
Abstract
Significance:
Iron is required for growth and is often redox active under cytosolic conditions. As a result of its facile redox chemistry, iron homeostasis is intricately involved with oxidative stress. Bacterial adaptation to iron limitation and oxidative stress often involves ferric uptake regulator (Fur) proteins: a diverse set of divalent cation-dependent, DNA-binding proteins that vary widely in both metal selectivity and sensitivity to metal-catalyzed oxidation.
Recent Advances:
Bacteria contain two Fur family metalloregulators that use ferrous iron (Fe2+) as their cofactor, Fur and PerR. Fur functions to regulate iron homeostasis in response to changes in intracellular levels of Fe2+. PerR also binds Fe2+, which enables metal-catalyzed protein oxidation as a mechanism for sensing hydrogen peroxide (H2O2).
Critical Issues:
To effectively regulate iron homeostasis, Fur has an Fe2+ affinity tuned to monitor the labile iron pool of the cell and may be under selective pressure to minimize iron oxidation, which would otherwise lead to an inappropriate increase in iron uptake under oxidative stress conditions. Conversely, Fe2+ is bound more tightly to PerR but exhibits high H2O2 reactivity, which enables a rapid induction of peroxide stress genes.
Future Directions:
The features that determine the disparate reactivity of these proteins with oxidants are still poorly understood. A controlled, comparative analysis of the affinities of Fur/PerR proteins for their metal cofactors and their rate of reactivity with H2O2, combined with structure/function analyses, will be needed to define the molecular mechanisms that have facilitated this divergence of function between these two paralogous regulators.
Iron's Double-Edged Sword
Iron is an essential element across all domains of life. It is used as a cofactor by enzymes involved in a wide array of metabolic processes spanning central metabolism, respiration, photosynthesis, nitrogen fixation, and DNA synthesis and repair (6). Bacteria are no exception to this, as is evident by their use of three major classes of iron-requiring proteins: (i) mono- and dinuclear iron enzymes, (ii) iron/sulfur cluster enzymes, and (iii) heme proteins. These three classes of iron-utilizing enzymes permeate many aspects of bacterial metabolism.
This need for iron in bacteria is ancient and dates from the earliest stages in the evolution of life >2 billion years ago. During this time, the Earth was an anoxic environment, which allowed iron to be maintained in its reduced, soluble ferrous (Fe2+) form (5). Iron was therefore highly bioavailable and found use as a cofactor for stabilizing proteins as “iron rivets” (39), for electron transfer reactions, and as a Lewis acid catalyst. However, as photosynthetic organisms became more prevalent, the gradual oxygenation of the Earth's atmosphere imposed a transition from anaerobic to aerobic conditions. This shifted the redox state of iron into its oxidized, weakly soluble ferric (Fe3+) form and led to the geologic deposition of massive amounts of iron in banded iron formations (5). These environmental changes imposed two new physiological challenges on early microbial life: iron starvation and oxidative stress.
The restriction on microbial growth that resulted from the depletion of dissolved iron from the ancient oceans persists to this day (97). As a result, bacteria evolved systems for iron homeostasis, which are often regulated by ferric uptake regulator (Fur) proteins (74). Iron homeostasis involves three distinct mechanisms (Fig. 1). First, bacteria synthesize high-affinity iron chelators (siderophores), their cognate uptake systems, and other transporters for iron import (6). Second, Fur often controls an “iron-sparing” response in which a Fur-regulated small RNA serves to inhibit the translation of nonessential iron requiring enzymes (82). Third, cells may replace iron-containing proteins with alternative, iron-independent proteins (87). For example, under iron limitation, many bacteria express flavodoxins as a substitute for iron-utilizing ferredoxins and manganese superoxide dismutase (MnSOD) in place of FeSOD (87). Collectively, these responses provide the cell with a viable iron economy despite the scarcity of the growth-limiting micronutrient.

The second chemical consequence of the rise of diatomic oxygen in the atmosphere was an increase in reactive oxygen species (ROS) such as hydrogen peroxide (H2O2) and superoxide radicals (O2
−). ROS are naturally generated in the environment in a variety of photosynthetic reactions and as metabolic by-products of flavoprotein autoxidation (62, 84). ROS can exert toxicity in several distinct ways, but one common theme is the oxidative inactivation of iron-containing enzymes, including both iron/sulfur-containing dehydratases and mononuclear iron enzymes (63). H2O2 reacts with ferrous iron through Fenton chemistry, and the resulting highly reactive hydroxyl radical can impair metabolism through DNA and protein damage, which can inevitably lead to cell death (63).
Similarly, superoxide inactivates iron-containing enzymes by oxidation of their required cofactors. In the case of mononuclear iron enzymes, oxidation and loss of iron have been shown to lead to mismetallation by zinc (Zn2+), which binds with higher avidity than iron (64). This has been observed in Escherichia coli, where ribulose-5-phosphate 3-epimerase and 3-deoxy-d-arabinoheptulosonate 7-phosphate synthase become mismetallated by Zn2+ on exposure to ROS, causing enzyme inactivation and subsequent blockage of specific metabolic pathways (64, 104, 105).
Aerobic bacteria are reliant on detoxification mechanisms to help counter the deleterious effects of ROS, by either directly detoxifying oxidants such as H2O2 or by decreasing intracellular labile iron to mitigate the effects of Fenton chemistry (61, 63, 77). H2O2 is removed enzymatically by catalase and alkyl hydroperoxide reductase (99), whereas superoxide is removed by SOD (63). Quantitative modeling in E. coli suggests that iron/sulfur-containing dehydratases and mononuclear iron enzymes are likely to be oxidized and repaired on a time scale of minutes under aerobic growth, even in cells having a normal complement of defensive enzymes (63, 104, 105). Any further increase in ROS requires the induction of defensive enzymes to an even higher titer (63). In E. coli, the induction of pathways for defense against ROS is controlled by the OxyR and SoxRS systems (63). Whereas OxyR senses H2O2 by disulfide bond formation, many other bacteria rely on a chemically distinct sensing mechanism involving metal-catalyzed oxidation of a regulator protein. The prototype for this class of regulators is the Bacillus subtilis PerR protein, a paralog of Fur (73).
