
Abstract
Significance:
Inflammation can be defined as a protective immune response against harmful exogenous and endogenous stimuli. Nevertheless, prolonged or autoimmune inflammatory responses are likely to cause pathological states that are associated with a production of inflammation-associated molecules along with reactive oxygen species (ROS). Kelch-like ECH-associated protein 1-nuclear factor erythroid 2-related factor 2 (KEAP1-NRF2) signaling provides a cell protection mechanism against oxidative insults when endogenous stress defense mechanisms are imbalanced. Understanding the roles of the KEAP1-NRF2 system in inflammation caused by various types of stimuli may aid in the development of new therapies.
Recent Advances:
There have been tremendous advances in understanding the mechanism by which the KEAP1-NRF2 pathway abrogates inflammation. In addition to the well-established ROS-dependent pathway, recent studies have provided evidence of the direct repression of the transcription of pro-inflammatory cytokine genes, such as IL1b and IL6 (encoding Interleukin-1β and Interleukin-6, respectively). Further, the expanding functions of NRF2 have elicited interest in the development of therapeutic modalities for inflammatory diseases, including multiple sclerosis and sickle cell disease.
Critical Issues and Future Directions:
Despite progress in the understanding of molecular mechanisms supporting the roles that NRF2 plays during inflammation, the relationship between NRF2 and other transcription factors and mediators of inflammation still remains ambiguous. Further studies are required to address the effects of functional polymorphisms in KEAP1 and NRF2 that modify susceptibility to specific disease-related inflammation. Comprehensive analyses in the future should explore tissue- or cell-type specific NRF2 activation to elaborate effects of NRF2 induction. Antioxid. Redox Signal. 00, 000–000.
Introduction
N

In the nucleus, NRF2 forms heterodimers with one of the small MAF (sMAF) proteins (41, 54). The NRF2-sMAF heterodimer complex then binds to DNA sequences that are referred to as antioxidant responsive elements (ARE) (66), electrophile responsive elements (EpRE) (19), or CNC-sMaf binding elements (CsMBE) (61) in the regulatory regions of target genes. Genes such as those encoding phase II detoxifying enzymes, including NAD(P)H-quinone oxidoreductase (NQO1), glutathione S-transferases (GSTs), and UDP-glycosyltransferases (UGTs), antioxidant enzymes such as glutamate-cysteine ligase (glutathione [GSH] production), HMOX-1 or heme oxygenase-1 (HO-1), thioredoxin reductase-1 (TXNRD1), thioredoxin, and ferritin (48), ABC transporters, and pentose phosphate pathway enzymes expression are induced (9, 29, 51, 52, 75).
Extensive analyses of Nrf2-deficient and/or Keap1-deficient animals demonstrate the cytoprotective functions of NRF2. Although Nrf2-null mice and rats grow normally, they are highly susceptible to oxidative and xenobiotic stress (36, 78). In contrast, Keap1 knockdown mice, in which NRF2 is constitutively activated, are resistant to the toxicity of xenobiotics (59, 77).
Anti-inflammatory Functions of NRF2
Inflammation can be defined as a protective immune response against harmful endogenous and exogenous stimuli. However, prolonged or autoimmune inflammatory responses cause a variety of detrimental states. Inflammation-associated molecules have been identified as endogenous inducers of NRF2. For instance, 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2), one of the end products of the cyclooxygenase-2 (COX-2) pathway, is predominantly and abundantly produced by macrophages (47, 71) and exerts potent anti-inflammatory effects. 15d-PGJ2 inhibits inflammatory transcription factors such as nuclear factor-κB (NF-κB) and strongly activates NRF2 by interacting with KEAP1 (37, 79). Notably, a genetic NRF2 deficiency in mice significantly counteracts the resolution of inflammation by 15d-PGJ2, indicating that NRF2 is essential for anti-inflammatory effects through 15d-PGJ2 (37, 53).
NRF2 suppresses inflammation through both ROS-dependent and ROS-independent mechanisms (Fig. 2). ROS are known to play significant roles in the progression of inflammatory disorders. Excessive ROS cause damage to lipids, proteins, and DNA, resulting in cell and tissue damage. Damaged cells release substances referred to as damage-associated molecular patterns (DAMPs), such as heat shock proteins and high-mobility group protein B1, which activate macrophages involved in the innate immune response (72). Activated macrophages release various inflammatory mediators and cytokines that induce a series of inflammatory responses. Therefore, the antioxidative function of NRF2 contributes to the elimination of ROS and subsequent inflammatory responses (81) (Fig. 2, right).
