
Abstract
Significance:
Reductionist studies have contributed greatly to our understanding of the basic biology of aging in recent years but we still do not understand fundamental mechanisms for many identified drugs and pathways. Use of systems approaches will help us move forward in our understanding of aging.
Recent Advances:
Recent work described here has illustrated the power of systems biology to inform our understanding of aging through the study of (i) diet restriction, (ii) neurodegenerative disease, and (iii) biomarkers of aging.
Critical Issues:
Although we do not understand all of the individual genes and pathways that affect aging, as we continue to uncover more of them, we have now also begun to synthesize existing data using systems-level approaches, often to great effect. The three examples noted here all benefit from computational approaches that were unknown a few years ago, and from biological insights gleaned from multiple model systems, from aging laboratories as well as many other areas of biology.
Future Directions:
Many new technologies, such as single-cell sequencing, advances in epigenetics beyond the methylome (specifically, assay for transposase-accessible chromatin with high throughput sequencing ), and multiomic network studies, will increase the reach of systems biologists. This suggests that approaches similar to those described here will continue to lead to striking findings, and to interventions that may allow us to delay some of the many age-associated diseases in humans; perhaps sooner that we expect. Antioxid. Redox Signal. 29, 973–984.
Introduction
A
In recent decades, work in laboratory-based genetic model organisms—the nematode Caenorhabditis elegans, the budding yeast Saccharomyces cerevisiae, the fruit fly Drosophila melanogaster, and the mouse Mus spp.—has demonstrated very large changes in lifespan from single mutations, or from combinations of small number of different mutations. This work illustrates the extreme plasticity of this phenotype in the laboratory in response to genetic alterations (3, 54, 56, 60, 76, 95, 101). Interestingly, in some cases, mutations of the same gene in all these species have extended lifespan, not only in invertebrate models but also in mammals (9, 39, 67, 68), suggesting conservation of phenotype over many hundreds of millions of years.
Obviously, if any of these interventions were shown to have a beneficial impact on human healthspan, the biological and social impact would be considerable. In fact, some pharmacological interventions are now moving into the testing phase in companion dogs (50, 104) and humans (7, 73).
The very nature of aging lends itself to a systems biology approach. Perhaps more than just about any other biological phenomenon, aging is notable for its diversity. Despite some common and evolutionarily conserved hallmarks of aging (72), we see tremendous variation in the consequences of aging, among tissues within a single individual, and among individuals and species. And one of the clear messages from the past 25 years of molecular studies of aging is that the molecular causes of aging are great in number and show highly complex interactions.
Our view of the causes and consequences of aging is illustrated in Figure 1. Some individuals are healthier agers than others. At a population level, variation in the risk of different age-related traits (the phenotypes, P) is shaped by variation in genes (G), the environment (E), and the interaction between those two factors, as well as age itself. We can illustrate this relationship using the classic quantitative genetic equation:
From an aging perspective, as we illustrate in Figure 1, we would add that age influences phenotypes, and does so in a way unique to each individual, determined by genetic and environmental factors. However, Equation [1] is agnostic about the mechanisms by which G and E shape P. We hypothesize that this gap between G and E and P can be bridged by omic analysis (in particular, the epigenome, transcriptome, proteome, metabolome, and microbiome). This conceptual model has, in turn, led us to recognize the power of a systems biology approach, one that embraces all domains, from genes and environment, to downstream aging-related phenotypes, and the intermediate omic levels that bridge the two.