The facile redox chemistry of ferrous iron requires that bacteria tightly regulate iron homeostasis and maintain an efficient mechanism to sense and respond to ROS such as H2O2. Defenses against both iron limitation and ROS are important in the physiology of many pathogens. During infection, the host restricts bacterial growth by sequestration of essential metal ions, including iron, and innate immune cells kill bacteria using ROS and reactive nitrogen species (RNS) (36).
This review will focus on the role of Fur family metalloregulators in coordinating these two interacting adaptive responses. A special emphasis will be placed on the biochemistry and genetics of two Fur family proteins in the gram-positive soil bacterium B. subtilis, which will be designated as FurBs and PerRBs. Comparison of the reactivity of these proteins and their orthologs with ROS and RNS reveals a wide range of sensitivity, and recent results have begun to shed light on how this reactivity may be tuned by changes in protein structure.
Fur Family Metalloregulators
The ferric uptake regulator (Fur) protein was first characterized in E. coli (FurEc) where it functions as an iron-responsive repressor. Initial genetic studies revealed that E. coli fur null mutants produced very high levels of siderophores (51, 53). Subsequent biochemical characterization demonstrated that FurEc is a dimeric, helix-turn-helix (HTH) containing DNA-binding protein that binds Fe2+ as corepressor (10, 11, 26). These observations, together with subsequent studies (89), led to a general model in which FurEc directly senses Fe2+ levels in the cell to maintain iron homeostasis.
Fur family metalloregulators are ubiquitous in bacteria and are commonly identified through the conserved HHHXHX2CX2C motif positioned at the hinge region between the metal-sensing C-terminus and the DNA-binding domain (40, 74). Although Fur proteins are most closely associated with iron homeostasis, it is now appreciated that the Fur family includes many proteins with distinct metal selectivities. Variations in the metal-binding sites have enabled the evolution of Fur family members that are activated by a diverse set of metals. For example, the Fur family includes sensors of zinc (Zur), manganese (Mur), and nickel (Nur) (3, 53, 74). In addition, a subset of the iron-utilizing Fur family proteins functions physiologically as sensors of intracellular H2O2 (PerR) (73, 74).
Given the broad metal-sensing scope of these regulators, it is not surprising that bacteria may possess multiple Fur homologs. For example, B. subtilis has three Fur family paralogs: a Zn2+-sensing Zur (45), an Fe2+-sensing Fur, and a peroxide-sensing PerR (16, 91). Interestingly, B. subtilis does not sense Mn2+ via a Fur family protein, but rather through MntR, a member of the diphtheria toxin regulator (DtxR) family (59, 96). Furthermore, it was recently reported that Bacillus licheniformis not only has Fur and Zur but also contains three PerR paralogs (68).
Paralogous Fur family members may evolve distinct metal selectivity by changes in the chemical nature and geometry of the metal-coordinating ligands at the metal-sensing sites (74). Furthermore, each sensor must have an affinity appropriate for detecting fluctuations in the intracellular buffered pool of free metal ions, as reviewed in detail elsewhere (49, 112, 113). However, Fur family regulators can bind to noncognate metals either as agonists or antagonists. The binding of noncognate metals to metalloregulatory proteins (mismetallation) can have deleterious consequences for regulators, just as it can for enzymes. For example, in E. coli and related proteobacteria, high levels of Mn2+ are toxic and mutations that inactivate Fur often arise conferring resistance to these elevated manganese levels (52). This suggests that mismetallation of Fur with Mn2+ may inappropriately repress Fe2+ uptake. Similarly, in B. subtilis, a modest increase in FurBs expression (in a perR null mutant) leads to mismetallation with Mn2+ and results in iron starvation (38, 78), and mismetallation of PerRBs with Zn2+ can lead to dysregulation of the PerR regulon resulting in heme intoxication (20).
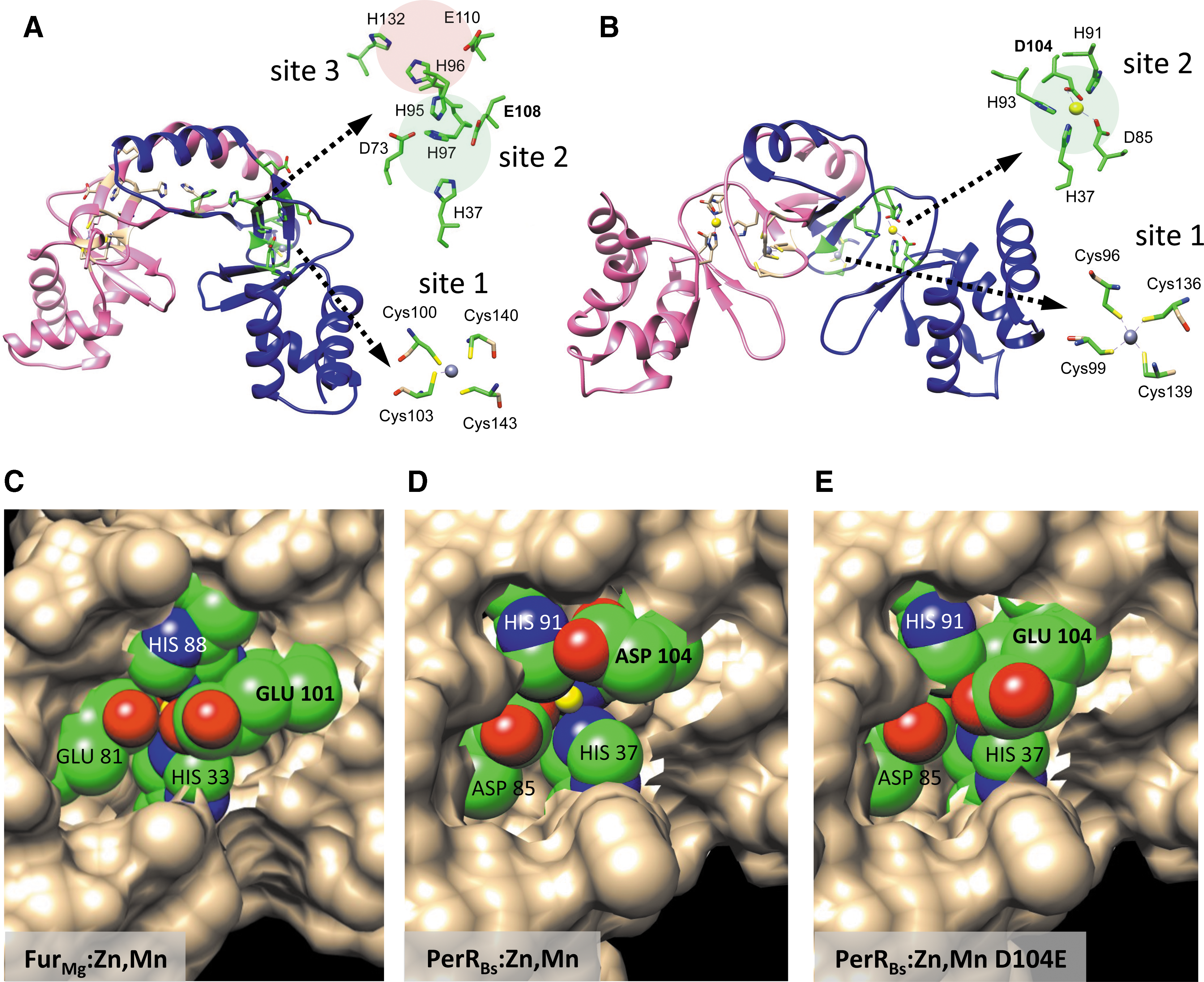
