
Abstract
Significance:
Nuclear factor kappa B (NF-κB) signaling is essential under physiologically relevant conditions. However, aberrant activation of this pathway plays a pertinent role in tumorigenesis and contributes to resistance.
Recent Advances:
The importance of the NF-κB pathway means that its targeting must be specific to avoid side effects. For many currently used therapeutics and those under development, the ability to generate reactive oxygen species (ROS) is a promising strategy.
Critical Issues:
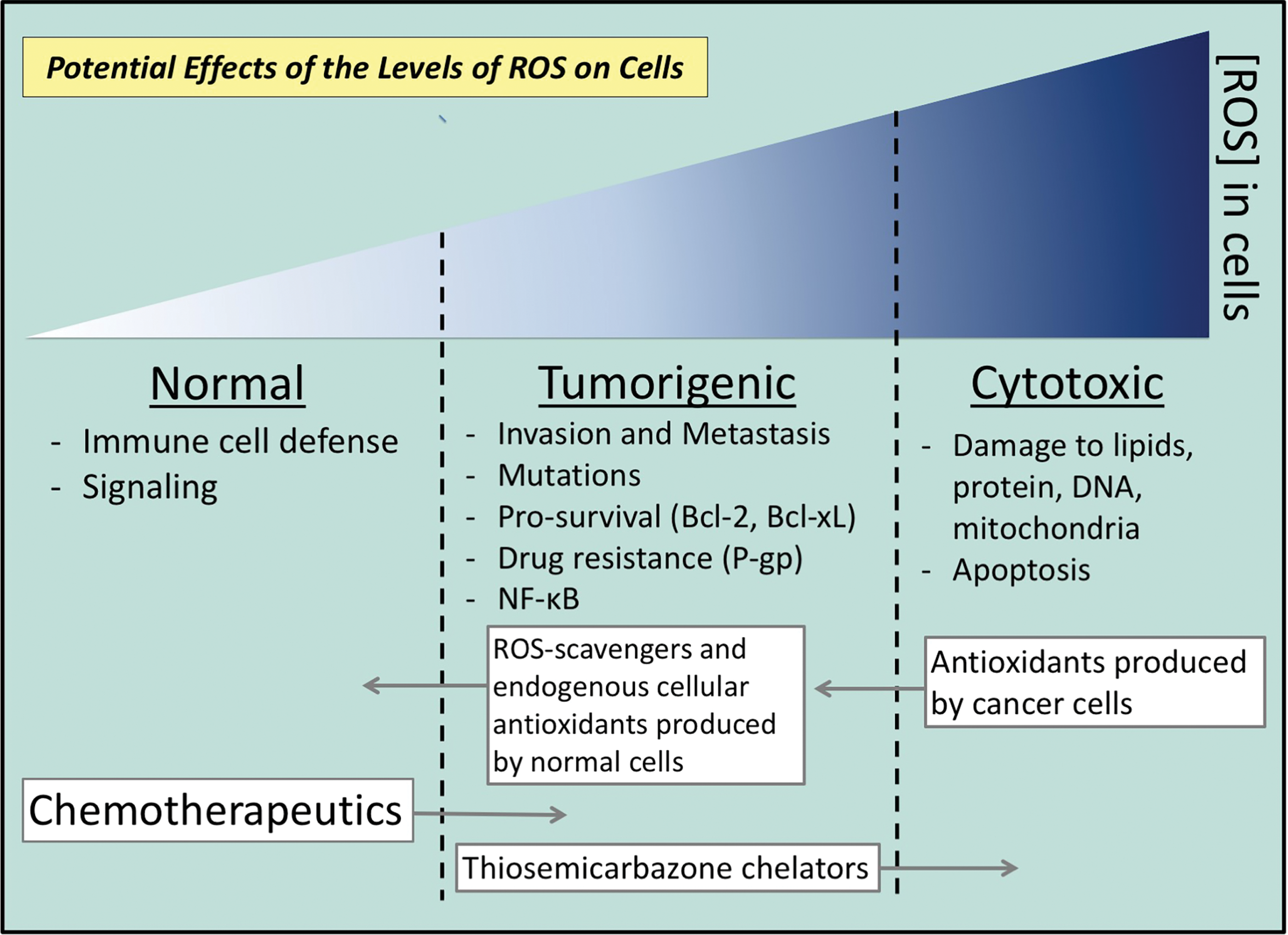
As cancer cells exhibit greater ROS levels than their normal counterparts, they are more sensitive to additional ROS, which may be a potential therapeutic niche. It is known that ROS are involved in (i) the activation of NF-κB signaling, when in sublethal amounts; and (ii) high levels induce cytotoxicity resulting in apoptosis. Indeed, ROS-induced cytotoxicity is valuable for its capabilities in killing cancer cells, but establishing the potency of ROS for effective inhibition of NF-κB signaling is necessary. Indeed, some cancer treatments, currently used, activate NF-κB and may stimulate oncogenesis and confer resistance.
Future Directions:
Thus, combinatorial approaches using ROS-generating agents alongside conventional therapeutics may prove an effective tactic to reduce NF-κB activity to kill cancer cells. One strategy is the use of thiosemicarbazones, which form redox-active metal complexes that generate high ROS levels to deliver potent antitumor activity. These agents also upregulate the metastasis suppressor, N-myc downstream regulated gene 1 (NDRG1), which functions as an NF-κB signaling inhibitor. It is proposed that targeting NF-κB signaling may proffer a new therapeutic niche to improve the efficacy of anticancer regimens.
Prevalence of Nuclear Factor Kappa B Constitutive Activation in Cancer
Cancer remains a leading global health issue (149). While recent advances have improved diagnostic and prognostic outcomes for many cancer patients, there is still a need for the enhancement of therapeutics for particularly aggressive cancer subtypes that do not respond to current treatment strategies (149, 318). Unfortunately, resistance to established chemotherapeutics plays a key role in promoting these poor prognostic outcomes for patients (135, 318). Thus, research into molecular facilitators of chemoresistance and tumor progression is imperative to discover new therapeutic approaches.
Of note, constitutive activation of the nuclear factor kappa B (NF-κB) pathway is evident in the majority of tumor cell lines, but is seldom found in normal cells with the exception of proliferating T cells, B cells, thymocytes, monocytes, and astrocytes (2). Aberrant NF-κB activity has been reported in a number of cancer types, including pancreatic (342), prostate (253), gastric carcinoma (284), renal (251), Hodgkin's lymphoma (17), multiple myeloma (94), leukemia (20, 111, 167), and breast cancer (308). On the contrary, NF-κB signaling is essential for the health of organisms, particularly in the event of an immune response (127, 191). NF-κB signaling facilitates the transcription of a large number of genes pertaining to cell development, proliferation, angiogenesis, inflammation, survival, and responses to stress (11, 12, 17, 72, 81, 159, 164, 174, 207). However, aberrant activation of this latter pathway also plays a role in tumorigenesis and drug resistance (4, 49, 69, 210).
The Canonical NF-κB Pathway
The NF-κB family comprises five homologous constituents each containing a Rel homology domain (RHD): c-Rel, RelA (p65), RelB, NF-κB1 (p50/p105), and NF-κB2 (p52/p100), which are synthesized in their proforms: p105 and p100, respectively (14, 40, 125). The latter proteins are subject to further proteolytic processing by the proteasome to generate p50 and p52 (14, 40, 125). The RHD functions to dimerize these proteins to generate NF-κB homodimers or heterodimers, while also functioning to facilitate their binding to cognate DNA elements to initiate transcription (143, 223).
