
Abstract
Significance:
The stress responsive transcription factor nuclear factor erythroid 2 p45-related factor 2, or NRF2, regulates the expression of many cytoprotective enzymes to mitigate oxidative stress under physiological conditions. NRF2 is activated in response to oxidative stress, growth factor signaling, and changes in nutrient status. In addition, somatic mutations that disrupt the interaction between NRF2 and its negative regulator Kelch-like erythroid cell-derived protein with CNC homology (ECH)-associated 1 (KEAP1) commonly occur in cancer and are thought to promote tumorigenesis.
Recent Advances:
While it is well established that aberrant NRF2 activation results in enhanced antioxidant capacity in cancer cells, recent exciting findings demonstrate a role for NRF2-mediated metabolic deregulation that supports cancer cell proliferation.
Critical Issues:
In this review, we describe how the NRF2-KEAP1 signaling pathway is altered in cancer, how NRF2 is regulated by changes in cellular metabolism, and how NRF2 reprograms cellular metabolism to support proliferation.
Future Directions:
Future studies will delineate the NRF2-regulated processes critical for metabolic adaptation to nutrient availability, cellular proliferation, and tumorigenesis. Antioxid. Redox Signal. 00, 000–000.
Introduction
N
The NRF2-KEAP1 complex
NRF2 levels are kept low in the absence of cellular stress through Kelch-like erythroid cell-derived protein with CNC homology (ECH)-associated 1 (KEAP1)-mediated NRF2 degradation by the proteasome. KEAP1 binds NRF2 as a dimer via the ETGE and DLG motifs of NRF2 (126) and recruits the E3 ubiquitin ligase CUL3, which ubiquitinates NRF2 leading to subsequent proteolysis. However, KEAP1 is oxidized by reactive oxygen species (ROS) at key cysteine residues under conditions of oxidative stress (133), causing a conformational change in the complex and loss of NRF2 ubiquitination (Fig. 1). Consequently, KEAP1 becomes saturated and newly translated NRF2 accumulates and translocates to the nucleus (8), where it heterodimerizes with small Maf proteins to bind to a specific antioxidant response element (ARE) in the promoters of target genes (52, 53). Binding of NRF2 to the ARE stimulates expression of enzymes necessary for detoxifying ROS, glutathione homeostasis, regeneration of NADPH, and other processes.
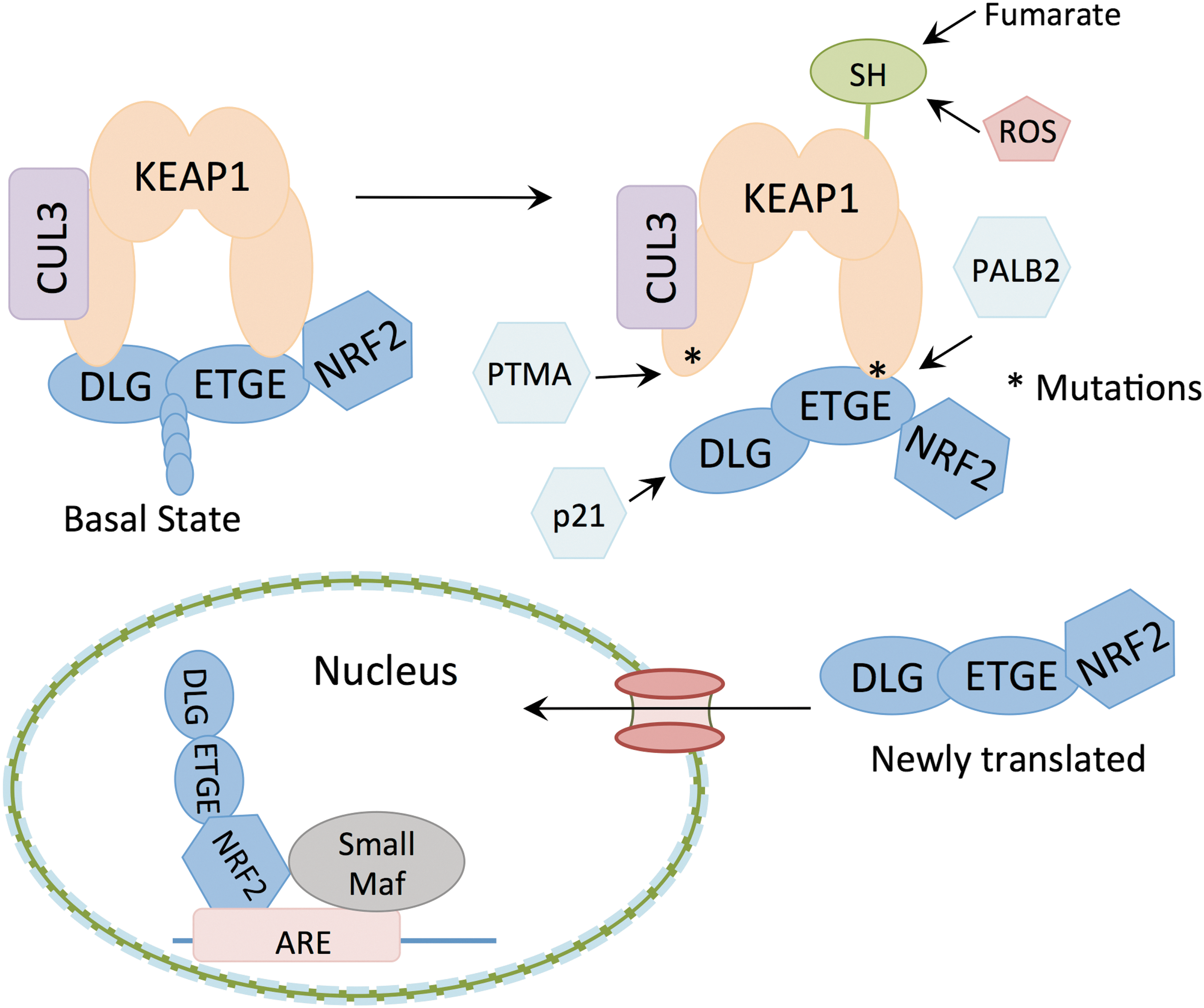
NRF2 is frequently deregulated in many human cancers due to the loss of NRF2-KEAP1 association. This is achieved through either mutation of NRF2 or KEAP1. NRF2 mutations cluster around the DLG (43% of mutations) or ETGE (57% of mutations) motifs, and result in diminished KEAP1 binding and ubiquitination. KEAP1 mutations are spread throughout the protein and result in loss of NRF2 degradation (38). NRF2/KEAP1 mutations occur frequently in certain cancer types, including lung squamous carcinoma where they are found in a third of cancer patients (19), and promote NRF2 activation and the growth of cancer cells (92). Accumulation of NRF2 in cancer cells is often a poor prognosis indicator, as NRF2 protects against oxidative stress and promotes anabolic metabolism, leading to a favorable environment for tumor growth. Furthermore, it has been shown that NRF2 serves as a protective agent against chemotherapy and radiotherapy (30, 114, 115). The importance of KEAP1 in controlling NRF2 activation is further highlighted by the early postnatal lethality of mice deficient in KEAP1, which present with elevated NRF2 target gene expression and NRF2-dependent hyperkeratosis of the esophagus and forestomach (134).
NRF2 activation by competitive binding
Activation of NRF2 is not limited to posttranslational modifications of KEAP1 or cancer mutations. NRF2 can be translocated to the nucleus due to competitive NRF2 or KEAP1 binding by other proteins. One such competitive binder is cyclin-dependent kinase inhibitor 1, or p21. The KRR domain of p21 binds the DLG motif of NRF2, thereby preventing KEAP1 from targeting NRF2 for proteasomal degradation (22). As a primary target for p53 activity, p21 is critical for mediating the cellular responses to p53 activation. p53 plays important functions in the cellular response to genotoxic stress; however, it is becoming more appreciated that p53 also plays a critical role in regulating metabolic homeostasis (12). Indeed, DNA damaging agents can promote NRF2 stabilization and transcriptional activity through p53-induced p21 expression (21). The role of the p21-NRF2 axis in the control of metabolism by p53 remains to be explored.