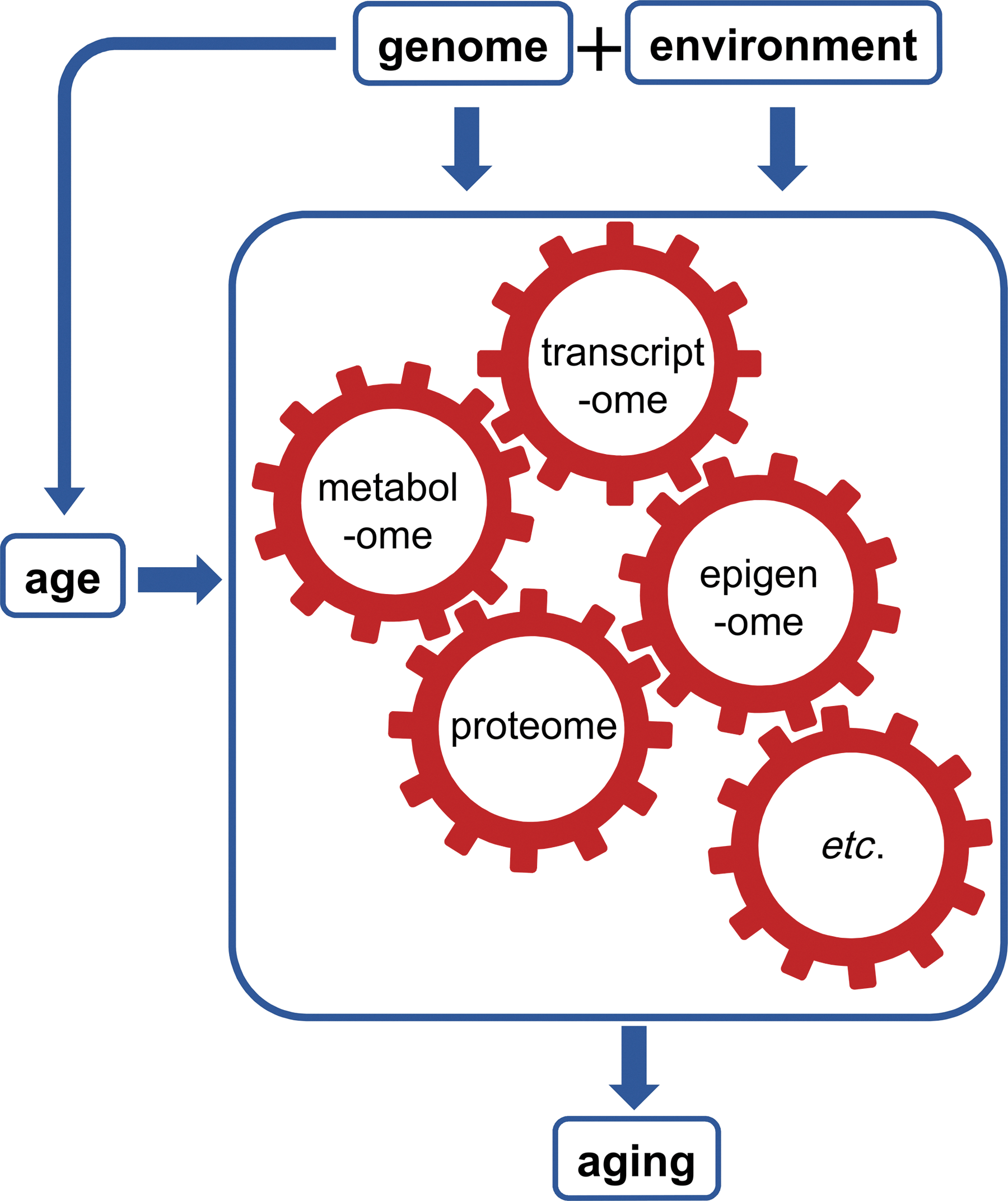
Here we explore ways that further progress in aging research can be made through systems biology. Systems biology, broadly, is defined by an integrated set of approaches that can include large-scale data sets, analysis at multiple levels of biological organization, computational models (often focused on construction and analysis of networks), and mathematical models. These approaches can be used in an attempt to integrate and connect findings from preceding reductionist work (40), as well as to generate novel mechanistic hypotheses in an iterative process (Fig. 2).