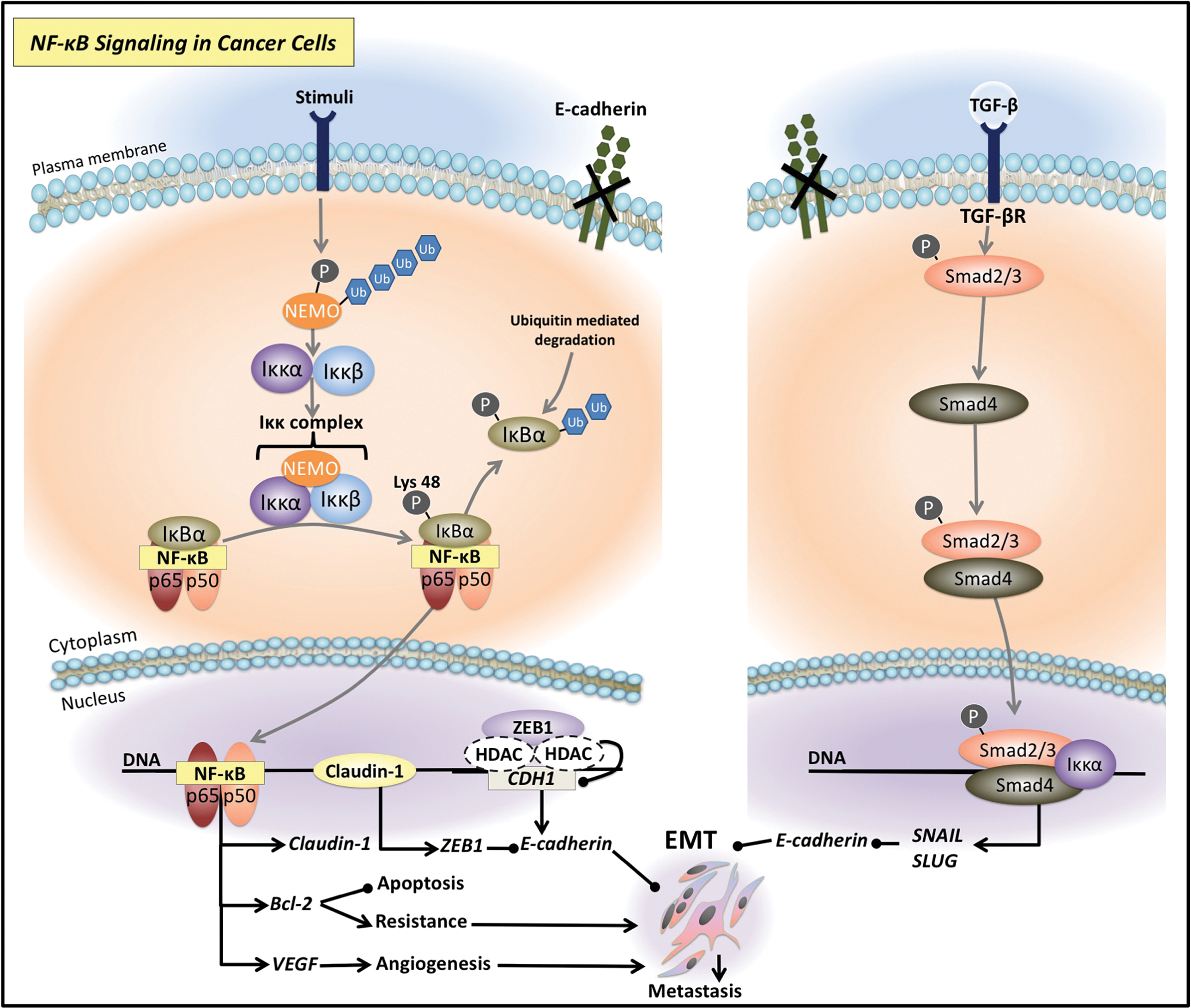
Constitutive activation of NF-κB signaling would be detrimental to health (158). As such, under physiological conditions, this pathway is tightly regulated, requiring post-translational modification (i.e., phosphorylation) of a cytosolic inhibitory molecule, namely IκBα/β/γ (IκB), to enter the nucleus [reviewed in Perkins (261)] (14, 18, 260) (Fig. 1). These inhibitory molecules are characterized by ankyrin repeats that complex to DNA binding domains of NF-κB dimers, rendering them transcriptionally inactive (218). In addition, these proteins also alter the steady-state localization of the NF-κB dimers primarily to the cytosol, although some shuttling to the nucleus still occurs, possibly to achieve basal transcriptional activity (25, 139, 199). Upstream activation is necessary to release the homodimeric/heterodimeric complexes from IκB, and this can occur through a plethora of signaling molecules and pathways, including tumor necrosis factor (TNF) (126), Smad-dependent and Smad-independent transforming growth factor-β (TGF-β) signaling (125), epidermal growth factor receptor (EGFR) (226, 321), phosphatidylinositol 3-kinase (PI3K), and its direct downstream effector, activated protein kinase B (AKT) (37, 317) (Fig. 1). Interestingly, NF-κB is an inducible factor and as its activity does not necessitate de novo protein synthesis, signaling can occur quickly following its induction (104).

Once activated upstream, NF-κB signaling begins with the phosphorylation of the regulatory subunit, NF-κB essential modifier (NEMO, also termed, Iκκγ) (38) (Fig. 1). Subsequent to this, NEMO undergoes K63-polyubiquitination, which enables the catalytic portions of the IκB kinase (Iκκ) complex, Iκκα and Iκκβ, to associate with NEMO, and be acted upon by activating kinases (38) (Fig. 1). Phosphorylation of the catalytic portion of the Iκκα/β component activates this complex, which may then use its inherent kinase activity to double phosphorylate the IκBα inhibitory protein on its substrate recognition sites (serine-32 and serine-36 for IκBα) (125, 157, 225, 320, 340, 356, 370). This event interferes with the capacity of IκBα to bind to the NF-κB subunit, which then leads to the targeting of IκBα by ubiquitination enzymes for subsequent degradation by the 26S proteasome (125, 157, 225, 320, 340, 356, 370) (Fig. 1).
Pertaining to cancer, inhibition of NF-κB signaling by expressing IκBα and using the pharmacologic NF-κB inhibitor, PS-341, can suppress the angiogenic potential and the metastasis of pancreatic cancer cells (102). On the contrary, the overexpression of the regulatory subunit, NEMO, was reported to occur in pancreatic cancer (38, 336). Intriguingly, NEMO is also rapidly upregulated in fibroblasts by ligands (i.e., tumor necrosis factor-α [TNF-α] that induce the epithelial-to-mesenchymal transition (EMT), suggesting an important functional role with respect to the EMT (15, 61).
As already mentioned, post-translational modification is an essential feature of NF-κB signaling, and thus, the inhibitory IκBα molecule must be targeted for ubiquitin-mediated degradation, to release the NF-κB transcriptional complex (125, 225). The free NF-κB dimer may then translocate into the nucleus and bind to NF-κB response elements (NRE) (112) (Fig. 1). This leads to the transcription of numerous genes, including those that regulate the following: (i) cellular growth [i.e., cyclin D1 (133), c-Myc (83, 113), C-X-C motif chemokine ligand 1 (CXCL1) (323)]; (ii) adhesion molecules [i.e., vascular cell adhesion molecule (VCAM) (145) and intracellular cell adhesion molecule (ICAM) (333)]; (iii) antiapoptotic factors such as B cell lymphoma 2 (Bcl-2), B cell lymphoma-extra (Bcl-x), and Bcl-2-related protein A1 (Bfl1/A1) (92, 186, 313, 365, 379); (iv) inflammatory response genes [i.e., interleukin-2 (IL-2) (181), IL-6 (179, 194), and IL-8 (179)]; (v) angiogenic factors such as vascular endothelial growth factor (VEGF) (56); and (vi) facilitators of the EMT such as vimentin, a mesenchymal gene upregulated during this process (165, 196), and oncogenic claudin-1, a tight junction (TJ) molecule that can also act as a transcription factor (44, 76, 125, 128, 130, 162, 205, 304) (Fig. 1). Interestingly, particular combinations of NF-κB dimers can induce differential gene expression (189). Thus, the complexity of the NF-κB signaling system relates to its varying preference for certain DNA binding specificities (189, 249, 353).
NF-κB and the EMT
The EMT process is significant to study in the context of cancer as it has been well documented to promote cell invasion and metastasis (79, 314, 339, 359, 363). The increased invasive potential arises from genotypic and phenotypic remodeling to cells that in turn are conducive to invasion and metastasis (79). Interestingly, a comprehensive study by Huber et al. reported the necessity of the NF-κB pathway as an inducer of the EMT process (141).
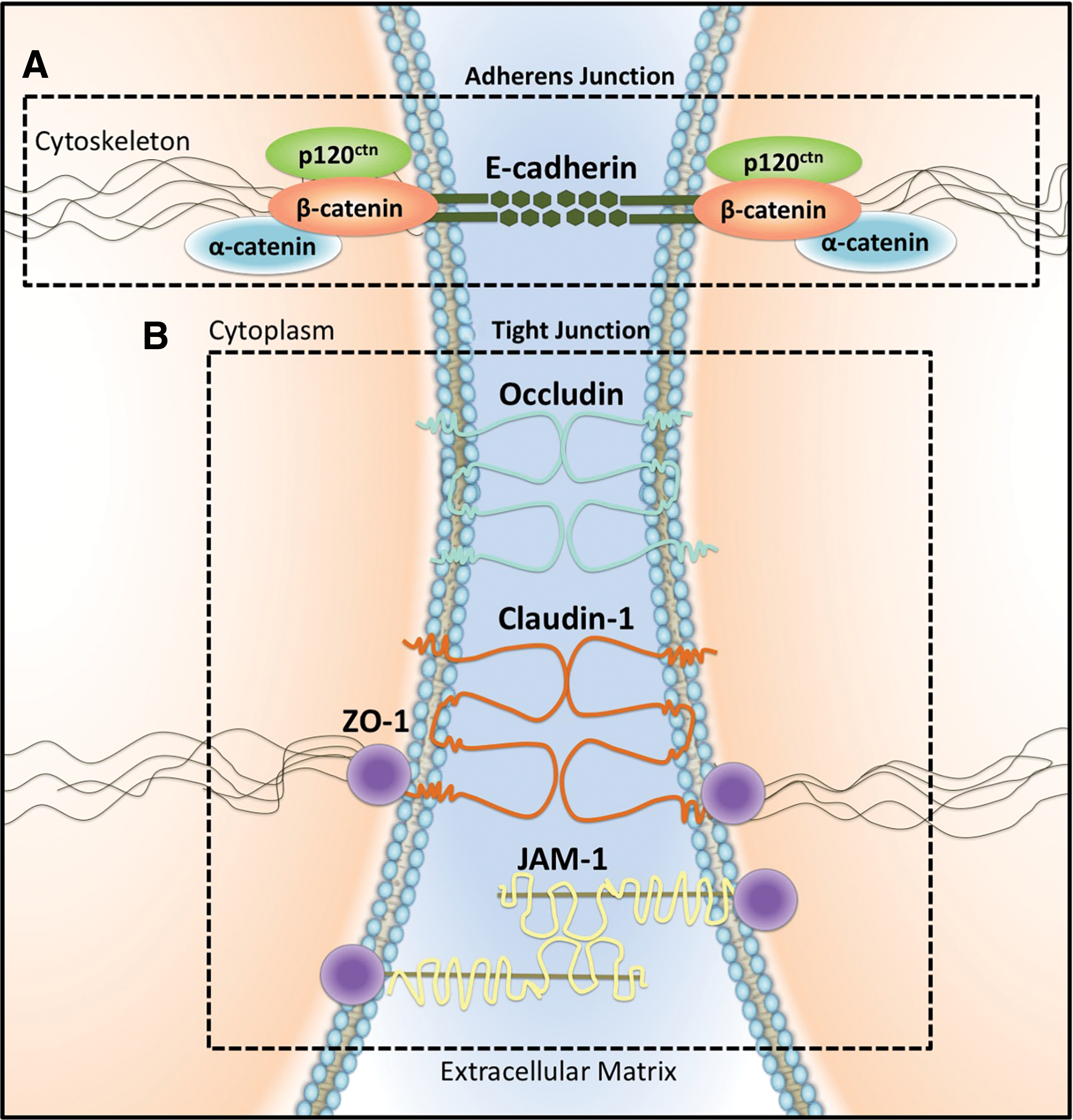
Hallmark features of the EMT typically include the genetic silencing of epithelial genes subsequently resulting in the loss of an epithelial phenotype (109, 217). The adherens junction (AJ) protein, E-cadherin, is critical for maintaining cell-to-cell adhesion and is downregulated during the EMT (1, 192, 206, 234). Indeed, E-cadherin has been described as a metastasis suppressor gene whose loss has been well documented in invasive tumor types (54, 262, 338, 364). This protein is a transmembrane spanning molecule that constitutes the AJ (122). These junctions contribute to cell polarity and cell-to-cell adhesion through calcium-dependent, homophilic binding of their extracellular domains on adjacent cells (1, 47, 122). In addition to E-cadherin, the AJ binding apparatus also comprises the catenins (α- and β-catenin) and p120ctn, whose interaction with the intracellular C-terminal tail of E-cadherin contributes to the AJ, while also associating with cytoskeletal filaments through α-catenin (1) (Fig. 2). Hence, in epithelial cells, E-cadherin is essential for maintaining the structural integrity of the epithelium, preventing cell disassociation, and thus, tumor cell metastasis (1, 41).