Another competitive KEAP1 binder is the autophagy adaptor sequestosome 1 (p62) (59). Autophagy is the catabolic process that cells undergo to break down proteins and intracellular organelles to maintain cellular homeostasis, remove buildup of toxic substances, and sustain metabolism in extreme instances. While in normal tissues autophagy serves to promote overall survival, autophagy can be both tumor promoting and tumor suppressive (106, 140, 142). It has previously been demonstrated that hypoxic tumor environments have higher basal autophagy levels than normal tissues (49), thereby promoting tumor cell survival, tumor growth, and metastasis. In a mouse model of K-Ras-driven lung cancer, autophagy deficiency caused by the deletion of Atg7 gene resulted in induction of p53, cell cycle arrest, and cell death (36). While the ability of autophagy to fuel tumor growth has been well established, recent data suggest that diminished autophagy may also promote tumorigenesis in certain contexts. p62 selectively flags targets for autophagic degradation but accumulates in situations of autophagy deficiency. The Keap1 interacting region of p62 competitively binds to KEAP1, thereby promoting NRF2 stabilization (59, 63). Indeed, liver-specific deletion of Atg7 leads to p62 accumulation, NRF2 activation, and hepatocellular carcinoma (HCC) (47). Furthermore, p62 deletion protects against liver tumorigenesis induced by carcinogens and diet, and ectopic p62 expression induces NRF2 activation and HCC (129). It is interesting to note that autophagy deficiency may be either tumor promoting or tumor suppressive, depending on the context, but the activation of NRF2 in both the liver, by autophagy deficiency, and the lung, by oncogenic MAPK signaling, is critical for tumor formation (27, 90). The effects of defective autophagy on NRF2 activation have not yet been explored in K-Ras mutant lung cancer.
Several recent findings have shed light on the interplay between the regulation of p62 by the nutrient status of the cell and activation of NRF2 by p62 (Fig. 2). First, p62 was found to be a key regulator of nutrient sensing in the mammalian target of rapamycin complex 1 (mTORC1) pathway (29). p62 interacts with mTOR and raptor in an amino acid-dependent manner and promotes the interaction of mTOR with Rag GTPases. This interaction was critical for the activation of mTORC1 by amino acids, but not other inputs such as insulin signaling. Second, it was shown that phosphorylation of p62 is critical for its interaction with KEAP1. Phosphorylation of p62 occurs on serine 351 in an mTORC1-dependent manner, which increases the affinity of p62 for KEAP1 (46). Importantly, expression of a phospho-mimetic S351E p62 mutant in HCC cells induced robust NRF2 activation and NRF2-dependent metabolic reprogramming (107). Collectively, these results establish that p62 is not only a signaling hub for mTORC1 activation but also regulated by mTORC1 to promote NRF2 activation. Furthermore, they suggest that NRF2 activation may respond to amino acid sufficiency.

However, studies have also shown that NRF2 responds to nutrient deprivation (60). Sestrin2, which inhibits mTORC1 signaling in the absence of adequate leucine, also promotes NRF2 activation by promoting KEAP1 degradation by p62 (7). Recently, the mechanism of this regulation was further refined in a study that identified that Sestrin2 promotes the association of p62 with ULK1. Following adenosine triphosphate (ATP) depletion, ULK1 was shown to induce significant phosphorylation of p62 in a Sestrin2-dependent manner (103). Furthermore, nutrient availability is not the only factor that regulates p62 phosphorylation by ULK1. ULK1 also phosphorylates p62 in response to proteotoxic stress to promote selective autophagic clearance of protein aggregates (69). The activation of NRF2 by p62 may serve very different functions in nutrient-replete conditions compared to nutrient-limited conditions, which remains to be explored. Furthermore, while these studies have focused on the role of p62-KEAP1 interaction in the cytoplasm, p62 may also play a nuclear role. p62 continuously shuttles from the nucleus to the cytoplasm (96). As NRF2 and KEAP1 also shuttle in and out of the nucleus via a KEAP1 and exportin-dependent nuclear export mechanism (122, 131), the regulation of this process by p62 and its phosphorylation is an interesting possibility.
Competitive KEAP1 binding can also occur in the nucleus. One such nuclear NRF2 activator is prothymosin alpha (PTMA), a small, ubiquitously expressed protein that promotes cellular proliferation and survival (51). This nuclear protein is abundant in tumor cells; however, the exact mechanism for its antiapoptotic properties is not fully understood. PTMA promotes NRF2 nuclear accumulation and transcriptional activity by competitively binding KEAP1 in the carboxyl-terminal half of KEAP1 (51). As described above, KEAP1 has been shown to shuttle from the cytosol to the nucleus (131) and PTMA may prevent NRF2 nuclear export by KEAP1. Similarly, the nuclear protein PALB2 has been shown to competitively bind KEAP1 (76). Like NRF2, PALB2 contains an extended ETGE-like KEAP1 binding motif, which competitively binds KEAP1 in the nucleus and prevents NRF2 from binding KEAP1, thereby promoting NRF2 nuclear accumulation and retention. NRF2 accumulation is also promoted by the ETGE motif containing metallopeptidase dipeptidyl peptidase 3 (DPP3), and Wilms tumor gene on X chromosome (WTX) (17, 39). The role of these proteins in NRF2 accumulation in cancer remains to be explored.
Regulation of NRF2 by Cellular Metabolism
NRF2 and PI3K signaling
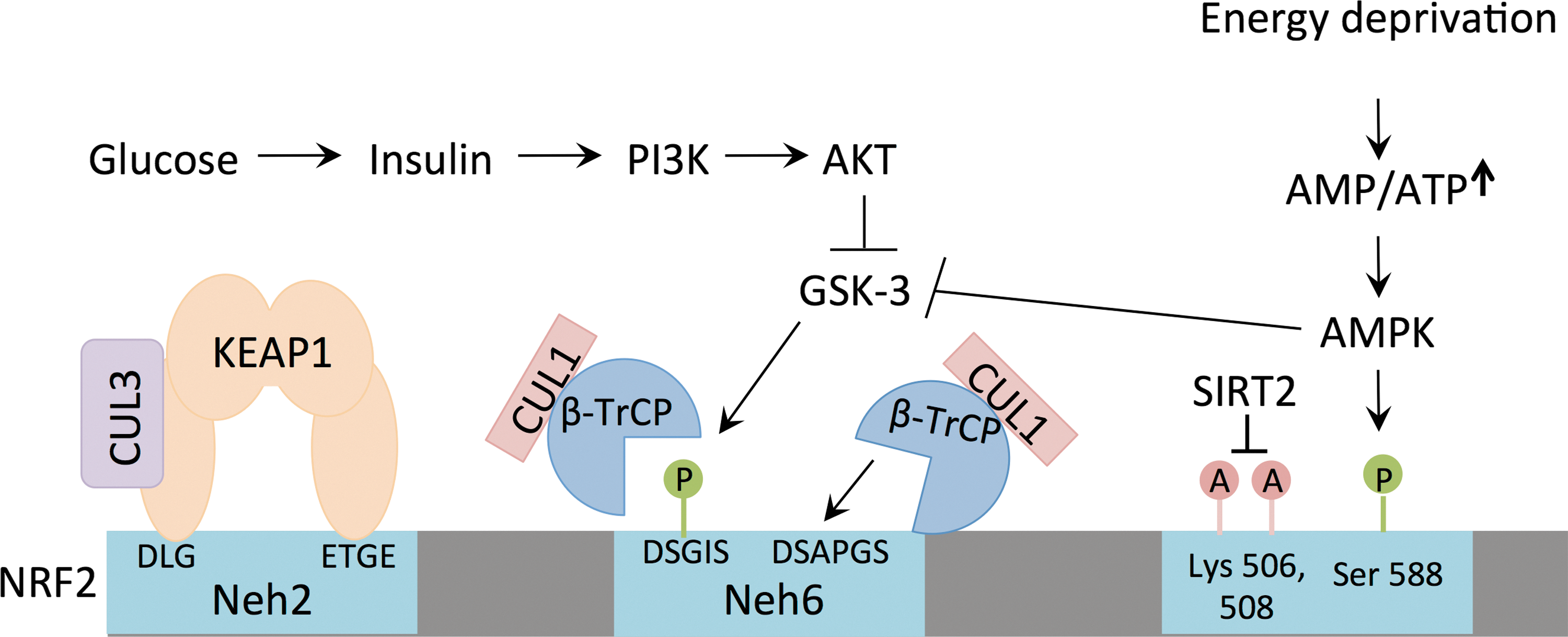
In addition to autophagy, the key insulin-responsive pathway in cells, the phosphoinositide-3-kinase (PI3K) signaling pathway, controls the regulation of NRF2 (132). In response to activation of the insulin or insulin-like growth factor receptor, PI3K catalyzes the phosphorylation of the lipid phosphatidylinositol 4,5-bisphophate (PIP2) to produce phosphatidylinositol 3,4,5-triphosphate (PIP3), a reaction that is reversed by the tumor suppressor phosphatase and tensin homolog (PTEN) (79). The generation of PIP3 is an important step in the activation of protein kinase B (also known as AKT) (33, 57), which mediates multiple downstream signaling events, including the inhibition of kinase glycogen synthase kinase-3 β (GSK3β) and transcription factor FOXO3a, and the activation of mTORC1 (132).