Structural studies have provided insight into how Fur family proteins sense metals and, in the case of PerR, respond to oxidants (73, 74, 79, 95), as well as into how they interact with DNA (32, 47). In general, Fur family proteins are homodimeric, and DNA-binding proteins comprised two main domains. The amino-terminal domain contains HTH motif and binds to DNA. The C-terminal domain is required for metal sensing and mediates dimerization. This metal-sensing domain typically contains two to three metal-binding sites (Fig. 2A). The first (site 1) is a structural metal-binding site comprising four cysteine residues that interact with a single Zn2+ atom (Cys4:Zn). This site is required for protein folding and dimerization for many Fur family proteins (4, 25, 72, 111, 115). However, some Fur homologs lack a structural zinc site. Such is the case for Pseudomonas aeruginosa Fur (FurPa), which only has one of the four conserved Cys residues, making the site functionally dispensable in vivo, and Pseudomonas putida, which lacks all four conserved Cys residues (76). The metal-sensing site in most Fur family proteins (site 2) is located in the C-terminal metal-binding domain and often bridges to the DNA-binding domain, thereby providing a mechanism for allosteric activation of DNA binding (40, 66). This is the key site for iron sensing by Fur and for peroxide sensing by PerR (73, 78). In some Fur proteins, including FurBs, there is also a third metal-binding site (site 3) of poorly defined function (33, 78).
Iron Utilizing Fur Family Regulators in B. subtilis: FurBs and PerRBs
B. subtilis has two Fur regulators that bind to iron: FurBs and PerRBs. Although these two proteins are closely related and bind to the same divalent cation, their functions are quite distinct. This is evident from the set of genes they control (13, 37, 56, 91). FurBs controls a large regulon that comprises ∼40 genes involved in iron homeostasis, consistent with its role as the primary sensor of iron limitation (13). PerRBs is involved in controlling expression of genes that help combat H2O2 stress, including genes that encode iron efflux and storage functions (21, 37, 48). Characterization of B. subtilis Fur and PerR illustrates an important feature of Fur family regulators: it is quite difficult to unambiguously assign function solely from inspection of protein sequence. Often, functional assignments can be best inferred by considering the set of genes regulated by each protein.
The FurBs regulon
The FurBs regulon can be divided into two main groups: iron acquisition genes and genes involved in the iron-sparing response. In the former, one subset is involved in siderophore biosynthesis. FurBs regulates the dhb operon, which encodes proteins involved in the synthesis of bacillibactin (BB) (13, 15, 46, 85). The second subset encodes ABC transporters involved in ferric siderophore uptake with specificity for BB and enterobactin (feuABC), ferrichrome (fhuBCDG), and schizokinen/arthrobactin (yfiZY/yfhA) (94). In addition, FurBs regulates ferric citrate uptake (yfmCDEF) and elemental iron uptake (efeOUB) (Fig. 1) (13, 88, 94). Second, Fur controls expression of a small RNA (FsrA) and three RNA chaperones (FbpA, FbpB, and FbpC). When derepressed, FsrA targets specific genes that code for selected iron enzymes, inhibiting their translation and thus effectively carrying out the iron-sparing response, which efficiently prioritizes iron utilization in the cell (Fig. 1) (44, 103). This system is functionally analogous to the similar iron-sparing response of E. coli mediated by the Fur-controlled small RNA RyhB (81 –83).
Fur recognizes genes within its regulon by binding to specific DNA operator sites (Fur boxes) located within the promoter region of its target genes. Originally, the Fur box was defined as a 19 bp inverted repeat (35), but subsequent studies demonstrated that a minimal-binding site is a heptameric inverted repeat motif (7-1-7): the 19 bp consensus corresponds to two, overlapping 7-1-7 motifs (12). The binding of Fur to these two overlapping binding sites has been visualized in crystals of Magnetospirillum gryphiswaldense Fur (FurMg) metallated with Mn2+ and bound to a Fur box DNA sequence (32). Remarkably, all three Fur paralogs in B. subtilis recognize very similar inverted repeat motifs, but there is minimal overlap in their target genes (42). Specific recognition takes place, in part, through a conserved arginine (Arg) residue located at position 61 (R61) of the FurBs DNA-binding domain (18). This residue recognizes the conserved guanine (G) and cytosine (C) bases located within the 7-1-7 motif (T
When B. subtilis cells experience iron depletion, the Fur regulon is derepressed. In the laboratory, iron depletion can be generated by treating cells with dipyridyl, an intracellular iron chelator, thus affecting the labile iron pool (13). As free iron levels drop in the cell, iron is no longer bound to Fur. The resultant conformational change leads to dissociation from DNA, thus allowing RNA polymerase to bind to the previously occluded promoter regions and initiate transcription. These Fur-regulated genes are thereby regulated directly by Fur binding, as confirmed by DNaseI footprinting (13). Some Fur family regulators may retain regulatory activity in the apo-form and bind to sites distinct from those bound by the metallated holo-protein. This type of more complex regulation has been described for Fur from both Campylobacter jejuni (17) and Helicobacter pylori (1).
