
Abstract
Significance:
Yamanaka and colleagues galvanized the field of stem cell biology and regenerative medicine by their generation of induced pluripotent stem cells. Evidence is emerging that activation of innate immune signaling is critical for efficient reprogramming to pluripotency and for the nuclear reprogramming occurring in transdifferentiation.
Recent Advances:
We have shown that innate immune signaling triggers a global change in the expression of epigenetic modifiers to enhance DNA accessibility. In this state of epigenetic plasticity, overexpression of lineage determination factors, and/or environmental cues and paracrine factors, can induce pluripotency, or can direct transdifferentiation to another somatic cell lineage. Accumulating evidence reveals that innate immune activation triggers the generation of reactive oxygen species and reactive nitrogen species, and that these free radicals are required for nuclear reprogramming to pluripotency or for transdifferentiation.
Critical Issues:
We have discovered a limb of innate immune signaling that regulates DNA accessibility, in part, by the action of free radicals to induce post-translational modification of epigenetic modifiers.
Future Directions:
It is of scientific interest and clinical relevance to understand the mechanisms by which free radicals influence epigenetic plasticity, and how these mechanisms may be therapeutically modulated. Antioxid. Redox Signal. 29, 205–218.
Introduction
N
Here, we review recent progress in our understanding of the mechanisms by which innate immune signaling and free radicals drive epigenetic plasticity and nuclear reprogramming. Innate immune activation causes changes in the expression of epigenetic modifiers and promotes post-translational alterations in epigenetic modifiers, so as to promote DNA accessibility. In this review, we also discuss new data for a role of reactive oxygen species (ROS) as well as reactive nitrogen species (RNS) to promote nuclear reprogramming to pluripotency (141) or transdifferentiation (93). Our data suggest that free radicals may participate in promoting the epigenetic plasticity required for nuclear reprogramming (93, 141).
Somatic Cell Fate Is Not Fixed: Nuclear Reprogramming to Induce Pluripotency
The nuclear reprogramming of a somatic cell to a pluripotent stem cell, by overexpression of four transcription factors (Oct4, Sox2, Klf4, and cMyc), was a Nobel Prize-winning discovery and galvanized the fields of stem cell biology and regenerative medicine (122, 123). These iPSCs resemble embryonic stem cells (ESCs) in that they have an extraordinary capacity for self-renewal, and may differentiate into any somatic cell of the three germ layers (endoderm, ectoderm, and mesoderm). Because they can be differentiated into any cell lineage, patient-derived iPSCs may be used to elucidate pathobiology of any somatic cell type; may facilitate high-throughput screening of therapeutic molecules; or may be used to generate progeny for cell therapy or tissue engineering.
The scientific foundation for the generation of iPSCs was laid by John Gurton 50 years ago, when he induced pluripotency by nuclear transfer. He removed the nucleus of a fertilized egg cell from a frog and replaced it with the nucleus of a cell taken from a tadpole's intestine. This modified egg cell grew into a new frog, proving that the mature cell nucleus could be reprogrammed into a totipotent cell capable of generating all cell lines (46, 47). These studies indicated that factors within the enucleated egg could act on the somatic cell nucleus to radically change its transcriptional program. Years later, the induction of pluripotency by ectopic gene expression of a defined set of transcriptional factors (Oct4, Sox2, Klf4, and cMyc) increased the feasibility of generating pluripotent stem cells; facilitated studies of the molecular mechanisms of cell plasticity; and transformed the field of regenerative medicine (Fig. 1) (122, 123). This work revealed the remarkable fluidity of somatic cell fate in response to the overexpression of master regulators of cell lineage.
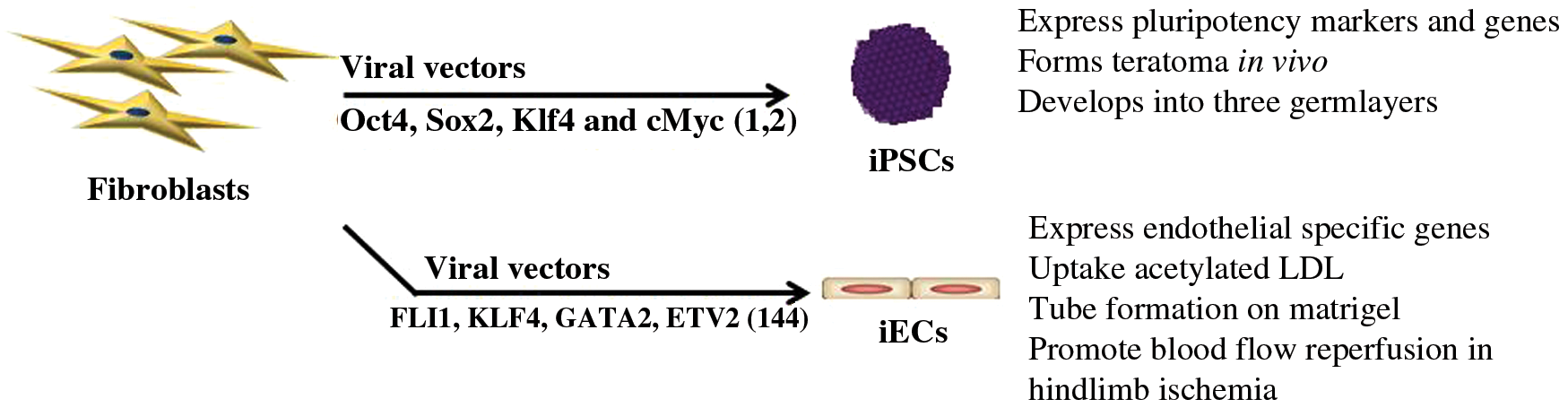
The iPSCs generated by nuclear reprogramming are able to self-renew, to differentiate into all three germ layers, and to any somatic cell type. Thus, they are functionally identical to ESCs, although they may bear some epigenetic traces of the somatic cell from which they were derived. It is now straightforward to generate iPSCs, and they can be readily used to study the mechanisms of pluripotency and differentiation. Furthermore, one can use patient-derived iPSCs to understand pathobiology. One may use somatic cells that are easily obtained (e.g., peripheral blood mononuclear cells), to generate the patient-specific iPSCs, and then differentiate these into cells affected by the disease that may not be so easily obtained (e.g., neurons). These iPSC derivatives may then be used in high-throughput screens for new therapeutic candidates (14, 25, 32, 54, 59, 84, 103, 104, 111, 136). Finally, iPSC technology makes it feasible to produce large amounts of therapeutic cells for regenerative engineering (23).