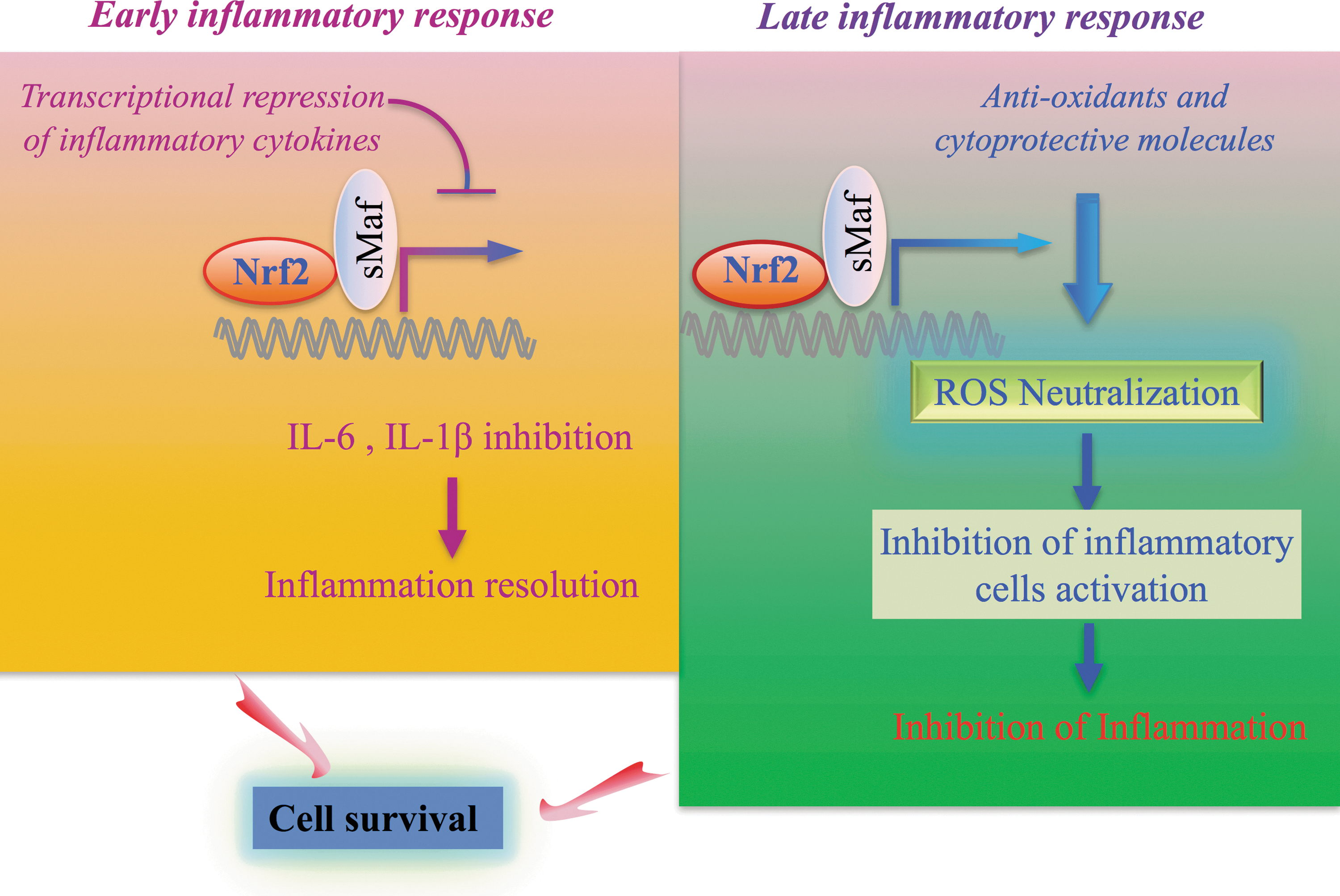
In addition to the anti-inflammatory function of NRF2 through the ROS elimination, we recently identified an ROS-independent anti-inflammatory effect of NRF2 (Fig. 2, left). Expression of pro-inflammatory cytokine genes such as IL6 and IL1b (encoding interleukin-6 [IL-6] and IL-1β) is quickly activated by pro-inflammatory transcription factors, including NF-κB (4), c-Jun (8, 82), and C/EBPβ (64). On the other hand, NRF2 directly inhibits transcription of these pro-inflammatory cytokine genes in an ROS-independent manner in macrophages (44). NRF2 binds to ARE within regulatory domains of these pro-inflammatory cytokine genes and inhibits the recruitment of RNA polymerase II to these loci, although the recruitments of pro-inflammatory transcription factors remain unchanged. Importantly, the suppression of pro-inflammatory cytokine genes occurs in an early phase, suggesting that NRF2 resolves inflammation quickly.