The tools applied in this endeavor range from mathematical approaches with a long history of success in other branches of science, now being applied to biology (1, 38, 110), to computational approaches that rely on relatively recent advances in computing power (36), among others. As aging, or any biological question, becomes more intensively studied, the number of interesting and potentially interconnected findings that may be incorporated into systems-level analyses increases. We may also find an increase in the number of questions that have proven difficult to answer using other, more reductionist, approaches. Aging research may now have reached a stage where it is very well positioned for fruitful application of systems biology techniques and approaches.
Both aging (6, 57) and systems biology (53, 96) are large and rapidly expanding fields, and reviewing each exhaustively would require a textbook-length treatment. Although systems biology as a discipline is newer than the biology of aging, it has been around since at least the 1960s (79). Widespread increases in interest in systems approaches were ushered in by an explosion of biological data that arose from enormous improvements in the throughput of DNA sequencing technology in the early 2000s, and aging has been considered a fruitful hypothetical target for systems approaches since at least that time (40). This is illustrated by the increasing number of systems biology/aging studies, as shown in Figure 3, which compares the number of PubMed entries by year for “aging/ageing,” “systems biology,” and both in combination, from 1950 to 2016. It clearly demonstrates a striking recent increase in systems biology-related publications, which is roughly proportionally reflected in the number of combined aging and systems biology-related publications.

To highlight the power and potential of systems biology of aging, instead of providing a comprehensive analysis of the many approaches that have been used, we have chosen instead to present a few very recent illustrative examples. These select studies serve as strong examples of what can be accomplished when we integrate systems biology and aging. In particular, we discuss three case studies that illustrate the power of systems biology to inform our understanding of aging through the study of (i) diet restriction, (ii) neurodegenerative disease, and (iii) biomarkers of aging.
Although the studies we present here are highly illustrative of the power of systems biology, we necessarily leave out much important work in this short review. In particular, although mathematical modeling plays a critical role in systems biology, the studies we describe here include computational models but not formal mathematical models. For reviews of more mathematical approaches to systems biology studies of aging, especially with respect to models of biochemical pathways, readers are encouraged to read reviews by Kirkwood (58) and, more recently, McAuley et al. (74). After our discussion of these examples, we briefly survey some of the state-of-the-art methods currently being applied by systems biologists generally, which might be worth applying to aging-related questions in the near future.
Aging Biology Is Poised to Benefit from Systems-Level Approaches
We have long had a strong conceptual framework to understand why aging evolved (35, 59, 61, 78, 113). Over the past two decades, researchers have also recognized that the mechanistic causes (19, 24, 56, 60, 69, 77, 98, 112) and consequences (72) of aging appear to be deeply evolutionarily conserved. Nevertheless, attempts to move from any of these pathways or genes in isolation to a fuller mechanistic understanding of aging and how it can be modulated have proved difficult. In this light, a systems-level approach, one that incorporates a large number of pathways into a single framework, could move us closer to a mechanistic understanding of aging.
Although we suggest here that the biology of aging may now be particularly well poised to benefit from systems approaches, applications of systems biology approaches to the study of aging are not new. Systems approaches have been used to investigate molecular networks associated with aging (63, 103, 115). Systems level thinking has also been used to dissect several specific facets of aging biology, such as telomere length (93), bioenergetics (105), proteotoxic stress (82), inflammation (14), epigenetic changes (88), mTOR signaling (21, 22, 102), and cellular senescence (28, 100). Several previous reviews also offer additional perspectives on the intersection of aging biology and systems biology at multiple stages during its development (16, 29, 58, 62, 81, 116).
Some Notable Recent Applications of Systems Approaches to Aging Biology
Dietary restriction
Caloric or dietary restriction is one of the earliest interventions shown to significantly increase lifespan (75), and subsequent work has shown multiple regimes of altered food intake that have dramatic effects on lifespan and healthspan in invertebrates (52) as well as mammals (2). Although many genetically identified nutrient sensing pathways appear to play a role in lifespan extension by dietary restriction (57), none of them appear to be completely responsible for these effects.