Although NRF2 turnover is generally thought to be mediated by engaging the KEAP1/CUL3 complex, GSK3β can also regulate NRF2 levels (Fig. 3). GSK3β phosphorylates NRF2 (108) to promote binding of beta-transducin repeat containing protein (βTRCP) followed by degradation by CUL1 (102). There are multiple lines of evidence that the PI3K pathway contributes to NRF2 activation in several contexts. First, PTEN inhibition has been shown to activate NRF2 in a KEAP1-independent manner. Forced activation of PI3K in the livers of mice due to PTEN deletion increased NRF2 nuclear accumulation and NRF2 target gene expression (85). Similarly, KEAP1- and PTEN-deficient livers demonstrate expansion of cholangiocytes, and combined loss of both PTEN and KEAP1 led to significantly greater NRF2 activation and dramatic cholangiocyte expansion, which is NRF2 dependent (123). Furthermore, NRF2 contributes to the tumorigenic potential of PTEN-deficient prostate cancer cells (105). Second, expression of an oncogenic AKT2E17K mutant promotes NRF2 activation and NRF2-dependent glutathione biosynthesis (68). Third, the protein levels of NRF2 in KEAP1-deficient mouse embryonic fibroblasts (MEFs) are sensitive to inhibitors of PI3K or AKT, suggesting that this pathway contributes significantly to NRF2 degradation (24). These studies demonstrate that the nutrient-responsive PI3K/AKT pathway regulates the stability of NRF2 in a KEAP1-independent manner.
NRF2 and the unfolded protein response
The unfolded protein response (UPR) also plays a key role in nutrient sensing (54). Glucose availability is a critical determinant for the efficiency of protein folding. Reductive protein folding in the ER requires a significant amount of energy, and glucose limitation reduces the amount of energy available for this process (54). Glucose is also required for protein glycosylation, which occurs concomitantly with protein folding. In response to defects in protein folding, cells activate the UPR, which is mediated by multiple kinases and transcription factors (54). The kinase PKR-like endoplasmic reticulum kinase/pancreatic eIF2a kinase (PERK) is critical for mediating the translation inhibition response of the UPR. Importantly, PERK also activates NRF2 by directly phosphorylating NRF2 to promote its dissociation from KEAP1 (25). Sestrin2 can also be transcriptionally induced by glucose starvation via the UPR through both NRF2 and ATF4 (28), which may contribute to NRF2 activation by glucose restriction. Because Sestrin2 and p62 are both transcriptional targets of NRF2, their induction may represent a feed-forward mechanism that promotes NRF2 activation and adaptation to starvation. Interestingly, ER stress does not always activate NRF2. The ER-resident E3 ubiquitin ligase Hrd1, which plays a role in the IRE1 branch of the UPR, mediates NRF2 ubiquitination in a KEAP1-independent manner (139). More work is needed to understand the differential regulation of NRF2 by the Hrd1 and PERK axes in response to ER stressors and nutrient availability.
NRF2 and AMPK
One of the most important pathways controlling energy homeostasis in the cell is the AMP-activated protein kinase (AMPK) pathway. In response to energy deprivation, the levels of ATP decrease and AMP increase, leading to the activation of AMPK (83). AMPK activation stimulates glucose and fatty acid uptake, increases flux through glycolysis and fatty acid β-oxidation, and activates autophagy, thereby increasing ATP levels and cell survival (83). This is accompanied by a concomitant suppression of ATP consuming processes, including fatty acid synthesis, gluconeogenesis, and protein translation (83). The activation of AMPK promotes NRF2 activation in multiple contexts (86, 148), and the mechanism for this interaction was delineated recently in a study that identified NRF2 phosphorylation on serine 558 by AMPK (50). Serine 558 is located in the nuclear export signal and Ser558-phosphorylated NRF2 demonstrates increased nuclear retention compared to unphosphorylated NRF2 (50). Because AMPK activation also promotes the phosphorylation and inhibition of GSK3β (44), which promotes the degradation of NRF2 via the βTRCP/CUL1 pathway, AMPK promotes NRF2 nuclear localization and stabilization through multiple mechanisms. These studies demonstrate that NRF2 activation is part of the cellular response to energy stress, and further studies are needed to examine the contribution of NRF2 to cellular adaptation and survival under nutrient-poor conditions.
Regulation of NRF2 by TCA cycle defects
In addition to the increase in NRF2 accumulation due to nutrient-responsive signaling pathways, it has recently been shown that NRF2 is activated by aberrant accumulation of tricarboxylic acid (TCA) cycle intermediates. Specifically, fumarate accumulation leads to an interruption in KEAP1-NRF2 binding (56). Fumarate covalently modifies KEAP1 at cysteine residues sensitive to modification by electrophilic compounds, thereby inducing a conformational change in KEAP1 and NRF2 stabilization (56). Mitochondrial fumarate hydratase (FH) catalyzes the reversible hydration of fumarate into malate during the TCA cycle and, interestingly, patients with hereditary leiomyomatosis and renal cell carcinoma (HLRCC) are born with heterozygous germ line mutations in the gene encoding FH (5). Loss of heterozygosity at the FH locus leads to an aggressive form of renal cancer known as type 2 papillary renal cell carcinoma (PRCC2) in HLRCC patients (94). Although it was initially hypothesized that hypoxia-inducible factor (HIF) may be the relevant driver of tumorigenesis in FH-deficient cells, renal cyst formation was found to occur independently of HIF activation (1, 95). These findings suggest that fumarate-mediated activation of NRF2 may instead provoke tumor formation in this context. The oncogenic role of NRF2 activation in PRCC is further supported by the significant frequency of both CUL3 and NRF2 mutations in this disease (93). However, the requirement of NRF2 for renal cyst formation in FH-deficient mice remains to be explored.
Interestingly, several NRF2 target genes have been shown to play a critical role in the viability and proliferation of FH-deficient kidney cancer cells. These include both heme oxygenase 1 (HMOX1) and ferritin genes (34, 55), although the mechanism and relatedness of these two genes are unclear. HMOX1 mRNA is induced in FH-deficient cells, and was found to be synthetic lethal with FH deficiency due to a failure to maintain mitochondrial NADH (34). Ferritin was also induced by FH deficiency in a NRF2-dependent manner, which promoted cellular proliferation (55). Furthermore, deletion of FH in the hearts of mice also results in NRF2 activation and promotes antioxidant response gene transcription and cardioprotection (6). While HMOX1 and ferritin genes are not solely regulated by NRF2, NRF2 activation may be part of the cells' natural response to fumarate accumulation, thereby promoting metabolic adaptation to support the function of the mitochondria by maintaining NADH and limiting iron accumulation.