Concomitant to the loss of an epithelial phenotype, mesenchymal genes are activated with molecules, such as N-cadherin, fibronectin, and vimentin, being upregulated to satisfy a mesenchymal phenotype (45, 192, 234). Such changes in cell phenotype manifest as dysfunctional intercellular adhesions, resulting in loosely arranged cells and dynamic cytoskeletal structures that are ideal for invasion (289). Indeed, studies have clearly demonstrated that NF-κB is required for the subsequent maintenance of a mesenchymal phenotype (141). In fact, it has been demonstrated that incubation of cells with p65-siRNA results in an increase in E-cadherin expression, concomitant with a decrease in the mesenchymal marker vimentin, which is a further testament to the significance of the contribution of NF-κB to the EMT (55).
Signaling through NF-κB is able to promote the EMT through the induced expression of claudin-1 (44, 130, 205). In normal noncancerous cells, the claudin family itself consists of over 24 members of relatively small-molecular-weight proteins that comprise the TJ apparatus (20–27 kDa) (332, 334). Essentially, TJs are supramolecular adhesive complexes, localized between two apical portions of adjacent lateral epithelial or endothelial plasma membranes (13, 123). In addition, TJ complexes, along with claudins, also contain occludins, junctional adhesion molecule 1 (JAM-1), and zonula occluden 1 (ZO-1) (144) (Fig. 2).
Importantly, TJ complexes comprise part of a physical barrier separating both the apical and basolateral fluid components (32). The location of the TJ allows it to serve a functional role as a selective filter, controlling the passage of fluids, molecules, and inflammatory cells through the paracellular aspect (32). In the presence of TJs, cells acquire a fixed position to both adjacent cells and the basement membrane (315). Importantly, by establishing cell-to-cell contact, TJ molecules maintain appropriate signaling between adjacent cells [reviewed in Steed et al. (311)]. Pertaining to this, growth factor receptors (i.e., TGF-βR) are located on the basolateral surface of cells, and thus, TJs prevent autocrine activity by regulating the passage of molecules (i.e., receptor ligands) (32, 68, 242, 362) (Fig. 3). Hence, cellular TJs may also prevent aberrant stimulation of potentially oncogenic TGF-β signaling in cancer [reviewed in Rahimi and Leof (271)]. Thus, TJ molecules represent a critical barrier that cancerous cells must surpass to successfully invade and metastasize.
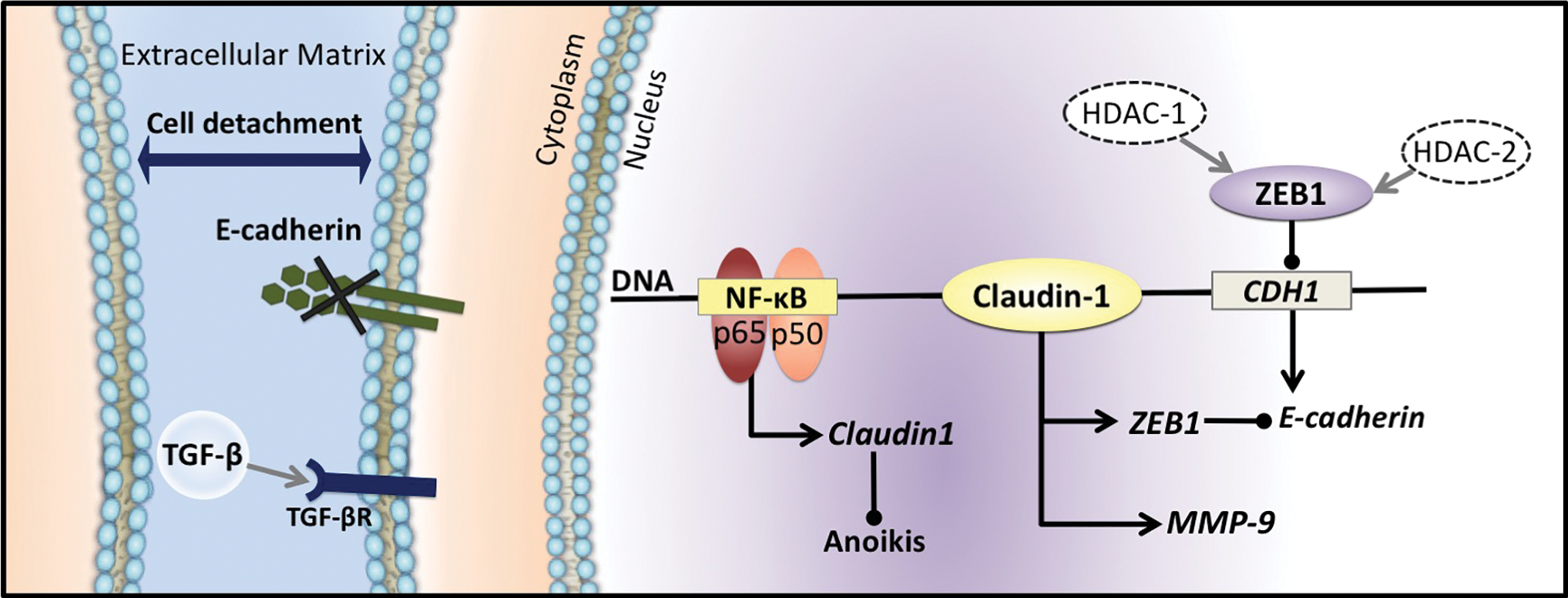
While membrane-associated claudin-1 exerts a participatory role as a TJ protein, recent evidence has demonstrated that within the context of cancer, claudin-1 serves to facilitate the EMT in a variety of ways (32, 76, 98, 243, 304) (Figs. 2 and 3). For instance, claudin-1 is able to positively regulate the expression and activation of matrix metalloproteinases (MMPs) (98, 118). In fact, claudin-1 expression has been linked to MMP-9 and may also operate in a synergistic manner with claudin-6 to accelerate MMP-2-induced invasion (98) (Fig. 3). The secretion of MMPs at the basolateral membrane enables the degradation of extracellular matrices (ECM) permitting the extravasation of mesenchymal cancer cells, which may then subsequently invade and metastasize to distal sites (21, 64, 244). Studies examining hepatocellular carcinoma implicate MMP-9 expression and activity as a result of NF-κB signaling (100).
Claudin-1 expression has also been correlated with a decrease in anoikis, a type of induced cell-mediated death following loss of contact to adjacent cells or detachment from the ECM (105, 303) (Fig. 3). Anoikis resistance allows cells to persist once detached and, for this reason, is intimately linked to the metastatic capabilities of tumor cells (105, 285, 366). In addition to claudin-1, NF-κB has also been involved in resistance to anoikis (285). In fact, using a specific NF-κB inhibitor, (−)-dehydroxymethylepoxyquinomicin (DHMEQ), it was demonstrated that this latter agent attenuated resistance to anoikis via its inhibitory effects on NF-κB (285).
The inhibition of claudin-1 expression has also been correlated with significant upregulation of E-cadherin expression (76, 211). A claudin-1 overexpressing cell line, SW480claudin-1, was shown to possess a markedly fibroblastic phenotype in addition to elevated levels of mesenchymal gene expression when compared to control cells (76). The capacity of claudin-1 to induce the EMT in this way may be due to the additional role of claudin-1 as a transcription factor, through which it enters the nucleus to upregulate epithelial gene repressors, such as zinc finger E-box-binding homeobox 1 (ZEB1) (5, 76, 103, 236, 304) (Fig. 3). Effectively, ZEB1 binds to the CDH1 promoter region encoding E-cadherin, which is followed by the recruitment of histone deacetylases (HDAC; HDAC-1 and HDAC-2) to the site, subsequently acting to physically inhibit the transcription of E-cadherin, leading to its genetic silencing (3) (Fig. 3). Consistently, ZEB1 has been linked to a reduction in E-cadherin gene expression, with E-cadherin levels inversely correlating with ZEB1 expression (3, 60, 169). The silencing of ZEB1 in the mesenchymal PANC-1 pancreatic cancer cell line was demonstrated to result in the restoration of E-cadherin expression, which is indicative of a reversal of the EMT (7).