From this starting point, Hou et al. have recently used a careful systems approach to add greatly to our understanding of dietary restriction (43) (Fig. 4). They measured the transcriptomes of cohorts of the nematode C. elegans, under ad libitum fed conditions, as well as under diet restricted (DR) and intermittent fasting (IF) conditions, both of which extend lifespan significantly. These transcriptomic measurements were repeated periodically, generating a transcriptomic time course for each treatment throughout the adult life of these organisms. These time courses were then used to cluster all transcripts (genes) using Bayesian Information Criterion-Super K Means clustering (117). They note an over-representation of known prolongevity factors among genes they observe to be upregulated by DR or IF, and of known antilongevity factors among those downregulated by DR or IF, consistent with the notion that DR and IF lead to extended lifespan through these genetic pathways.

To ask which genes might be expected to mediate the observed DR and IF responses, the authors used extended Deletion Mutant Bayesian Network (eDM_BN) analysis, an adaptation of their previous Deletion Mutant Bayesian Network analysis (66). They compared 73 genetic perturbations with their measurements of calorically restricted and IF worms, identifying nine genes whose perturbation closely mirrored the effects of DR or IF on the worm transcriptome: rheb-1, aak-2, xbp-1, tax-6, glp-1, daf-16, dpy-10, gas-1, and ogt-1.
Several of these genes have previously been shown to have some relationship with dietary restriction, and some have been shown to have relationships with one another (e.g., glp-1 and daf-16) (44). These genes were grouped into three clusters, and additional analysis based on eDM_BN incorporating modENCODE TF chromatin immunoprecipitation sequencing data (17) (TF-expanded eDM_BN), as well as CoCiter (89), also resulted in the same three clusters. These implicate three pathways—rheb-1/let-363, aak-2/tax-6/xbp-1, and daf-16/glp-1, as distinct modules involved in the transcriptome level changes observed upon DR or IF.
The authors then constructed aak-2(o/e), daf-2(e1370), and rsks-1(ok1255) worms, and used these, as well as daf-2(e1370), rsks-1(ok1255), and tax-6(RNAi) worms, to ask what happened when all three of the modules they identified were perturbed simultaneously. In general, as they moved from single genetic perturbations to double and then triple, the transcriptomes of these worms began to more closely resemble those of DR or IF worms. They modeled the kinetics and steady states of their network model and concluded that the triple module-perturbed animals should show a shift in the network steady state toward a longevity state. This model predicts that making a small perturbation in all three modules should have a larger effect on lifespan than a much larger perturbation in a single module, as was observed.
As the triple-perturbed worms were insensitive to dietary restriction, they conclude that perturbing these three modules recapitulates essentially all of the DR phenotypes. Finally, they asked whether new individual genetic phenotypes might be uncovered by their approach. For example, they predicted a lifespan interaction between aak-2(o/e) and xbp-1(RNAi) as suggested by their placement into the same module, and found that xbp-1 is necessary for the extended lifespan of aak-2(o/e).
This work has important implications for attempts to phenocopy lifespan and healthspan extension by DR using drugs or genetic manipulation. Based on these findings that perturbation of three distinct modules is needed to recover most of the lifespan extension caused by caloric restriction or intermittent fasting, we might need to simultaneously modulate multiple modules to recover the full phenotype of this intervention in the absence of caloric restriction. These findings of multiple necessary modules and their identities would be hard to arrive at by a nonsystems approach. This study provides a compelling example of the kind of progress that systems approaches will continue to bring to aging biology.