Regulon derepression can take place in one of two general ways. As iron levels fall, all members of the regulon may be derepressed simultaneously, or instead as a stepwise (graded) response where different subsets of genes are derepressed according to the level of stress the cell is experiencing. For example, the B. subtilis Zur regulon is derepressed in three distinct stages as bioavailable zinc levels decline (101). A similar effect is observed in the Streptomyces coelicolor Zur (102).
Fur regulons may also respond to gradual iron depletion by the sequential derepression of various adaptive mechanisms, but the details are still emerging. The mechanisms underlying this sequential response are also not yet well understood, but may be related to variations in the architecture of the Fur-binding sites. For example, at some sites, Fur may function as a single dimeric protein and bind an isolated 7-1-7 motif (12). At other sites, Fur may bind to the classic 19 bp consensus as two, opposed dimers (binding on opposite faces of the DNA helix) (32, 47). Finally, Fur is known to have a propensity to bind to extended regions of DNA, and this cooperativity may be facilitated by protein/protein interactions (19, 29, 31, 41, 71). Differences in both the number and affinity of Fur-binding sites presumably help tune both the magnitude of the induction and the responsiveness of each target operon to the level of iron depletion.
Structural insights into Fe2+ sensing by Fur
The activity of FurBs is regulated by its specific interaction with Fe2+. As noted above, FurBs is representative of those Fur family members with a structural Zn2+ site and two additional metal-binding sites (Fig. 2A). Purification of FurBs in the presence of metal chelators yields the stable, dimeric apo-protein containing only the structural Zn2+ ion (FurBs:Zn) (78). Addition of Fe2+ (or other divalent cations) activates DNA binding with a stoichiometry of two Fe2+ atoms per monomer. The site 2 metal-sensing site is required for iron sensing. In addition, FurBs contains a third site (site 3) near the dimerization interface. In vitro, Fe2+ first binds to site 3, followed by subsequent binding of the metal to site 2 (78). Although both site 2 and site 3 are required for DNA binding, site 2 is the major iron-sensing site; binding to site 3 has a relatively modest effect (∼7-fold) on DNA-binding affinity compared to that achieved by binding to site 2 (∼150-fold) (78).
The precise mechanism by which these two metal-sensing sites interact to trigger the conformational changes that activate DNA binding by Fur remains unclear (74, 78). It is possible that the sequential binding of Fe2+ to site 3 and then site 2 helps to fine-tune the expression of genes within the Fur regulon, or to help FurBs avoid inappropriate activation by mismetallation. For example, sensing of Mn2+ by the DtxR family protein MntR involves the sequential loading of Mn2+ into two neighboring sites in each monomer, which is thought to provide a mechanism to increase the selectivity of metal responsiveness (86). Alternatively, site 3 may play a largely structural role in stabilizing the dimeric repressor. Indeed, it has not even been established whether or not Fe2+ is the metal that occupies site 3 in vivo.
Under normal physiological conditions, Fur binds specifically to Fe2+, and other divalent metal ions do not elicit repression of the Fur regulon. The concentration of free iron required to trigger DNA binding by FurBs is defined by the Kd for Fe2+ binding (∼1 μM) in vitro, which thereby provides an estimate of the buffered concentration of free Fe2+ in the cell. A nearly identical affinity (∼1.2 μM) has been measured for FurEc (89).
However, in vitro studies reveal that FurBs can also be activated by Mn2+, although with a lower binding affinity (K d of ∼24 μM) (78). Typically, the cellular concentration of free Mn2+ is maintained at a level too low to mismetallate FurBs. Mn2+ levels are regulated by MntR, which both represses uptake and activates efflux of Mn2+ and has an Mn2+ affinity (K d ∼6 μM) several fold higher than that of FurBs (K d ∼24 μM). Interestingly, under conditions where FurBs is overexpressed (as is the case in a perR null strain), FurBs is now constitutively activated by physiological levels of intracellular Mn2+ (78). In addition, FurBs has also been shown to exhibit some cross talk with Cd2+ or Zn2+ in the presence of sufficiently high concentrations of these metals (90), but it is unknown if this is mediated by direct binding of these cations to FurBs or indirectly. The high selectivity of FurBs for Fe2+ in vivo is remarkable in light of the many genetic studies suggesting that mismetallation of Fur by Mn2+ leads to dysregulation of the Fur regulon in many proteobacteria, including E. coli and Salmonella enterica (52, 60).
The PerR regulon
PerRBs was originally identified as a Fur family protein required for the adaptation of B. subtilis to H2O2 stress (16, 119). The PerRBs regulon comprised two groups of genes: (i) those involved in the direct detoxification of ROS and (ii) those that contribute to iron homeostasis. The former group consists of katA (vegetative catalase) and ahpCF (alkyl hydroperoxide reductase), both of which encode enzymes involved in the direct detoxification of hydrogen peroxide (H2O2) (16, 22, 37, 63). The heme biosynthesis operon (hemAXCDBL) also falls under this category, since induction of heme biosynthesis supports the activity of the abundantly expressed, heme-dependent catalase KatA (38) (Fig. 1). The second group of PerR-regulated genes encodes a Dps-like miniferritin involved in iron sequestration (MrgA) (21, 23) and a P1B4-type ATPase involved in ferrous iron efflux (PfeT) (Fig. 1) (48). Finally, PerR also controls fur and autoregulates its own expression (41, 54). The observation that PerRBs specifically regulates H2O2 detoxification and iron sequestration (MrgA), efflux (PfeT), and import (indirectly through Fur) is consistent with a primary role in minimizing the damage elicited by iron-catalyzed Fenton chemistry.