Several pathologic models involving the KEAP1-NRF2 pathway provide evidence of the roles of NRF2 in the alteration of acute inflammatory signaling. For instance, Nrf2-deficient mice exhibit severe cigarette smoke-induced lung inflammation compared with similarly exposed wild-type mice (32). In the case of oxidative stress-induced inflammation, ROS enhance the translocation of Toll-like receptors (TLRs) to lipid layers, where they interact with other factors, activating downstream signals (56, 63). In the next few sections, we will describe the contributions of NRF2 to multiple sclerosis and sickle cell disease (SCD).
The Involvement of NRF2 in Multiple Sclerosis
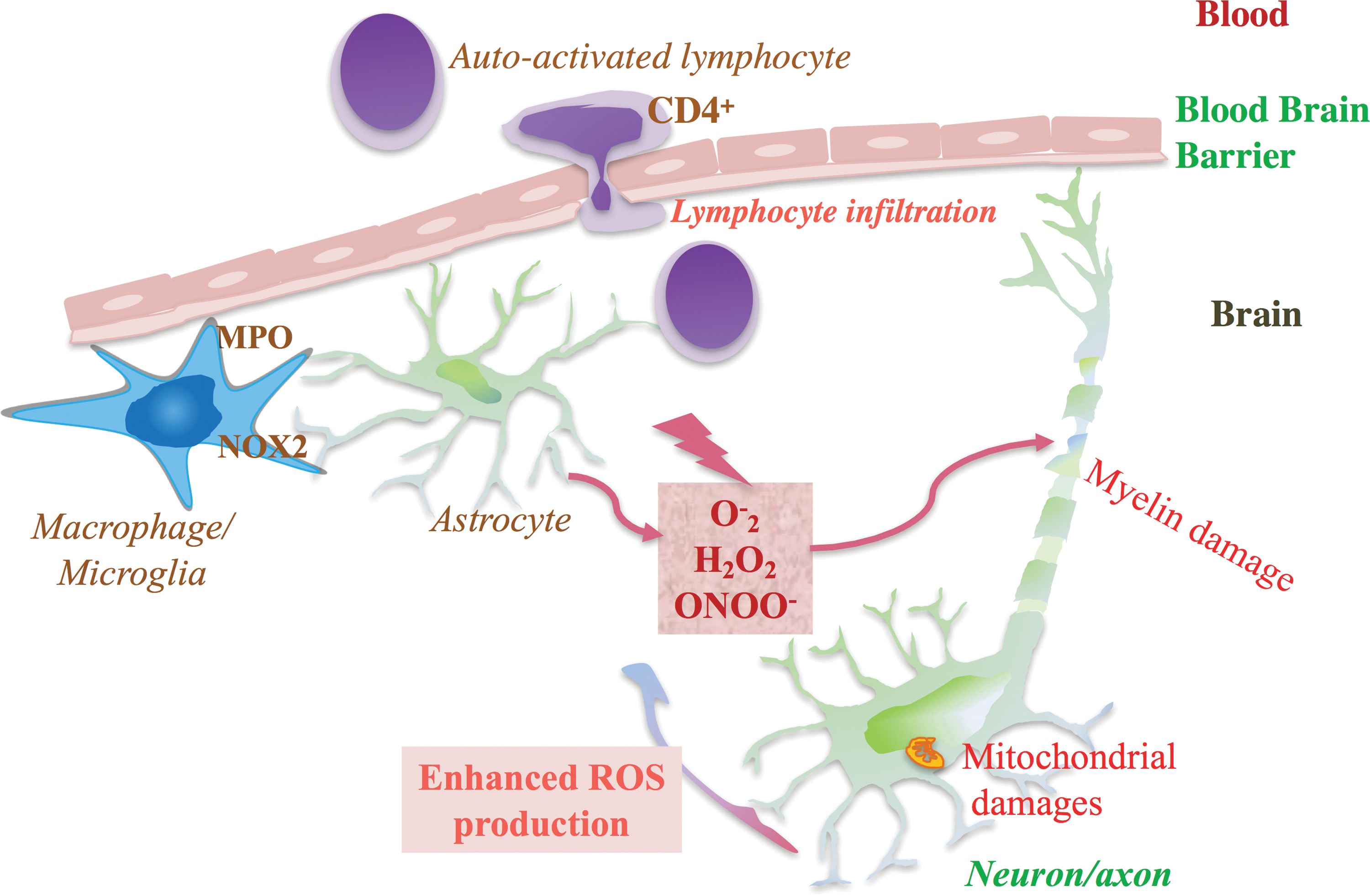
Multiple sclerosis is a chronic inflammatory demyelinating disease of the central nervous system (CNS), characterized by peri-venous infiltration of lymphocytes and macrophages in the brain parenchyma. Moreover, the loss of myelin and the fatty tissue surrounding and protecting nerve fibers sustain neuronal damage (20). The pathology is initiated by the migration of auto-reactive lymphocytes that are activated by myelin-like antigenic peptides through the blood-brain barrier (BBB) (11). Activated CD4+ lymphocytes recruit other activated cells such as CD8+ lymphocytes and resident macrophages, and trigger a series of cell lesions, including destruction of the myelin sheath, oligodendrocyte damage, axon injury, and glial scar formation (20). Clinically, the patterns of CNS injuries differ from one patient to another due to various lesions in the white matter. Predominant axonal injuries in multiple sclerosis are likely correlated with permanent functional deficits (58).