A Missing Link in Nuclear Reprogramming: Activation of Innate Immunity
Mouse (123) and human iPSCs (hiPSCs) (122) were first generated using retroviral or lentiviral vectors to overexpress the four Yamanaka factors (Oct 4, Sox 2, Klf4, and cMyc) in murine and human fibroblasts. Although the Yamanaka factors are master regulators of the core pluripotency network, we discovered that the viral vectors that carried the Yamanaka factors also contributed to the reprogramming process (74). Indeed, when the Yamanaka factors are administered in the absence of a viral vector (i.e., in the form of cell-permeant peptides), they are not effective at inducing nuclear reprogramming to pluripotency. This lack of efficacy is despite the fact that the cell-permeant Yamanaka factors can bind to their consensus sequence; can enter the cell nucleus; and can even rescue iPSCs where one of the Yamanaka factors is knocked down (74). We discovered instead that the lack of efficacy of the cell-permeant Yamanaka factors was related to their inability to activate innate immunity. However, when the cell-permeant Yamanaka factors are delivered together with an activator of innate immunity, they now had the expected transcriptional effects (74).
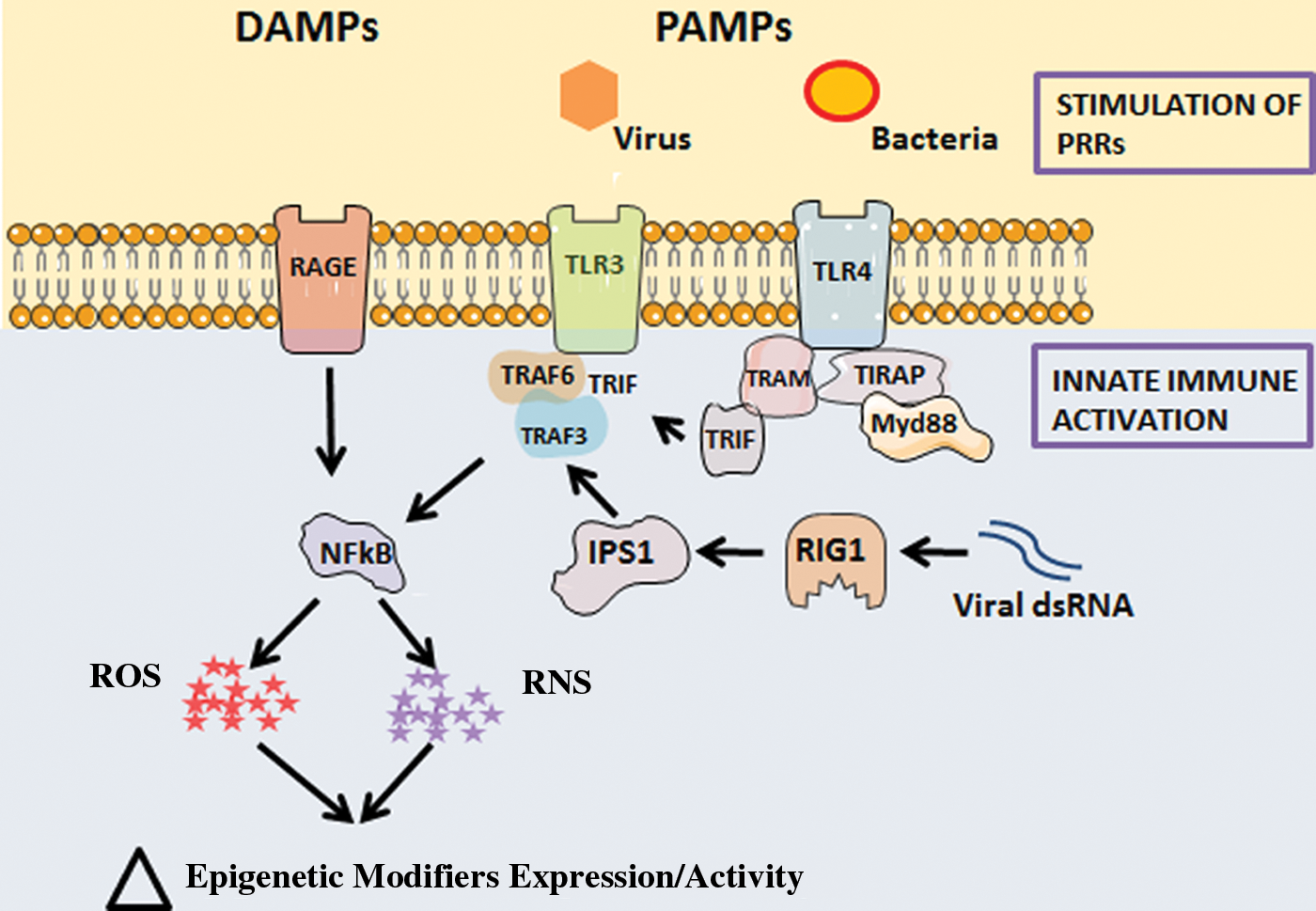
We found that during reprogramming using the Yamanaka method, the toll-like receptor (TLR) 3 is activated (Fig. 2). Indeed, genetic abrogation of TLR3 signaling markedly impaired reprogramming to pluripotency using the Yamanaka approach (74). The TLR signaling pathway was also required for other reprogramming methods, such as when using modified messenger RNA (mRNA) encoding the Yamanaka factors. In another approach to generating iPSCs, we used mouse embryonic fibroblasts (MEFs) carrying a doxycycline-inducible cassette encoding the Yamanaka factors (dox-inducible MEFs). The exposure of the cells to doxycycline generates murine iPSCs within about 2–3 weeks. The yield of iPSCs was increased by stimulating the cells with the TLR3 agonist polyinosinic-polycytidylic acid (polyI:C). By contrast, inhibition of innate immune activation using an NFκB decoy reduced the yield of iPSCs. In this regard, we observed that in the absence of polyI:C, there was a basal activation of NFκB in the MEFs, perhaps explaining the observation that MEFs are easily reprogrammed to pluripotency (74).

Subsequently, we observed that innate immune signaling caused widespread alterations in the expression and activity of epigenetic modifiers (74). These changes include a profound downregulation of epigenetic factors that enforce suppressive epigenetic marks, such as Dot1L and members of the histone deacetylase family. Conversely, innate immune signaling increases expression of members of the histone acetyltransferase family, favoring an open chromatin configuration. We observed that TLR3 stimulation during reprogramming enhanced H3K4 trimethylation (H3K4me3, active histone marker) and reduced H3K27 trimethylation (H3K27me3, repressive histone marker) at the Oct4 and Sox2 promoters (74). These changes of histone markers on promoters of pluripotent genes are consistent with a transcriptionally active state.