Neurodegenerative disease
Aging is the single greatest risk factor for a wide range of diseases, but the effect is seen most dramatically for neurodegenerative diseases (Fig. 5). This has led to a sustained interest among aging researchers in neurodegenerative diseases. The most commonly diagnosed and most intensively studied of these is Alzheimer's disease (AD) (34). Total annual healthcare costs for AD and related diseases are now estimated to exceed $200 billion in the United States, and could rise to $1 trillion by 2050 (23). We have now identified multiple human genes associated with both early onset (genetic or familial) and late onset (sporadic) Alzheimer's disease (LOAD), often using genome-wide association studies (GWASs) (20, 32, 64, 85), but a complete mechanistic understanding of this disease has yet to emerge. The need for systems approaches in AD has been recognized, and ongoing systems-level National Institute on Aging-supported initiatives such as Accelerating Medicines Partnership-Alzheimer's Disease target discovery and preclinical validation project (8, 83) and Model Organism Development and Evaluation for Late-onset AD have been funded with this in mind.

Mukherjee and colleagues (84) recently used a versatile gene-based test for genome-wide association study (VEGAS) analysis (70), as well as a dense module search (DMS) approach (47) to synthesize human protein–protein interaction (PPI) data and GWAS data. Briefly, the VEGAS approach uses a weighted sum of single nucleotide polymorphism chi-squared tests, corrected for linkage disequilibrium. Per-gene empirical p-values are determined by simulation-based permutations, stopping when a value of 1 × 10−6 is reached, corresponding to a genome-wide false discovery rate of 0.05. In the DMS approach, the authors mined human interactome PPI data, and used DMS GWASs to identify statistically significant (improbable) networks of genes by exhaustively examining the combined effect of multiple genes in this interactome. They then tested the resulting predictions using both transgenic C. elegans models of Aβ toxicity, and human brain expression level data. This multimethod, multiorganism systems biology approach proved fruitful and may illustrate the types of approaches that will become increasingly prevalent. A schematic diagram of their approach is shown in Figure 6.

The VEGAS approach corresponded well with previously published GWAS analyses and yielded an additional three genes and four pseudogenes. The DMS approach, which integrates additional information from human PPI data, provided a top module with 33 genes, 16 of which were on chromosome 19, leaving open the possibility of linkage disequilibrium with APOE, the strongest known genetic determinant of Alzheimer's risk. Of the remaining 17 genes, four had previously been associated with increased risk of LOAD in GWAS analyses. The authors focused on the 13 novel genes not on chromosome 19: ALB, EGR1, HLA-DRA, CHRNA2, MYC, NDUFS3, UBC, SLC25A11, C1QBP, KRT14, ICT1, ATP5H, and APP.
Of these, four had predicted C. elegans orthologs, indicated in parentheses: NDUFS3 (nuo-2), UBC (ubq-1 and ubq-2), ATP5H (atp-5), and EGR-1 (egrh-1). Ribonucleic acid interference knockdown of all five of these predicted orthologs had significant effects on Aβ toxicity phenotypes. Knockdown of ubq-1 and ubq-2 significantly accelerated Aβ toxicity, whereas knockdown of nuo-2, atp-5, and egrh-1 delayed it, in various transgenic worm models. In examining human transcriptomic data comparing diagnosed LOAD patients with patients with other neurological diseases, reliable transcript levels were available for 11 of the 13 novel genes (excluding CHRNA2 and KRT14). Of these, four or 36% (NDUFS3, SLC25A11, ATP5H, and APP) were differentially expressed in the temporal cortex of LOAD patients, compared with 14% of all measured probes, and two more (UBC and C1Q8P) were differentially expressed in the cerebellum.
This work demonstrates the ability of systems approaches to generate and validate entirely novel genes of interest and leverage existing large data sets. The results expand our understanding of the biology of AD and of targets for possible intervention, and similar approaches should be expected to continue to add to our understanding of this complex and important disease.
These strengthened connections between known C. elegans genes and AD point out an interesting future direction for this work. C. elegans has been used extensively as a model of aging more generally, and many genes and pathways have been identified that alter aging in worms, hif-1, hsf-1, etc. Similarly powerful models to study the impact of Aβ and tau also exist in the fruit fly, Drosophila melanogaster (11, 114). Much could potentially be learned from both of these invertebrate models by studying how these pathways alter Aβ toxicity phenotypes and interact with the Alzheimer's related genes described in this study.