NRF2 and SIRT2
Recently, NRF2 has also been shown to be modified following sirtuin-mediated changes in acetylation at the C-terminus of NRF2. Sirtuins are a class of proteins characterized by their NAD-dependent deacetylase or mono-ADP-ribosyltransferase activity (35). Sirtuins have been implicated in a variety of cellular processes, including cellular metabolism, inflammation, and tumorigenesis (35). Unlike other sirtuin family members, the function of the cytoplasmic member sirtuin 2 (SIRT2) is less well understood. A recent study, however, suggests that SIRT2 plays a role in cellular iron homeostasis through NRF2 signaling (143). Iron is required for many cellular processes required for proliferation, including oxygen transport, ATP synthesis, and DNA synthesis (14); however, free iron is also a source of injury and must be tightly regulated in the cell. Thus, the amount of free iron (Fe2+) in the cell is kept very low due to its ability to directly catalyze the generation of hydroxyl radicals via the Fenton reaction. The hydroxyl radical is significantly more reactive than other ROS such as superoxide and hydrogen peroxide and can cause significant damage to lipids, DNA, RNA, and protein (127). In excess, this can lead to cellular death via apoptosis or an iron-dependent form of death known as ferroptosis. SIRT2 homozygous knockout MEFs were found to contain significantly less nonheme iron than wild-type MEFs and restoration of iron levels was dependent on the deacetylase activity of SIRT2. SIRT2 was also found to inhibit the nuclear localization and activation of NRF2, and NRF2 mediated the regulation of iron content downstream of SIRT2 through transcription of ferroportin-1 (FPN1). SIRT2 directly deacetylates NRF2 at lysines K506 and K508, thereby promoting NRF2-KEAP1 association and NRF2 degradation (143). The activation of NRF2 in response to iron is not well studied, and may represent a major part of the cellular adaptation to changes in cellular iron that extends beyond regulation by SIRT2.
NRF2 Directs Anabolic Metabolism in Cancer
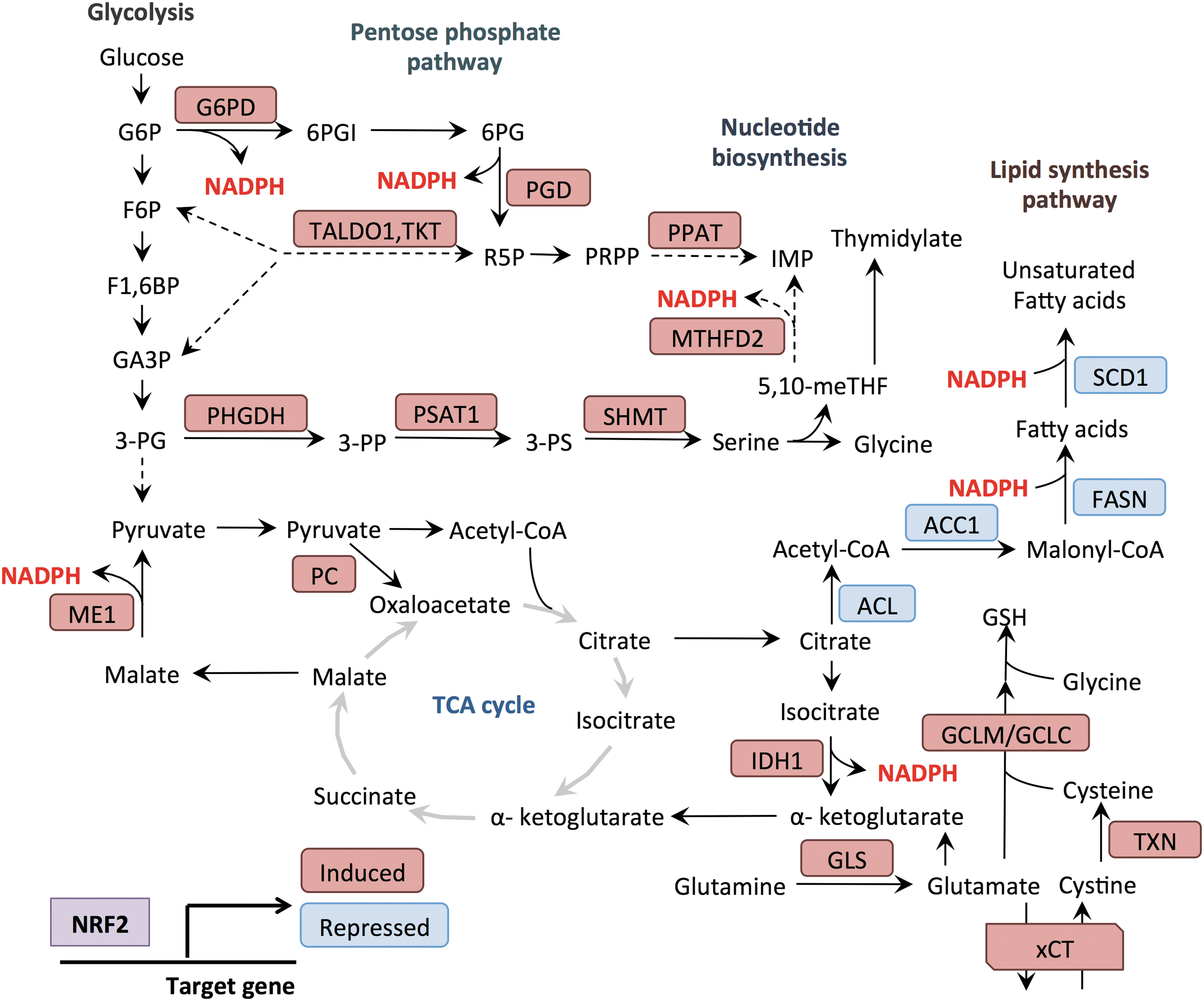
In the above section, we have described how NRF2 is activated by a variety of metabolic changes in cells. In this section, we discuss how NRF2 promotes anabolic metabolism to meet the metabolic demands for cellular proliferation. Proliferating cells require a constant supply of biosynthetic precursors for the generation of proteins, lipids, and nucleotides to facilitate their growth and division. Recent studies have demonstrated that NRF2 induces metabolic reprogramming by regulating the expression of metabolic enzymes, thereby directing metabolic intermediates toward anabolic processes (Fig. 4).
NRF2 and the PPP
Glucose is the major source of cellular energy and its metabolites serve as substrates for biosynthetic processes. Glucose enters the cells via glucose transporters and is metabolized through glycolysis to pyruvate, while also providing other metabolic intermediates that are diverted into other biosynthetic pathways, such as the PPP. The PPP consists of two branches: the oxidative branch and the nonoxidative branch (97). These two branches provide metabolic flexibility and serve to adapt to the changing demands of the cell for both ribose-5-phosphate and NADPH. The oxidative branch consists of three irreversible reactions and is inhibited by NADPH (65). Entry into the oxidative arm is catalyzed by glucose-6-phosphate dehydrogenase (G6PD), which metabolizes the glycolytic intermediate glucose-6-phosphate to 6-phosphogluconolactone. The nonoxidative arm consists of multiple reversible reactions and serves to funnel intermediates between glycolysis and the PPP (97). The reversible nature of this arm allows cells with a high demand for nucleotides to synthesize adequate ribose-5-phosphate (R5P), even in the presence of elevated NADPH. R5P contributes the sugar group to nucleotides, and ultimately forms the sugar backbone in DNA. R5P is metabolized to phosphoribosyl-pyrophosphate (PRPP) by PRPP synthase, and PRPP is used for both purine and pyrimidine nucleotide syntheses, as well as nucleotide salvage pathways. Purine nucleotide de novo synthesis begins with PRPP, and the purine ring is built through subsequent reactions incorporating glycine, two 10-formyl THF, two glutamine, and one aspartate molecules. In contrast, pyrimidine nucleotide synthesis starts from carbamoyl phosphate and aspartate, and PRPP is incorporated later on formation of the pyrimidine ring structure.