The retinoic acid-inducible gene 1 (RIG-1)-like receptors (RLRs) also detect the presence of viral RNA to initiate inflammatory signaling (66, 138). The RLR proteins comprise RIG-I, MDA5, and LGP2 in mammalian cells. Interferon-beta promoter stimulator 1 (IPS-1; i.e., mitochondrial antiviral-signaling protein) is a key protein in the chain of signaling molecules needed for RLR-mediated generation of interferon in response to a pathogen (67). When we knocked down this key intermediary protein, we observed a marked reduction in the yield of iPSCs during nuclear reprogramming (114). In unpublished studies, we also find that low concentrations of bacterial lipopolysaccharide (LPS; a TLR4 agonist) facilitate nuclear reprogramming. These studies suggest that it is a general characteristic of innate immune activation to set in motion a cascade of events that result in epigenetic plasticity.
Transflammation: The Role of Innate Immune Signaling in Nuclear Reprogramming
“Transflammation” is the term that we have coined to describe the process by which innate immune activation enhances epigenetic plasticity (90, 92). When a cell is exposed to pathogens or damage, pattern-recognition receptors (PRRs) activate innate immune signaling to facilitate epigenetic plasticity. The phenomenon of transflammation is required for efficient nuclear reprogramming to pluripotency (74) as well as in transdifferentiation to other cell lineages (115).
Our data suggest that transflammation plays a role on the front lines of host defense against infiltrating pathogens. Pathogens present pathogen-associated molecular patterns (PAMPs) to receptors on the surface of mammalian cells (1, 60, 87, 95). PAMPs are highly conserved features displayed by large families of pathogens (87). PAMPs bind to PRRs to activate innate immune response. The PRRs include the TLRs (2, 88, 89) as well as cytosolic PRRs, which include nucleotide-binding oligomerization domain-like receptors (NLRs) (65), and RLRs (138). Generally speaking, TLRs form the first line of defense by detecting pathogens on the cell surface and in endosomes, whereas RLRs and NLRs recognize microbial elements in the cytosol (13, 124). In addition, TLRs at the cell surface may detect damage-associated molecular patterns (DAMPs) (96), such as those presented by a necrotic cell (29).
Activation of the PRRs triggers signaling cascades that lead to the activation of AP1, ERK, NFκB, p38, and/or other antiviral signaling pathways through IRFs (41). Activation of these pathways generates cytokines and chemokines that initiate the inflammatory response to a microbe, foreign body, or cell debris (90). Our work indicates that this same innate immune signaling triggers widespread alterations in the activity and expression of epigenetic modifiers so as to promote DNA accessibility and phenotypic fluidity. This process of transflammation facilitates the phenotypic fluidity that a cell requires to respond to a pathogenic challenge or injury (90).