Auto-reactive lymphocytes may initiate the lesions of multiple sclerosis, but it is also possible that impairment of the mechanism opposing electrophile accumulation maintains the progression of the pathology. A reduced level of antioxidants in the serum of patients affected by multiple sclerosis supports the hypothesis of an imbalance between oxidants and antioxidants (46). In fact, several reports have shown that ROS significantly contribute to the pathology of multiple sclerosis (83, 90). The primary sources of ROS are macrophages and activated microglia. These cells produce a critical amount of ROS, which can induce demyelination, oligodendrocyte death, and axonal degeneration. In addition, mitochondrial damage within neurons is another significant source of ROS that contributes to axonal injury (Fig. 3).
The first insight regarding the contribution of NRF2 activation to the treatment of multiple sclerosis emerged from studies on fumaric acid esters, which are NRF2 activators (1, 60). In 2006, a pilot study revealed that an oral formulation of fumaric acid esters, which is known to be effective in patients with psoriasis, reduces the number of lesions in multiple sclerosis patients (68). Later, a more efficient and safer oral formulation of dimethyl fumarate (BG-12) was shown to reduce relapse rates and improve lesions in phase II and III studies (18, 22, 40). In 2013, dimethyl fumarate was approved for the treatment of multiple sclerosis by the U.S. Food and Drug Administration (FDA) and the European Medicines Agency (EMA). In 2017, the Japanese Pharmaceuticals and Medical Devices Agency (PMDA) also approved the use of dimethyl fumarate in Japan.
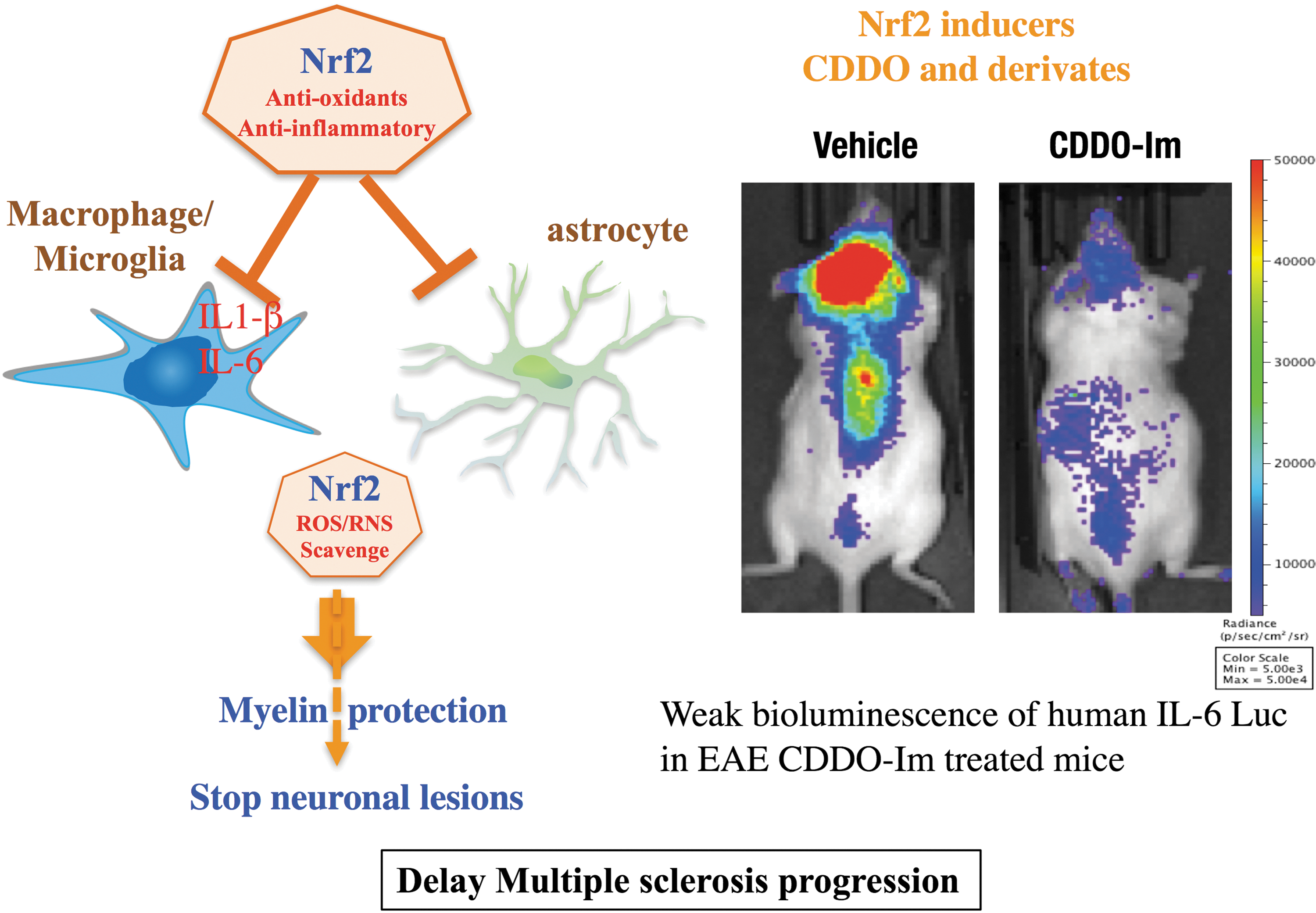