In concordance with the link to NF-κB facilitating the EMT, a range of studies have supported the link between NF-κB and the expression of ZEB1 (60, 176, 294, 358). Notably, in cancer cells where NF-κB is constitutively active, ZEB1 was observed to be highly expressed, with these cells also exhibiting classical features of the EMT (59, 60, 214). In fact, it has been reported that the EMT process necessitates ZEB1 expression (214). In addition to facilitating the EMT, ZEB1 is also believed to facilitate cell cycle progression of progenitor cells through direct transcriptional repression of cyclin-dependent kinase inhibitors, p21 and p15INK4b, and may enhance evasion of apoptosis by cancer cells via acting as a repressor of the p53 homologue, p73, subsequently avoiding the classical DNA damage checkpoint (35). Hence, it is evident that ZEB1 expression exerts a critical functional role in tumorigenesis, with its overexpression being identified in a number of human cancers, including uterine cancer (309), pancreatic cancer (33, 348), osteosarcoma (295), lung cancer (222, 372), liver cancer (378), gastric cancer (250), colon cancer (371), melanoma (350), prostate (109), hepatocellular carcinoma (124), and breast cancer (84).
In cancer cells, a key NF-κB signaling molecule, namely Iκκα, has been shown to mediate the EMT process via the downregulation of epithelial genes, such as E-cadherin (31) (Fig. 4). In fact, in addition to classical TGF-β signaling involving the Smad proteins that upregulate the E-cadherin repressors, SNAIL and SLUG (8, 129), Iκκα was demonstrated to modulate canonical TGF-β-Smad signaling (31) (Fig. 4). Using a pancreatic cancer model, TGF-β induced Smad3 and Iκκα complexation (31), which bound DNA and regulated the transcription of SNAIL and SLUG (31, 224, 299, 343). In fact, SNAIL and SLUG genetically silence the E-cadherin molecule (42, 46, 119, 257) (Fig. 4), and high levels of these transcriptional repressors have been reported in a number of cancer types, including pancreatic (137), breast (65), ovarian (180), gastric (46), lung (298), and bladder tumors (367).
The EMT process has been linked to drug resistance via the promotion of stem cell-like properties (376). Of note, the silencing of NF-κB p65 inhibited the ability of the anticancer agent, gemcitabine, to increase the proportion of stem cell-associated markers (i.e., CD24, CD44, and CD133) (376). As NF-κB signaling is able to markedly affect the EMT, the major first step in metastatic progression and potentially drug resistance, anticancer agents that are able to downregulate this latter signaling pathway are important to consider for cancer treatment (32, 55, 76, 98, 304, 376).
NF-κB is activated by chemotherapeutic agents and irradiation leading to chemoresistance
In addition to its EMT-promoting effects, NF-κB is also imperative to investigate as a target for antitumor therapy, as a variety of drugs used to treat cancer and other conditions, including 7-ethyl-10-hydroxycamptothecin (SN38), taxol, doxorubicin, daunorubicin, etoposide, vincristine, vinblastine, ara-C, anthralin, AZT, ciprofirate, cisplatin, haloperidol, methamphetamine, phenobarbital, tamoxifen, and camptothecin, as well as gamma irradiation, activate this latter pathway (29, 252, 267, 324). This incidental activation of NF-κB reduces the efficacy of these agents by promoting tumorigenesis and antiapoptotic effects, and thus, conferring drug resistance (4, 94, 163).
Interestingly, anticancer agents activate NF-κB signaling by affecting a number of molecules integrally involved in the NF-κB pathway. For example, the topoisomerase inhibitors, SN38 and doxorubicin, activate NF-κB through their stimulating effects on the Iκκ complex, promoting the nuclear translocation of the NF-κB subunit and activation of NF-κB target genes (29) (Fig. 5). As discussed in the Canonical NF-κB Pathway section, the kinase activity of the Iκκ complex is imperative for phosphorylating the inhibitory IκBα subunit (14, 18, 260). Once phosphorylated, IκBα is degraded and this enables the release of the NF-κB heterodimer, which enters the nucleus (14, 18, 260). Doxorubicin has also been found to bypass the Iκκ complex to phosphorylate this inhibitory IκBα molecule (324) (Fig. 5). Indeed, these latter investigators demonstrated that IκBα degradation induced by doxorubicin does not require the classical serine-32 and serine-36 phosphorylation or the PEST domain of IκBα (324). Furthermore, IκBα degradation was partially blocked by the PI3K inhibitor, LY294002, and was mediated by the proteasome (324). In addition, doxorubicin-induced cell death in Iκκα/β (−/−), murine embryonic fibroblasts, was enhanced by inhibition of NF-κB activation by blocking proteasomal activity (324). These results indicate an additional pathway of activating NF-κB during doxorubicin treatment and provide a mechanistic basis for the observation that proteasome inhibitors can be implemented as doxorubicin adjuvants (324).
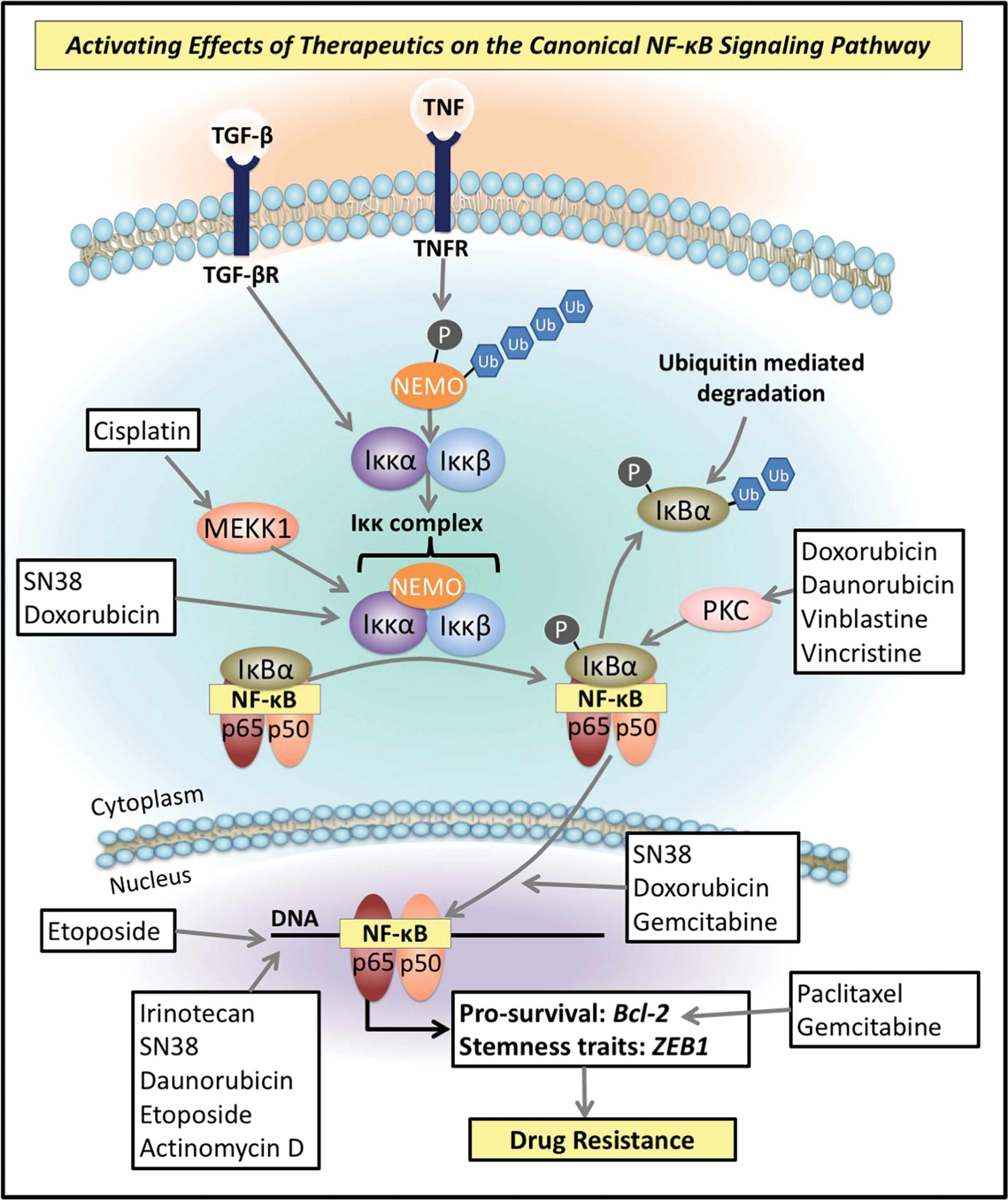
Moreover, doxorubicin, daunorubicin, vinblastine, and vincristine have been found to phosphorylate the inhibitory IκB molecule via promoting the activity of protein kinase C (PKC) (71) (Fig. 5). PKC is a calcium- and phospholipid-dependent protein kinase involved in the regulation of cell growth through signaling pathways such as NF-κB and is also implicated in enhancing drug resistance (146, 219). Interestingly, induction of Src tyrosine protein kinase signaling cascades by reactive oxygen species (ROS) can lead to the activation of PKC (239). Thus, potentially these agents may affect PKC activation and induce NF-κB activation via this mechanism.
The chemotherapeutic agent gemcitabine, which is currently considered to be the “gold standard” for the treatment of pancreatic cancer, has been reported to increase nuclear translocation of the NF-κB p65 subunit (75, 296) (Fig. 5). In addition, etoposide has also been reported to enhance the DNA binding activity of NF-κB p65 (6) (Fig. 5). Furthermore, the agents, irinotecan, SN38, daunorubicin, etoposide, and actinomycin D, are able to induce double-stranded DNA breaks that have been found to increase NF-κB activation (264) (Fig. 5). Other chemotherapeutics are also able to indirectly mediate their effects on NF-κB through the activation of other signaling pathways that converge onto NF-κB signaling (282). For instance, cisplatin activates mitogen-activated protein kinase kinase kinase 1 (MEKK1), a common mitogen-activated protein kinase (MAPK) kinase, that is subsequently able to activate prosurvival NF-κB (282) (Fig. 5). Activation of NF-κB has also been identified as a key mediator of cisplatin resistance (175, 316).