NRF2 promotes nucleotide synthesis in multiple ways. First, it controls the direct transcriptional regulation of the PPP. NRF2 directly regulates the expression of the enzymes of the oxidative branch of the PPP, including G6PD as well as phosphogluconate dehydrogenase (PGD) (85, 116). NRF2 also regulates the nonoxidative branch through the transcriptional regulation of transketolase and transaldolase 1 (TALDO1) (85, 116). By regulating both branches, NRF2 can simultaneously promote the production of R5P for nucleotide synthesis and NADPH production for antioxidant defense and biosynthetic reactions. Beyond the regulation of PPP reactions, NRF2 supports purine base synthesis by the indirect regulation of phosphoribosyl pyrophosphate amidotransferase (85), which catalyzes the addition of a nitrogen group from glutamine onto PRPP, thus initiating the creation of the purine ring. NRF2 also indirectly regulates the transcription of methylenetetrahydrofolate dehydrogenase 2 (MTHFD2) (85), a folate cycle enzyme that generates 10-formyl THF, two molecules of which are incorporated at positions C-2 and C-8 in the purine ring. This coordinate regulation of R5P production and downstream steps facilitates the production of nucleotides by NRF2.
NRF2 and amino acid metabolism
Cysteine metabolism
NRF2 coordinates the metabolism and intracellular concentrations of cysteine (Fig. 5). Cells obtain cysteine from either de novo synthesis or uptake from the extracellular space via amino acid transporters (66). De novo cysteine synthesis occurs from methionine and serine through the reverse transsulfuration pathway (82, 141). In the first step of de novo cysteine synthesis, cystathionine b-synthase catalyzes the synthesis of cystathionine from homocysteine and serine (82). Cystathionine is subsequently hydrolyzed by cystathionine γ-lyase to produce cysteine, α-ketobutyrate, and ammonia (82). We and others have observed that the transsulfuration pathway only slightly contributes to intracellular cysteine pools in cultured cells (26, 82); however, its contribution to tumor cysteine pools in vivo remains to be determined. Indeed, methionine can contribute significantly to cysteine synthesis in tissues and cysteine is considered a nonessential amino acid in vivo due to its production via the transsulfuration pathway (120).
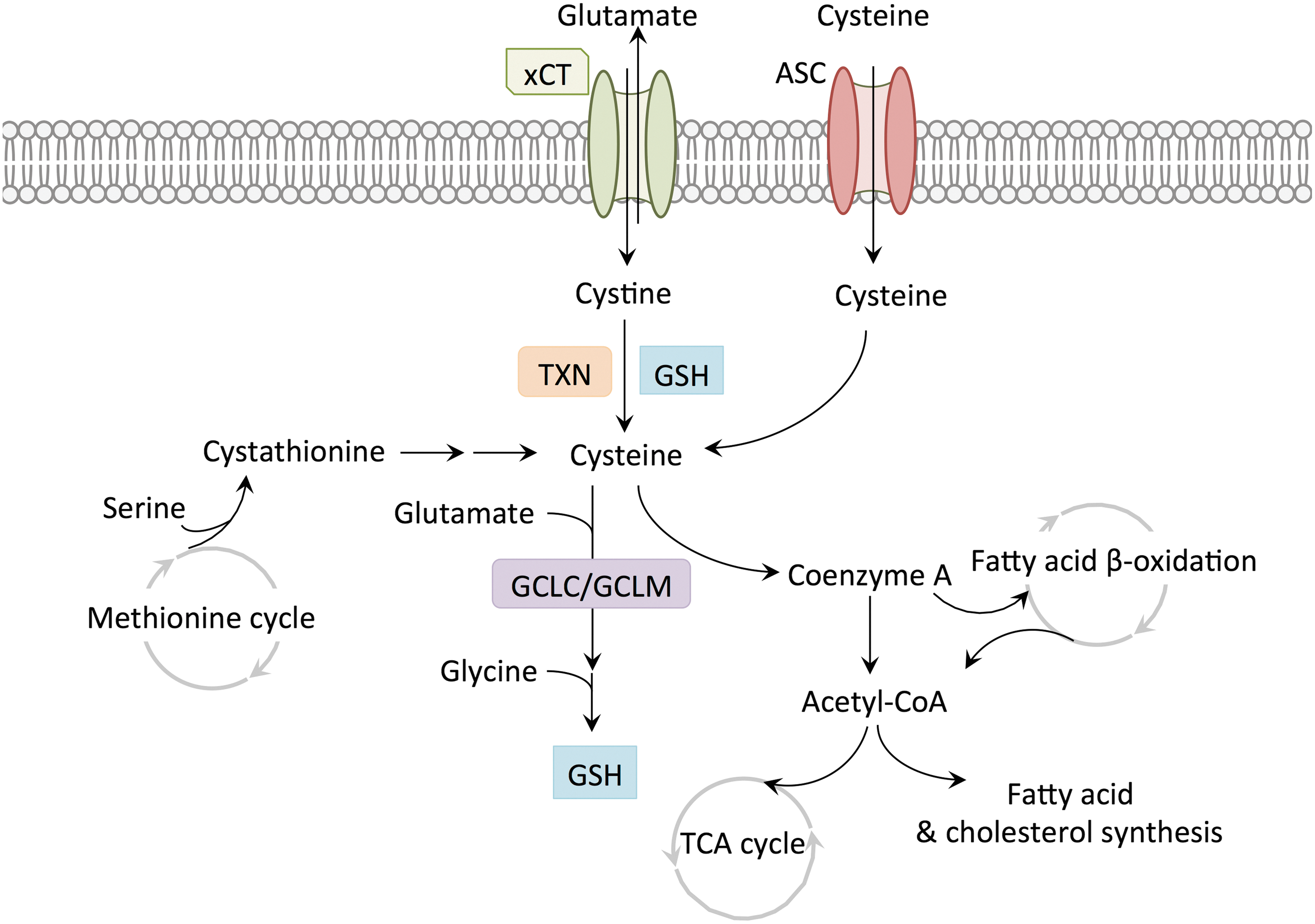
Cysteine uptake occurs via the alanine–serine–cysteine (ASC) amino acid transport system in its reduced form or via the cystine/glutamate antiporter system xc − in its oxidized form, cystine. NRF2 regulates the expression of xCT, the light chain subunit of xc −, through binding to the antioxidant/electrophile response element (ARE/EpRE) within the xCT promoter, thereby regulating the levels of intracellular cysteine (109). Within the cell, cystine is reduced to cysteine by the glutathione or thioredoxin antioxidant systems, which are transcriptionally controlled by NRF2. Through the regulation of both xCT and these antioxidant programs, NRF2 may facilitate cysteine uptake through multiple mechanisms. Glutathione (GSH) is a potent antioxidant in cells and reduces ROS through the reactions catalyzed by glutathione peroxidase and glutathione S-transferase. GSH is a tripeptide and synthesized by conjugation of cysteine and glutamate by the action of glutamate/cysteine ligase (GCL) (84), and the subsequent conjugation of glycine by glutathione synthetase. GCL consists of a catalytic subunit (GCLC) and a modifier subunit (GCLM), and NRF2 transcriptionally regulates both GCLC and GCLM genes (87, 149). The oxidized GSH is reduced by glutathione reductase. Importantly, the availability of cysteine is the rate-limiting factor of GSH biosynthesis (73). Cystine to cysteine conversion is also achieved by the thioredoxin (TXN) system where TXN reduces the cystine disulfide bonds and thioredoxin reductase (TXNRD1) reduces oxidized TXN using NADPH as an electron donor. Both TXN and TXNRD1 are transcriptional targets of NRF2 (40, 80). By regulating both the transport and metabolism of cystine, NRF2 increases the availability of cysteine for downstream processes.
NRF2 can also indirectly regulates cystine transport. The NRF2 target activating transcription factor 4 (ATF4) induces expression of xCT via the amino acid response element sequence in the xCT promoter (110). ATF4 is a transcription factor that plays a critical role in the cellular response to multiple stresses, including amino acid deprivation. Following amino acid deprivation, the accumulation of uncharged tRNAs leads to the activation of the kinase general control nonderepressible 2, which phosphorylates eukaryotic initiation factor 2α (eIF2α) (137), leading to general translation inhibition but enhanced translation of ATF4 (37). However, the upregulation of ATF4 is not limited to stress conditions. The translation of ATF4 is regulated by mTOR in an eIF2α-independent manner (11). Furthermore, NRF2 regulates ATF4 through transcriptional upregulation in NRF2/KEAP1 mutant nonsmall-cell lung cancer cells (26). In addition, it has been demonstrated that NRF2 and ATF4 can cooperatively regulate the expression of xCT in T24 human bladder carcinoma cells on proteasome inhibition (146).