Orally administered dimethyl fumarate is rapidly converted into monomethyl fumarate (87). Mass spectroscopy analyses revealed that monomethyl fumarate modifies the Cys151 residue of KEAP1 and activates NRF2 (50). Several studies using an animal model of multiple sclerosis known as experimental autoimmune encephalomyelitis (EAE) (23, 27) have provided evidence that NRF2 activation improves the symptoms of multiple sclerosis. Activation of NRF2 through the administration of dimethyl fumarate and a triterpenoid inducer of NRF2, 2-cyano-3,12-dioxo-oleana-1,9(11)-dien-28-oic acid (CDDO)-trifluoroethyl-amide (TFEA), was shown to alleviate the course of the disease (2, 50). In addition, Nrf2-deficient mice exhibit severe symptoms of EAE that cannot be rescued via the administration of CDDO-Im (imidazole). This finding confirms the underlying significance of NRF2 in neuroprotection against inflammation and oxidative stress (Fig. 4) (25, 39). NRF2 overexpression in neural cells exacerbates ROS/reactive nitrogen species disposal, protecting the CNS from oxidative damage. Although it was recently shown that dimethyl fumarate could also protect against EAE in Nrf2−/− mice (69), Nrf2-mediated activity of dimethyl fumarate against multiple sclerosis has been supported through multiple lines of evidence. The discovery of NRF2 modulators as therapeutic drugs for multiple sclerosis will potentially expand the use of NRF2 inducers for the treatment of other inflammatory diseases.
Heme-induced Inflammation in SCD and NRF2
SCD is an inherited disorder caused by mutations in the β-globin gene (33). Hemoglobin tetramers containing mutated β-globin polymerize in red blood cells (RBCs) and generate rods that change the shape of RBCs to a sickle-like shape. Since sickle-shaped RBCs are prone to intravascular hemolysis, chronic hemolytic anemia and the impeded heme-scavenging system are some of the characteristics of SCD. These factors promote an establishment of an oxidative microenvironment, which, in turn, further enhances the production of ROS through ischemia-reperfusion in the microvascular sector (49).