Innate immune activation increases ROS signaling (101, 106, 137, 141). Furthermore, ROS are known to activate NFκB (11, 35). Reciprocally, NFκB promotes Nox2 expression in MEFs, which subsequently increases the generation of ROS (3). Intriguingly, we observed that ROS signaling is activated at the onset of nuclear reprogramming. Furthermore, we found that an optimal level of ROS signaling is required to generate iPSCs (141).
Free Radicals and Cell Signaling
As well known to the readership of this journal, free radicals have one or more unpaired electrons and as such are highly and rapidly reactive (16, 31, 48, 56, 108, 126). The role of ROS and RNS in cell signaling and defense has been intensively studied. ROS refer to reactive chemical species containing oxygen and are generated in several enzymatic and nonenzymatic reactions (79, 108). The most common ROS in biological systems include superoxide (O2 −), hydroxyl radical (HO•) (27), and nonradical species such as hypochlorous acid (HOCl), hydrogen peroxide (H2O2), and ozone (O3). Generation of ROS occurs in the mitochondria (complex I and III) as well as in the cytosol through the action of NADPH oxidases, lipoxygenases, xanthine oxidase, and cytochrome P450 (49, 97). At low or moderate levels, free radicals have a role in cellular signaling (27), in both physiological and pathophysiological conditions (38). Excessive generation of ROS may contribute to pathobiology of multiple diseases (108). ROS level is tightly regulated by antioxidant processes mediated by superoxide dismutase, catalase, peroxiredoxins, thioredoxin, glutathione peroxidase (GPX), and glutathione reductase.
RNS include nitrogen-derived molecules such as nitric oxide (NO•), peroxynitrite (ONOO−), nitrogen dioxide (•NO2), and dinitrogen trioxide (N2O3) (31, 119). Endogenous NO is generated from the metabolism of
The concentration of NO dictates its biological effects. At picomolar concentration, it reacts with ferrous (Fe2+) heme proteins to activate soluble guanylyl cyclase (sGC) (6). At moderate concentrations, it reacts with lipid peroxide and thiol groups to form S-nitrosothiols, and at higher concentrations, it reacts with oxygen or ROS in oxidative reactions (7, 44, 53). Posttranslational modification by NO includes nitration or S-nitrosylation of proteins. Protein nitration occurs when a nitro (NO2) group covalently bonds to the tyrosine residue of protein. (58) The covalent modification by NO of a thiol (R-SH) group of a cysteine residue is called protein S-nitrosylation (82). S-nitrosylation is an important, reversible post-translational modification involved in cell signaling, which we find is also involved in promoting epigenetic plasticity in the transdifferentiation of fibroblasts to endothelial cells (93).
The Role of Free Radicals in Innate Immunity
ROS are downstream effectors of innate immune signaling (26, 43, 70, 102). A major source of ROS in activated neutrophils and macrophages is NADPH oxidase (5, 61). The ROS generated by these inflammatory cells further amplify the inflammatory response by causing the generation of additional DAMPs (18). For example, DAMPs may be generated by the ROS-induced oxidation of extracellular matrix proteins such as heparan sulfate (69). Within the cell, ROS mediate both negative and positive feedback loops to control NFκB activation and ROS generation (11, 15, 22, 78, 112) (summarized in Fig. 3).

Evidence that RNS such as NO might participate in the immune response arose in the early 80s when it was shown that plasma nitrite and nitrate levels increase during infection, suggesting increased endogenous production of NO (45). Subsequently, it was shown that IL-2 increased plasma NOx levels in patients and facilitated antitumor response in mice (52). Stimulation of membrane-bound or cytosolic PRRs by PAMPs activates NFκB to generate type I interferons such as IFN-α/β. In addition, NFκB binds to its consensus sequence on the promoter of iNOS, to induce its expression in macrophages and other cell types, which cells in turn generate substantial quantities of NO to contribute to immune response (68).
NO may modify the function of other proteins via S-nitrosylation of cysteine residues (30). Elements of the signaling cascades that lead to activation of NFκB, and the transcriptional factor itself, are modulated by S-nitrosylation. For the most part, S-nitrosylation of these elements (e.g., MyD88, CD40, IKKβ, p50, and p65) inhibits inflammation. By contrast, inflammation may be exacerbated by S-nitrosylation of surfactant protein-D and Src (51). S-nitrosylation of apoptosis signal regulation kinase 1 (ASK1) inhibits its binding to, and phosphorylation of, its substrates (107). ASK1 modulates both NFκB and apoptotic pathways downstream of TLR2 and significantly regulates the signaling pathway involving TLR4-TRAF6-p38 MAPK (86). Caspase-1 can cleave the TIR domain-containing adaptor protein TIRAP thus regulating both TLR2- and TLR4-mediated MyD88-dependent pathways (94). The proteolytic activity of caspase-1 is suppressed by S-nitrosylation of one cysteine residue within its enzymatic active site (83). It is evident that S-nitrosylation is a key post-translational modification of elements of the innate immune signaling pathway.