Cysteine can be metabolized by several pathways to form GSH, pyruvate, taurine, and coenzyme A (CoA) (118). While the importance of cysteine as a biosynthetic precursor of GSH is well understood, the roles of other cysteine-consuming processes during tumorigenesis are underinvestigated. CoA is synthesized from cysteine, pantothenic acid (vitamin B5), and ATP. CoA reacts with carboxylic acid and functions as the major acyl group carrier in intermediary metabolism (112). Acetyl-CoA provides carbon to the TCA cycle, which is the central process in energy metabolism, and to the fatty acid synthesis pathway. As pyruvate enters the TCA cycle, it is decarboxylated to acetyl-CoA by pyruvate dehydrogenase. Following condensation with oxaloacetate to form citrate, this acetyl group enters the TCA cycle to produce reducing equivalents in the form of NADH and FADH2 for ATP synthesis by the electron transport chain. CoA is regenerated by citrate synthetase to accept another acetyl group from pyruvate dehydrogenase. Fatty acid β-oxidation is also dependent on CoA molecules for activation and cleavage (20). Cytoplasmic long-chain fatty acids must be converted into fatty acyl-CoA molecules before being transported into the mitochondria by the carnitine shuttle (20). Within the mitochondrial matrix, fatty acyl-CoA undergoes repeated cleavage of two-carbon units, which combine with CoA molecule to produce acetyl-CoA, which enters the TCA cycle (20). Thus, CoA is a critical molecule for the activity of the TCA cycle and ATP production in the mitochondria.
CoA also participates in the synthesis of both lipid families, fatty acids and cholesterol (88, 104). Lipid synthesis is an essential anabolic process by which cancer cells synthesize cell membranes and store energy. However, mitochondrial acetyl-CoA is not available for biosynthesis reactions, which take place in the cytosol. Therefore, mitochondria must export citrate to the cytosol for the generation of cytosolic acetyl-CoA by ATP citrate lyase (ACL) (10). Fatty acid and cholesterol synthesis both start from cytosolic acetyl-CoA. In the first step of the fatty acid biosynthesis pathway, acetyl-CoA carboxylase (ACC1) converts acetyl-CoA to malonyl CoA, and the sequential elongation of two-carbon units by fatty acid synthase (FASN) generates palmitate, which in turn is modified by elongases and desaturases forming various saturated and unsaturated fatty acids (104). Fatty acid biosynthesis is a highly NADPH-consuming process. Cholesterol biosynthesis is initiated by hydroxymethylglutaryl-CoA (HMG-CoA) synthase, which catalyzes the condensation of acetyl-CoA and acetoacetyl-CoA, and the resulting HMG-CoA is reduced to mevalonate and undergoes a series of NADPH-dependent reactions to generate cholesterol, as well as intermediates for the synthesis of heme, ubiquinone, and protein prenylation (88). Thus, CoA is a critical molecule for both catabolic and anabolic processes in proliferating cells. By increasing the availability of cysteine for CoA biosynthesis NRF2 may also support CoA-dependent metabolic processes in cells, and warrants further study. Furthermore, the relative contribution of cysteine to CoA biosynthesis in cancer versus normal tissues is unknown.
Serine and glycine metabolism
The metabolism of serine and glycine makes profound contributions to several metabolic processes in cancer (Fig. 6). These amino acids serve as precursors for the synthesis of many macromolecules, including cysteine, choline, creatine, bile salts, porphyrins, glutathione, and others (71). Furthermore, serine and glycine provide one-carbon units to folate cycle intermediates, which support purine and thymidine syntheses, and the methionine cycle (71). Cells can modulate nucleotide metabolism, NADPH levels, methylation reactions, and sulfur metabolism through the activity of these cycles (71). Cells obtain serine and glycine through their uptake via amino acid transporters or de novo synthesis. De novo synthesis occurs from the glycolytic intermediate, 3-phosphoglycerate (3-PG), which is metabolized by phosphoglycerate dehydrogenase (PHGDH) to form 3-phosphohydroxypyruvate (3-PP). Subsequently, phosphoserine aminotransferase 1 (PSAT1) converts 3-PP to 3-phosphoserine (3-PS) in a transamination reaction that requires glutamate as the amino group donor (141). 3-PS is then hydrolyzed, generating serine (141). Metabolism of serine to glycine, which is catalyzed by serine hydroxymethyltransferases (SHMT1 and 2), transfers one-carbon units to tetrahydrofolate, resulting in the formation of 5,10-methylene tetrahydrofolate (5,10-meTHF) (141). Subsequently, 5,10-meTHF is used to produce thymidylate (141). In addition, methylenetetrahydrofolate reductase (MTHFR) converts 5,10-meTHF to 5-methyl-THF, which transfers its methyl group to the methionine cycle, producing S-adenosylmethionine (SAM) (141). Alternatively, methylenetetrahydrofolate dehydrogenases 1 and 2 (MTHFD1 and MTHFD2) convert 5,10-meTHF into 10-fomylTHF, which is incorporated into purines (141). Glycine can also donate one-carbon units to the folate cycle via the glycine cleavage system in the mitochondria, and is incorporated into the purine ring (141).
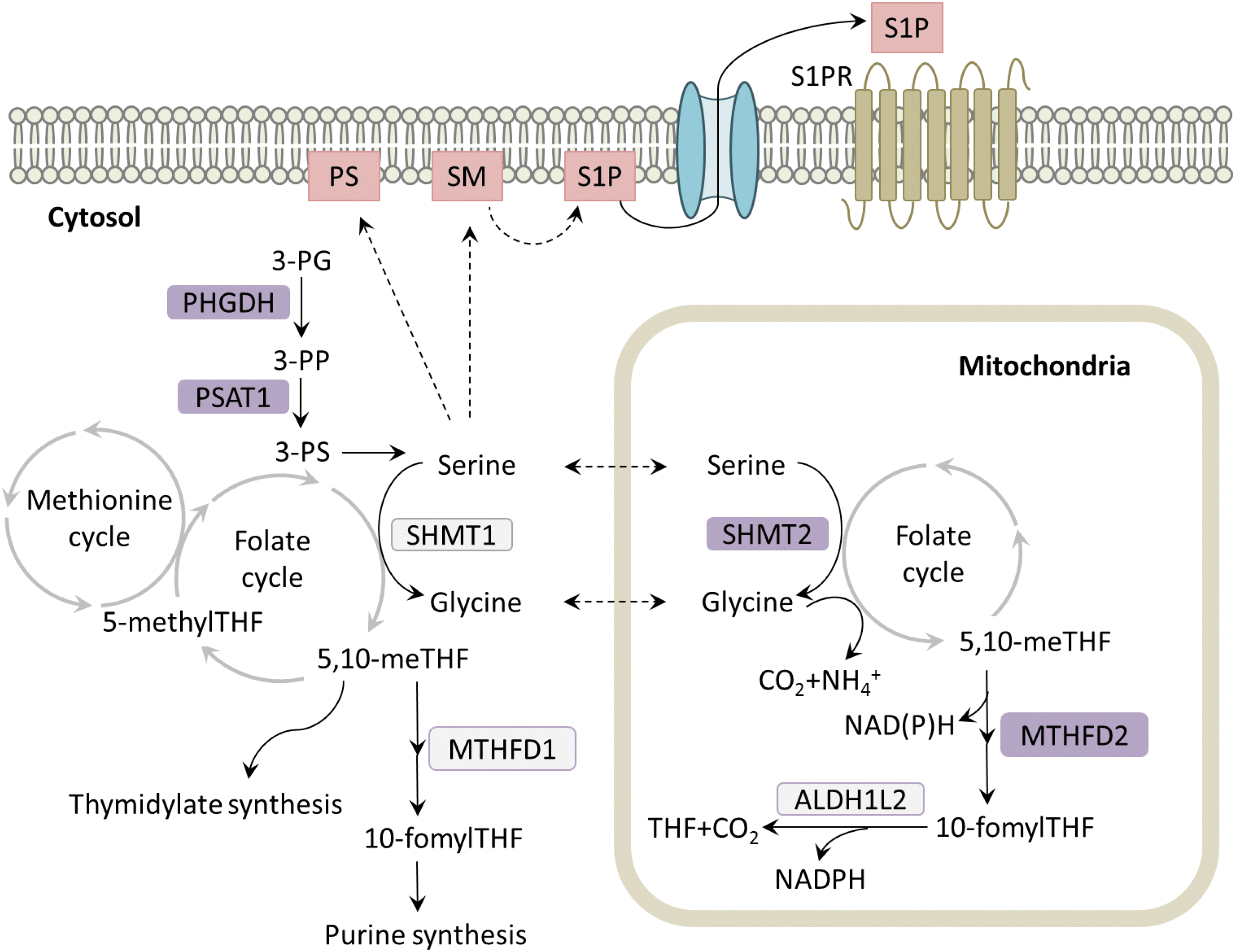
Recently, we found that NRF2 regulates the expression of the serine/glycine biosynthesis enzymes PHGDH, PSAT1, and SHMT2, through ATF4 (26). By profiling a large panel of nonsmall-cell lung cancer cell lines for serine biosynthesis activity by 13C-glucose metabolite tracing, we identified that KEAP1/NRF2 mutant cell lines displayed a significant capacity for serine and glycine biosynthesis and an independence from exogenous sources of these metabolites (26). Serine and glycine production by NRF2 via ATF4 was used to support glutathione biosynthesis, purine biosynthesis, thymidine production, and NADPH levels (26). Our study joins a growing body of literature regarding the importance of serine and glycine metabolism for tumor growth. Indeed, the first and rate-limiting enzyme in serine biosynthesis, PHGDH, is commonly amplified in melanoma and breast cancer (72, 100). Our findings corroborate multiple other studies demonstrating that serine supports one-carbon metabolism and the methionine cycle of cancer cells, thereby supporting glutathione production (77), nucleotide production (62), and NADPH production via the folate cycle (32, 144). Interestingly, serine has also been shown to influence the levels of SAM for DNA and RNA methylation (78), which was not influenced by NRF2 in lung cancer cell lines (26).
