
Abstract
Significance:
Numerous studies have demonstrated the actions of reactive oxygen species (ROS) as regulators of several physiological processes. In this study, we discuss how redox signaling mechanisms operate to control different processes such as neuronal differentiation, oligodendrocyte differentiation, dendritic growth, and axonal growth.
Recent Advances:
Redox homeostasis regulates the physiology of neural stem cells (NSCs). Notably, the neuronal differentiation process of NSCs is determined by a change toward oxidative metabolism, increased levels of mitochondrial ROS, increased activity of NADPH oxidase (NOX) enzymes, decreased levels of Nrf2, and differential regulation of different redoxins. Furthermore, during the neuronal maturation processes, NOX and MICAL produce ROS to regulate cytoskeletal dynamics, which control the dendritic and axonal growth, as well as the axonal guidance.
Critical Issues:
The redox homeostasis changes are, in part, attributed to cell metabolism and compartmentalized production of ROS, which is regulated, sensed, and transduced by different molecules such as thioredoxins, glutaredoxins, peroxiredoxins, and nucleoredoxin to control different signaling pathways in different subcellular regions. The study of how these elements cooperatively act is essential for the understanding of nervous system development, as well as the application of regenerative therapies that recapitulate these processes.
Future Directions:
The information about these topics in the last two decades leads us to the conclusion that the role of ROS signaling in development of the nervous system is more important than it was previously believed and makes clear the importance of exploring in more detail the mechanisms of redox signaling. Antioxid. Redox Signal. 28, 1603–1625.
Introduction
R
The proper balance between oxidants and reductants confers a specific redox state to the cells, under which is allowed the correct functioning of redox-sensitive pathways that regulate different processes, such as neuronal differentiation and axonal growth. Alteration of the elements that control this redox state has severe consequences for the development of the brain. For example, glutathione peroxidase 4 (GPx4)-deficient mice die in utero as they fail to initiate gastrulation. Likewise, silencing of mitochondrial GPx4 during in vitro embryogenesis disturbs the segmentation of rhombomeres 5 and 6 during hindbrain development and induces cerebral apoptosis (17). Similarly, mice deficient in thioredoxin reductase 1 and 2 die by E10 and E13, respectively (139). The selective deletion of thioredoxin reductase 1 in the nervous system induces motor behavior disorders, explained by a reduction in the proliferative precursor cells in the external granule cell layer of the cerebellum, which impairs fissure formation and laminar organization of the cerebellar cortex (139). In addition, a mutation of the mitochondrial superoxide dismutase (MnSOD) in Drosophila produces abnormal brain morphology, aberrant axonal targeting, and increased mitochondrial ROS levels (21).
During postnatal development of the cerebellum, different members of the NOX family are expressed, which might contribute to the transitory production of ROS in particular regions of the developing cerebellar cortex (24). These ROS are required for normal cerebellar development since inhibition of ROS leads to changes in cerebellar folium formation as well as an alteration in motor behavior (24). These studies exemplify the importance of the proper control of the redox state during nervous system development.
The function of different proteins can be controlled by reversible oxidation of amino acids, particularly cysteine and methionine. Cysteine amino acids are the most important sensors within the redox-sensitive molecules (160). It is accepted that hydrogen peroxide acts as a second messenger since this molecule possesses favorable characteristics for mediating redox signaling events. For example, hydrogen peroxide is relatively stable and a poor oxidant of most biological molecules compared with other ROS such as superoxide anion and hydroxyl radical. It is transported across membranes through aquaporins (14), although its diffusion in the cytosol is very limited due to the strength of the antioxidant network (69, 97), which makes the hydrogen peroxide signal very localized.
It is well accepted that hydrogen peroxide, directly or indirectly, oxidizes cysteine thiol groups, however, this capacity to oxidize thiol groups is strikingly limited (95) and only a small number of proteins are known to react directly with hydrogen peroxide (166). Hydrogen peroxide can directly oxidize the thiol group (R-SH) of cysteines when it is in the form of thiolate (R-S−), producing sulfenic acid (R-SOH). The sulfenic acid, commonly presented as sulfonate (R-SO−), can rapidly react with an RSH to form an intramolecular disulfide (R-SS-R) or mixed disulfides with other molecules such as glutathione (R-SS-G). Further oxidation leads to the formation of sulfinic (R-SO2H) and sulfonic (R-SO3H) acids, which are considerably more difficult to reduce and generally are considered irreversible (37, 52, 98, 166). The reduction of protein disulfides is executed by the thioredoxin/thioredoxin reductase system, while the reduction of glutathionylated proteins is mediated by the glutathione/glutaredoxin system (166). In certain cases, such as some peroxiredoxins, sulfinic acid can be reduced by sulfiredoxin and sestrin (75).
These properties confer on proteins the ability to act as redox switches, that is, the ability of the thiol group to sense a redox signal that is transduced to effectors to get a cellular response in a process that is reversible (31). Many of the actions of hydrogen peroxide are mediated through the regulation of thiol redox sensor proteins such as thioredoxins, peroxiredoxins, glutaredoxins, and nucleoredoxin. As is described in the present review, such modifications of the cysteine residues affect the functioning of different proteins and represent a major regulatory mechanism to control various processes during development of the nervous system. In this study, we describe the mechanisms by which ROS regulate several processes that take place during nervous system development.
Neuronal Differentiation
Neural stem cells (NSCs) are self-renewing cells that have the potency to differentiate into neural cell types that include neurons and glial cells. NSCs are allocated in neurogenic regions in the ventricular and subventricular zones during development. In the adult, NSCs are localized in the forebrain subventricular zone of lateral ventricles and the subgranular zone of the dentate gyrus in the hippocampus (34, 96). Coincidently, it is in the neurogenic regions of the developing and adult brain where ROS reach elevated levels (24, 91, 105, 152). ROS control multiple aspects of the differentiation process in cortical NSCs. For example, when cortical neural NSCs are selected according to their levels of ROS, it is found that cells with higher levels of ROS differentiate into neurons, while cells with lower levels of ROS differentiate into astrocytes, oligodendrocytes, and other types of neurons. In addition, in this preparation, during neuronal differentiation, antioxidant treatment alters the proportion of different types of cortical neurons (152).
This pioneering study suggests that ROS are determinants of the NSC fate. In agreement with this idea, there are different studies describing redox signaling events that take place during neuronal differentiation and maturation. ROS govern the differentiation process at different levels by regulating redox-sensitive molecules, controlling in this way different signaling pathways. It is now generally accepted that the differentiation process of NSCs is dependent on the redox state, which is determined by metabolism, balance of redox couples, antioxidant capacity, and pro-oxidant elements. The redox state becomes relevant in the neurogenic process as early as the establishment of committed proliferative cells until neuronal maturation, which includes dendritic and axonal development. In the following sections, we present evidence that supports an important role of the redox state as well as redox-sensitive molecules in the neuronal differentiation process of NSCs.
Metabolism
Metabolic changes
NSC metabolism relies on glycolysis and β-oxidation, which changes toward an oxidative metabolism during neuronal differentiation to meet the high-energy requirements of neurons (5, 84, 86, 144, 172). During glycolysis, glucose is converted to pyruvate, which is used in the tricarboxylic acid (TCA) cycle to produce NADH and FADH2, which are subsequently used in oxidative phosphorylation to produce ATP. Important changes in regulatory enzymes of these pathways occur to allow the transition from a glycolytic metabolism used by NSCs to a mitochondrial electron transport chain and oxidative phosphorylation, primarily used by neurons. Indeed, this is not a consequence of the differentiation process, instead it is a requirement to achieve terminal differentiation as is the case of Drosophila neuroblasts (66). In the rodent hippocampus, these changes appear as early as the transition from NSCs to intermediate progenitor cells (IPCs), during which the expression of mitochondrial complexes increases (10) (Fig. 1). Ablation of the mitochondrial transcription factor A significantly decreases the mitochondrial membrane potential and ATP production, which produces a profound loss of IPCs and immature neurons (10). These studies support the idea that stage-specific metabolic programs are functionally related to different developmental stages of the differentiation process of NSCs.
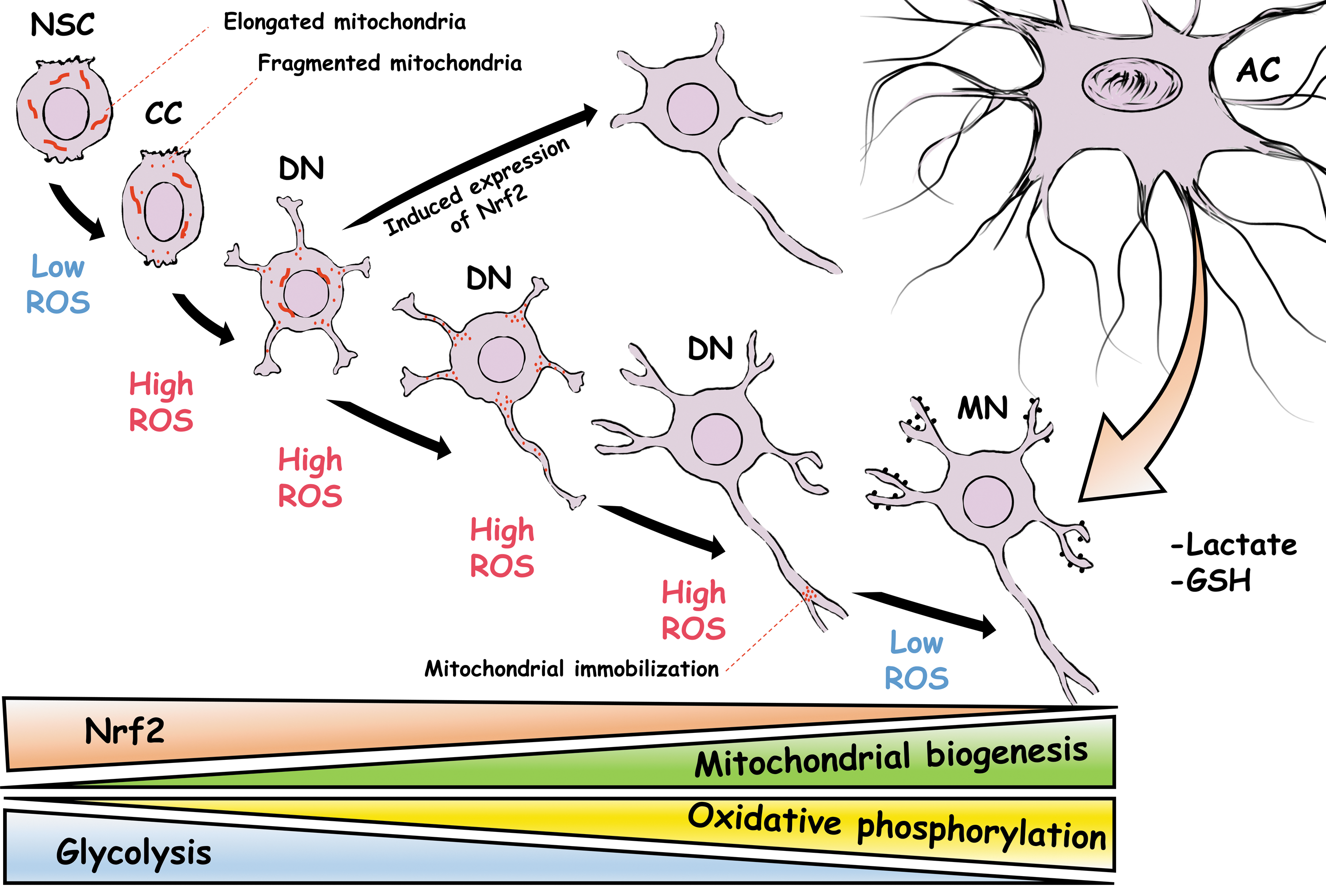
The transition between different metabolic programs might convey a change in the redox state of differentiating neurons since mitochondrial oxidative phosphorylation produces high amounts of ROS (155). In agreement with this idea, during the differentiation of cerebellar granule neuron precursors (CGNPs), there is a transient increase in the levels of ROS (115) and there is a decrease in the glycolytic pathway, which is determined by downregulation of the enzyme hexokinase 2 (49, 50). Elimination of hexokinase 2 leads to an altered migration of CGNPs, attributed to premature differentiation of these cells (49, 50). Furthermore, inducing neuronal differentiation by reprogramming astrocytes is accompanied by an increased production of ROS that leads to lipid peroxidation, which is prevented when the cells are only allowed to use the glycolytic pathway to produce ATP, although the differentiation process is also completely inhibited (46). The augmented levels of ROS in these examples might be mediated, in part, by the partial contribution of oxidative metabolism and the activity of other producers of ROS.