Early genetic studies revealed that B. subtilis peroxide stress genes were regulated by both metal ions and by H2O2, and this regulation required the same cis-acting operator site and the same trans-acting factor (22). This coordinate regulation is explained by the role of PerRBs as a metal-dependent repressor (strongly activated by either Fe2+ or Mn2+). Like other Fur family proteins, PerRBs contains a structural Zn2+ (site 1) and metal-sensing site 2 (Fig. 2B). PerRBs is highly sensitive to H2O2 inactivation when in its Fe2+-cofactored form (designated PerR:Zn,Fe). In response to H2O2 stress, the mrgA, katA, and pfeT genes are all highly induced (55) (Fig. 3). The ahpC and hemA promoters are also modestly induced by H2O2, and PerRBs also exhibits a weak (twofold to threefold) repression of fur and perR (43) (Fig. 3).

Operator sites for PerR-regulated genes (Per boxes) are very similar to those for Fur (42). They both align as 7-1-7 motifs and have a high sequence similarity with the exception of thymine (T) and adenine (A) at the −6 and +6 positions, respectively, of the two heptamers (T
Structural insights into H2O2 sensing by PerR
Much like FurBs, PerRBs is a dimeric DNA-binding protein with two distinct functional domains: a DNA-binding domain and a sensing domain. PerRBs binds two metal ions per monomer (72, 73, 78, 79, 108), instead of three as noted for FurBs (78) (Fig. 2A, B). The first is a structural Zn2+ atom necessary for structural integrity and protein dimerization. This Zn2+ is bound tightly to PerRBs in a cysteine pocket (Cys4:Zn) and remains intact even with high concentrations of peroxides. Indeed, this Zn2+ is often retained in the protein structure even during denaturing sodium dodecyl sulfate/polyacrylamide gel electrophoresis (SDS-PAGE) (72). The binding of a second metal to the metal-sensing site 2 leads to activation of DNA binding. In cells, PerR can be metallated by either Fe2+ or Mn2+ simply by altering the availability of metals in the growth medium (43, 56). In most iron-rich media, PerRBs is bound with Fe2+ and, in this state, is responsive to H2O2 (56). However, addition of excess Mn2+ can shift PerR into the Mn2+-activated form (PerR:Zn,Mn) and in this state, peroxide stress genes are tightly repressed (Fig. 3).
Typically, peroxide sensing is mediated by redox active cysteines, in sensors such as OxyR in E. coli (116) and OhrR in B. subtilis (75, 106). These are metal-independent peroxide sensors that detect ROS via the oxidation and subsequent formation of reversible disulfide bonds (8, 14, 34, 107). This contrasts with PerRBs, which senses H2O2 by metal-catalyzed oxidation through site 2 (73). Binding of H2O2 to Fe2+ in PerR:Zn,Fe is proposed to generate a localized hydroxyl radical that modifies one of two His residues (H37 or H91) in site 2 to 2-oxo-histidine (Fig. 4) (73, 98, 109). Recently, an alternative mechanism has also been suggested in which the initial bound H2O2 undergoes heterolytic cleavage releasing water and generating an Fe(IV) oxo intermediate (100). His oxidation may also occur on direct reaction of PerR:Zn,Fe with O2, and the presence of O2 also stimulates the modification by H2O2 (Fig. 4) (100). Interestingly, these two oxidizing agents appear to use distinct modification pathways: reaction with H2O2 targets H37 and H91 at about equal rates, whereas reaction with O2 selectively leads to H37 modification (100) (Fig. 4).

Regardless of the precise pathway, oxidized PerR loses its iron cofactor and the resulting conformational changes trigger its release from DNA and exposes specific signature residues targeted by the LonA protease (Fig. 3) (2). Thus, after regulon derepression occurs, oxidized PerRBs is degraded thereby preventing the accumulation of nonfunctional protein (2). Oxidation of PerRBs also leads to derepression of the autoregulated perR gene, which presumably helps re-establish repression once the newly synthesized PerR:Zn,Fe protein is no longer consumed by reaction with H2O2. Transcriptomic studies demonstrate that derepression of the PerR regulon in response to H2O2 is transient and repression is rapidly re-established (55). This is likely important since constitutive derepression of the PerR regulon, such as seen in a perR null mutant, imposes a very high iron demand on the cell due to the high levels of catalase that are produced (38, 78). This iron deficiency imposed in a perR null mutant is also observed in B. licheniformis, where overexpression of fur, rather than katA, is the main reason why cells cannot grow in media not supplemented with iron (70).
Metal-dependent changes in PerRBs repressor activity
In addition to Fe2+, PerRBs also binds Mn2+ to generate PerR:Zn,Mn. This form of PerR also represses the PerR regulon but is peroxide insensitive and fails to derepress the PerR regulon in response to H2O2 (43, 73). The reason why PerRBs binds to both Fe2+ and Mn2+ is not well understood: in B. subtilis the PerR regulon does not control any manganese-related genes. In contrast, E. coli OxyR, which also senses H2O2, directly activates the expression of the NRAMP family manganese importer MntH in response to H2O2 stress (7, 37, 80). This is not the case in B. subtilis, where manganese homeostasis is maintained independently of PerRBs, via MntR (59). The repression of the PerRBs regulon by Mn2+ may have resulted as a corollary of comparatively high affinity of PerR for Fe2+: PerRBs binds Fe2+ tighter than FurBs (79), and this may have also led to a high Mn2+ affinity. This high Fe2+ affinity ensures that the peroxide stress response is not induced by mild iron depletion, which would be maladaptive and impose a high iron demand through the increased synthesis of heme and catalase and the expression of proteins mediating iron sequestration (MrgA) and efflux (PfeT). Hence, PerR may have sacrificed metal selectivity to help increase its specificity to respond to H2O2 stress rather than fluctuations in metal availability.
