfully to block both the activation of COX2 and the downregulation of filaggrin (101).
The skin as the body's outermost protective cover is permanently exposed to exogenous agents, including air pollution as well as various pathogens (bacteria, viruses, and fungi). Contact activates the immune system and causes an inflammatory reaction in the skin (18, 91, 136). NADPH oxidases are integral and essential components of these processes. However, in the event of chronic activation, a reduction in the activity of NADPH oxidases has been shown to have a protective effect that limits tissue damage.
For instance, Lee et al. (101) showed that skin inflammation in response to air pollution is triggered by NOX2-derived ROS, which caused an increase in COX2/PGE2 levels and activated several redox-sensitive signaling pathways. The inhibition of NOX2 reduced inflammation and barrier defects. The activation of NADPH oxidases has also been reported in several viral infections, including hepatitis C virus, rhinovirus, dengue virus, and HIV (23, 42, 133, 166). In particular, NOX2 has been linked to the antiviral defense in epithelial cells through the regulation of the COX2/PGE2 axis (106). In this model, NOX2 silencing decreased TNFα-mediated phosphorylation of p38, c-jun, and NF-κB as well as reducing levels of COX2 and PGE2, thereby causing an increased susceptibility for viral infections (106). Recurrent and severe bacterial and fungal infections in CGD patients do not leave any doubt about the contribution of NOX2 in the defense against bacterial and fungal infections. Increasing evidence suggests that both the direct effects of NADPH oxidase-derived phagocytotic ROS on pathogens and ROS-dependent signaling contribute to the cellular host defense mechanisms (29, 76). In addition, the abovementioned examples show that the modulation of the activities of NADPH oxidases may be highly beneficial in preventing and treating a variety of inflammatory skin diseases.
NADPH Oxidase Inhibitors in Research and Therapy of Skin-Related Diseases
NADPH oxidases have many important functions in normal cellular homeostasis, the cellular stress response, as well as playing a central role in a wide range of pathologies (19). The design of highly effective isoform-specific inhibitors is not only important from a therapeutic viewpoint but there is also an increasing need for target-specific molecular tools for scientific purposes [for recent reviews on NADPH oxidase inhibitors, see Altenhöfer et al. (5), Cifuentes-Pagano et al. (41), and Teixeira et al. (176)]. Many of the inhibitors used so far do not target NADPH oxidases directly, but concern upstream signaling pathways; or they function as ROS scavengers (79). Most publications to date based at least part of their findings regarding functional aspects of NADPH oxidases on the use of two compounds: diphenyleneiodonium (DPI) and apocynin. However, these chemicals are only of limited benefit for research purposes as their mode of action is neither NADPH oxidase specific nor fully understood (3, 5, 74). Another aspect of NADPH oxidase research is that many conclusions are drawn from measuring ROS production without being able to identify the source exactly (79). Recently, NS1, an NADPH analog, showed promising results in reducing ROS and the induction of apoptosis in melanoma cells (154). Finally, the isoform specificity of antibodies needs to be ensured by using mRNA knockdown experiments, as this is often not clear from published results.
The fact that NADPH oxidases require different subunits (Fig. 1) in addition to having distinct spatiotemporal expression and activity patterns should make it possible to design isoform-specific inhibitors. The small-molecule inhibitors that are currently available can roughly be separated into three groups: (i) ROS scavengers such as epselen and plumbagagin; (ii) inhibitors of flavin-bound enzymes, including NADPH oxidases, such as fulvene-5 or VAS2870; and (iii) NADPH oxidase, but not isoform-specific inhibitors such as GKT37831 or celastrol. For many, the exact mechanism of action and hence their target specificity still need to be determined (5, 41). Currently, there are only a few publications to have used these inhibitors in skin models. GKT137831, one of the most promising candidates, was shown to reduce ROS production after UVB treatment in a mouse model and led to decelerated aging and a reduction in the development of SCC tumors (77, 145). This inhibitor has also been used successfully in preclinical fibrotic disease models and could be a good candidate for the treatment of skin fibrosis (13). Celastrol was shown to reduce the phosphorylation of the ERK1/2 MAP kinases and to have an antiallergenic effect on mouse skin (94). It effectively reduces the activity of several NADPH oxidase isoforms within minutes, which is potentially the underlying reason for the reduction in ERK phosphorylation (78).