During the neuronal differentiation process, a majority of glycolytic genes as well as glucose transporters 1 and 3 are downregulated. In contrast, most of the TCA genes and the genes encoding for proteins pertaining to the pyruvate dehydrogenase complex remain at the same level (179). In particular, the levels of two protein supporters of glycolysis, hexokinase 2 and lactate dehydrogenase A, practically disappear upon differentiation (179). Conversely, inducing constitutive expression of hexokinase 2 and lactate dehydrogenase A results in reduced pyruvate entry into the TCA cycle, which causes massive cell death only in the differentiating neurons (179). The changes that occur during neuronal differentiation are in agreement with the idea that neurons use the astrocyte-derived lactate in oxidative phosphorylation to produce ATP (99, 150). Nevertheless, this idea is still a matter of debate. Despite the glycolytic metabolism, NSCs possess functional respiratory complexes capable of efficiently driving the electron transport chain and oxidative phosphorylation, which is probably determined by the high expression of the proteins UCP2 and IF1 (84) that are implicated in the suppression of oxidative phosphorylation (133, 175).
In contrast to the early stages of differentiation, during terminal differentiation of primary cortical neurons, there is an increase in glycolysis, glucose uptake, and mRNA levels of glucose transport 3 and phosphofructokinase 1 (3). The impairment of glycolysis with 2-deoxy-D-glucose, a competitive inhibitor, diminishes cortical neuron differentiation by ∼75% and neurons are arrested at stage 2 of differentiation (3). It is expected that cortical neurons possess a lower glycolytic rate than astrocytes. These neurons, but not astrocytes, downregulate the enzyme 6-phosphofructo-2-kinase/fructose-2, 6-bisphosphatase-3 (Pfkfb3) through the ubiquitin–proteasome pathway (63). Pfkfb3 produces fructose-2, 6-bisphosphate, that is, the most potent activator of phosphofructokinase 1, which is a master regulator of glycolysis (15). Interfering with the degradation of Pfkfb3 in cortical neurons increases glycolysis and reduces glucose consumption through the pentose phosphate pathway by 50%, which normally is double than that through glycolysis (63). The pentose phosphate pathway regenerates NADPH used by many thiol-based redox systems such as thioredoxin reductase and glutathione reductase to produce reduced thioredoxin and reduced glutathione (31). Upregulation of Pfkfb3 changes a fraction of glucose 6-phosphate directed to the pentose phosphate pathway toward glycolysis, which results in a reduction in levels of reduced glutathione, increased levels of ROS, and apoptotic cell death (63).
Mitochondrial dynamics
Other conditions related to the number, integrity, and dynamics of mitochondria also exert a modulatory function over the differentiation process of NSCs. For example, differentiated neurons possess higher levels of mitochondrial proteins related to oxidative metabolism in comparison with their precursor cells (3, 151). In cortical NSCs, neuronal differentiation is accompanied by an increased frequency of mitochondrial superoxide flashes, which are spontaneous bursts of superoxide generation triggered by transient openings of the mitochondrial permeability transition pore (mPTP) (159). Pharmacological inhibition of the mPTP or scavenging of mitochondrial superoxide inhibits superoxide flashes and neuronal differentiation (68). During neuronal differentiation of hippocampal NSCs, there is an increase in the number of mitochondria and ROS levels (158). Although mitochondrial biogenesis increases on a per cell basis, it seems that mitochondrial density remains unchanged during neuronal differentiation (179) and mitochondrial distribution is not homogeneous. For example, the arrest of mitochondrial mobilization at specific sites of the axon directs axon branching (23, 141) and filopodium formation during axonal development (83) (Fig. 1).
Mitochondrial dynamics and morphology play a determinant role in the self-renewal capacity and fate decisions of NSCs through the regulation of ROS levels, which control the nuclear developmental program of these cells (84). This role is beyond ATP production and depends on the regulation of mitochondrial fission and fusion. NSCs in the ventricular zone possess mitochondria with an elongated morphology, which changes toward to a fragmented state in committed neural progenitor cells in the subventricular zone (84). Disruption of mitochondrial dynamics, by silencing of the membrane fusion proteins mitofusin 1 and 2 or OPA1, produces mitochondrial fragmentation, resulting in a profound inability of cells to form neurospheres in vitro and increasing the number of cells committed to a neuronal fate in vivo. Concomitantly, the metabolic profile of these cells switches toward an oxidative metabolism with increased levels of the complex I and reduced levels of superoxide dismutase (SOD) 1, which is accompanied by increased levels of mitochondrial and cytoplasmic ROS required to modify the nuclear transcription profile of committed progenitor cells (84) (Fig. 1).
Likewise, in hippocampal NSCs, neuronal differentiation is accompanied by profound changes in mitochondrial mass, shape, and distribution. Silencing of dynamin-related protein 1 (Drp1), a mitochondrial fission protein, drastically impairs the survival, neuronal differentiation, and dendritogenesis of the cells (143). It seems that Drp1 controls neuronal development by promoting the distribution of mitochondria. In line with this, overexpression of Drp1 in hippocampal differentiating neurons increases fragmented mitochondria in the dendrites at the expense of the mitochondrial content in the soma and accelerates neuronal maturation (143). Furthermore, a dominant negative of Drp1 impairs the dendritic arborization of cerebellar Purkinje neurons by preventing the movement of mitochondria from the soma (Fig. 1) (41).
Antioxidant regulation
Nrf2
The antioxidant capacity of NSCs is different from mature neurons and it is regulated during the differentiation process. NSCs are more resistant to oxidative insults and possess lower levels of ROS and higher levels of antioxidant enzymes than differentiated neurons (100, 101). Several changes occur during this transition, among which regulation of the transcription factor NF-E2-related factor 2 (Nrf2) seems to play a significant role in the differentiation of NSCs. The binding of Nrf2 to the antioxidant response element induces the transcription of major antioxidant molecules, including superoxide dismutase, catalase, glutathione peroxidase, and glutathione among others, which are determinants of control of the redox state of the cells (108). Loss of Nrf2 in NSCs from the subventricular zone induces a deficit in the survival, proliferation, and neuronal differentiation of these cells (22). During the differentiation process of NSCs, the mitochondrial complex 1 produces ROS that activates Nrf2, which is a determinant of the increase in the expression of proneural transcription factors and the protein botch (84), an inhibitor of notch, and NSC self-renewal (27). In this way, mitochondrial ROS regulates the transcription program of NSCs, which leads to the neuronal fate commitment of precursor cells.
During nervous system development, the levels of glutathione drastically change. By E10, NSCs possess similar levels of glutathione in different areas of the developing nervous system; however, by P5, most neurons drastically decrease their glutathione content (12). Since glutathione is a major determinant of the redox state of cells (134), it is expected that changes in the regulation of this molecule play an important role during the neuronal differentiation process. Weakening of the antioxidant capacity in neurons occurs gradually and can be observed in neurogenic regions of the adult brain. For example, progenitor cells in the subventricular zone and developing neurons in the subgranular zone contain more glutathione than complete differentiated neurons localized in the neighboring granular layer of the hippocampus (145).
One possible reason for the decline in neuronal antioxidant capacity could be that this condition may allow the proper function of different redox signaling pathways that take place during the differentiation process. In consequence, neurons rely more on antioxidant support provided by astrocytes. It is known that the levels of Nrf2 in cortical neurons are 100- to 1000-fold lower than in astrocytes (9), which might reflect differences observed in the glutathione content between neurons and glial cells (129). The reduction of Nrf2 levels occurs during brain development, specifically during the neuronal differentiation process.
Cultured cortical neurons express higher levels of Nrf2 mRNA at the initial stage of the differentiation process in comparison with later stages of differentiation. Similarly, the cerebral cortex at P0 also contains higher levels of Nrf2 mRNA than the adult cerebral cortex (13), indicating that at some point during neuronal differentiation, there is a marked reduction of Nrf2 expression, weakening their antioxidant capacity (13). This weakening occurs through an epigenetic repression of the Nrf2 promoter, which is accompanied by a reduction in the expression of the Nrf2 target genes Hmox1, Srxn1, xCt, Cat, and Gclc (13). Forced expression of Nrf2 during later stages of neuronal development diminishes the postsynaptic density protein 95, as well as the dendritic length and branching, and delays the electrophysiological maturation of the developing neurons (13). This evidence suggests that weakening the antioxidant capacity at very early stages of differentiation by downregulation of Nrf2 allows proper neuronal development. Nevertheless, neuroblastoma cells upregulate Nrf2 upon the induction of neuronal differentiation, which is required for the differentiation process (Fig. 1) (28, 29, 178). Furthermore, reprogramming of astrocytes to generate neurons is importantly enhanced by improving the antioxidant response, which is probably mediated, in part, by upregulation of Nrf2 (46).
Peroxiredoxins
Although it seems that the differentiation process requires the weakening of the antioxidant capacity, not all antioxidant components are downregulated. For example, oxidoreductases such as thioredoxins, peroxiredoxins, glutaredoxins, and nucleoredoxin are required to mediate redox reactions during the neuronal differentiation and maturation processes (18, 56, 87, 102, 170, 171). During neuronal differentiation, there are several genes that are upregulated compared with the precursor cells. These genes include, but are not limited to, genes related to energy metabolism and genes related to redox homeostasis. Among the upregulated genes related to redox homeostasis are the peroxiredoxins (Prxs) 1, 2, 3, 4, and 6 (35). Prxs are highly efficient hydrogen peroxide elimination enzymes with catalytic efficiencies comparable with glutathione peroxidase and catalase (69, 168), are relatively abundant, and represent primary sensors for hydrogen peroxide signaling in most cells (125). There are six Prxs divided into three classes, typical 2-Cys (Prx1-4), atypical 2-Cys (Prx5), and 1-Cys (Prx6) (60). Hydrogen peroxide oxidizes the redox-active peroxidatic cysteine of typical 2-Cys Prxs, forming an intermolecular disulfide with the resolving cysteine of a second molecule of Prx, which can be reduced by thioredoxin (60).