The implications of the NRF2-serine biosynthesis pathway for cellular proliferation may extend beyond glutathione and nucleotides. Serine is also required for production of membrane lipids, including phosphatidylserine (PS) and sphingolipids. PS is synthesized at the mitochondrial-associated membrane (MAM), a specialized region of endoplasmic reticulum that is enriched in enzymes involved in lipid metabolism (119). PS synthase 1 and 2 catalyze the displacement of the choline and ethanolamine groups of phosphatidylcholine and phosphatidylethanolamine, respectively, by serine to form PS (119). Once PS is generated, it is transported from the MAM to the plasma membrane and is predominantly localized to the inner leaflet (64). The negative charged head group of PS interacts with the polycationic domain of intracellular proteins, such as protein kinase C and phospholipases C and D, leading to their membrane recruitment and activation. While the inner leaflet membrane placement of PS promotes intracellular signaling, PS localization to the exterior leaflet of the plasma membrane is a classic feature of cellular apoptosis. PS externalization serves to flag dying cells for phagocytosis (111), but can occur in viable cells as well. In the tumor microenvironment, tumor cells and blood vessel endothelial cells express PS on the exterior leaflet of the plasma membrane to facilitate immune evasion (13). Interaction of immune cells with PS-expressing tumor cells induces the secretion of anti-inflammatory cytokines such as IL-10 and TFG-β, thereby creating a local immunosuppressive environment (13). Moreover, TGF-β prevents dendritic cell maturation, and the resulting immature dendritic cells fail to express costimulatory molecules required for proper antigen presentation. In addition, chemo- and radiotherapy increase PS exposure in tumors. The contribution of NRF2 and serine to PS metabolism and downstream processes remains to be explored.
Serine also contributes to sphingolipid synthesis. Sphingolipid metabolites function as signaling molecules, and sphingosine-1-phosphate (S1P) signaling promotes tumor growth and survival. De novo synthesis of sphingolipids starts at the ER with the condensation of serine and palmitoyl-CoA by serine palmitoyltransferase, which is the rate-limiting step in sphingolipid synthesis (15). Ceramide is generated in several subsequent reactions and transported to the Golgi apparatus from the ER. Ceramide can be further converted to sphingomyelin and glycosphingolipid, which are then transported to and incorporated in the plasma membrane (15). Plasma membrane sphingomyelin is degraded to sphingosine, which is then phosphorylated by sphingosine kinase (SK) to form S1P (101). S1P is released from the cells via transporter proteins. In the extracellular compartment, S1P can bind to five S1P receptors, which are G protein-coupled receptors, in both autocrine and paracrine manners, thereby initiating several signaling pathways involved in regulating the growth and migration of cancer cells (61). Importantly, several human cancers exhibit increased expression of SK1, which is correlated with tumor grade and reduced patient survival (101). While the role of NRF2-dependent serine production in the biosynthesis of PS and sphingolipids and in downstream processes remains to be firmly established, NRF2-regulated serine/glycine metabolism may not only confer independence of extracellular serine to tumor cells but may also shape diverse protumorigenic processes such as oncogenic signaling and immune evasion.
Glutamine metabolism
Glutamine plays critical roles in cancer metabolism as a source of both nitrogen and carbon. Glutamine is transported into cells through numerous transporters such as the ASCT2 antiporters or SNAT cotransporters (99). Glutamine is the most abundant amino acid in the circulation and glutamine consumption by cancer cells in culture is very high (31). Cancer cells utilize glutamine to fuel TCA cycle anapleurosis. In mitochondria, glutaminase converts glutamine to glutamate. NRF2 regulates the metabolism of glutamine in several ways. First, NRF2 regulates the expression of glutaminase, which converts glutamine to glutamate in the mitochondria (2). Glutamate can be further metabolized by glutamate dehydrogenase or aminotransferases to α-ketoglutarate, which enters the TCA cycle. NRF2 regulates the expression of malic enzyme 1 (ME1), which generates pyruvate and NADPH from cytoplasmic malate, as well as the expression of isocitrate dehydrogenase 1 (IDH1), which generates α-ketoglutarate and NADPH from cytosolic isocitrate, both derived from the TCA cycle (41). Thus, NRF2 activation may confer a greater dependence on glutamine anapleurosis due to the metabolism of TCA cycle intermediates by these alternative pathways. In addition to glutaminase, the anapleurotic enzyme pyruvate carboxylase, which replenishes oxaloacetate from pyruvate, has been identified as an NRF2 regulated gene (43), although the contribution of this enzyme to NRF2-regulated metabolism has not been established. Interestingly, glutamate production from glutamine may also serve to support glutathione production by increasing the availability of glutamate for GCLC, similar to cysteine and glycine as described above. Indeed, treatment of KEAP1 mutant A549 cells with NRF2 siRNA decreases the incorporation of glutamate into glutathione (85).
Beyond its anapleurotic role, glutamine supports multiple anabolic processes in cells. Glutamine can contribute to the biosynthesis of both purines and pyrimidines by supplying both carbon and nitrogen atoms. Glutamine provides the amide group in three and two enzymatic steps of the purine and pyrimidine biosynthesis pathways, respectively. Glutamine can also indirectly provide the carbon skeleton for purine and pyrimidine synthesis through aspartate, which can be generated by transamination of oxaloacetate that is produced, through several steps of the TCA cycle, from glutamine-derived α-ketoglutarate.