Although it is still unclear whether oxidative stress is a consequence or cause of SCD pathology, several hypotheses have been proposed to determine the origin of ROS in SCD. One hypothesis incriminates enzymatic (NADPH, xanthine oxidase) (88) and non-enzymatic pathways (auto-oxidation of sickle hemoglobin) that provoke oxidative stress (26). An alternative hypothesis implicates iron-mediated Fenton chemistry reactions catalyzed by denatured heme moieties bound to the RBC membrane (7) that subsequently lead to ROS generation. Indeed, SCD patients exhibit low levels of antioxidants such as vitamins and glutathione peroxidase compared with non-SCD patients (3, 57). Heme release from RBCs during hemolysis is also evident in some pathologic events in the murine model of SCD (84). Animal models of SCD treated with hemopexin and carbon monoxide show a diminished vaso-occlusion process and decreased heme-induced inflammation, confirming the hypothesis of impaired antioxidant mechanisms in SCD (5).
Recently, we and others have demonstrated that genetic and pharmacological induction of NRF2 relieves SCD symptoms in mouse models (6, 42). By crossing Keap1 knockdown mice and Townes SCD mice (89), we generated SCD mice with systemic constitutive NRF2 stabilization. In this model, NRF2 upregulates the expression of superoxide dismutase and catalase, which are critical for ROS removal. As described in the Anti-inflammatory Functions of NRF2 section, NRF2 has been reported to regulate inflammation through the increased expression of antioxidant genes that reduce ROS levels (80) and the suppression of pro-inflammatory cytokine gene expression. We conclude that NRF2 relieves SCD by alleviating inflammation.
As in Nrf2-deficient macrophages, heme activates TLRs in the same manner as LPS (lipopolysaccharide), resulting in the production of adhesion molecules and inflammatory cytokines in SCD mice (5, 45). Similar to the genetic induction of NRF2, NRF2 activation by the drug CDDO-Im in SCD mice also represses the expression of IL-6, IL-1β, and IL-18 in the lungs and vascular cell adhesion molecule 1 (VCAM-1) and P-selectin in the aortas of SCD mice compared with control mice (42). Since Nrf2 regulates inflammation through transcriptional inhibition of the pro-inflammatory cytokine Il6 and Il1b genes (44), these observations clearly demonstrate that NRF2 is a crucial regulator of heme-induced inflammation in SCD (Fig. 5). These results further imply that by ameliorating the oxidative microenvironment and inflammation, NRF2 protects organs from other ischemia/reperfusion-based injuries and permanent damage.