Two Prxs regulate the differentiation of motor neurons in the spinal cord through different redox reactions that control the localization and function of glycerophosphodiester phosphodiesterase-2 (GDE2) (170, 171). GDE2 is a transmembrane protein that contains a glycerophosphodiester phosphodiesterase ectodomain (GDPD) that is key to conduct motor neuron differentiation (127) by acting through the inactivation of RECK, which downregulates Notch to induce differentiation of motor neuron progenitors (119). During development, Prxs1–6 are expressed in ventral regions of the spinal cord (126). Prxd4 is expressed in motor neuron progenitors localized in the ventricular zone and early differentiated neurons localized in the intermediate zone, but not in fully differentiated neurons localized in the lateral marginal zone (171). The levels of oxidants vary in these regions, being the progenitor cells in the ventricular zone, those with the highest content of oxidants, followed by early differentiated neurons (171).
Prx4 regulates the timing of neuronal differentiation by regulating the activity of GDE2 in a process that involves oxidation of GDE2 in the GDPD domain at Cys 340 and Cys 467 (171). Overexpression of GDE2 triggers premature neuronal differentiation in the spinal cord, which is inhibited by coexpressing Prx4, but not when Cys 340 or Cys 467 of GDE2 is substituted by serine or when Cys 239 of Prx4 is substituted by serine (171). The oxidation of Cys 340 and Cys 467 occurs in the endoplasmic reticulum and this oxidation inhibits the transport of GDE2 to the plasma membrane. This is mediated by oxidized Prx4 since treatment of primary motor neuron cultures with hydrogen peroxide increases the pool of oxidized dimers of Prx4 and diminishes the levels of GDE2 surface expression, whereas antioxidant conditions do the opposite (171). However, when primary motor neuron cultures are obtained from Prx4-null mice, hydrogen peroxide treatment does not diminish the levels of GDE2 surface expression (171).
The direct oxidation of GDE2 by Prx4 is also supported by the observation that the embryos lacking Prx4 develop premature motor neuron differentiation by an increased activity of GDE2 (171). It is expected that deficiency of Prx4 leads to increased levels of hydrogen peroxide; however, basal or increased levels of hydrogen peroxide are not able to modify the activity of GDE2 in the absence of Prx4. It has been proposed that Prxs mediate redox signaling through two distinct, but not exclusive, models. The peroxide floodgate model proposes that the hyperoxidation of Prxs inactivates its own function and, by this way, hydrogen peroxide can be locally accumulated to oxidize a target protein (168). This localized inactivation of Prxs can also be induced by phosphorylation as in the case of Prx1 (167). The signal peroxidase model contemplates that Prxs act as hydrogen peroxide sensors and transmit their oxidation state to other proteins by thiol–disulfide exchange (125). The last model is certainly the case for Prx4. In this model, hydrogen peroxide oxidizes Prx4, and subsequently, Prx4 oxidizes Cys 340 and Cys 467 of GDE2, which interferes with its activity and localization, regulating, in this way, progression of the neuronal differentiation process in the spinal cord (Fig. 2).

Once GDE2 is localized at the cell surface, its activity is a target of another redox regulation, although the regulation in this location is elicited by Prx1. Prx1 is expressed in motor neuron progenitors in the ventricular zone before GDE2 expression, but overlaps with GDE2 in the intermediate zone cells and with fully differentiated neurons in the marginal zone (170). Silencing of Prx1 reduces the number of differentiated neurons, but does not alter the number of progenitor cells (170). This effect of Prx1 is mediated by the redox regulation of GDE2. GDE2 is usually inactive due to the presence of an intramolecular disulfide. Prx1 directly activates GDE2 by reducing the intramolecular disulfide formed between Cys 25 and Cys 576 of GDE2, which bridges the N- and C-terminal domains of GDE2 in the intracellular space (170). Overexpression of GDE2 elicits premature motor neuron differentiation; however, when the levels of GDE2 are not sufficiently high, the overexpression of GDE2 fails to conduct differentiation. Overexpression of Prx1 with suboptimal levels of GDE2 induces premature motor neuron differentiation, whereas overexpression with suboptimal levels of GDE2 that does not form the disulfide between Cys 25 and Cys 576 increases motor neuron differentiation in the spinal cord and gains independence from Prx1 (170). Therefore, reduction of this disulfide by Prx1 promotes differentiation, which implies the possibility that oxidation of Cys 25 and Cys 576 might be a direct control of GDE2 function, confirming a cascade of different redox signaling events that take place to control the differentiation process. Interestingly, the evidence presented in this study describes a new role of Prx1 as protein disulfide reductase, which is distinct from its function as an antioxidant enzyme. These studies demonstrate a fine redox regulation of GDE2 by Prxs that are required to control the timing of the differentiation process in the spinal cord (Fig. 2).
In a model of embryonic stem cells (ESCs), Prxs control the temporal progression of the neuronal differentiation process. During neuronal differentiation, there is an increase in the levels of ROS and Prx2 and a decrease in the levels of Prx1 (87). Knockdown of Prx1 or Prx2 in ESCs further increases the higher levels of ROS and accelerates the time of the differentiation process (87). The effect of ROS on differentiation is through activation of JNK since increased levels of ROS in Prx1 or Prx2-null ESCs promote a further phosphorylation of JNK and neuronal differentiation, which can be inhibited by NAC or JNK inhibition (87). The expression of Prx1 is controlled by the stemness-related transcription factor, Oct4, since silencing of Oct4 reduces the levels of Prx1, but not the levels of Prx2 (87). It is interesting that redox modifications regulate the activity of Oct4. The oxidation of Oct4 leads to diminished activity, while reduction of Oct4 by thioredoxin increases its activity (56). Furthermore, the activity of JNK can be regulated by ROS by modifying the interaction between JNK and the glutathione S-transferase π (GSTπ). The complex formed between these proteins inhibits JNK activity (2). ROS promotes the formation of one disulfide between Cys 47 and Cys 101 of GSTπ and also promotes the formation of one disulfide between two Cys 47s of two GSTπ monomers, preventing the interaction with JNK and allowing its activity (56). Thus, Prx1/2 contributes to the suppression of JNK by reducing ROS levels. Upon the differentiation stimulus, ESCs increased the levels of ROS to activate the JNK cascade, inducing ESC stemness loss by downregulation of Oct4, which controls the expression of Prx1. Then, Prx2 might control the levels of ROS once the differentiation process is initiated. This supports the idea that the redox state of the cell is a determinant of cell fate by controlling the function of different proteins involved in the differentiation process (Fig. 3).

Nucleoredoxin
Another oxidoreductase protein involved in neuronal differentiation is nucleoredoxin (Nrx), a member of the thioredoxin family of proteins, which possess structural characteristics similar to thioredoxin (43). Nrx is an important regulator of WNT/β-catenin signaling (42), which is a major pathway during embryonic development and is a key component in the neuronal differentiation process of human NSCs (58). Neuronal differentiation can be induced by growth factor removal in the cell line ReNcell VM, which is a model of human NSCs. In these cells, growth factor depletion activates the WNT//β-catenin pathway independently of WNT in a process that depends on ROS (58, 128). Upon growth factor depletion, calcium is released from the endoplasmic reticulum stores through the IP3 receptor type 1, then a fraction of this calcium is loaded into the mitochondria through the calcium uniporter within the first hour of the differentiation process, inducing the transitory generation of mitochondrial ROS (128). The increase of ROS levels induces the accumulation of nuclear β-catenin, promotes the transcription of the gene MAP2, and leads to neuronal differentiation. Silencing the IP3 receptor type 1 or the mitochondrial calcium uniporter reduces the levels of ROS and MAP2 mRNA (128). In addition, treatment of differentiating cells with NAC reduces the number of differentiated neurons by 50%, indicating that mitochondrial ROS induce neuronal differentiation via β-catenin (128) (Fig. 4).
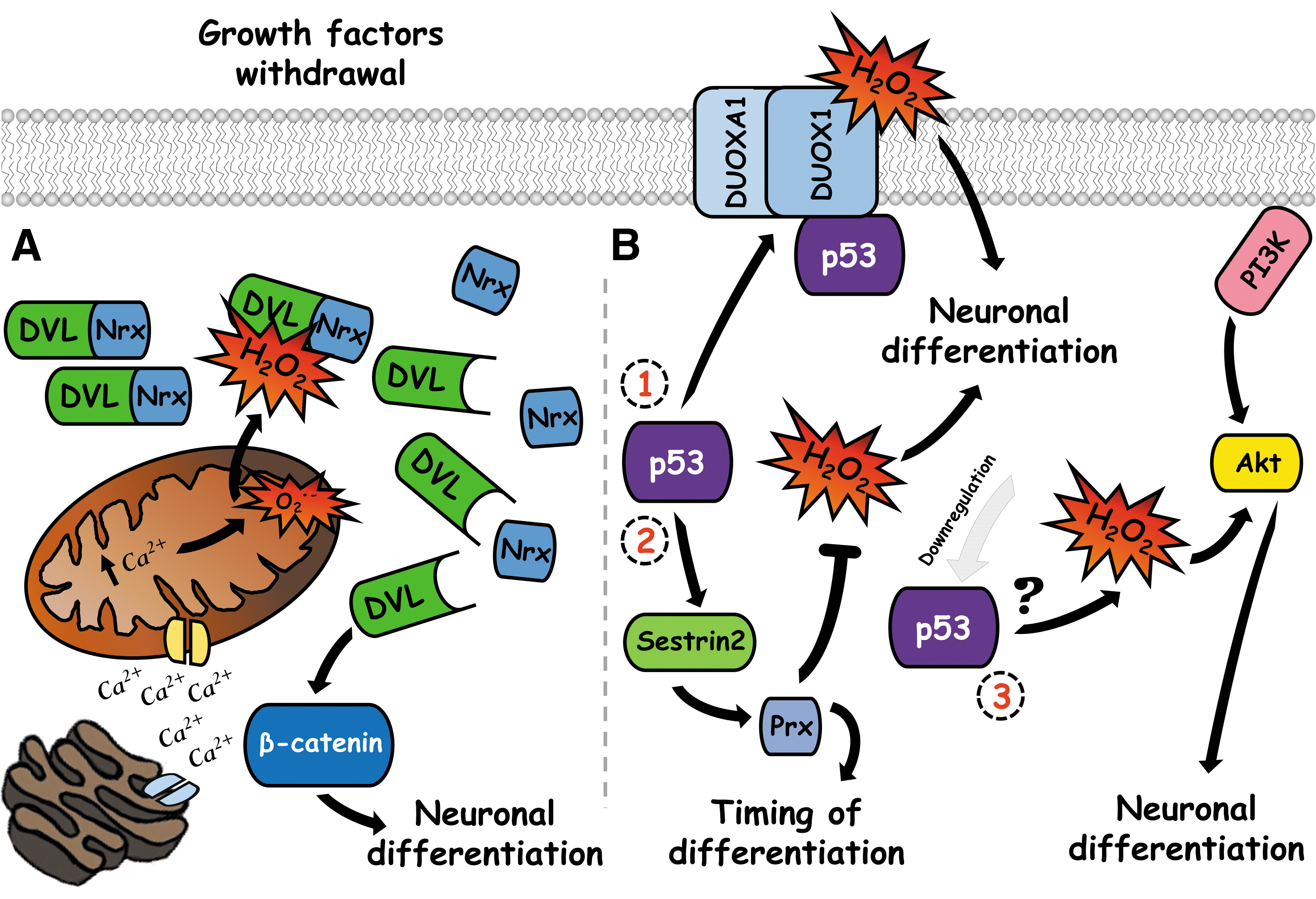
The mitochondrial ROS activate the β-catenin pathway by controlling levels of the regulatory protein disheveled (DVL). This protein transduces the signal from the WNT receptor, leading to stabilization of β-catenin and activation of the transcription factor TCF (107). Under basal conditions, the PDZ domain of DVL binds to the Cys 205 and Cys 208 of Nrx, forming a complex in the cytoplasm that maintains a pool of inactive DVL protected from ubiquitin-mediated degradation, which can activate the WNT/β-catenin pathway (42, 44). Mitochondrial ROS or exogenous hydrogen peroxide inhibits the interaction between DVL2 and Nrx in differentiating neurons, allowing a transitory increase in the levels of DVL2 that induces the β-catenin pathway and subsequently neuronal differentiation (58, 128). During the differentiation process, there is a transient increase in the levels of Nrx (128), which is probably required to allow the proper control of DVL levels and β-catenin activity. While these events take place within the first hours of differentiation, at more advanced stages of neuronal development, Prx1 is upregulated, and Prx4 is downregulated in comparison with undifferentiated cells (65). This suggests that the switch in the expression of these proteins is probably involved in the differentiation process of the cell line ReNcell VM (Fig. 4).