Downstream targets of NF-κB contributing to treatment resistance
The activation of NF-κB signaling in response to many current chemotherapies has been demonstrated to play a major role in the development of therapeutic resistance (29, 121, 190, 252, 263, 269, 324, 346). These effects are largely due to the prosurvival effects of NF-κB downstream signaling, including the activation of numerous proteins that promote cancer progression and inhibit apoptosis (90). One such downstream target of NF-κB is the proto-oncogene, Bcl-2 (90). Bcl-2 has been implicated in treatment resistance due to its prosurvival activity (106, 163) (Fig. 5). Both paclitaxel and gemcitabine enhance Bcl-2 expression and this was found to occur via NF-κB signaling (90) (Fig. 5).
As discussed above in the NF-κB and the EMT section, NF-κB has been linked to the high expression of the epithelial gene repressor, ZEB1 (60, 176, 294, 358) (Fig. 5). The overexpression of ZEB1 has also been associated with a prosurvival and therapy-resistant phenotype (35, 233, 274, 305, 373, 374, 377) (Fig. 5). More recently, ZEB1 has been shown to facilitate drug resistance, in part, through its transcriptional downregulation of the microRNA-200 (miRNA-200) family (30). Such miRNAs are short noncoding sequences that offer a means of post-transcriptional regulation of messenger RNA (mRNA) expression through the binding of cognate mRNA sequences (241). In this case, ZEB1 acts as a repressor of both miR-200 and miR-203, which are involved in the suppression of stemness traits (30, 349). Thus, in the presence of ZEB1 expression, an EMT-associated stemness phenotype eventuates (30, 349). Stemness traits are facilitators of chemoresistance, as they enable cells to readily adapt to survive the cytotoxicity generated by anticancer agents (51, 292, 341, 375). The re-expression of miR-200 family members has been found to contribute to a partial resensitization to chemotherapeutic agents, paclitaxel, vincristine, and epothilone B, highlighting their potential and significance in resistance (7, 63, 349).
The suppression of NF-κB has been consistently reported to sensitize tumor cells to chemotherapeutic agents and ionizing radiation (27, 48, 117, 142, 201, 360). For instance, chemopreventative agents such as curcumin and resveratrol, which block NF-κB activation, were found to promote apoptosis of cancer cells (39, 151). Thus, these chemopreventative agents enhanced the efficacy of chemotherapeutics and irradiation therapy (62, 120, 151, 240). In the case of human gastric cancer cells, cisplatin resistance was reversed by inhibiting NF-κB, using the naturally derived O-methylated flavone, wogonin, complexed to Pt(IV) (49). The wogonin-Pt(IV) prodrug is able to circumvent chemoresistance via its negative regulation of casein kinase 2 (CK2)-mediated NF-κB induction (49). Importantly, CK2-mediated NF-κB activation is believed to be the major cause of cisplatin resistance (49). Mechanistically, CK2 acts as a serine/threonine protein kinase, with recent data suggesting CK2 is able to activate the Iκκα kinase leading to the phosphorylation of IκBα on serine-32/serine-36 and plays an additional role in the transactivation of the p65 NF-κB subunit (49, 93, 287).
External stress stimuli, such as ROS, can also promote activation of NF-κB and have also been implicated in resistance to therapy (107, 239, 288) (Fig. 6). A number of chemotherapeutics as well as radiation can induce the generation of ROS and have been reported to confer drug resistance through activation of NF-κB signaling (53, 230, 231). In fact, ROS generated by radiation therapy initiates mitochondrial apoptosis (347, 354) (Fig. 6). However, high ROS levels produced via radiation in the mitochondria indirectly activate NF-κB-mediated expression of the antioxidant enzyme, superoxide dismutase 2 (SOD2), that curtails cytotoxic ROS induced by irradiation and chemotherapeutics (70, 73, 110, 231, 347) (Fig. 6). This response prevents mitochondrial injury and cytochrome c release, protecting cells from apoptosis (74, 300, 347, 355). Hence, the induction of SOD2 by NF-κB has been linked to radioadaptive and chemotherapy resistance in gastric cancer, lung cancer, prostate cancer, adenocarcinomas, and breast cancer (52, 58, 74, 114, 134).
The production of ROS has also been found to promote cancer cell survival in glioblastoma tumors via the activation NF-κB signaling, subsequently inducing the expression of antiapoptotic gene, Bcl-xL, promoting cancer cell survival and chemoresistance (53) (Fig. 6). The ROS-associated upregulation of the drug efflux pump, P-glycoprotein (P-gp), markedly involved in multidrug resistance (MDR), has been linked to activation of the NF-κB (67, 228, 327) (Fig. 6). In addition, in cancer cells, high and low glucose levels have been reported to markedly increase ROS levels, subsequently conferring drug resistance via the upregulation of P-gp (290).
Molecular Effects of Redox-Active Agents
Redox homeostasis is essential for normal cellular growth, metabolism, immunity, and survival (276). The term ROS encompasses small oxygen-containing species that comprise the hydroxyl radical (OH•), superoxide anion radical (O2 •−), hydroxyl ion (OH−), hydrogen peroxide (H2O2), singlet oxygen (1O2), and ozone (O3) (22, 306). Exogenously, ROS may be increased in cells through ultraviolet radiation, ionizing radiation, drugs, aging, or in the context of inflammation and injury (279). Intrinsically derived ROS can be produced by the mitochondrial electron transfer chain (204), through myeloperoxidase (MPO) activity in phagocytes (280), and by NADPH oxidase (NOX) enzymes (22). Low levels of ROS are required to participate as signaling molecules mediating crucial processes, including cellular proliferation, migration, and apoptosis (34, 198, 237, 276). In addition, ROS may elicit an antipathogenic role by stimulating immune cells to induce a potent innate immune response (91).
Scavengers of ROS and endogenous cellular antioxidants are also produced by the cell, including glutathione peroxidase (GPX), catalase (CAT), SOD, enzymes of the ascorbate-glutathione (AsA-GSH) cycle, including ascorbate peroxidase (APX), mono-dehydroascorbate reductase (MDHAR), dehydroascorbate reductase (DHAR), and glutathione reductase (GR), which function to defend against and regulate ROS levels by counterbalancing production (227, 247) (Fig. 6). A tight regulation of the homeostasis between ROS production and scavenging is imperative to allow enough ROS to participate in molecular signaling, while also curtailing the harmful effects of oxidative stress (160, 268, 276) (Fig. 6). When these systems become saturated and ROS are not counterbalanced effectively, the result is the cumulative and irreparable damage of proteins, lipids, and DNA (268, 276) (Fig. 6). Such damage may lead to cell death at high levels, or potentially mutagenesis and carcinogenesis at sublethal levels (88, 96, 278, 301, 302, 322) (Fig. 6).
NF-κB and ROS interaction
Activation of NF-κB signaling by ROS
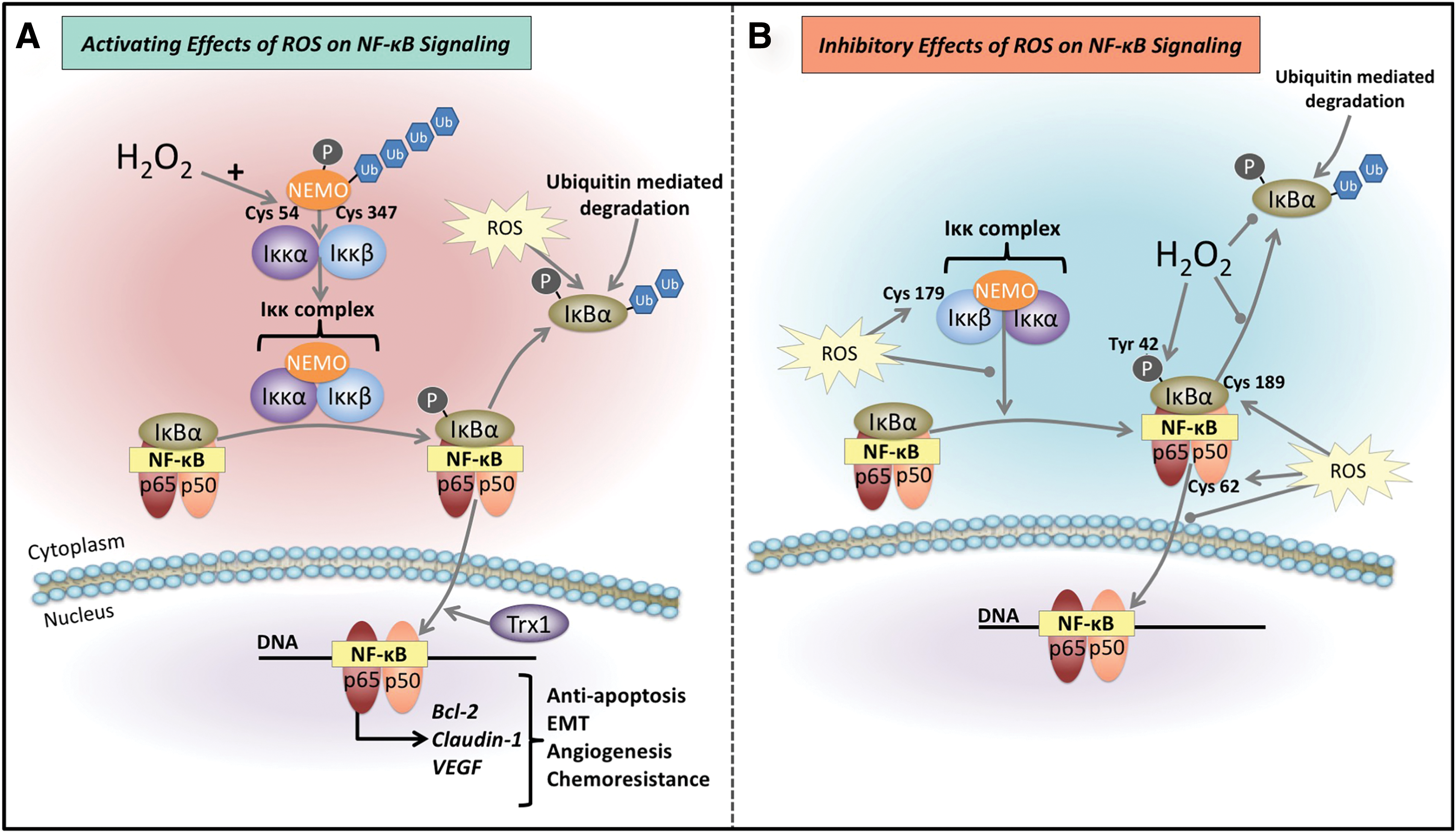
ROS have been found to directly interact with the NF-κB signaling pathway and have also been found to be important for its full activation, which is redox dependent (288) (Fig. 7A). As detailed previously, the Iκκ complex requires the complexation of NEMO to the catalytic portions of the Iκκ complex, Iκκα and Iκκβ, (38), which then uses its inherent kinase activity to phosphorylate the IκBα inhibitory protein (38). Interestingly, H2O2 is able to potentiate the complexation of NEMO dimers by introducing disulfide bonds between adjoining cysteine residues −54 and −347 (131) (Fig. 7A). In addition, ROS have been demonstrated to increase p-IκBα levels thus enabling more NF-κB dimers to enter the nucleus (153) (Fig. 7A). Once in the nucleus, the redox-regulated protein, thioredoxin-1 (Trx1), may facilitate NF-κB/DNA binding, through its reaction with cysteine residues of the DNA binding domain (95, 153, 197) (Fig. 7A).