More than half of the dry cellular mass is protein; consequently, protein synthesis represents a significant consumer of cellular amino acid pools (45). The mTOR complex is a major regulator of protein translation as well as a cellular sensor of amino acid availability. Both glutamine and leucine regulate the activity of mTORC1, however, they do so through different mechanisms. While glutamine activates mTORC1 through ADP ribosylation factor 1, leucine induces mTORC1 activation through Rag guanosine triphosphatases (GTPases) (48). NRF2 controls amino acid availability through ATF4 expression, which regulates de novo amino acid biosynthesis as well as the transcription of amino acid transporters such as ASCT2 and LAT1 (135, 136). By inducing ATF4, NRF2 may promote mTORC1 signaling by increasing glutamine uptake, which remains to be explored. Glutamine can directly activate mTOR, or promote leucine uptake via LAT1, which is an obligate exchanger that utilizes glutamine (91). Recently, it was demonstrated that NRF2 promotes the translation of mRNAs by regulating the intracellular redox state (23). The regulation of amino acid availability and mTORC1 activity by the NRF2-ATF4 pathway to further support translation is an interesting possibility.
Asparagine metabolism
Asparagine is synthesized de novo from aspartate and glutamine via a transamidation reaction that is catalyzed by asparagine synthetase (ASNS) (9). We observed that NRF2 controls the binding of ATF4 to the ASNS promoter (26), suggesting that NRF2 regulates the cellular capacity for ASNS. Synthesis of asparagine can provide an advantage to tumor cells under conditions in which the supply of nutrients is limited due to inadequate vascularization and perfusion. Importantly, ATF4 expression is required for cell survival in the absence of nonessential amino acids (145). Asparagine and ASNS are capable of rescuing the survival of cells following ATF4 depletion, arguing that asparagine is the key effector of ATF-dependent amino acid homeostasis under starvation conditions (145). However, asparagine is not metabolized to other metabolites within cells (128). Why is asparagine so critical under starvation conditions? The answer may be that cells possess an asparagine sensor that determines whether cells undergo adaptation or apoptosis under starvation. Thompson and colleagues found that asparagine was sufficient to suppress the glutamine depletion-induced apoptosis without replenishing TCA cycle intermediates or other nonessential amino acids (147). Thus, the regulation of the ATF4-ASNS pathway by NRF2 may represent a prosurvival pathway that promotes tumor adaptation to starvation.
NADPH synthesis
NADPH is the reduced form of nicotinamide adenine dinucleotide phosphate (NADP+) and is required as a coenzyme for various anabolic reactions such as lipid (fatty acid and cholesterol) and dNTP synthesis. For example, synthesis of one molecule of the fatty acid palmitate requires 14 molecules of NADPH and cholesterol biosynthesis requires 26 molecules (75). Furthermore, the reduction of nucleotide triphosphates to deoxynucleotide triphosphates by ribonucleotide reductase utilizes NADPH as the ultimate electron donor (113). NADPH is also consumed in the production of proline, with two molecules of NADPH consumed per proline molecule produced from glutamate (3). Indeed, an analysis of NADPH consumption in proliferating cells revealed that fatty acid metabolism demonstrates the highest NADPH consumption, followed by dNTP synthesis and then proline synthesis (32).
Therefore, availability of NADPH is important for biosynthesis pathways that are required for growth, and NADPH production is thought to be rate limiting for proliferation (130). NRF2 supports the maintenance of the cellular NADPH pool by upregulating NADPH-generating enzymes while suppressing the NADPH-consuming processes (41, 138). NRF2 activates the cytosolic NADPH production through regulation of four main enzymes: G6PD and PGD in the oxidative arm of PPP; IDH1, which catalyzes the oxidative decarboxylation of isocitrate to α-ketoglutarate; and ME, which metabolizes malate to pyruvate. To conserve NADPH for antioxidant functions, NRF2 downregulates the expression of genes encoding lipogenic enzymes, including ACL, ACC1, FASN, and stearoyl‐CoA desaturase (SCD1), to suppress NADPH consumption by the lipogenic process (138). NRF2 can also contribute to mitochondrial NADPH production via the folate cycle (26). Oxidation of 5,10-meTHF to 5,10-methenyl THF by the NRF2-regulated enzyme MTHFD2 contributes to NADPH production in the mitochondria, although whether it is direct or through the activity of mitochondrial enzyme ALDH1 L2 is still unclear (32, 67, 141). Collectively, these NRF2-regulated processes support NADPH generation, although the inhibition of lipid synthesis may be a barrier to proliferation that is relieved through the mutations of other oncogenes or tumor suppressors during tumorigenesis.
Iron metabolism
As described in the introduction, NRF2 plays a key role in cellular iron homeostasis. Due to its potential to cause oxidative injury, free iron levels are kept very low. However, cells need iron for proliferation, so the uptake and storage of iron are tightly regulated. First, NRF2 controls the storage of iron through the regulation of ferritin. NRF2 regulates the expression of ferritin heavy and light chains (FTH1 and FTL) (98), which immediately bind free iron on its entry into the cell, thereby preventing its reaction with oxygen. Furthermore, NRF2 induces the expression of the iron export FPN1 (89, 125), which is negatively regulated by the ARE-binding transcriptional repressor Bach1. Consistent with this observation, Bach1−/− mice demonstrate iron deficiency anemia under conditions of iron depletion (58). NRF2 also promotes the transcription of HMOX1 (4), and the iron released by HMOX1 promotes the translation of FPN1 (81). Furthermore, NRF2 has been shown to play an important role in the resistance to ferroptosis (121), suggesting that the regulation of iron homeostasis is an important component of the NRF2 transcriptional program.
NRF2 also regulates the turnover of heme. Heme is required for electron transport, oxygen transport, ROS detoxification, signaling, and other functions. NRF2 controls the expression of heme oxygenase (4), which metabolizes heme to iron and biliverdin. NRF2 also regulates biliverdin reductase to metabolize biliverdin to bilirubin (138), which is excreted as waste. Paradoxically, NRF2 also regulates heme biosynthesis. The first step in heme biosynthesis requires amino acid glycine, the production of which is NRF2 regulated through the ATF4-serine biosynthesis pathway (26). Furthermore, ferrochetalase is a direct NRF2 target gene that is responsible for the final step in heme biosynthesis (18). Heme turnover is critical for the cellular response to stress (124) and the dual regulation of synthesis and degradation likely plays an important role in both the cellular protective function and metabolic regulation by NRF2.
The regulation of iron metabolism by NRF2 may also play an important role in cellular signaling. The iron-binding protein pirin is a direct transcriptional target of NRF2 (16) and regulates the NFkB pathway (70). Although the regulation of pirin by NRF2 has not been shown to play a role in iron metabolism, pirin modulates cellular metabolism through the inhibition of pyruvate dehydrogenase in Serratia marcescens (117). Although this function has not been demonstrated in mammalian cells, it raises the interesting possibility that pirin is a cellular sensor that modulates cellular metabolism in response to free iron and/or oxygen levels.
Conclusions and Future Directions
NRF2 is activated by many nutrient-sensing pathways, including the PI3K/AKT/mTOR pathway, autophagy/p62 pathway, UPR, AMPK, SIRT2, and defects in the TCA cycle. However, the role of NRF2 in the cellular response to changes in nutrient availability is incompletely understood. Further work is needed to elucidate the seemingly paradoxical activation of NRF2 by both nutrient limitation and availability, and to unravel which NRF2-regulated processes, if any, promote cellular adaptation under these conditions. Furthermore, NRF2 is emerging as a major regulator of cellular metabolism in cancer cells and we are just beginning to identify the metabolic processes that NRF2 controls. Furthermore, NRF2 regulates the transcription of metabolic enzymes via both direct and indirect regulation of their expression, which may have far-reaching implications for cellular metabolism. In addition, NRF2 controls the intracellular concentrations of multiple amino acids, such as cysteine, serine, and glycine, and many feed into metabolites that play key roles in pathways that promote proliferation, survival, and immunosuppression. While one must be cautious about drawing conclusions from noncancer studies, the regulation of these pathways by NRF2 during tumorigenesis is an exciting future direction that remains to be explored.
Footnotes
Acknowledgments
We thank Florian Karreth and Nathan Ward for critical editing of the article, and Yun Pyo Kang for editing of the figures. G.M.D. is supported by the Pancreatic Cancer Action Network Pathway to Leadership Award and the American Cancer Society's Institutional Research Grant.