Pro-oxidant regulation
NOX2
The levels of ROS are strictly controlled in time, concentration, and intracellular localization, which are conditions that define the specificity of ROS to control the differentiation process without compromising the integrity of the cell. NOX enzymes represent major sources of ROS in the cells (11) and are implicated in the physiology and pathology of the nervous system (106, 140). There are seven NOX homologs, NOX1-5 and DUOX1/2, which produce superoxide anion and hydrogen peroxide as their primary biological function (11). The best known homolog is NOX2, this enzyme requires at least five subunits to function, two cytosolic adapters (p47phox and p40phox), one activator protein (p67phox), Rac, and the membranal subunit p22phox (90).
In the adult, NOX2 is highly expressed in the dentate gyrus and the subventricular zone in comparison with other areas of the brain (105). ROS produced by NOX2 modulates neurogenesis in these regions by promoting proliferation of NSCs (33, 105, 173) through the PI3K/Akt pathway, which involves the oxidative inactivation of PTEN (91). In KO NOX2 mice, there are fewer proliferating cells in the subventricular zone and fewer differentiated neurons in the olfactory bulb, which might be the result of an altered migration since a large number of BrdU-positive cells remain in the subventricular zone (91). This can also be explained by a deficiency in the differentiation process since neurosphere cultures derived from KO NOX2 mice produce significantly fewer neurons than those obtained from wild-type animals. Nevertheless, neuronal differentiation might also be due to a decrease in the NSC pool since NOX2 induces the self-renewal capacity of NSCs (91, 105). Thus, the evidence suggests that NOX2 acts at different levels since differentiating NSCs deficient in either NOX2 or p22phox express lower levels of neuronal markers, generate few neurons, and the differentiating neurons possess a reduced length of the developing neurites (105). It is not known how NOX2 regulates the differentiation process; however, it could be through the PI3K/Akt/mTOR pathway since this pathway finely controls the neuronal differentiation of NSCs (94, 176). Furthermore, as it was mentioned, ROS produced by NOX2 during the proliferative phase activate this pathway. Finally, this pathway is differentially regulated by distinct gradients of hydrogen peroxide since direct oxidation of Akt could either increase or decrease its activity, whereas further increased levels of hydrogen peroxide oxidize the phosphatases PTEN and PP2a, leading to an increased phosphorylation of Akt (148).
NOX4
NOX4 regulates the differentiation of NSC populations. This enzyme is constitutively active (136) and produces hydrogen peroxide as its main product (109). It was reported that the levels of NOX4 mRNA in proliferating NSCs obtained from the subventricular zone are exceedingly elevated in comparison with NOX1 and NOX2 (118). In this study, when neuronal differentiation was induced, the levels of NOX4 mRNA substantially increase during the first 2 days of differentiation, and at day 5, NOX4 is only expressed in differentiating neurons. Silencing of NOX4 or an antioxidant treatment greatly reduces the number of differentiated neurons (118). Likewise, in neural crest stem cells, the bone morphogenetic protein (BMP) induces neuronal differentiation probably through NOX4 (93). NOX4 is the only NOX enzyme expressed in these cells and NOX inhibition reduces the levels of ROS and blocks neuronal differentiation, while silencing of NOX4 leads to the apoptotic death of these cells (93). Although NOX4 seems to be required for neuronal differentiation of neural crest stem cells, the neural crest-derived peripheral nervous system normally develops in NOX4 KO mice (93). The relative contribution of different NOX enzymes to the differentiation process is not entirely clear, and there is no information about any specific NOX enzyme able to compensate the function of another NOX during the differentiation process in knockout models. However, these studies provide significant evidence that implicates NOX4 in the differentiating process of different NSC populations.
DUOX1
Another member of the NOX family, DUOX1, and its maturation factor, DUOXA1, are modulators of the neuronal differentiation process. DUOX1 generates hydrogen peroxide, is regulated by calcium, and requires the formation of a heterocomplex with its maturation factor DUOXA1 to be active (11). DUOXA1 is expressed in neurogenic regions during development and it is highly expressed in NSCs (82). DUOX1 and DUOXA1 form a complex with the transcription factor and tumor suppressor p53 in NSCs, which modulates neuronal differentiation (116). p53 is involved in the loss of stemness as well as the beginning of differentiation and its function is redox regulated at different levels (38, 117). In telencephalon NSCs, knockdown of p53 induces the production of more ROS than wild-type cells, which induces neuronal differentiation through the PI3K/Akt pathway, without affecting proliferation of precursor cells (39). The absence of p53 causes downregulation of different antioxidant components, among them, sestrin 2 is critical for restoring the redox state in KO p53 NSCs (39), indicating that p53 regulates the timing of neuronal differentiation by controlling the antioxidant capacity of NSCs (Fig. 4).
In addition, P53 also controls the expression of DUOX1 and DUOXA1 and induces differentiation by controlling the expression and interaction of these proteins. In the embryonic carcinoma P19 cell line, the overexpression of p53 in these cells upregulates the expression of DUOX1 and DUOXA1 and induces the formation of the complex between these proteins (116). In the absence of p53, although DUOX1 and DUOXA1 are expressed, these proteins do not form a complex (39), indicating that p53 might function as an adapter protein to allow interaction between these proteins. In NSCs, the interaction between DUOX1, DUOXA1, and p53 declines in terminal differentiated neurons as well as the levels of DUOX1 and DUOXA1 (116). The neuronal differentiation of P19 cells can be induced by retinoic acid, which is greater when p53 is expressed in these cells (116). In addition, overexpression of DUOXA1 induces neuronal differentiation of P19 cells in the absence of retinoic acid and accelerates the differentiation process in retinoic acid treatment (82). The overexpression of DUOXA1 also increases the amount of ROS generated in undifferentiated cells, which is prevented by NOX inhibition (82). Antioxidant treatment in overexpressing DUOXA1 cells slightly reduces the differentiation induced by retinoic acid, while silencing of DUOXA1 partially reduces the levels of ROS and diminishes the differentiation process induced by retinoic acid (82). This suggests that a part of the effects of the complex DUOX1, DUOXA1, and p53 on the neuronal differentiation process is mediated by hydrogen peroxide produced by DUOX1 (Fig. 4).
NOX and CD38
The differentiation of ESCs is regulated by ROS produced by NOX through its regulation by the protein CD38, a multifunctional membrane enzyme that is the main ADP-ribosyl cyclase for synthesizing cyclic adenosine diphosphoribose (cADPR) from NAD (57). cADPR induces calcium release through the ryanodine receptor (45), which might contribute to the differentiation process since calcium is relevant in processes such as dendritic arborization and axonal pathfinding (130). At the stage of embryoid body formation, the expression of CD38 increases (162); however, during the neuronal differentiation process, the expression of CD38 diminishes (161). Silencing of CD38 reduces neuronal differentiation, while overexpression of CD38 increases the percentage of differentiated cells (161). During neuronal differentiation, the levels of ROS increase, which are markedly reduced by silencing of CD38 and conversely increased by overexpression of CD38 (161). Similarly, NOX activity is reduced by silencing of CD38 and increased by overexpression of CD38 (161). Thus, CD38 is upstream of NOX activity, which is required to promote neuronal differentiation.
ROS are the mediators of CD38 actions required for neuronal differentiation since exogenous hydrogen peroxide significantly mitigates the negative effects of CD38 knockdown on neuronal differentiation (161). It is important to mention that silencing of CD38 increases the levels of NAD/NADH ratio, but does not alter mitochondrial function measured by ATP production, membrane potential, and calcium content (161), suggesting that CD38 regulates ROS levels through NOX and not by mitochondria. It is not clear how CD38 controls the function of NOX; however, the regulation of NOX activity is not related to the levels of p47phox, gp91phox, and NOX4 (161). Interestingly, internalization of CD38 is required to produce cADPR for subsequent calcium signaling, and the internalization of CD38 is promoted by the reduction of one disulfide formed by Cys 119 and Cys 201 of CD38. This probably allows the formation of a mixed disulfide with other molecules of CD38, which is required for internalization (59). In line with this, the ROS produced by NOX1 induces the internalization and activity of CD38 (59) and CD38 colocalizes with p22phox (161), suggesting that the relationship between NOX activity and CD38 might be bidirectional.
Differentiation of PC12 cells and neuroblastoma cells
Different models have been extensively used to unravel the molecular mechanisms that govern neuronal differentiation. The first evidence describing the regulatory effects of ROS in neuronal differentiation came from studies based on cell lines, such as PC12 cells and neuroblastoma cells. Neuroblastoma cells such as SH-SY5Y cells can develop a functional and morphological phenotype upon exposure to several molecules such as retinoic acid, quinolinic acid, and docosahexaenoic acid in a process dependent on ROS produced by NOX (62, 110, 169). On the other hand, PC12 cells treated with nerve growth factor (NGF) develop neurites and express different neuronal markers, including βIII-tubulin, GAP-43, and neurofilament L, among others (78, 123), which are also dependent on ROS produced by NOX.