NRF2 Inducers as Therapeutic Agents for Inflammatory Diseases
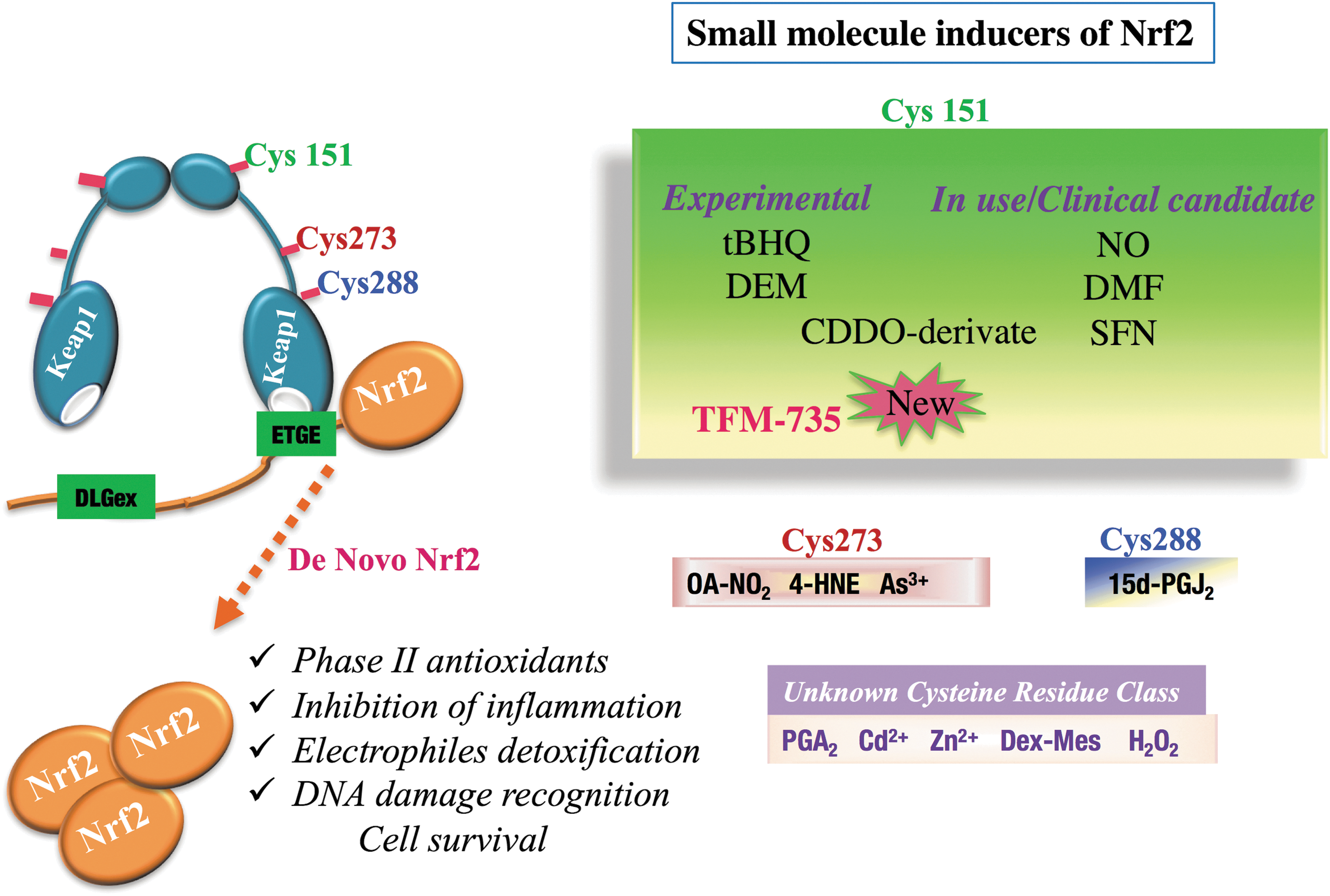
Several classes of chemical compounds can actively compromise the KEAP1-NRF2 interaction and activate NRF2 (Fig. 6). Some of these compounds are used in the agro-alimentary industry as food preservatives, and others are used intensively in animal models as experimental therapies for cancer chemoprevention. The latter group includes phenolic antioxidants (butylated hydroxytoluene [BHT], butylated hydroxyanisole [BHA]) and dithiolethiones (oltipraz, D3T) (55, 65, 86). Since NRF2 inducers exhibit potent anti-inflammatory properties, some of these inducers are candidates for clinical trials, such as isothiocyanates (sulforaphane), fumaric acid (dimethyl fumarate), CDDO, and their derivatives.
Isothiocyanates are metabolites of the natural plant components glucosinolates, which are widely found in cruciferous vegetables, including cabbage, horseradish, and radishes. The concentration of isothiocyanates varies among plants and plant parts. In broccolis, a higher concentration of glucosinolates is found in young plants or 3-day-old sprouts (17). A naturally active form of isothiocyanates is obtained from the hydrolysis of glucosinolates by the enzyme myrosinase, a product of cell membrane lysis, when vegetables are chewed or chopped (91). The absence of myrosinase in humans does not stop the conversion of glucosinolates into isothiocyanates because the microflora of the gastrointestinal tract facilitates the conversion of glucosinolates into isothiocyanates or more active metabolites (12, 70).
Glucoraphanin is a natural precursor of isothiocyanates, which actively form sulforaphane as a consequence of myrosinase activity. Pharmacokinetic studies conducted on human subjects who had ingested either standard broccoli (mixture made from standard cultivated broccoli) or a super broccoli mixture (mixture of broccoli that contain high concentrations of isothiocyanates) (21) revealed that sulforaphane and its metabolites reached maximal concentrations after 1.5 or 2 h and then returned to the baseline level after 24 h (21). Sulforaphane directly interacts with cysteine 151 of KEAP1 and induces detoxifying enzymes such as GSTs and NQO1 (13, 31). In addition, sulforaphane mitigates inflammatory infiltrates and decreases MMP-9, the enzyme responsible for the degradation of the extracellular matrix of the BBB in multiple sclerosis (2, 24, 34).