Small peptide inhibitors seem to be most promising from a scientific and therapeutic viewpoint owing to their specificity (41). However, their drawback is their relatively low stability and a low gastrointestinal absorption rate, which can make administration quite inconvenient for the patient (28). Nevertheless, for the treatment of skin conditions, delivery through topical application is a possibility. For NOX1, two peptide inhibitors are available: NOXA1ds and InhNOX1 (77, 145, 147). NOXA1ds mimics an 11-amino-acid stretch of the interaction domain of the NOXA1 activator subunit with the NOX1 core unit. It inhibits ROS production by NOX1 by about 95% without affecting any other isoforms (147). Despite its commercial availability, it seems that it has not been used widely as yet. InhNOX1 targets the interaction between NOXA1 and NOXO1. In particular, it consists of seven amino acids of the proline-rich domain of human NOXA1 and was shown to inhibit ROS production by NOX1 by almost 100% (145). InhNOX1 has also been shown to reduce UV sensitivity, tumor development, and the growth of existing tumors in XPC knockout mice post-UVB irradiation. Its low cytotoxicity makes it a good candidate for incorporation into topical treatments. NOX2 peptide inhibitors encompass only NOX2ds-tat to date. However, it is highly effective and inhibits NOX2 exclusively (43). It mimics a nine-amino-acid region of the cytosolic B-loop between transmembrane helices 2 and 3 of the catalytic core unit (Fig. 1). It has been successfully used in many in vitro and in vivo disease models, but not in relation to skin or skin disorders. Two peptides have been designed for NOX5 inhibition targeting its C-terminal EF hand but have not yet been thoroughly tested for specificity (180). To our knowledge, there are no specific inhibitors for NOX3, NOX4, or DUOX1/2 so far.
Another alternative would be to target pathways in which NADPH oxidases are embedded. Indeed, for many pathologies, the goal should be to re-establish normal cellular homeostasis rather than attempt to achieve complete enzymatic inactivation. This could prevent the undesirable activation of redundant or alternative pathways often encountered with targeted therapy. It should be emphasized that owing to their functions as both upstream modulators and downstream effectors in various signaling pathways, NADPH oxidases can regulate numerous biological processes. This means they can act at different stages of a disease as discussed in the sections “Melanoma” and “NOXs and Their Role During Wound Healing”. However, to be able to modulate the activity of NADPH oxidases appropriately for the prevention and treatment of a specific disease, our knowledge about the precise roles of this enzyme family in skin physiology and pathophysiology needs considerable improvement.
Conclusions
In recent years, we have come a long way in understanding the roles that NADPH oxidases play in normal cellular homeostasis and during responses to endogenous and exogenous stress stimuli. Even though our knowledge of the regulation and the exact roles of NADPH oxidases in the skin is not complete and the findings even appear contradictory, it is clear that they are indispensable for normal skin functioning. They fulfill essential roles during epidermal stratification and barrier formation, wound healing, the defense against invading pathogens, and in the response to chemical and physical mutagens (19). Moreover, they are involved in the development of autoimmune and chronic inflammatory skin diseases, chronic wounds, barrier defects, aging, and skin cancers (30, 77, 96, 101, 167, 194). Embedded in intricate signaling networks, NADPH oxidases can both respond to different stressors and modulate cellular responses by using an ROS thresholding mechanism. However, the details of the exact roles that distinct isoforms play in skin under physiological and pathophysiological conditions are yet to be determined. For this reason, it is crucially important not only to have accurate genotypic information about cells and cell lines but also to describe culture conditions more carefully. Stress response networks are sensitive to changing conditions, as are the expression and activity patterns of NADPH oxidases. Furthermore, the availability of better and more specific molecular tools should facilitate research efforts in the future. For example, the development of isoform-specific small-molecule and small-peptide inhibitors in recent years, many of which have not yet been widely tested in research, will be beneficial not only for scientific purposes but also for the prevention and treatment of several skin disorders and skin cancers.
Footnotes
Acknowledgments
The authors acknowledge the patients' support group “Les Enfants de La Lune.” H.R.R. gratefully acknowledges support from the ARC “Association pour la Recherche sur le Cancer,” the Institut National du Cancer “INCA_6654,” and FR TransBioMed. H.R. was financed by grants from “La ligue contre le cancer.”