In SH-SY5Y neuroblastoma cells, the induction of differentiation by retinoic acid and docosahexaenoic acid is mediated by ROS. Retinoic acid induces the expression of the neuronal marker MAP2 and induces neurite elongation (110). Retinoic acid treatment induces the expression of p47phox, a cytosolic adapter subunit of NOX2, and induces its translocation to the membrane, which is required to promote neurite elongation through PKCδ (110). Retinoic acid also induces upregulation of antioxidant genes promoted by Nrf2 (28), which promotes neurite outgrowth since silencing of Nrf2 compromises neuronal differentiation (178), probably by promoting cell survival (29). In addition, overexpression of glutaredoxin 2 promotes neurite outgrowth (18). This suggests that the proper balance between oxidants and reductants is a critical factor for the differentiation process of these cells.
Docosahexaenoic acid treatment induces neurite outgrowth in SH-SY5Y cells through ERK1/2 in a process that is dependent on hydrogen peroxide since treatment with catalase reduces ROS levels, ERK1/2 activation, and neurite outgrowth (169). In other neuroblastoma cells such as N2a neuroblastoma cells, serum starvation, forskolin, and cAMP induce neurite outgrowth and mitochondrial biogenesis through ERK1/2 in a process dependent on ROS (154). In these cells, antioxidant treatments reduce ROS levels, mitochondrial biogenesis, and neurite outgrowth, while MEK inhibition only reduces mitochondrial biogenesis and neurite outgrowth (154), indicating that ROS act upstream of ERK1/2 activation.
In PC12 cells, the process of differentiation begins with the activation of the neurotrophin receptor TrkA by NGF, which leads to the activation of PLCγ-PKC-Raf and the subsequent activation of the signaling pathway Ras-Raf-MEK-ERK. The sustained phosphorylation of ERK is necessary and sufficient to induce the acquisition of neuronal-like characteristics of PC12 cells. The TrkA receptor also activates the PI3K-Akt pathway, which promotes cell survival (78, 123). ROS regulates neuronal-like differentiation of PC12 cells at different levels. For example, staurosporine, a nonspecific protein kinase inhibitor, induces differentiation dependent on ROS through the activation of Rac1 and NOX (85). Furthermore, hyperoxia and xanthine/xanthine oxidase induce the formation of neurites through the sustained activation of ERK1/2 in a process dependent on ROS (80). The activation of ERK1/2 mediated by ROS is also elicited by the activation of another tyrosine kinase receptor, the Erb-4 receptor. The activation of ErbB-4 by neuregulin induces sustained production of ROS and the activation of the Ras-Raf-MEK-ERK pathway and subsequent differentiation of PC12 cells (53). The activation of Ras and ERK1/2 is dependent on ROS since an antioxidant treatment inhibits the activation of Ras, ERK1/2, and differentiation (53), indicating that ROS acts upstream of Ras. Interestingly, a dominant negative of Ras inhibits the ROS produced in response to neuregulin (53), which suggests a positive feedback mechanism to regulate ROS levels and the activation of ERK1/2 (Fig. 5).
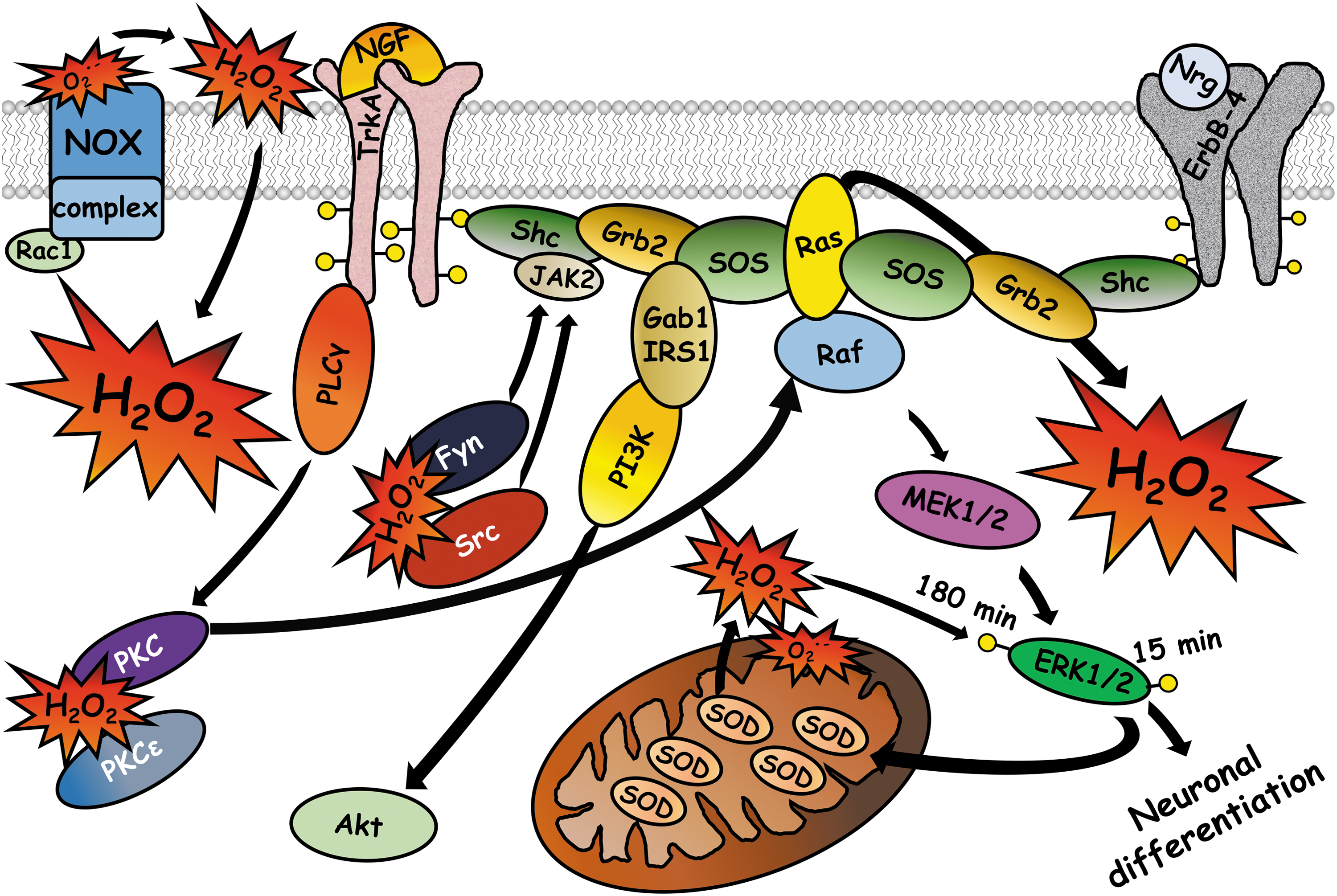
ROS regulates the activation of ERK1/2 at different points of the signaling cascade. ROS induce phosphorylation of TrkA through activation of PLCγ and PI3K, produce inhibition of protein tyrosine phosphatases, and induce the formation of a receptor complex with the scaffold proteins Shc, Grb2, and Sos, which are indispensable for activation of the MAPK pathway (77). Although it is not clear how ROS induce the formation of the complex formed between the TrkA receptor and the scaffold proteins, it is possible that hydrogen peroxide induces the activation of ERK1/2 by Src through the Ras-Raf pathway (4) and by Fyn through the Jak2-Shc-Grb2-Sos-Ras pathway (1). Interestingly, neurite outgrowth induced by NGF is mediated by Fyn (32), while Src is required for neurite outgrowth induced by cAMP, but not by NGF (113). However, it is possible that ROS regulate Src in this model since Src function can be regulated through oxidation of different cysteines. Oxidation of Cys 277 induces the formation of a mixed disulfide with another monomer of Src, which causes partial inactivation (81), while oxidation of Cys 245 and Cys 487 forms an intramolecular disulfide that induces Src activation (51). Therefore, these kinases might be involved in the differentiation of PC12 cells conducted by ROS, Fyn being one of the possible targets of ROS (Fig. 5).
Different sources of ROS are involved in differentiation processes induced by NGF in PC12 cells, NOX enzymes and mitochondria being the major components (1, 180). NGF induces a peak of ROS after 10 min of treatment. This production of ROS is inhibited by DPI, suggesting that NOX is the source of ROS production induced by NGF at this time (147). Posteriorly, NGF treatment gradually reduces the expression of NOX2 at 48 h, with concomitant increase in the expression of NOX1 (73). After 72 h, there is a second peak of ROS, which is produced by NOX1, since silencing of NOX1 abolishes this peak (73). After 48 h of NGF treatment, inhibition of ROS by NOX inhibitors or by antioxidant treatment increases the neurite length and the expression of βIII-tubulin (73). These results suggest that ROS produced by different sources at different stages of the differentiation process play different roles. The first peak of ROS, most probably generated by NOX2, is critical to initiate neurite differentiation, while the second peak of ROS, which is produced by NOX1, negatively regulates this process.
NGF treatment also induces the expression of the antioxidants glutathione peroxidase and catalase after 3 days in PC12 cells (132). This regulation of antioxidant systems allows the cells to contend with the hydrogen peroxide chronically produced by NGF (74). However, the role of antioxidant systems in PC12 differentiation is not completely understood. For example, an increase in thioredoxin levels promotes differentiation in a similar way as NGF (67) and MnSOD controls the differentiation induced by NGF by inducing phosphorylation of ERK1/2 (20). NGF induces the phosphorylation of ERK1/2 from 5 to 30 min, which is crucial to induce the expression and activity of MnSOD that is required to induce a second peak of ERK1/2 phosphorylation 180 min after NGF treatment (20). Overexpression of MnSOD induces differentiation, while silencing of MnSOD reduces the transcription and phosphorylation of ERK1/2 (20). The mechanism by which MnSOD induces the phosphorylation of ERK1/2 at 180 min is through the conversion of superoxide anion into hydrogen peroxide, which is indispensable to induce the long-term phosphorylation of ERK1/2, since hydrogen peroxide degradation by a glutathione peroxidase mimetic inhibits the second peak of ERK1/2 phosphorylation and the differentiation process is completely inhibited (20) (Fig. 5).
One possible mechanism of action to activate ERK1/2 is through oxidative regulation of PKC. The redox catalyst CoCl2 causes differentiation of PC12 cells through the oxidation of PKCɛ. CoCl2 promotes the translocation of PKC from the cytosol to the membrane and the capacity to bind phorbol esters (54). The exposure of cells to CoCl2 oxidizes 7 of the 18 cysteines detected in PKC and exposure to higher concentrations of CoCl2 induces the oxidation of 3 more cysteines and the inactivation of PKC (54). In this study, the MEK inhibitor PD98059 substantially decreases the activation of ERK1/2, CREB, and the differentiation induced by CoCl2 (54). These results suggest that PKCɛ couples the oxidative signal to the final activation of ERK1/2 and the possible existence of more targets susceptible to oxidative modifications that are upstream of MEK. Finally, NGF treatment induces ROS generation that activates PKCɛ during the first minutes (54), indicating that a redox modification in the regulatory domain of PKCɛ is a critical event in the neuronal differentiation induced by NGF (Fig. 5).