ROS and Reprogramming to iPSCs
Although pluripotent stem cells are characterized by glycolytic metabolism and low generation of ROS, we have recently discovered that the initial phase of nuclear reprogramming of somatic cells to iPSCs begins with a burst of ROS generation (141). This oxidative activity becomes attenuated in the later stage of nuclear reprogramming by an upregulation of antioxidant enzymes.
During reprogramming to iPSCs, both the intensity and kinetics of ROS signaling are critical for efficient nuclear reprogramming (summarized in Fig. 4) (141). Specifically, ROS signaling is required during the early stages of reprogramming to pluripotency, whereas ROS generation is opposed at later stages of reprogramming by an upregulation of antioxidant enzymes.

We generated iPSCs from MEFs carrying a doxycycline-inducible cassette of the Yamanaka factors (141). In the early phase (0–7 days) of reprogramming to iPSCs, there is an increase in ROS generation as assessed by dihydroethidium (DHE), which is associated with an upregulation of NADPH oxidase family members Nox1 and Nox2. Notably, p22phox knockdown or knockout significantly reduced reprogramming efficiency. To confirm the importance of ROS generation to reprogramming, we used ROS scavengers (EUK134, Epselen, or Mito-TEMPO) or inhibitors of NOx (diphenyleneiodonium and apocynin). These studies revealed that iPSC generation was compromised by scavenging or inhibiting the generation of ROS in the early phase of nuclear reprogramming. By contrast, in the later stage of reprogramming (>7 days), ROS scavengers or NOx inhibitors did not affect reprogramming. Instead, the later stage of reprogramming was characterized by a fall in intracellular ROS levels, associated with an increase in the expression of antioxidant enzymes. The antioxidant genes (Nrf2, Sod1, Sod2, Gpx4) began to increase in the early phase of reprogramming and plateaued at a higher level of expression in the later stage, although some (Gpx1 and Gpx2) increased throughout the reprogramming process (141).
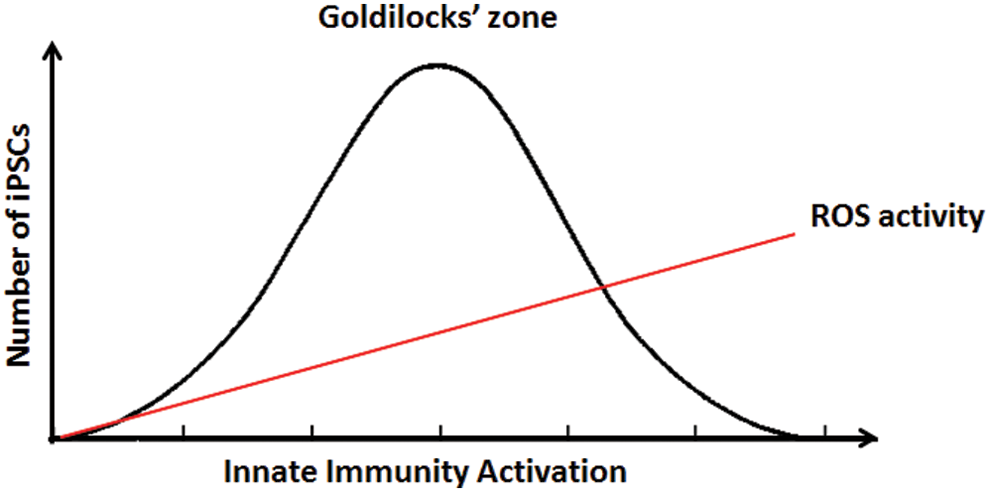
Unexpectedly, genetic or pharmacological measures to increase ROS generation actually impaired reprogramming to pluripotency (141). An MEF cell line overexpressing Nox2 generated fewer iPSCs. Furthermore, the addition of buthionine sulfoximine (BSO; for the first 12 days of reprogramming) also reduced iPSC yield. Because BSO inhibits the synthesis of glutathione, it would be expected to increase intracellular ROS. Furthermore, the administration of exogenous H2O2 (for the first 12 days of reprogramming) at higher doses in a physiological range reduced the yield of iPSCs, although at lower doses of H2O2, there was a tendency for iPSC generation to be enhanced. Taken altogether, our studies indicate that an optimal increase of ROS levels is necessary for efficient reprogramming. Indeed, there appears to be “Goldilocks zone” of optimal innate immune activation and ROS generation in the early phase of nuclear reprogramming (Fig. 5). Furthermore, at later stages of reprogramming, ROS levels decline as antioxidant mechanisms are increased. In the mature iPSC, oxidative phosphorylation is attenuated with a shift to glycolytic metabolism (141).
The reduction in ROS levels in later stages of reprogramming provides for reduced oxidative stress in the mature iPSC (141). As also reported by others, we find that iPSCs have lower levels of ROS by comparison to the MEFs from which they are derived. Specifically, flow cytometry using DHE or dichlorodihydrofluorescein diacetate (CM-H2DCFDA) indicated that ROS levels were 50% lower in iPSCs by comparison with parental MEFs. We confirmed these results using a redox-sensitive green fluorescent protein. We noted that in the iPSCs, NADPH oxidase family members (Nox1, Nox2, Nox3, and Nox4) are downregulated, while antioxidant genes (Sod1, Sod2, Gpx1, Gpx2, Gpx3, and Gpx4) are upregulated compared with the parental MEFs (141).
