
Abstract
Significance:
Particulate matter (PM) air pollution is a leading cause of global cardiovascular morbidity and mortality. Understanding the biological action of PM is of particular importance in improvement of public health.
Recent Advances:
Both fine (PM <2.5 μM) and ultrafine particles (<0.1 μM) are widely believed to mediate their effects through redox regulated pathways. A rather simplistic graded ramp model of redox stress has been replaced by a more sophisticated understanding of the role of oxidative stress in signaling, and the realization that many of the observed effects may involve disruption and/or enhancement of normal endogenous redox signaling and induction of a potent immune-mediated response, through entrainment of multiple reactive oxygen species (ROS).
Critical Issues:
The molecular events by which pulmonary oxidative stress in response to inhalational exposure to air pollution triggers inflammation, major ROS (e.g., superoxide, hydroxyl radical, nitric oxide, and peroxynitrite) generated in air pollution exposure, types of oxidative tissue damage in target organs, contributions of nonimmune and immune cells in inflammation, and the role of protective proteins (e.g., surfactant, proteins, and antioxidants) are highly complex and may differ depending on models and concomitant disease states.
Future Directions:
While the role of oxidative stress in the lung has been well demonstrated, the role of oxidative stress in mediating systemic effects especially in inflammation and injury processes needs further work. The role of antioxidant defenses with chronic exposure will also need further exploration. Antioxid. Redox Signal. 28, 797–818.
Background
T
Air Pollutant Considerations
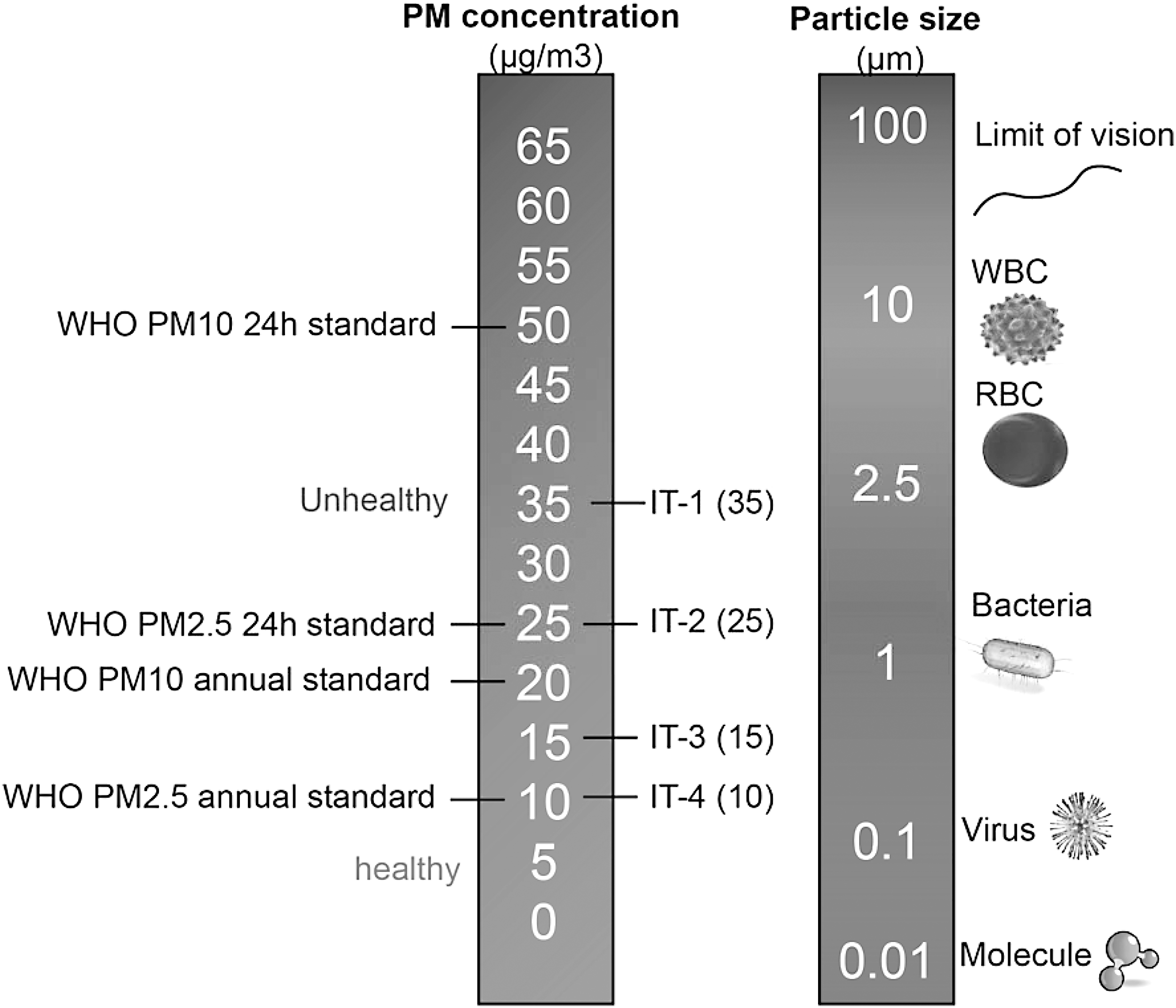
Airborne particulates constitute a heterogeneous complex mixture, differing based on source and varying with time, and atmospheric conditions (18). A number of factors affect the toxicity of particles, including size, shape, structure, surface reactivity, solubility, biopersistence, and “leachable” components (114, 122, 123, 139). Considering these factors, classification of particles based on physicochemical attributes is not easy and for lack of better ways, the most common method to characterize PM is based on size (Fig. 1). The most important size-fractions are particulate matter <10 μM (PM10), particulate matter <2.5 μM (PM2.5), and particulate matter <0.1 μM (PM0.1; also called ultrafine particles, UFPs). Coarse