Nitric oxide
Nitric oxide is an important determinant of NSC differentiation. During nervous system development, the expression of the neuronal nitric oxide synthase (nNOS) increases progressively from the E13 to P2 and it is regulated by the brain-derived neurotrophic factor (BDNF) (26). A nitric oxide donor or BDNF treatment reduces the proliferation of NSCs and promotes neuronal differentiation, which is suppressed by inhibition of nNOS (26). Interestingly, in the adult, nitric oxide inhibition promotes proliferation of NSCs in the subventricular zone and impairs neuronal differentiation (26). Similarly, in neural precursor cells from the hippocampus, nitric oxide promotes neuronal differentiation (120). During neuronal differentiation, the production of nitric oxide and the levels of nNOS increase markedly and silencing of nNOS inhibits the neuronal differentiation process (120). The proposed mechanism is that nitric oxide produced by the nNOS activates PKCα, which in turn inactivates GSK3β, inhibiting proliferation of precursor cells and promoting neuronal differentiation (120).
Oligodendrocyte Differentiation
ROS also promote the differentiation of oligodendrocytes. These cells arise from a distinct precursor cell that progresses through different stages of maturation characterized by the expression of specific markers (8). In NSCs obtained from the subventricular zone, exogenous hydrogen peroxide induces neuronal differentiation and oligodendrocyte differentiation (124). The action of hydrogen peroxide is mediated by upregulation of the histone deacetylase, sirtuin2, which promotes differentiation (124). In oligodendrocyte progenitor cells obtained from the optic nerve, the redox state of cells determines the balance between self-renewal and differentiation (138). Progenitor cells with high levels of ROS are differentiated to a greater extent than progenitors with low levels of ROS (138). Additionally, antioxidant treatment reduces the number of differentiated cells, while treatment with pro-oxidant conditions increases the number of differentiated cells (138), indicating that the redox state of the cell modulates the differentiation process rather than being a consequence of the differentiation process.
Oligodendrocytes are generated with different temporal patterns in all regions of the central nervous system. This can be observed in progenitor cells isolated from the optic nerve, optic chiasm, and cerebral cortex, which generate oligodendrocytes at different times (111). Interestingly, there is a correlation between the temporal patterns of differentiation and the redox state of the cells. Optic nerve progenitors are the more oxidized cells and generate oligodendrocytes faster than progenitors isolated from the optic chiasm and the cerebral cortex (111). On the other hand, progenitor cells from the cortex are the most reduced cells and differentiate into oligodendrocytes more slowly than progenitors from the optic nerve and the optic chiasm (111).
NOX enzymes regulate the differentiation of oligodendrocytes. NOX2-deficient NSCs generate a reduced number of oligodendrocytes than control cells (105), and in the cell line MO3-13 (which exhibits molecular characteristics of human oligodendrocyte precursor cells), exogenous hydrogen peroxide induces oligodendrocyte differentiation through activation of ERK1/2 and CREB (1). Hydrogen peroxide activates ERK1/2 and CREB through PKC activation. This is based on the fact that hydrogen peroxide induces phosphorylation of PKC and that PMA induces differentiation through the activation of ERK1/2 (1). Furthermore, silencing or inhibition of PKC inhibits the effects of hydrogen peroxide treatment (1). Both treatments—hydrogen peroxide and PMA—upregulate the expression of NOX3 and NOX5, and silencing of these enzymes inhibits the differentiation induced by hydrogen peroxide and PMA (1). Since silencing of NOX3 has the same effect as silencing of NOX5, it seems that these enzymes do not play redundant functions. In line with this idea, the silencing of NOX5 downregulates the expression of NOX3, but silencing of NOX3 does not modify the expression of NOX5 (1). Therefore, it is possible that NOX3 is a downstream effector of PKC and NOX5. It is interesting that ROS activates PKC and that PKC induces ROS production, conforming a positive feedback that induces the sustained phosphorylation of ERK1/2 and the activation of CREB, which leads to oligodendrocyte differentiation.
Axonal and Dendritic Growth
Multiple redox signaling events take place in the axonal growth cones during axonal growth and guidance processes. These processes are orchestrated by cytoskeletal dynamics that are regulated by extracellular cues and intracellular signaling events; their importance relies on the proper conformation of the neuronal polarity and the subsequent establishment of neural circuits (7). It is well known that cytoskeletal elements are highly susceptible to be regulated by redox signaling events (47, 163) and some redox-regulated processes have been described to take place in the developing axon. Two main components have been described in the redox regulation of signaling cascades in the developing axon, one is the molecule interacting with CasL (MICAL) and the other one is NOX.
Axonal guidance
Mossy fiber lamination in the hippocampus
The neuronal growth cone is a highly motile structure situated at the tips of axons and dendrites. This structure senses environmental cues through receptors located at the plasma membrane and transduces the signal through different signaling pathways that regulate cytoskeletal and growth cone dynamics by controlling the growth, retraction, and direction of neuronal processes (70). One of the proteins involved in regulation of the cytoskeleton in response to extracellular cues belongs to the family of MICAL proteins, which are enzymes that catalyze redox reactions to control cytoskeletal dynamics through their flavin monooxygenase activity (55, 71, 72).
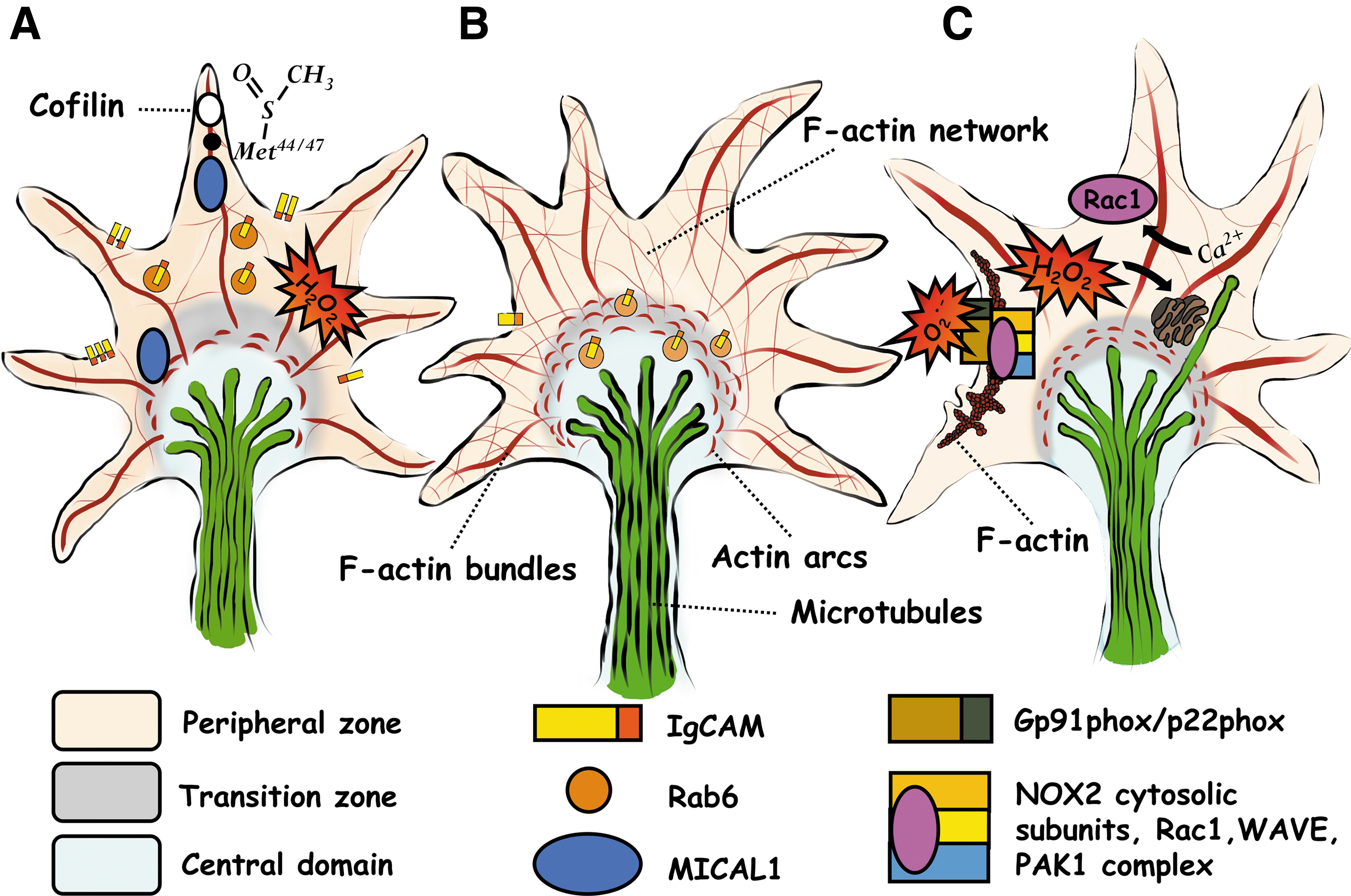
MICAL1 controls the establishment of the mossy fiber axons from granule cells into two laminae on either side, the basal or the apical dendrites of the CA3 pyramidal neurons in the dentate gyrus (156). Neuronal circuit assembly depends on the cell surface expression of different proteins at the growth cone, such as the cell adhesion molecules of the immunoglobulin superfamily (IgCAMs) (79), whose cell surface levels are essential to establish the mossy fiber lamination (64). MICAL1 KO mice form thick fasciculate bundles that are aberrantly positioned between the CA3 pyramidal neuron somas, instead of being positioned on either side of the dendrites (156). Two IgCAM KO mice, the neural cell adhesion molecule (NCAM), and the close homolog of L1 (CHL1) exhibit similar defects in the lamina-specific mossy fiber connections as in the MICAL1-deficient mice (156).
MICAL1 deletion reduces the cell surface expression of NCAM and CHL1 and produces an aberrant accumulation of the Rab6 secretory vesicles containing NCAM at the central domain of the axonal growth cone, where it induces an aberrant F-actin organization, showing an increase in the F-actin content (156). It is known that MICAL1 induces F-actin disassembly through direct oxidation of Met 44 and Met 47 (71), which increases the binding of cofilin, an F-actin disassembly factor (55). The reaction is mediated by hydrogen peroxide produced by MICAL rather than by direct hydroxylation of methionine residues (157).This is a reversible process and is mediated by the activity of methionine-R-sulfoxide reductase B1 (92). Therefore, the aberrant accumulation of F-actin in the growth cones might be explained by the lacking redox regulation of these residues.