Inhibition of NF-κB signaling by ROS
Conversely, ROS have also been reported to inhibit NF-κB signaling (168, 255, 275) (Fig. 7B). For instance, Iκκ complex kinase activity, which is important for the phosphorylation of the IκBα subunit, is also reported to be inactivated by ROS (225, 275, 370). This is potentially mediated through the oxidation of cysteine-179 of the Iκκβ constituent of the complex (275) (Fig. 7B). Exogenous sources of H2O2 have also been demonstrated to affect the inhibitory IκBα subunit (43, 286, 287, 319). The putative mechanism of p-IκBα degradation entails the phosphorylation of serine-32 and serine-36 and this consequently leads to proteasome-mediated degradation (125, 157, 225, 320, 340, 370). However, in the presence of exogenous H2O2, phosphorylation of tyrosine-42 residue and potentially other residues may also prevent the degradation of this molecule, thus inhibiting NF-κB transcriptional complex release (43, 286, 287, 319) (Fig. 7B). It has also been reported that ROS can attenuate NF-κB signaling through glutathionylation of cysteine-189 of the IκBα protein (161) (Fig. 7B). This glutathionylation results in the stability of this molecule, preventing its degradation via the proteasome, therefore promoting the inhibitory complex (161) (Fig. 7B). In addition, ROS are also able to directly oxidize cysteine-62 of the p50 subunit, which inhibits the DNA binding ability of NF-κB dimers (328) (Fig. 7B).
ROS, resistance, and cancer progression
Interestingly, current chemotherapeutics and radiation, both widely used for the treatment of cancer, also generate ROS that at high concentrations are cytotoxic to cancer cells (108, 230, 231, 352) (Fig. 6). However, at sublethal levels, the ROS produced by these agents have been found to promote invasion and metastasis and may thus contribute to drug resistance, ultimately limiting the success of treatment, as described above (77, 245, 270) (Fig. 6). MDR is considered a major hurdle dictating the efficacy of cancer treatment, with ∼90% of cancer recurrence caused through MDR genes (66, 87, 116, 178, 216, 258, 265, 337). The resistance occurs as a result of inherent and/or acquired resistance to the therapeutic applied (10, 97, 101, 138). Of interest, there is mounting evidence to support the contribution of redox activity to the molecular events leading to MDR (108, 254, 330). Of note, as cancer cells already exhibit high levels of ROS, they are more vulnerable to further increases (232, 237, 254, 281). Importantly, high ROS levels are antitumorigenic and result in apoptosis, and thus, this vulnerability provides a potential therapeutic niche (232, 248, 331) (Fig. 6).
Considering its significant role in cell signaling and the NF-κB pathway, ROS have also been implicated in cancer progression (237, 254) with NOX-derived ROS considered to be the predominant source of oxidative stress in cancer cells [for review see Moloney and Cotter (232) and Roy et al. (281)]. Moreover, elevated levels of ROS are reported in the majority of cancer cell lines when compared to normal cells and have been strongly implicated in metastasis and the ability of tumor cells to be refractory to treatment (254). Indeed, ROS are thought to play a critical role in carcinogenesis via their ability to induce genetic mutations (150), while also mediating various signaling cascades pertaining to survival, proliferation, antiapoptosis, neovascularization, invasion, and extravasation and metastasis (34, 182, 198, 237, 276).
Moreover, reports also indicate that selective pressure on cancer cells induce an adaptive antioxidant response to reduce the high levels of ROS and oxidative stress generated either by the immune response or by anticancer agents (77, 108, 330). Indeed, cancer cells have the capacity to balance ROS to achieve levels that are not cytotoxic, but that can stimulate tumorigenic signaling (77, 108, 188, 220, 272). That is, ROS can induce the following: (i) MAPK signaling; (ii) extracellular signal-regulated kinase (ERK) signaling; (iii) JUN N-terminal kinase (JNK) signaling; (iv) cyclin D1 expression; and (v) inactivate the tumor suppressor phosphatase and tensin homologue deleted on chromosome 10 (PTEN) (77, 108, 188, 220, 272). Ultimately, the capacity of the tumor cell to evade ROS cytotoxicity reduces the efficacy of ROS-generating anticancer agents, or ROS-mediated defense by the immune response (108).
The genomic instability and DNA damage inflicted by ROS in cancer have also been implicated in drug resistance and are associated with patient relapse (259) (Fig. 6). Potentially, these genetic alterations may provide a means of drug-resistant variants evolving to induce heterogeneity within the cancer cell population, rendering treatments ineffective and enabling disease progression (82, 97, 235, 335).
Targeting ROS and NF-κB
Adjuvant antioxidant and pro-oxidant therapy
There are probably a multitude of redox-dependent mechanisms that are able to contribute to drug resistance in cancer. Considering this, redox-active drugs (antioxidants and pro-oxidants), or inhibitors of cancer cell antioxidant defense (ROS, resistance, and cancer progression section), could be a valuable therapeutic strategy to “tip the balance” in favor of normal cellular ROS activity or proapoptotic cytotoxicity (67, 108, 229, 330) (Fig. 6).
Interestingly, antioxidant therapy, including agents such as pyrrolidine dithiocarbamate (PDTC) and vitamin E used in conjunction with 5-fluorouracil (5-FU) and doxorubicin, was found to enhance the cytotoxicity of these agents in vitro against colorectal cancer cells (28). This effect was reported to occur through induction of the potent cell cycle inhibitor, p21WAF1/CIP1, resulting in a significant reduction in colorectal cancer cell growth (28). Thus, one therapeutic strategy could entail the coadministration of antioxidants with chemotherapeutic agents for the treatment of cancer (Fig. 8). Indeed, such evidence supports the great potential of targeting ROS in cancer for more effective therapeutics. Of note, antioxidants have previously been found to inhibit NF-κB activation, and as discussed in the Downstream targets of NF-κB contributing to treatment resistance section, this latter pathway is known for its contribution to resistance (26, 132, 140, 187, 345). Thus, antioxidants may reduce the levels of tumorigenic ROS required by cancer cells to incite oncogenic pathways and prosurvival activity (Figs. 6 and 8). However, as antioxidants possess low specificity for the targeted site, further development is required for their application as a cancer therapeutic (154, 307). One such development is the potential of antioxidant therapy delivered as nanoparticles, to facilitate more targeted delivery in cancer treatment (307).
Metal chelation therapy
There are also pro-oxidant agents capable of producing very high levels of targeted ROS to proffer cytotoxicity as a means of targeting cancer. Of note, thiosemicarbazone chelators bind intracellular iron and copper to form redox-active complexes that have the appropriate electrochemistry to enable ROS generation (277, 368). These agents have the capacity to produce cytotoxic ROS, in a manner that induces selective anticancer activity (147, 291, 312) (Fig. 6). This arises from the tumors overt metabolic needs that require increased uptake of nutrients, including iron and copper (85, 195, 246). Both iron and copper are capable of producing ROS, such as O2 •− and •OH (155, 368). The cycling between the ferrous [Fe(II)] and ferric [Fe(III)] states, and cupric [Cu(II)] and cuprous [Cu(I)] states, enables the activity of these metal ions as electron donors or acceptors (155).
By way of Fenton chemistry, ferrous iron or cuprous copper can react with H2O2, giving rise to the cytotoxic OH• (166, 326). Thus, with greater amounts of these respective metals, cancer cells are more susceptible to metal chelation and will also be subject to greater levels of thiosemicarbazone-metal complex-induced ROS (99, 156, 184, 256, 277, 369). The generation of ROS initiates oxidative damage by reacting with biological targets that induce lysosomal membrane permeabilization and tumor cell death (19, 24, 148, 208).