Although NADPH oxidase family members are important for ROS generation during nuclear reprogramming, it is known that iPSCs can still be generated from fibroblasts derived from patients with chronic granulomatous diseases where NADPH oxidase activity is severely impaired (12, 143). It is possible that in this case, the source of ROS for iPSC generation comes from other cytosolic or mitochondrial sites. Indeed, it appears that mitochondrial-derived ROS play a role in iPSC generation. We found that mito-TEMPO (which preferentially scavenges ROS generated by the mitochondria) (141) modestly reduced iPSC yield.
The mechanisms by which an increase in intracellular ROS facilitates iPSC generation remain unclear. However, it is well known that an increase in ROS levels can increase NFκB activation. As described above, NFκB activation by innate immune signaling plays a key role in nuclear reprogramming.
It is also reported that free radicals generated during reprogramming may cause oxidative DNA damage and p53 activation, which hindered efficient iPSC generation (81). Antioxidant supplements have also been reported to not alter the efficiency of reprogramming to iPSC (36, 62) but reduce genomic aberrations (62). Interestingly, vitamin C, a well-known antioxidant, enhanced reprogramming efficiency independent of its antioxidant property (36, 120). Although these observations seem opposed to ours, the conflict may be more apparent than real, and explained by the phenomenon of the “Goldilocks zone” that we have described. Specifically, insufficient or excessive activation of innate immune signaling and free radical generation may each impede nuclear reprogramming.
iPSCs and ESCs Maintain Low ROS Status
Although activation of ROS signaling is required for the induction of pluripotency, once iPSCs are generated, ROS generating enzymes are downregulated (141). Indeed, it is well recognized that iPSCs exist in a state of glycolysis, as opposed to the somatic cells from which they are derived, which favor oxidative phosphorylation (39, 134). The somatic (differentiated) cells substantially utilize mitochondrial oxidative phosphorylation to generate adenosine triphosphate (ATP) (28, 39). By contrast, pluripotent stem cells (ESCs and iPSCs) preferentially generate ATP by glycolysis (39, 110), which is reflected by smaller and fewer mitochondria (39, 110), less ATP production, and restrained mitochondrial activity (4, 39, 110). Notably, iPSCs maintain low ROS level compared with somatic cells from which they are derived (141), propagate better in low-oxygen conditions (50), and are prone to apoptosis and growth arrest in excessive ROS condition (132). This is consistent with ESCs, which also adopt glycolytic ATP as a major energy source (21, 71) and contain low copy number of mitochondrial DNA (19).
ROS signaling plays an important role in stem cell homeostasis and lineage commitment, which has been reviewed previously (9, 116). Reduced electron transport chain coupling in pluripotent stem cells lessens the production of ROS (24, 113, 139, 140). The increased expression of antioxidant enzymes also maintains intracellular levels of ROS at a low level in pluripotent stem cells (17, 141). By maintaining a low level of ROS generation, pluripotent stem cells limit the risk of cellular and genomic damage during self-renewal and prevent differentiation (98, 113, 141). As pluripotent stem cells differentiate into somatic cells, mitochondrial superoxide production and cellular levels of ROS increase in association with an augmentation of mitochondrial biogenesis. Concordantly, there is a reduction in the expression of major antioxidant genes despite increased oxidative stress (113). During ESC differentiation, decreased levels of NADH as well as reduced glutathione are consistent with an increase in the overall cellular oxidation state (129, 135).
Patient-specific iPSC-derived cells recapitulate the disorders of ROS signaling that may be characteristic of a disease pathobiology (20, 128). For example, hiPSCs, from patients with myoclonus epilepsy associated with ragged-red fibers, may be differentiated into cardiomyocyte (CM)-like cells or neural progenitor cells, each of which exhibit abnormal mitochondrial ultrastructure and increased ROS levels (20). CMs derived from hiPSCs of Barth syndrome (BTHS) patients showed increased ROS levels, whereas suppression of ROS rescued the aberrant metabolic, sarcomeric, and contractile phenotypes of BTHS hiPSC-CMs (128).
Other Forms of Nuclear Reprogramming
Transdifferentiation also requires nuclear reprogramming to facilitate the metamorphosis of a somatic cell into a somatic cell of a different lineage. The process of transdifferentiation may occur in pathological conditions, such as in patients with chronic esophageal reflux and acid injury to the esophagus. In this condition, the chronic injury is associated with so-called Barrett's esophagus where the squamous epithelial cells of the esophagus undergo a premalignant alteration, transforming into cells reminiscent of the intestinal columnar epithelium (121). Transdifferentiation also plays a role in atherosclerosis, for example, when vascular smooth muscle cells transdifferentiate into macrophage-like cells that imbibe lipid and contribute to the growth of plaque (37). Whereas these are examples of pathological transdifferentiation, the same underlying epigenetic mechanisms may serve therapeutic purposes. Therapeutic transdifferentiation is a novel therapeutic avenue under development, through which somatic cells that may contribute to the pathobiology are transformed into cells that may restore homeostasis, for example, as in the therapeutic transdifferentiation of fibroblasts into endothelial cells, to generate a microvasculature that can enhance perfusion in the setting of ischemia (115).