In this model, the monooxygenase domain of MICAL1 is responsible for the aberrant F-actin levels and vesicle localization. This is supported by rescue experiments in MICAL1 KO neurons with MICAL1 transfection, which normalized F-actin levels and vesicle distribution, while transfection of the enzymatically inactive monooxygenase domain of MICAL1 did not rescue the aberrant phenotype (156). These experiments demonstrate that MICAL1 controls the transport of IgCAMs through redox regulation of F-actin dynamics, which allows the proper establishment of hippocampal circuits (Fig. 6).
Redox regulation of CRMP2
Neurons establish synaptic contacts during development with specific targets in a complex process regulated by extracellular cues that guide axons. Semaphorins constitute a family of membrane-associated and secreted proteins involved in axonal growth cone guidance and cellular processes that are required for the generation of specific neural circuits (121). Their mechanisms of action require the activation of receptor proteins of the plexin and neuropilin families. These receptors induce the activation of the GTPase domain of plexin, coupling specific signaling pathways that regulate repulsive axon guidance and cell migration through downstream molecules such as protein kinases, GTPases, and cytoskeleton-associated proteins (121). In general, semaphorins are chemorepulsive proteins, although they can also act as chemoattractants and can regulate axon branching and dendritic pruning (89). Semaphorin3A and semaphorin3F mediate axonal repulsion through the flavin monooxygenase domain of MICAL (122, 149). The effect of semaphorin3A on the growth cone collapse occurs through the regulation of cytoskeletal dynamics by the collapsin response mediator protein 2 (CRMP2) (137).
CRMP2 is a member of a family of microtubule assembly factors. This protein binds to tubulin heterodimers and promotes microtubule polymerization (40). CRMP2 is regulated post-translationally by phosphorylation through cyclin-dependent kinase 5 (CDK5) at Ser 522, which acts as a priming site for glycogen synthase kinase 3 (GSK3) phosphorylation at Thr 509, Trh 514, and Ser 518 (19, 174). Such phosphorylations reduce CRMP2 affinity for tubulin heterodimers, thereby reducing microtubule polymerization at the distal part of the axons and, in this way, inducing axon retraction. It has been demonstrated that redox modification of CRMP2 controls its function during the axonal growth cone collapse (18, 48, 102) and in the reversible retraction of oligodendrocyte processes (36).
In dorsal root ganglion neurons, phosphorylation of CRMP2 by CDK5 and GSK3 promotes microtubule disassembly, leading to the growth cone collapse in a process that involves different redox signaling events. Morinaka et al. (102) found that phosphorylation of CRMP2 is precisely regulated by an oxidative modification of CRMP2 through MICAL1 and MICAL3. Semaphorine3A induces the generation of hydrogen peroxide through MICALs in the axonal growth cone, oxidizing CRMP2 at Cys 504 and inducing the formation of a homodimer (102). Since CRMPs bind to the monooxygenase domain of MICAL (135), the most possible scenario is that MICALs directly oxidize CRMP2. The disulfide formed between two molecules of CRM2 is a target of Trx1, which reduces this bond by forming a transitory mixed disulfide between the Cys 32 of thioredoxin 1 (Trx1) and the Cys 504 of CRMP2. This promotes the specific phosphorylation of CRMP2 by GSK3 and the subsequent growth cone collapse (102). Cys 504 of CRMP2 is also crucial for semaphorin3A signaling during radial migration of cortical neurons since the transfection of CRMP2 with the substitution of Cys 504 for Ser induces migration defects that resemble those seen in neuropilin1 and plexin KO (102) (Fig. 7).
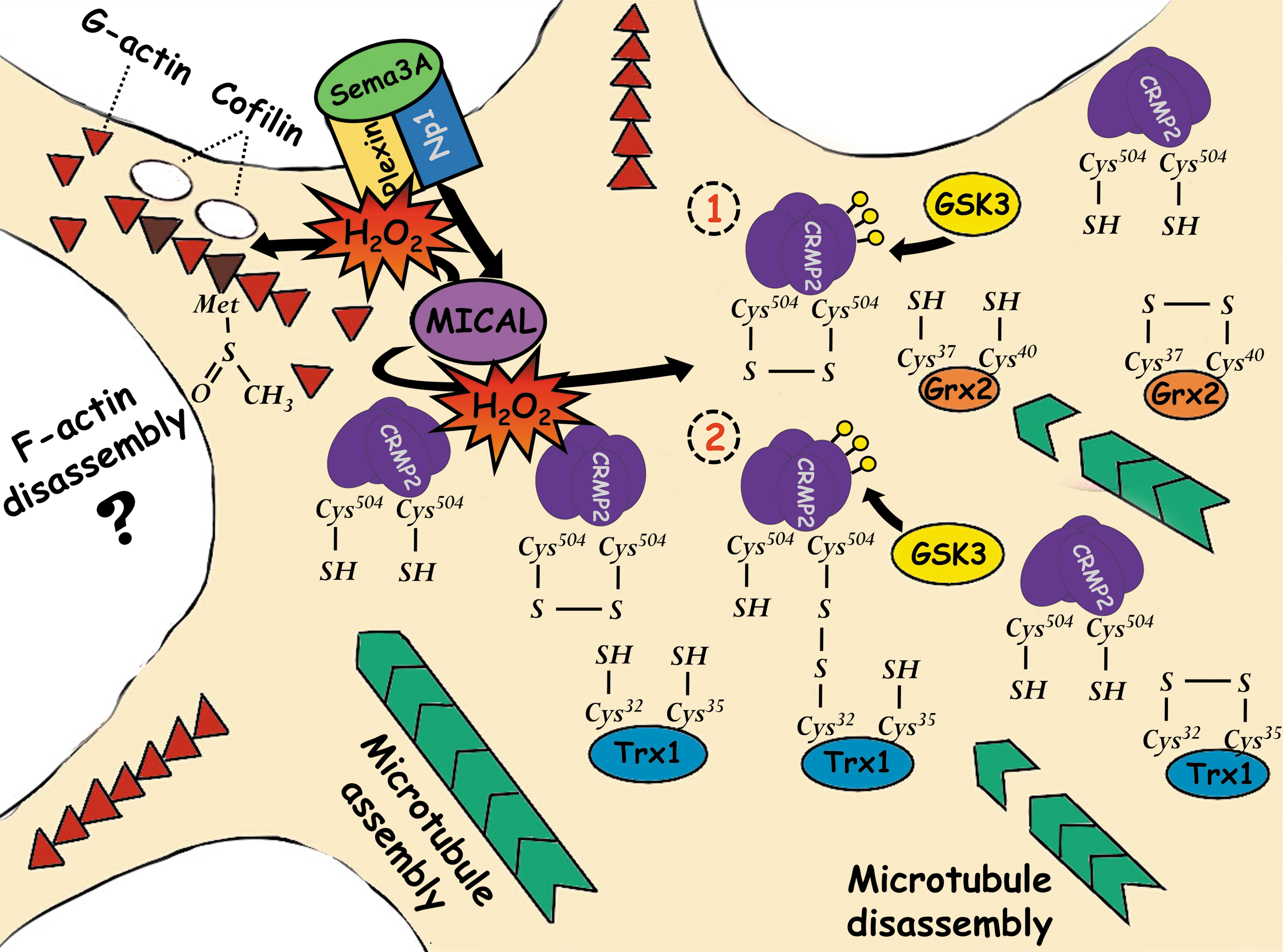
It has been suggested that a stable bond between Trx1 and CRMP2 is unlikely since active cysteines have different pKa values and the Cys 35 of Trx1 would rapidly reduce the disulfide formed. Therefore, an alternative model with Grx2 has been proposed (18, 48). Whether this assumption is correct or not, the study by Morinaka et al. (102) provides important evidence demonstrating the involvement of Trx1 in the semaphorin3A response since silencing of Trx1 reduces both the growth cone collapse and CRMP2 phosphorylation. Besides, the inhibition of thioredoxin reductase suppresses the repulsive response in a gradient of semaphorin3A. These experiments do not exclude the possibility that the levels of reduced Trx1 serve to balance the redox state, allowing proper redox signaling events to take place, rather than directly interact with CRMP2, although in this study, direct interaction between these proteins was detected (Fig. 7).
An alternative model of redox regulation of CRMP2 is exemplified in the studies by Brautigam et al. (18) and Gellert et al. (47). These studies propose that glutaredoxin 2 (Grx2) reduces CRMP2 in a nonstable interaction and, in this way, regulates the actions of CRMP2. In zebrafish, Grx2 controls axonal growth through a redox modification of CRMP2 (18). Grx2 KO embryos display a severe reduction of axonal tracts, which is evidenced by a reduction of brain commissures and major longitudinal axonal tracts, as well as the branching points and the length measured of individual axons (18). The effects of Grx2 are mediated by CRMP2 since knockdown of CRMP2, as well as the knockdown of Grx2 are rescued by injection of CRMP2 mRNA, which shift the oxidized pool of CRMP2 to reduced CRMP2 (18). These experiments demonstrate that the correct redox balance of CRMP2 allows proper axonal growth and the establishment of neural circuits (Fig. 7).
The proposed mechanism of Grx/CRMP2 interaction is based on the idea that low activity of Grx2 leads to an increase of oxidized CRMP2 and this oxidation produces a conformational change of CRMP2. In the presence of Grx2, the oxidized cysteines can be reduced by a process that does not form a relatively stable mixed disulfide between these molecules (18). The quaternary structure of CRMP2 is homotetrameric, this structure is not changed whether CRMP2 is reduced or oxidized, whereas oxidation of CRMP2 causes the formation of one disulfide between the Cys 504s of two CRMP2 monomers in the tetrameric complex (48). The formation of this disulfide leads to conformational changes of CRMP2 that are time dependent and reversible. Whereas oxidation increases the ellipticity of the CRMP2 complex, reduction leads to a decrease of ellipticity between 210 and 220 nm, which induces the exposition of hydrophobic regions (18, 48) (Fig. 7).
Then, the oxidized CRMP2 is reduced by Grx2 in a dithiol reaction mechanism, where the most reactive cysteine of the active site (Cys 37) performs a nucleophilic attack on the mixed disulfide of CRMP2 and the new intermediate mixed disulfide is immediately attacked by the second cysteine in the active site (Cys 40) (48). Gellert et al. (47) found that oxidized CRMP2 can be reduced by Grx2 and Trx1 without forming a stable mixed disulfide between these proteins. In this model, it is proposed that a redox switch controls CRMP2 conformation and phosphorylation. On the other hand, Morinaka et al. (102) found that oxidized CRMP2 is reduced by Trx1, forming a transitory mixed disulfide that induces CRMP2 phosphorylation. These studies exemplify redox mechanisms that control CRMP2 phosphorylation and activity and the importance of this regulation during nervous system development (Fig. 7).