The binding of either Fe2+ or Mn2+ may additionally allow PerR to function as a ratiomeric sensor of the relative iron and manganese levels. For example, when the level of Mn2+ is much higher than Fe2+ in the cell, the threat posed by H2O2 may be significantly reduced, thereby justifying a reduced induction of PerR regulon genes. Furthermore, there is suggestive evidence that the PerR:Zn,Fe and PerR:Zn,Mn forms of the repressor may differentially affect some genes (43) (Fig. 3). For example, in resuspension experiments, either Mn2+ or Fe2+ enabled repression of some PerR-regulated genes (katA, ahpCF, mrgA), whereas only Mn2+ elicited repression at the pfeT, per, and fur promoters. The basis for this Mn2+ selectivity is presently unknown, but two models have been suggested. First, PerR:Zn,Fe and PerR:Zn,Mn may be qualitatively different: they may have slightly different conformations that affect their DNA-binding selectivity. Second, the difference may be primarily quantitative: the much more stable PerR:Zn,Mn form may accumulate in cells to a higher active concentration allowing interaction with weaker operators within the PerR regulon (Fig. 3). Regardless of the mechanism, these observations are relevant when we consider below PerR orthologs that were reported to be specific for Mn2+ as a corepressor.
Structural differences that tune H2O2 sensitivity (it is the little things that count)
Despite the overall structural similarity between FurBs and PerRBs, they differ greatly in their sensitivity to H2O2. PerRBs is highly sensitive to H2O2 with a second-order rate constant for protein oxidation of ∼105 M−1 s−1 when associated with iron (73). This is comparable to the reported sensitivity of OxyR, which responds to submicromolar levels of intracellular H2O2 (9). As a result, isolation of unoxidized PerR:Zn is technically challenging and is best achieved by purification from cells grown with high Mn2+ (or in medium amended with iron chelators) to prevent in vivo oxidation, lysis, and purification in the presence of EDTA to remove any Fe2+ from the protein, and exclusion of thiol reducing agents (which can facilitate reduction of Fe3+ to Fe2+) (73, 100). The high sensitivity of PerRBs to H2O2 inactivation is appropriate for its role in regulating detoxification functions.
In contrast with PerR, Fur proteins are generally less sensitive to H2O2 inactivation, which is likely adaptive since the derepression of iron uptake functions in response to elevated H2O2 levels would likely be deleterious to the cell. Compared to PerRBs, FurBs is relatively easy to purify in an active state, although care is needed to prevent the adventitious binding of metals to the sensing site(s) (15). As discussed in more detail below, Fur proteins from a variety of organisms can be activated in vitro by addition of Fe2+ as a corepressor, even in aerobic buffers.
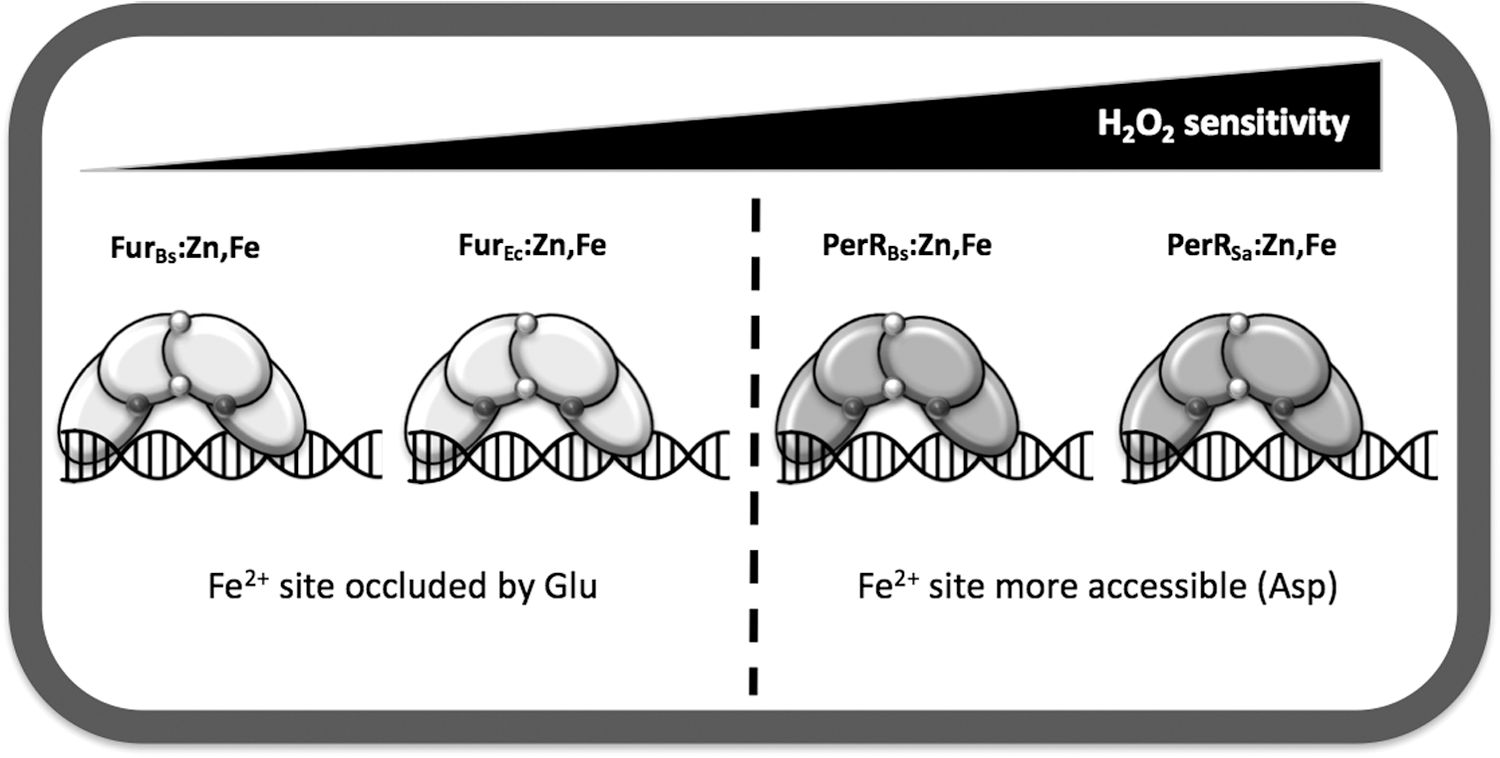
Recent results have begun to provide insights into how PerRBs and FurBs display such different sensitivities to H2O2. Both PerRBs and FurBs have a histidine-rich binding pocket (site 2) that coordinates Fe2+ (15, 54, 74). However, an aspartate (Asp) residue in PerRBs (position 104) replaces a glutamate (Glu) in FurBs (position 108) as the Fe2+ ligand (95). Mutational studies demonstrate that the PerRBs D104E and FurBs E108D mutants still bind Mn2+ with near-normal affinities. However, the sensitivity of each protein to metal-catalyzed oxidation of the neighboring histidine ligands (H37 and H91 in PerR and the equivalent residues in Fur) was significantly reduced. This was assessed using electrospray ionization mass spectrometry analysis to monitor His oxidation in proteins overexpressed and purified from aerobically growing E. coli, which generally maintains steady-state levels of H2O2 in the range of 50 nM. Under these conditions, ∼63% of the recovered PerR was oxidized, with a much lower value noted for Fur (4%). Significantly, oxidation of the PerRBs D104E mutant was substantially reduced (to 3%) whereas that for FurBs E108D was somewhat increased (to 14%) (95). These results were corroborated by in vitro analyses of protein oxidation by H2O2.