Transdifferentiation is also known as direct reprogramming. This metamorphosis of a somatic cell into one of a different lineage requires global alterations in the epigenetic and transcriptional signature of the cell (34). The first experimental reprogramming of a somatic cell nucleus was achieved by cell fusion (10). For example, fusion of a myocyte with a hepatocyte caused the hepatocyte to express muscle proteins (10). The effect of cell fusion may be mimicked by overexpression of specific lineage determination genes. For example, much of the effect achieved by fusion of a liver and muscle cell can be recapitulated by the overexpression in hepatocytes of MyoD, a transcription factor specific for muscle cells (130). Subsequently, a variety of specific lineage factors have been overexpressed to induce direct reprogramming (23), for example, of fibroblasts into endothelial cells (77), cardiac muscle cells (99), or neurons (118).
Whereas transdifferentiation has long been known to participate in mammalian development, its role in the adult mammal has only recently been defined. The use of lineage tracing methods has facilitated the investigation of transdifferentiation in adult mammalian physiology and pathobiology. The process of endothelial-to-mesenchymal transition (EndoMT) is a pathological transdifferentiation contributing to fibrotic disorders. Specifically, EndoMT is an aberrant transdifferentiation of endothelial cells to fibroblasts, which is associated with a loss of capillary density and an increase in fibrosis (109). This process may play a role in myocardial and renal fibrosis. Intriguingly, recent evidence (125) suggests that the reverse transition, mesenchymal-to-endothelial transition, may be a physiological adaptation to ischemia that participates in angiogenesis. Specifically, in an experimental model of myocardial ischemia, physiological reprogramming of fibroblasts to endothelial cells contributes to an increase in capillary density that normalizes myocardial perfusion (125). Thus, it is quite likely that modulation of transdifferentiation might be a novel therapeutic avenue. Indeed, the forced expression of lineage-determining factors in vivo has been used to generate a therapeutic cell type (CMs or pancreatic β cells). Although the in vivo yield is low, these transdifferentiated cells are associated with improvements in organ function (cardiac or pancreatic function, respectively) in experimental models (57, 142). Small-molecule-based methods are an alternative to gene therapies, and might be a more feasible clinical approach, with promising results in vitro (33, 72).
Therapeutic transdifferentiation in vivo should be restricted in space and time to avoid off-target effects. This might be done by locally administering DNA (in plasmids or viral vectors) or mRNA (using a nanoparticle vector) encoding the transdifferentiation factors into the tissue to facilitate the reprogramming of tissue cells (e.g., fibroblasts) to a beneficial cell type (e.g., endothelial cells). Therapeutic reprogramming of cardiac mesenchymal cells into endothelial cells might be of benefit in the setting of an acute ischemic injury to the heart. Cardiac fibroblasts that are proliferating and migrating to the zone of myocardial damage could be reprogrammed into endothelial cells that would expand the microcirculation (90). These reprogrammed endothelial cells would thus supply the nutrition and the niche for myocardial regeneration rather than fibrosis.
Most investigators have induced direct reprogramming using viral vectors encoding transcriptional factors regulating the desired cell type (90). Perhaps a more feasible approach from regulatory and operational perspectives would be an RNA or small-molecule strategy that would obviate the concerns related to integration of foreign DNA into the patient's genome. We have developed a new strategy for nuclear reprogramming based on our discovery of transflammation, in which epigenetic plasticity is facilitated by innate immune signaling (115).
Innate Immune Signaling, RNS, and Transdifferentiation
Viral vectors encoding lineage determination factors have been used to transdifferentiate a cell of one lineage into another (55, 127, 131). Our realization that innate immune signaling was required to generate iPSCs caused us to wonder if transflammation may also be involved in transdifferentiation of somatic cells. We believe that it is likely that innate immune signaling is required for the induction of transdifferentiation using viral vectors encoding lineage determination factors. If this is true, it might be possible to induce transdifferentiation by activating innate immunity with a small-molecule agonist, rather than a viral vector (so as to increase epigenetic plasticity) together with small molecules or recombinant proteins that promote endothelial lineage. Indeed, recently we showed that fibroblasts could be effectively transdifferentiated into endothelial cells with a formulation that included a TLR3 agonist and endothelial growth factors (115). We primed the fibroblasts with the TLR3 agonist polyI:C to induce epigenetic plasticity, and then added small molecules and endothelial growth factors to promote endothelial lineage (VEGF, BMP4, FGF, 8-Br-cAMP, and a TGFβ receptor antagonist) (115). Using this protocol, we observed that some of the treated fibroblasts transdifferentiated into endothelial cells. These induced endothelial cells (iECs) resembled authentic endothelial cells rather than the parental fibroblasts from which they were derived. The iECs manifested the typical surface markers of endothelial cells. Furthermore, the iECs were capable of endothelial processes such as nitric oxide synthesis, the incorporation of acetylated low-density lipoprotein (LDL), and the formation of networks in Matrigel. The transcriptome of the iECs and that of authentic endothelial cells was almost identical and quite different from that of parental fibroblasts. Furthermore, we observed the expected epigenetic marks on typical endothelial genes (115) (e.g., increased amounts of activating marker H3K4me3 and reduced amounts of the repressive marker H3K27me3 on CD31 gene promoter).