Biomarkers of aging
Finally, a longstanding interest among aging researchers has been the reliable generation of biomarkers of aging. These could, for example, more rapidly suggest whether or not an intervention appears to be slowing aging, without waiting for a cohort of experimental organisms or human patients to live out their entire lifespans. Thirty years ago, the National Institutes of Health created an initiative to identify biomarkers of aging (4). Such efforts have often been criticized (18), and at least until recently, these initiatives showed little success. With the advent of high-dimensional and relatively inexpensive molecular phenotypes, systems biology approaches may at last be able to provide us with meaningful biomarkers of aging (so-called biological clocks), measures that could predict future risk morbidity and mortality even in apparently healthy individuals. For example, a series of recent studies suggest that epigenetic marks (specifically DNA methylation) have promise as a predictor of biological age (10, 37, 42).
In humans, wherein the largest differences in lifespan among modern cohorts are modest relative to the changes in survival we can demonstrate in model organisms, it can be difficult to distinguish a chronological clock (which measures years lived so far) from a biological clock (which would predict years still to live). To clearly discern the two requires that we consider individuals that have been alive for the same amount of time (same chronological age) but have a very different remaining lifespan and healthspan (biological age). Recent work from Wang et al. has applied systems approaches to demonstrate epigenetic signatures that differ between control mice and mice that age more slowly (107).
The authors analyzed 107 mouse liver methylomes from mice aged 0.2–26 months, and used an ElasticNet statistical regression approach (118) to train an age predictor, using a subset of 148 CpG sites, regions in the genome that can be highly methylated, and whose methylation status can influence rates of transcription. Four-way cross-validation using the original data set, and application of the predictor to 50 novel methylome samples of either 2- or 22-month-old wild-type mice, both showed excellent correlation between predicted age and chronological age. The resulting model was then applied to the methylomes of three cases of mice with increased lifespan and delayed aging. These included Prop1df/df dwarf mice (5), as well as rapamycin-treated mice and calorically restricted mice (80).
The methylome-based model predicted a lower chronological age for all three of these groups of mice, and in all three cases the difference between the predicted epigenetic lifespan of the long-lived mice and their chronological lifespan was statistically significant, whereas epigenetic lifespan and chronological lifespan were not significantly different for the control mice that were not long lived (Fig. 7). Looking at the first principal component from a principal components analysis of long-lived and control methylomes, or at the trends of the most significantly changed CpG islands selected in their model, led to the conclusion that changes in methylation over time were generally less pronounced in long-lived mice than control mice that were not long lived, as might be expected. In humans, Steve Horvath's epigenetic clock analysis based on the methylome indicates that tissues do not age at identical rates (42), a finding supported by recent analysis of molecular biomarkers in different tissues of an annual killifish, Notobranchus guentheri (25). Based on Horvath's findings, Wang et al. (107) note that extension of their current liver-specific epigenetic model will eventually allow us to ask whether some tissues or cell types have an epigenetic age that diverges more or less from their chronological age than others, in long-lived mouse models. This and other similar recent work suggests that the time is near when we will be able to assay the effect on meaningful biomarkers of interventions that delay aging rapidly and cheaply, which would promise to greatly enhance the pace of this type of research.

Systems Biology Includes a Powerful and Rapidly Evolving Set of Tools that can be Applied to Questions in Aging Biology
Systems biology approaches are often characterized by the analysis of very large, multidimensional data sets, with thousands to millions of data points. Moreover, these data might represent highly disparate traits—genome, epigenome, transcriptome, proteome, microbiome, and metabolome—each with different ways of being described. Typically, the number of traits and trait interactions greatly outnumbers the samples collected, creating tremendous challenges in the collection, curation, and analysis of data.