These mutational studies support a model in which the H2O2 reactivity of the bound Fe2+ can be quantitatively modulated by single amino acid changes in site 2 that affect coordination geometry. Structural studies of PerR:Zn,Mn revealed a penta-coordinate site 2 with three His and two Asp residues (32, 66). Assuming Fe2+ binds similarly to Mn2+, this leaves a sixth coordination site available for interaction with incoming H2O2. Structural modeling supports the notion that substitution of the PerR Asp104 ligand with the longer Glu side chain allows formation of a weak bidentate interaction between Glu and Fe2+ that occludes access of H2O2 and thereby minimizes protein oxidation (95) (Fig. 2C–E). The resultant Fe2+-binding site can be considered to have a “5 + 1” coordination mode as proposed previously for Fur based on Mössbauer and X-ray absorption spectroscopy (65). Recent structural studies of M. gryphiswaldense Fur indicate that Mn2+ (bound as an iron surrogate) is indeed hexacoordinate in this protein, with three His and two Glu ligands (32).
Collectively, these results support a simple model in which PerR proteins bind Fe2+ with five ligands and one open coordination site that can interact with H2O2, whereas Fur proteins use six ligands and the interaction of H2O2 with Fe2+ is sterically impeded. It would be revealing to determine if these same types of steric access effects influence the reactivity of bound Fe2+ with other molecules. In addition to H2O2, PerR can also be inactivated by O2 (100) and nitric oxide (NO) inactivates FurEc by direct nitrosylation of the bound Fe2+ (27, 28). Transcriptomic studies indicate that NO, as well as sodium nitroprusside, also inactivates both PerRBs and FurBs under both aerobic and anaerobic conditions (92). Finally, it has been shown in vitro that Fe2+ bound to Fur can be oxidized by K3Fe(CN)6 (89). It would be interesting to test the ability of these various redox agents to interact with PerRBs and FurBs and the corresponding mutants affecting the Fe2+ ligands. However, it is likely that it will ultimately be necessary to account for second coordination sphere effects and allosteric coupling to fully understand metal selectivity and peroxide reactivity, as noted in biophysical studies of other metalloregulators (49).
PerR and Fur Orthologs Across Species: Key Directions for Further Work
The revelation that varying a single amino acid in the Fe2+ coordination sphere of Fur/PerR type proteins can modulate H2O2 sensitivity (Fig. 2) is an important first step in understanding how these regulators have evolved distinct physiological roles. As evident from the literature, it is likely that there is a wide variation in H2O2 sensitivity among Fur and PerR orthologs in other organisms, and we are yet to tackle the complex problem of understanding how this variation has emerged. Certainly, variations in protein structure and the metal coordination environment are important factors, but the most relevant data relate to H2O2 sensitivity in vivo, and variations in the cellular environment are also likely important in interpreting the observed differences. As noted above, it is clear from numerous studies that Fur and PerR proteins can function with either Fe2+ or Mn2+ as corepressors. However, these two forms cannot be considered as equivalent. This is important when considering in vitro studies that often use Mn2+ as a cofactor to activate DNA binding, even for Fur proteins that function physiologically to sense Fe2+. With these caveats in mind, we here review recent insights into the effects of H2O2 and other oxidants on Fur and PerR function across a variety of bacteria, and identify some key questions to guide future research.
PerR orthologs with varying susceptibility to oxidation
PerR orthologs have been identified in a wide range of bacteria and seem to exhibit significant variation in their sensitivity to oxidation. However, comparative sensitivities are difficult to derive, given the differences in experimental parameters cited across the literature, including differences in media and treatment protocols for each organism tested. For example, the facultative anaerobe Staphylococcus aureus regulates peroxide stress using a PerR ortholog designated PerRSa (67% identical to PerRBs). Initial reports indicated that PerRSa repressed its target genes (e.g., katA, ahpCF, mrgA) only in the presence of Mn2+ (24, 58, 67). Indeed, in Fe2+-amended medium, the expression of katA was as high as in a perR null mutant. Although these studies led to the suggestion that PerRSa is functionally distinct, and is specialized to use Mn2+ as corepressor, this seems unlikely since this form (PerR:Zn,Mn) would not be able to respond to peroxide using the mechanism established for PerRBs (Fig. 4).
An alternative explanation is that under the aerobic growth conditions used, PerRSa is oxidized by endogenously generated H2O2 and only the Mn2+ form is able to accumulate to levels needed to effect repression. Support for this model is provided by recent in vivo and in vitro studies that allow a direct comparison of PerRSa and PerRBs (67). These studies reveal that the apparent lack of Fe2+-dependent repressor activity for PerRSa reflects a hypersensitivity to protein oxidation under aerobic culture conditions: Fe2+ is an effective corepressor under microaerobic conditions and the cell is poised to respond to increased oxygen availability and the resultant production of ROS (67). Biochemical studies also indicate that PerRSa maintains its ability to bind to both Fe2+ and Mn2+, and that PerRSa:Zn,Fe is more sensitive to H2O2 than either PerRBs or B. licheniformis PerR (PerRBl) (67, 69). This increased sensitivity likely confers this facultative anaerobic pathogen with the necessary fine-tuning to detect lower levels of peroxides.