As discussed in the Molecular Effects of Redox-Active Agents section, ROS have the potential to induce cell death through the initiation of reactions with important biomolecules concerning the lysosome, mitochondrion, lipid membranes, and DNA (19, 155). The ROS activity proffered by the iron chelator, 3-aminopyridine-2-carboxaldehyde (Triapine®) (Fig. 9A), has been demonstrated to damage the ribonuclease reductase (RR) enzyme (266). Of note, RR activity is the rate-limiting step of DNA synthesis, catalyzing the conversion of ribonucleotides into deoxyribonucleotides, with its inhibition resulting in the arrest of cells at the G1/S interface (266, 293, 325). However, a number of significant adverse effects were reported following administration of Triapine, including neutropenia, hypoxia, hypotension, methemoglobinemia, and abnormal electrocardiograms (9, 361). Hence, these problems demonstrate a need for more selective and effective agents.
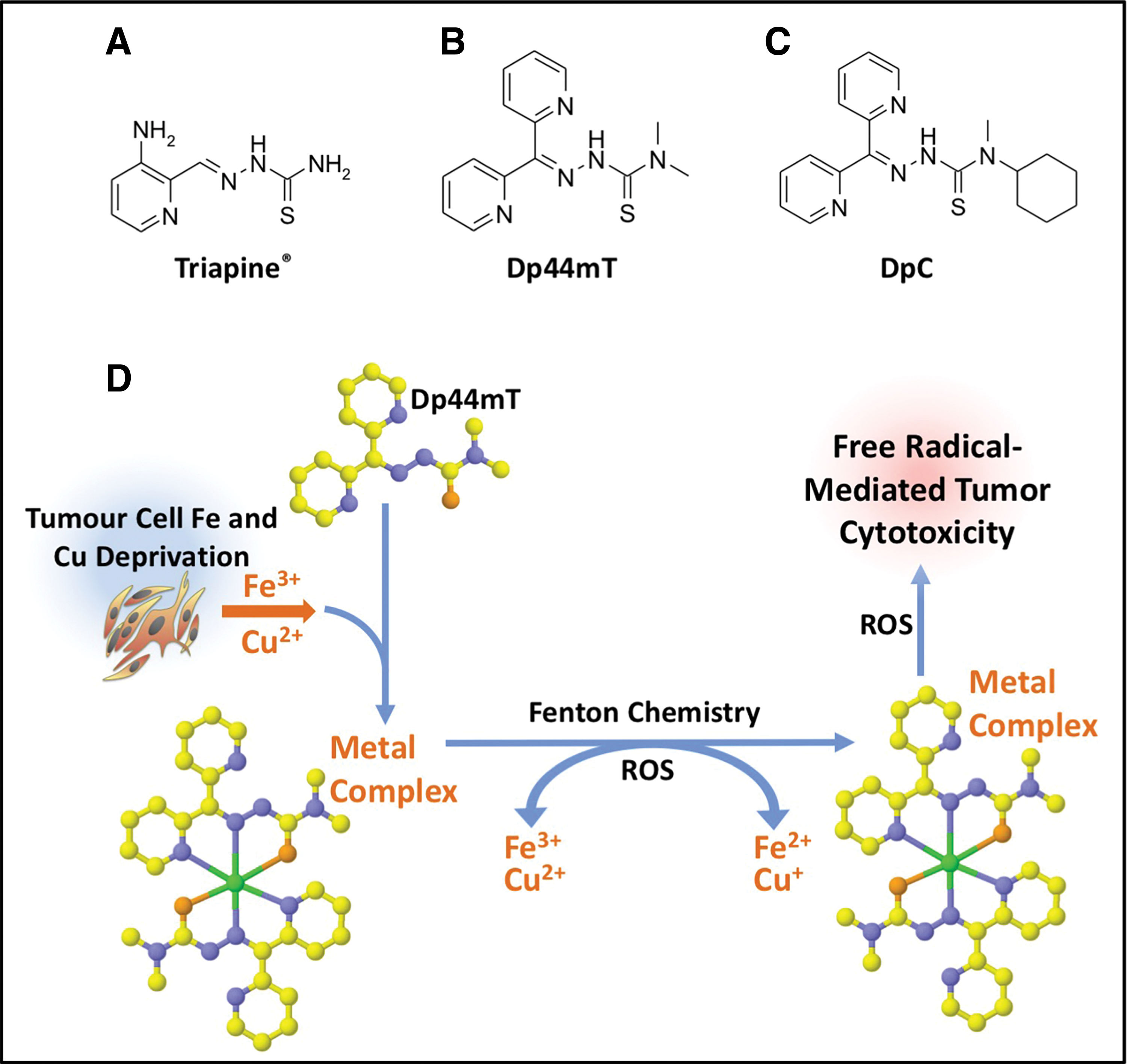
As such, the di-2-pyridylketone thiosemicarbazone (DpT) series were developed, including the lead agents, di-2-pyridylketone 4,4-dimethyl-3-thiosemicarbazone (Dp44mT) (351, 369) and di-2-pyridylketone 4-cyclohexyl-4-methyl-3-thiosemicarbazone (DpC) (115, 170, 173, 209) (Fig. 9B, C). Due to the lysosomotropic properties of both these agents, they accumulate in lysosomes, where they bind iron and copper released via the process of autophagy, forming redox-active complexes, which elicit lysosomal membrane permeabilization and potent cytotoxicity (148, 208, 290, 310).
Both Dp44mT and DpC demonstrated marked antitumorigenic activity when compared to conventionally used chemotherapeutic agents for the treatment of pancreatic cancer (namely, 5-FU and gemcitabine) (170). Moreover, DpC was also found to be active by both the oral and intravenous routes, which was an advantage over Dp44mT that was only active via the intravenous route (209). In light of these findings, the pursuit of DpC for the treatment of cancer appears to be a highly promising and viable strategy, with DpC recently entering Phase I clinical trials for advanced and resistant cancers (NCT02688101) (148, 156). The success of thiosemicarbazone chelators is attributed to (i) their binding to transition metals (i.e., copper and iron) generating ROS (148, 256) (Fig. 9D); and also (ii) their capacity to induce a number of molecular effects that are antitumorigenic and antimetastatic, that is, their ability to upregulate the metastasis suppressor, N-myc downstream regulated gene 1 (NDRG1) via a mechanism involving hypoxia inducible factor-1α (HIF-1α)-dependent and hypoxia inducible factor-1α-independent processes (171, 172, 183, 184) (Fig. 10).
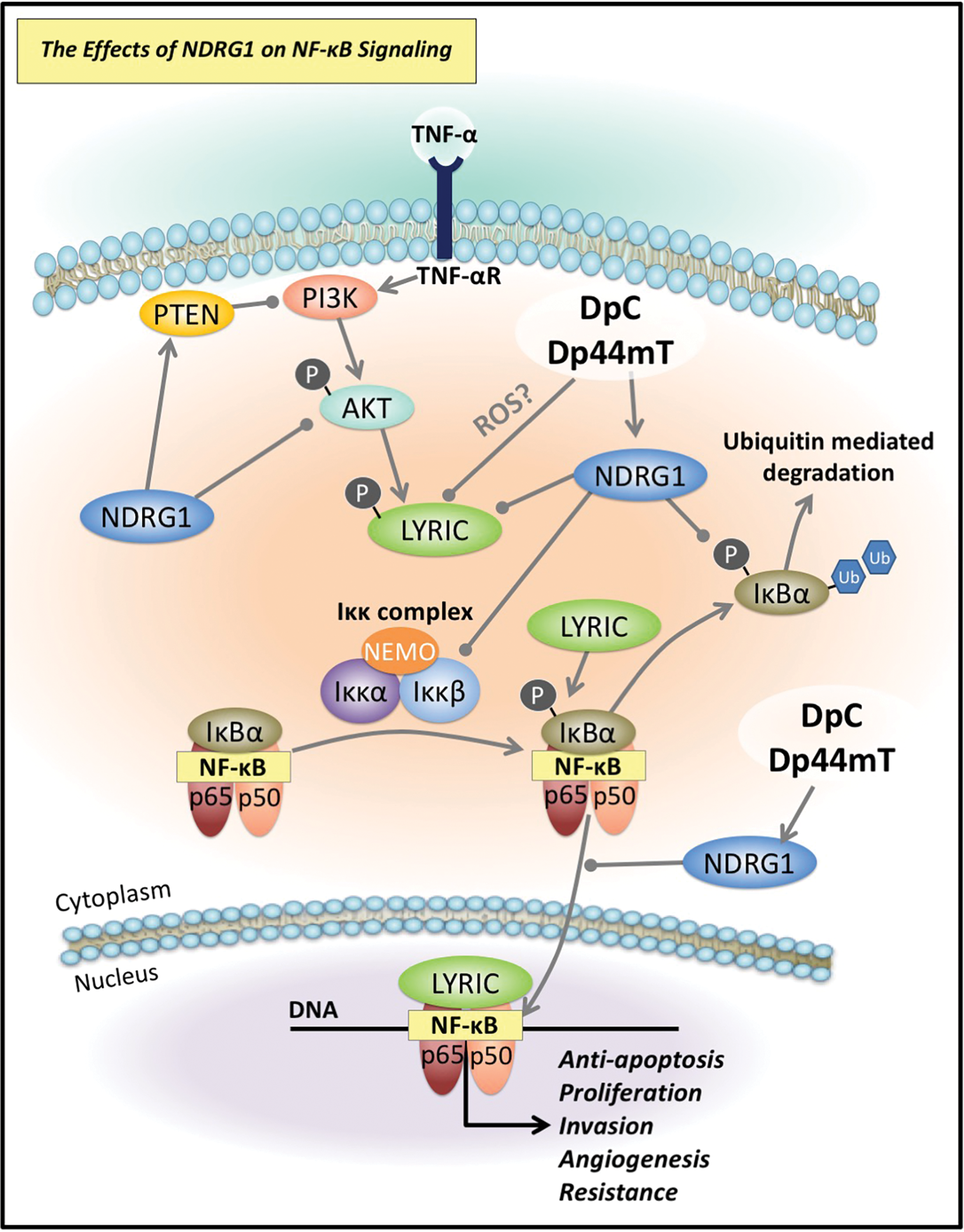
Of pertinence to this review, Dp44mT and DpC were recently demonstrated to significantly inhibit NF-κB signaling (357). This is mediated, in part, through attenuating the expression of the oncogene lysine-rich carcinoembryonic antigen-related cell adhesion molecule 1 coisolated protein (LYRIC) by these agents, which plays a functional role in activating the NF-κB pathway (357) (Fig. 10). LYRIC activates the NF-κB pathway by promoting degradation of the inhibitory IκBα bound to NF-κB, enabling nuclear translocation of the active p50/p65 NF-κB heterodimer (86, 193, 283) (Fig. 10). The ability of Dp44mT and DpC to inhibit LYRIC was demonstrated to occur via their ability to upregulate the metastasis suppressor, NDRG1 (357) (Fig. 10).