Studies conducted in animals revealed that sulforaphane can not only induce detoxifying enzymes and phase II detoxifying enzyme genes but also further mitigate inflammation, vaso-occlusion, organ damage, and retinopathy in SCD. A phase I study using sulforaphane confirmed the induction of HO-1 and NQO1 with good tolerance and few side effects related to the administration of sulforaphane to SCD patients (16). Although NRF2-dependent genes are induced by the prolonged consumption of sulforaphane, there are no significant changes in fetal hemoglobin levels (16).
Triterpenoids are derivatives of oleanolic acid. These small molecules hinder NRF2 degradation through covalent binding to cysteine 151 of KEAP1 (10), similar to isothiocyanates. Natural triterpenoids are used in traditional Asian medicine due to their anti-inflammatory characteristics, and they exist in various forms, such as oleananes and ursanes. (73). Sporn and colleagues have been developing synthetic triterpenoids from oleanolic acid. In contrast to natural triterpenoids, which show weak anti-inflammatory activity and are required at high concentrations, these synthetic compounds inhibit cellular growth and induce cellular arrest and apoptosis in cancer cells at lower levels. Their anti-inflammatory activity is correlated with their ability to produce cytoprotective enzymes due to the presence of Michael acceptors at the critical position (14, 15). Among triterpenoids, CDDO induces phase II detoxifying enzyme genes, plus HO-1 and enzymes involved in glutathione synthesis, both in vitro and in vivo. Further, this anti-inflammatory activity was supported by the interferon-γ, tumor necrosis factor-α-dependent induction of inducible nitric oxide synthase (iNOS), and COX-2 during inflammation in mouse macrophages (30, 74). The members of the CDDO family include CDDO-Me (methyl ester or bardoxolone methyl), CDDO-MA (methyl-amide), CDDO-Im, and CDDO-TFEA. These molecules protect cells and repress inflammation in an NRF2-dependent manner at nanomolar concentrations.
Recently, our team identified a novel NRF2 inducer, TFM-735, that modifies Cys151 (28). This Nrf2 inducer was tested for its anti-inflammatory properties and its capacity to delay the onset and inhibit the progression of multiple sclerosis in EAE mice. Anti-inflammatory properties of TFM-735 ability have been supported by the observations, including those to inhibit the production of IL-6 and IL17 and to repress luciferase activity in Wim-6 transgenic EAE mice (28).
Pharmacological Use of NRF2 Chemical Inducers
Several small molecules show the ability to induce NRF2 through the binding of one or more cysteines in Keap1 in cell and animal experiments at a low concentration. However, in humans, the effective dosage should be well tolerated and cause as few side effects as possible. It is worth mentioning that genetic overexpression of NRF2 may be beneficial via enhanced NRF2 downstream functions such as detoxification, cytoprotection, cancer chemoprevention, anti-inflammation, and cell survival. In contrast, excessive NRF2 activation exerts harmful effects, especially during the developmental stage. Keap1-null mice die at weaning because of hyperkeratosis in the esophagus (85). Recently, by deleting Nrf2 in the esophagus of Keap1-null mice, we generated mice with hyper-activated NRF2 throughout the entire body, except for the esophagus (76). Even though the mice did not show hyperkeratosis of the esophagus and survived until adulthood, they developed polyuria and bilateral hydronephrosis due to a water reabsorption defect caused by reduced aquaporin 2 levels in developing renal tubular cells. The mice also showed growth retardation independent of renal tubular defects, suggesting that other tissues are also affected by excessive NRF2 activation. Based on these studies, the timing and dose of NRF2 inducer treatments should be carefully considered, especially in pregnant women and children.
Concluding Remarks
The KEAP1-NRF2 pathway is an attractive target for drug discovery and development since NRF2 not only protects cells from toxic oxidative microenvironments but also has the ability to control the inflammatory response. The transcription factor NRF2 is the master regulator of the antioxidant response; considering that the oxidative environment is either a trigging factor or a consequence of human diseases, NRF2 is a good candidate for anti-inflammation therapy.
Footnotes
Acknowledgments
The authors thank the laboratory members for the constructive discussions. This work was supported, in part, by KAKENHI 15H02507 (M.Y.), 24799957 (M.S.), and 16H06639 (N.K.L.) from Japan Society for the promotion of science, AMED-P-CREATE (M.Y.), the Naito Foundation (M.Y.), the Takeda Science Foundation (M.Y.) and the platform Project for supporting in Drug Discovery and Life Science Research (Platform for Drug Discovery, Informatics, and Structural Life Science) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT), and AMED (M.Y.).