Notably, the generation of iECs required activation of innate immunity. In the absence of priming with the TLR3 agonist polyI:C, the endothelial growth factors alone did not transform the parental fibroblasts, which retained their original phenotype. As further evidence, in TLR3-deficient fibroblasts, iEC generation was reduced; moreover, these iECs were incompletely reprogrammed. Specifically, these iECs manifested an impairment of typical endothelial processes such as network formation on Matrigel and incorporation of acetylated LDL. The transcriptional profile of the TLR3 KD iECs was also consistent with incomplete generation of an endothelial program (115).
Furthermore, we found that RNS mediated the innate immune activation of epigenetic plasticity (93). Innate immune activation induces iNOS expression via NFκB (73, 133). Subsequently, the generation of NO leads to the activation of sGC, which in turn generates the second messenger cGMP. In addition, the generated NO may covalently bind to cysteine residues of proteins, which S-nitrosylation may alter protein structure, function, and binding partners (30). Prior reports have suggested that the activity of epigenetic modifiers such as histone deacetylase 2 might be modulated by S-nitrosylation (105). Furthermore, it has been reported that iNOS may undergo nuclear translocation in malignant (75) and normal cells (42, 64). However, the physiological significance of a nuclear location of iNOS is not clear. Screening assays indicated that several other epigenetic modifiers are capable of being S-nitrosylated (40, 76, 117). It is not known how S-nitrosylation affects the behavior of these epigenetic modifiers.
The Role of iNOS in Transdifferentiation
We found that iNOS plays a critical role in transdifferentiation of fibroblasts to endothelial cells in vitro (93) (Fig. 6). During innate immune activation, we find that iNOS is upregulated during transdifferentiation and is associated with increases in intracellular NO, with increased generation of protein S-nitrosylation. More notably, we found that both knockdown of iNOS in human fibroblasts and knockout of iNOS in murine fibroblasts nearly abrogated the transdifferentiation process. Similarly, NOS antagonists and inhibitors of innate immune signaling impaired the generation of iECs (93).
In addition, we noted that during transdifferentiation, iNOS underwent nuclear translocation. We further demonstrated that iNOS binds to RING1A, an important protein within the polycomb repressive complex 1 (PRC1). The binding of iNOS to RING1A is associated with S-nitrosylation of RING1A and its dissociation from the chromatin. This effect would be expected to reduce the repressive influence of PRC1, and thus favor an open chromatin configuration (93). Indeed, we observed a decrease in suppressive markers (H3K9me2 and H3K27me3) in the promoter region of CD144, a gene that is active in endothelial cells.
Related to these findings, a sequence motif recognition site for iNOS binding has recently been described (63). A conserved I/L-X-C-X2-D/E motif is a target for an inducible heterotrimeric S-nitrosylase complex that consists of iNOS, S100A8, and S100A9. Intriguingly, a preliminary bioinformatic search of the protein sequences for all known epigenetic modifiers reveals this sequence on 20% of the nuclear epigenetic modifiers. Accordingly, S-nitrosylation may regulate the activity of epigenetic modifiers. Curiously, RING1A lacks this motif, so there may be a variant consensus sequence for iNOS binding, which could further increase the number of potential epigenetic targets of S-nitrosylation. This physical association of iNOS with epigenetic modifiers may provide specificity for its action. This notion is supported by our finding that we were not able to rescue iEC generation in iNOS−/− MEFs using the NO donor SIN1.
Protein denitrosylation refers to the removal of NO groups from the thiol side chain of cysteine. Importantly, protein S-nitrosylation can be actively denitrosylated through enzyme-mediated processes (8) such as S-nitrosoglutathione reductase and thioredoxin reductase. We speculate that inhibition of denitrosylase would enhance intracellular level of protein S-nitrosylation and thus may enhance transdifferentiation efficiency.
Perspectives and Significance
Transflammation describes a phenomenon by which activation of innate immune signaling increases DNA accessibility so as to favor phenotypic fluidity. This process may be seen as an adaptation to the challenge represented by a pathogen or local damage. Transflammation requires the generation of RNS and ROS. These free radicals alter cytoplasmic signaling to favor transcriptional events mediated by NFκB, mediating global changes in the expression of epigenetic modifiers. In addition, RNS-mediated S-nitrosylation of epigenetic modifiers alters their activity so as to favor an open chromatin state. Based on our unpublished work, there appears to be a “Goldilocks zone” of innate immune activation and ROS signaling for optimal nuclear reprogramming. Insufficient or excessive activation of innate immunity is each associated with impaired nuclear reprogramming. These observations lead us to speculate that there may also be an optimal zone of innate immune signaling in tissue regeneration in vivo. Some degree of inflammatory signaling seems necessary for tissue healing in humans. For example, suppression of inflammatory signaling (as in people receiving intensive steroid therapy) might explain the observation that wounds heal poorly in these individuals. On the contrary, excessive activation of inflammatory signaling (as observed in “fixed inflammation” circumscribing a diabetic foot ulcer) may also slow the rate of healing. We intend to comprehensively characterize the epigenetic and transcriptional determinants of transflammation, with the hope that this work will provide new therapeutic avenues to enhance regenerative therapies.
Footnotes
Acknowledgment
This work was supported, in part, by grants to J.P.C. from the National Institutes of Health (U01HL100397 and RC2HL103400).