There has been expanding evidence to indicate that NDRG1 signaling may negatively regulate the NF-κB pathway (202, 212, 357) (Fig. 8). Expression of NDRG1 has been reported to lead to a marked reduction in tumor angiogenesis through the inhibition of the NF-κB signaling pathway, whose downstream effectors include VEGF, a well-established promoter of angiogenesis (221, 297). It has been reported that NDRG1 inhibits the internuclear actions of NF-κB by attenuating levels of the upstream molecules, Iκκβ and p-IκBα (136) (Fig. 10). In this way, NDRG1 effectively prevents degradation of the inhibitory IκBα molecule, and thus, this prevents the consequential release of the active NF-κB dimer (125, 225) (Fig. 10). Interestingly, a knockdown of the NDRG1 gene was found to promote the EMT of colorectal cancer cells, amplifying the phosphorylation of NF-κB (212).
Of note, NF-κB may be activated upstream by other oncogenic signaling pathways widely implicated in tumorigenesis, including PI3K/AKT and Wnt signaling (16, 177, 185, 329). Interestingly, NDRG1 is able to inhibit activation of PI3K/AKT signaling by decreasing p-AKT (activator of LYRIC) and c-Myc levels, while also increasing expression of the tumor suppressor, PTEN, which acts as an antagonist to PI3K/AKT signaling (80, 171, 357) (Fig. 10). In addition, NDRG1 has an inhibitory effect on Wnt signaling through the modulation of β-catenin levels and localization, inhibiting its phosphorylation at serine-33/-37 and threonine-41 and its nuclear translocation (152, 203). Of note, nuclear β-catenin is able to indirectly lead to the activation of NF-κB signaling (16). Importantly, the inhibitory effects of NDRG1 on these latter pathways have been shown to be pharmacologically induced by Dp44mT and DpC (80, 152, 171, 203).
Interestingly, NDRG1 levels correlated with a significant decrease in drug-resistant lung cancer cells (200). Furthermore, the inhibition of NDRG1 expression resulted in the acquisition of an EMT phenotype and an increase in these cells' resistance to cisplatin (200). In addition, in colorectal cancer cells, the silencing of NDRG1 induced the stem cell-like properties that have been established as key factors for enabling chemoresistance (57, 213, 273, 344). As Dp44mT and DpC have the capacity to upregulate NDRG1 (170, 184, 357) and inhibit NF-κB p65 nuclear translocation (357) (Fig. 10), there may also be potential for these agents to combat drug resistance by this mechanism.
Conclusions
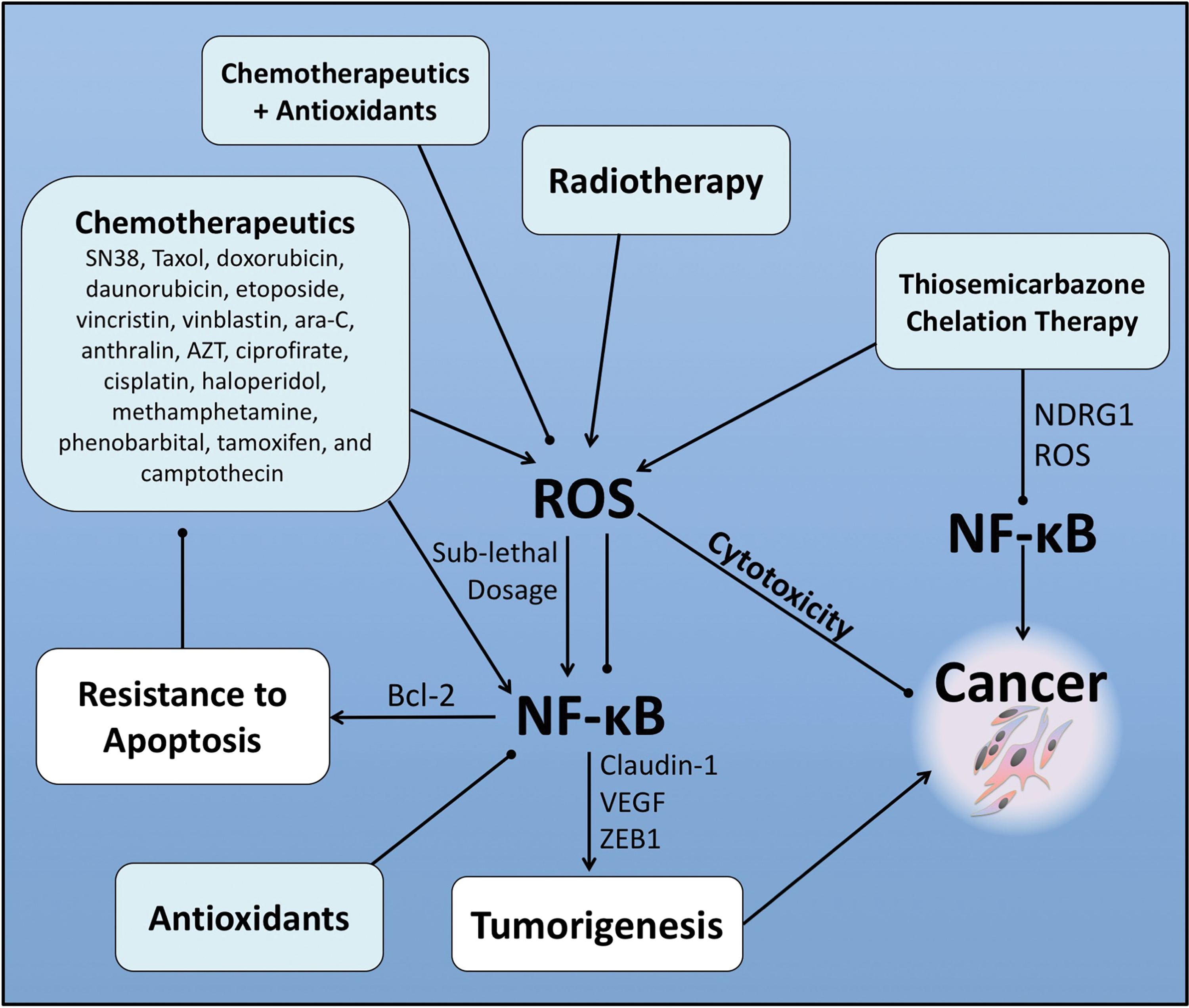
NF-κB is a major chemoresistance-related prosurvival factor (Fig. 8). Many human cancers have high levels of constitutive NF-κB activity, which can be further induced by anticancer drugs. As discussed, ROS elicit a number of effects on NF-κB signaling and is important for (i) activation of NF-κB signaling; (ii) inhibition of NF-κB signaling when at higher levels; and (iii) ROS-induced cytotoxicity resulting in apoptosis (153) (Fig. 8). Many cancer treatments currently used activate NF-κB (see the NF-κB is activated by chemotherapeutic agents and irradiation leading to chemoresistance section), and in so doing may potentially confer resistance (Fig. 8). As discussed, combinatorial approaches may prove an effective strategy to reduce NF-κB activity alongside conventionally used therapeutics that utilize ROS cytotoxicity to kill cancer cells (Fig. 8).
In addition, novel anticancer agents that generate large amounts of ROS may play an important role in attenuating the activation of NF-κB and promoting apoptosis of cancer cells. Such agents include the thiosemicarbazones, Dp44mT and DpC, which inhibit NF-κB signaling via upregulation of the metastasis suppressor, NDRG1 (Fig. 8). Indeed, targeting NF-κB signaling using the advantageous effects of ROS could reduce the incidence of drug resistance and enhance the efficacy of currently used chemotherapeutics (Fig. 8).
Footnotes
Acknowledgments
We appreciated comments on the article before submission from Drs. Danuta Kalinowski, Michael Huang, and Sumit Sahni of the Molecular Pharmacology and Pathology Program, Department of Pathology and Bosch Institute, University of Sydney. L.F. sincerely appreciates an Australian Postgraduate Award from The University of Sydney. Z.K. acknowledges the support of the National Health and Medical Research Council (NHMRC) of Australia for a Peter Doherty Early Career Fellowship and RD Wright Fellowship. Z.K. also kindly thanks the Cancer Institute of New South Wales for an Early Career Fellowship and Fellowship. Z.K. appreciates a Young Investigator PdCCRs Grant jointly funded by Cancer Australia and the Cure Cancer Australia Foundation. D.R.R. sincerely appreciates Project Grants and a Senior Principal Research Fellowship from the NHMRC.
Author Disclosure Statement
D.R.R. is a stakeholder in Oncochel Therapeutics, LLC and Pty. Ltd. that is developing the thiosemicarbazone, DpC, for the treatment of advanced and resistant cancer.