To address the challenges of understanding the statistical and biological significance of systems biology data, we need methods that can incorporate standardization on open formats (97). Systems biologists have embraced this concern and proposed or adopted multiple open formats for the exchange of data and models, for example, the systems biology markup language (SBML) (46), as well as CellML (71) and NeuroML (33). Multiple searchable repositories now use SBML, including the BioModels database of quantitative models (48) and the Reactome pathway database (27).
These open formats for describing data are equally important when it comes to software for analyzing data. It is critical that we have openly available software that allows biologists to apply modern computational techniques to their data. Several useful stand-alone systems such as COPASI (41), the complex pathway simulator, have been developed for systems biology (31). As with file formats, many advantages accrue to users of open source software that allows contributors to freely improve, add, edit, and modify the tools that they use (92), and most systems biology tools fall into this category. The Cytoscape framework is a powerful general-purpose platform emphasizing network-based approaches, with a large and growing set of add-ons that greatly extend its core functionality (94). GeneMANIA (108), which exists as a stand-alone tool or a Cytoscape App, allows users to easily construct and explore networks based on multiple biological features, whereas STRING (99) focuses specifically on PPI.
Perhaps the most rapidly evolving set of tools for computational work by biologists centers around the R programming language (90). Like Cytoscape, R seamlessly utilizes user-contributed packages that add to its core functionality, and there are many R packages that are specific to systems biology as well as many others covering multiple statistical and machine-learning techniques of interest to the systems biologist. Many of these packages are hosted at the Comprehensive R Archive Network. Among these are systems biology packages implementing Bayesian active learning strategy (87) and modeling and simulation of stochastic kinetic biochemical network models (111). Another centralized repository of biology-specific R packages is hosted by the Bioconductor project (45), and Bioconductor hosts several systems biology-related R packages. These include a metagenomic pipeline for systems biology (15), a graphical user interface for exploratory analysis of systems biology data (65), metaheuristics for global optimization in systems biology (26), flexible identification of differentially expressed subpathways using RNA-seq experiments (106), constraint-based modeling using metabolic reconstruction databases (30), and a SBML interface to R (91), among many others. Although users may understandably be drawn to the powerful mathematical methodologies or aging-specific use cases, the most important tools to master to most effectively utilize a framework like the R programming language are probably the bread-and-butter data transformation, collation, visualization, and import functions. These tools quickly make large and unwieldy data sets useful and interpretable. Readers new to R are strongly encouraged to familiarize themselves with this type of essential R package sooner rather than later (109). All of the R packages already mentioned are summarized in Table 1.
Conclusions
In the past two decades or so, we have moved from a study of aging based largely in physiology, comparative biology, and evolutionary theory, which are still very strong components of ongoing research, to one that now includes multiple, diverse genetic, and pharmacological interventions, many with very large effects on lifespan and healthspan. These interventions are often broadly conserved genetically, suggesting further utility in pointing toward additional human interventions in time. Although we do not understand all of the individual genes and pathways that affect aging, as we continue to uncover more of them, we have now also begun to synthesize existing data using systems-level approaches, often to great effect. The three examples noted here all benefit from computational approaches that were unknown a few years ago, and from biological insights gleaned from multiple model systems, from aging laboratories as well as many other areas of biology. These data sets and computational tools are both improving at a rapid and accelerating rate. Many new technologies, such as single-cell sequencing, advances in epigenetics beyond the methylome (specifically, assay for transposase-accessible chromatin with high throughput sequencing), and multiomic network studies, will also increase the reach of systems biologists. This suggests that approaches similar to those described here will continue to lead to striking findings, and to interventions that may allow us to delay some of the many age-associated diseases in humans, perhaps sooner that we expect.
Footnotes
Acknowledgments
DELP was supported in part by funding from the NIH (AG049494 and CA211160) and the National Science Foundation (DMS1561814).