PerR orthologs from other facultative aerobes and strict anaerobes may have similarly increased sensitivity to oxidation and may thereby function for survival under aerobic conditions (aerotolerance). Such is the case for Clostridium acetobutylicum PerR (PerRCa), which controls genes important for survival in the presence of H2O2 or when exposed to air (57). The sulfate reducing, obligate anaerobe Desulfovibrio vulgaris Hildenborough also may contain a highly sensitive PerR (114). In this organism, cells exposed to air for 30 min were more tolerant to subsequent challenge with H2O2. Similarly, PerR in the microaerophilic food pathogen C. jejuni is important for adaptation to aerobic conditions (50).
It is presently unclear whether PerR functions during the transition from anaerobic to aerobic conditions by sensing endogenously produced H2O2 or whether O2 itself suffices (100). Indeed, recent results indicate that O2 can modify PerRBs directly (100). Since this modification proceeds by a chemically distinct pathway (Fig. 4) and at least with PerRBs leads to a notable increase in the ratio of His37 to His91 oxidation (100), it may be possible to determine whether O2 is the true signal for PerR orthologs that function to sense the transition from anaerobic to aerobic growth conditions.
Fur orthologs with varying sensitivity to oxidation
Fur orthologs have also been identified from many different bacteria, and subject to the same caveats noted above, they seem to display a wide variation in their sensitivity to oxidation. Insights into the relative susceptibility to oxidation can be obtained by comparisons of the susceptibility of Fur proteins to inactivation in vitro and in vivo, although in both cases detailed side-by-side comparisons have generally not been done, and few studies have directly addressed the issue of sensitivity to ROS.
The vast majority of biochemical studies of Fur proteins have as their major goal the demonstration that Fur acts directly at a specific target promoter region, and for this purpose it is common to metallate Fur with the more redox stable Mn2+ rather than the more relevant Fe2+ cofactor. Nevertheless, there are several reports of activation of DNA binding by Fe2+ in vitro (1, 15). This contrasts with PerRBs, which is rapidly inactivated if Fe2+ is added to air-saturated buffer (73). Indeed, the original demonstration that FurEc functions as a direct sensor of Fe2+ was obtained by addition of Fe2+ to a coupled in vitro transcription/translation reaction (10) and subsequent DNA footprinting studies demonstrating activation of Fur binding by Fe2+ (30). Similarly, H. pylori Fur was active in vitro when reconstituted with Fe2+ as monitored by DNAse I footprinting (1, 31). Since DNA-binding activity was monitored aerobically in these studies, this suggests that the iron metallated form of Fur proteins is often relatively stable, even in air-saturated buffers. In one of the few side-by-side comparisons to date, incubation of anaerobically reconstituted FurBs with bound Fe2+ with a single molar equivalent (50 μM) of H2O2 led to a modest level of protein oxidation (23%), but notably less than that see for PerRBs under identical conditions (66%) (95).
We can also glean insights into the reactivity of Fur proteins to ROS from the large number of transcriptome and proteome studies of cellular responses to redox stress. For example, treatment of B. subtilis with either a low (8 μM) or high (58 μM) dose of H2O2 strongly induces the PerR regulon, but had only a modest effect, and only at the higher concentration, on Fur-repressed genes (55). However, a second study did find substantial derepression of the Fur regulon in cells treated with 58 μM H2O2 as well as by superoxide stress (93). Early studies also indicated that the E. coli Fur regulon was relatively insensitive to H2O2. Treatment of cells growing in rich medium with 1 mM H2O2 for 10 min strongly induced the OxyR-mediated stress response, but did not derepress the Fur regulon (118). In contrast with this result, an E. coli strain (hpx -) lacking all three major peroxide detoxification enzymes (katG, katE, and ahpCF) was found to derepress the Fur regulon in minimal medium. In this strain, endogenously generated H2O2 accumulates to levels between 500 nM and 1 μM, suggesting that this level is sufficient for oxidative inactivation of Fur (110). The authors further show that this derepression is not observed in rich medium due to the OxyR-dependent upregulation of Fur synthesis, which increases Fur levels from ∼5000 to 10,000 molecules per cell (117). The nature of the oxidative inactivation of FurEc is unclear and could result from oxidation and loss of the bound Fe2+ corepressor, with or without accompanying protein oxidation. It has been noted, however, that oxidation of FurEc bound with Fe2+ to the Fe3+ bound state did not appreciably decrease DNA binding (89), supportive of a model in which metal-catalyzed oxidation is required for protein inactivation.
Conclusions and Perspectives
Iron sensing and oxidative stress are intricately intertwined. The major physiological effects from ROS often involve the inactivation of iron-dependent enzymes. We have here reviewed how B. subtilis used two functionally distinct members of the Fur family of metalloregulators, FurBs and PerRBs, to coordinate the adaptive responses to iron limitation and peroxide stress, respectively. These proteins provide an attractive model system to begin to understand how protein structural changes can modulate both the metal selectivity and the sensitivity to H2O2 and other oxidants in this family of regulators. Fur and PerR proteins are widespread among bacteria, yet available evidence suggests that they vary markedly in their sensitivity to redox active compounds. These proteins seem to occupy a continuum of reactivity from highly sensitive (PerR proteins from facultative aerobes and anaerobes) to relatively nonreactive (some Fur proteins) (Fig. 5). Unfortunately, controlled side-by-side comparisons of redox reactivity are rare. PerR proteins are typically inactivated by a wide variety of redox stresses (O2, H2O2, HOCl, NO), and a subset of these agents, although sometimes only at much higher levels, may also affect Fur activity. Recent studies are beginning to shed light on some of the key differences in the metal-sensing sites that modulate redox sensitivity, but it is clear that much work remains to be done.
Footnotes
Acknowledgments
The authors thank Dr. Pete Chandrangsu from the Helmann Laboratory for his critical insight during the preparation of the article. This work was supported by a grant from the NIH (R35GM122461) to J.D.H.