Axonal growth
The coordinated regulation of different elements of the cytoskeleton is essential for proper neural growth cone dynamics. It is believed that redox regulation of cytoskeleton has a major role in the axonal growth process. One extensive model employed to study the redox regulation of cytoskeletal dynamics is Aplysia neurons. These neurons possess growth cones that are 5–10 times larger than typical vertebrate growth cones, which facilitate the visualization of cytoskeleton dynamics (146). In these neurons, axonal growth cone dynamics is regulated by ROS that accumulate at the axonal growth cone. These ROS are required for normal axonal growth since antioxidants, NOX inhibitors, and lipoxygenase inhibition decrease the levels of ROS and diminish axonal growth (103). This regulation is mediated by the action of ROS on the dynamics of the actin cytoskeleton since physiological levels of ROS promote the assembly and protrusion of actin filaments, and a reduction of ROS levels reduces the ruffling activity at the transition zone, inhibiting actin assembly at the leading edge and promoting arc contractibility and disassembly of actin filaments (103).
One primary source of ROS involved in actin remodeling is NOX. NOX2 responds to cues from the extracellular environment to regulate axonal growth cone dynamics. In Aplysia neurons, an NOX2-type enzyme is expressed uniformly in a punctate way in the peripheral domain and is concentrated at the central domain, while p40phox exhibits a punctate distribution that is very similar to the F-actin bundles in the peripheral domain (104). The localization of both proteins is dependent on F-actin since cytochalasin B treatments that disrupt F-actin bundles reduce the levels of p40phox and NOX2 in the peripheral domain (104). When these neurons were grown in beds of Aplysia cell adhesion molecule (apCAM), which is known to evoke growth, there was a significant increase in the colocalization between NOX2 and p40phox only in areas where apCAM was restrained (104). On the other hand, activation of NOX2 induces ROS accumulation in the peripheral domain of the axonal growth cone, while pharmacological inhibition of NOX2 impairs axonal growth, decreases the F-actin content in the transition zone, and reduces filopodial actin bundles and lamellipodial actin networks in the peripheral zone (104). Together, these results demonstrate that ROS produced by NOX regulate actin dynamics during axonal growth and that this regulation is not unidirectional since actin dynamics might regulate NOX activity by controlling the localization of the NOX complex and its activity (Fig. 6).
In axonal growth cones of hippocampal neurons, NOX inhibition diminishes the number, length, and lifetime of filopodia in the axonal growth cone and reduces the activity of Rac1 and Cdc42 (165). This suggests that NOX activity regulates actin dynamics in the axonal growth cone, at least in part, through the regulation of Rac1 and Cdc42. These proteins act as molecular switches that cycle between an inactive GDP-bound state and an active GTP-bound state (142). Since these proteins have a redox-sensitive motif comprising the Cys 18 and a nucleotide-binding phosphoryl-binding loop, the regulation by NOX can be direct through oxidation of Cys 18, which promotes their activation by increasing the rate of nucleotide dissociation (142). This regulation of the actin cytoskeleton by NOX promotes axonal growth and allows the proper establishment of neuronal polarity since NOX inhibition impairs these processes (165). The use of gp91ds-tat and VAS2870, two selective inhibitors of NOX2, as well as the dominant negative of p22phox, which inhibit the activity of different NOX enzymes, had the same effect on neuronal polarity and axonal growth, suggesting that NOX2 could be the NOX homolog responsible for these processes.
It is well known that Rac proteins are activators of some members of the NOX family (11), for this reason, it is expected that Rac1 controls axonal growth cone dynamics through regulation of the actin cytoskeleton and NOX activity. However, this occurs in a complex manner. In Aplysia neurons, Rac1 promotes calcium release from the endoplasmic reticulum stores through the production of ROS (177). Calcium modulates different cytoskeletal elements and membrane dynamics in the growth cone. Their effects are pleiotropic on growth cone motility and depend on the concentration range. For example, normal motility requires optimal levels of calcium and higher or lower ranges of concentration inhibit axonal growth (61). Therefore, regulation of calcium levels is a major point of control during the axonal growth process. When the constitutively active form of Rac1 is injected into Aplysia neurons, the levels of calcium are elevated in the axonal growth cones in response to serotonin (177). This condition promotes the movement of microtubules and endoplasmic reticulum Ca2+ stores toward the peripheral domain of the axonal growth cone (177). The constitutively active form of Rac1 also promotes ROS formation in response to serotonin, while the dominant negative form has the opposite effect (177). Antioxidants completely blocked the increase of calcium release evoked by serotonin in the presence of the constitutively active form of Rac1, and conversely, the dominant negative form of Rac1 restored normal calcium release when a low concentration of hydrogen peroxide was added (177). The effect of ROS is dependent on IP3 receptors since blockade of these receptors with xestospongin C prevents calcium release in the presence of hydrogen peroxide (177), which is in line with other studies illustrating that ROS oxidize IP3 receptors, which induce calcium release (76) (Fig. 6).
In hippocampal neurons, the blockade of IP3 and ryanodine receptors impairs the establishment of neuronal polarity and diminishes axonal growth (164). Conversely, short stimulation of calcium release through ryanodine receptors by 4-CMC induces Rac1 activity and increases the axonal growth, while NOX2 inhibition blocks calcium release, Rac1 activity, and axonal growth (164). When NOX2 activity was induced by expressing p47phox in hippocampal neurons, the production of hydrogen peroxide increased and the axons of these neurons were 50% longer (164). This effect is reduced by the blockade of ryanodine receptors (164). This indicates that a part of the effects of NOX2 activity on axonal growth is mediated by calcium release through ryanodine receptors. Interestingly, the relationship between NOX2 activity and the calcium released through ryanodine receptors is bidirectional since the stimulation of these receptors with 4-CMC induces NOX2 activation dependent on Rac1 (164). This suggests a fundamental role of NOX2 promoting axonal growth and the establishment of neuronal polarity through calcium regulation (Fig. 6).
In developing cerebellar granule neurons, NOX2 activity promotes axonal growth since in NOX2 KO neurons, this process is slightly impaired (115). Besides, the expression of NOX2 is restricted to specific regions of the developing axon and developing dendrites. These regions correspond to growth cones, filopodia, and branching points and also correspond to the structures that accumulate the highest concentration of hydrogen peroxide (115). The production of hydrogen peroxide is restricted in a spatiotemporal manner since hydrogen peroxide production starts before the formation of the filopodium and this production persists only during the time the filopodium exists, suggesting that the high content of hydrogen peroxide might regulate these structures through regulation of the cytoskeleton (115). During the time the axon and dendrites are growing, ROS and NOX2 mRNA levels increase and afterward they decrease. In addition, during this time, glutathione depletion induces an aberrant formation of the axon, high accumulation of hydrogen peroxide, and cell death (115), suggesting that glutathione controls proper hydrogen peroxide signaling during axonal development.
These examples illustrate an intricate relationship between hydrogen peroxide signaling and regulation of cytoskeletal dynamics. The flavin monooxygenase activity of MICAL is essential to control the proper targeting of different axonal projections during development. MICAL promotes F-actin disassembly through direct oxidation of Met 44 and Met 47 (71), increasing the binding of cofilin (55). Furthermore, MICAL controls microtubule polymerization by oxidation of CRMP2. On the other hand, NOX2 regulates F-actin content in the transition zone and peripheral zone, which is probably mediated through regulation of Rac and Cdc42 activity, as well as by regulation of calcium content. It is interesting to note that all these elements are closely related. First, NOX2 activity regulates F-actin dynamics, and F-actin might control NOX2 activity by translocating p47phox and other cytoskeletal regulatory elements such as WAVE and PAK1 (142, 153) to the plasma membrane. Second, NOX2 activity controls calcium release from the endoplasmic reticulum, which induces Rac1 activity. Rac1 activity then promotes NOX2 activity.
Furthermore, it is possible that NOX2 negatively regulates actin polymerization in the axonal growth cone. For example, in another preparation, NOX2 can oxidize G-actin at Cys 374, inducing S-glutathionylation and preventing actin polymerization, which is reverted by actin deglutathionylation through Grx1 (131). Actin can be oxidized, S-nitrosylated, and S-glutathionylated at Cys 374 and oxidized at Met 44, 47, 178, 190, and 355, which affects G-actin polymerization and the stability and disassembly of F-actin (47). Finally, the effect of NOX2 on actin cytoskeleton might also be controlled by its localization, which could define the specific targets of hydrogen peroxide. Whether NOX2 is only expressed at the plasma membrane or it is expressed at endosomal vesicles has not been described.
Dendritic growth
Dendrites are specialized structures that receive signals from the axons and are the primary sites for synapse formation. The structure of dendrites is crucial for neuronal function and there are some e-studies demonstrating that the formation of these structures can be regulated by redox state. In cerebellar granule neurons, the developing dendrites accumulate a high content of hydrogen peroxide in their axonal growth cones (115). In sympathetic neurons, BMP-7 induces dendritic growth through NOX2 (25). Neurons treated with BMP-7 develop dendrites, whose complexity, measured by number, length, and branching points, was affected by antioxidant treatments and silencing of NOX2 and NOX inhibition (25).
Dendritic development is regulated by some trophic factors such as BDNF. It was reported that BDNF increases the levels of nitric oxide in the cytoplasm and the nucleus, which is completely inhibited by specific inhibition of the nitric oxide synthase neuronal (112). On the other hand, nitric oxide induces the S-nitrosylation of different proteins in the cytoplasm and the nucleus. In that regard, BDNF induces S-nitrosylation of histone deacetylase 2 (HDAC2) at Cys 262 and Cys 274, which triggers the dissociation of HDAC2 from chromatin, thereby facilitating the acetylation of histones H3 and H4 and the expression of CREB target genes (112). In cortical neurons, BDNF controls dendritic development through the S-nitrosylation of HDAC2 (112). When neurons are transfected with a mutant of HDAC2 that cannot be nitrosylated at Cys 262 and Cys 274, there is a decrease in the length of the dendrites and a slight decrease in dendritic branching even when dendritic growth is stimulated with depolarizing concentrations of potassium chloride (112). Since nitric oxide is detected in the nucleus in response to BDNF, S-nitrosylation of HDAC2 might occur directly through nitric oxide that freely diffuses into the nucleus; however, a series of transnitrosylation events of different proteins in the cytoplasm and the nucleus might conduct S-nitrosylation of HDAC2 (114). These studies demonstrate that dendritic growth is also regulated by reactive oxygen and nitrogen species.
Conclusion
The experimental evidence presented in this review sought to demonstrate that ROS and nitric oxide act as critical signals for the control of various physiological processes, particularly in different aspects of nervous system development. It remains clear that the levels of ROS are not the only aspect that determines their actions in these processes but also other factors, including the context of redox couples and localization of the produced ROS, are major determinants of their actions. These characteristics evidence the intricate control of redox signaling during nervous system development. Moreover, some studies also demonstrate that the interactions of ROS with target proteins may occur in different residues of the protein, which confers specificity of their signaling actions. Data shown here not only indicate that several proteins can be directly oxidized and regulated by ROS, but it is also clear that a fundamental mechanism point of control in the regulation of other proteins requires the participation of an oxidoreductase domain that is present in Prxs, Trxs, Grxs, and Nrx. Thus, ROS are crucial for the regulation of multiple processes during nervous system development, whose target proteins are diverse, as are the mechanisms by which they exert their actions.
Footnotes
Acknowledgments
This work was partially supported by grants from DGAPA-PAPIIT, UNAM, México (IN210716), and CONACYT, México (179234).