particles typically originate from natural sources such as dust from earth cover, road and tire abrasion, construction work, and agricultural sources. This component may also contain substantial amounts of lipopolysaccharides. The fine and ultrafine fractions tend to be dominated by anthropogenic sources such as power, combustion, mining, and other industrial sources. Size is an important determinant of locus of deposition (upper vs. lower airways) and toxicity, as the smaller particles have a larger surface-to-mass ratio and also compositionally are replete with reactive components (121, 122, 139). UFPs carry an abundance of soluble components on the particle surface, such as transition metal ions and organic compounds, including polycyclic aromatic hydrocarbons (PAHs) that may mediate systemic effects (122 –124). PAHs are a group of hydrocarbons—organic compounds that are produced from the burning of organic substances such as coal, oil, gasoline, trash, tobacco, wood, and charcoal-broiled meat. They are catalyzed by cytochrome P450 and dihydrodiol dehydrogenase and generate ROS (92, 126). Exposure to PAHs has been implicated in CV disease related to smoking and environmental exposures (134). Source apportionment studies demonstrate that the organic carbon fraction and sulfate had the strongest evidence for associations with the CV disease endpoints, with much weaker evidence for elemental carbon and silicon (12, 17, 99, 131, 169).
Current challenges in interpretation of studies of air pollution-mediated toxicity
Clarifying the mechanisms by which particles trigger inflammatory and redox pathways are at the core of particle toxicology and biological effects. There are several difficult questions pertaining to better understanding of the role of ROS in the pathogenesis of air pollution-mediated effects. These are enumerated below and are considered throughout the article.
(i) Contribution of endogenous cellular sources of ROS: ROS may originate not only directly from the particles, but also from various intracellular sources. The best evidence for the contribution of endogenous cellular sources at least in animal models is proof that manipulation of ROS pathways through knockout or other models modulates effects of air pollution exposure (31, 63, 89, 136, 178, 184).
(ii) Delineating contributions of ROS from immune cells versus nonimmune cells: The precise delineation of the role of ROS from immune-mediated cells to nonimmune cells such as endothelial and epithelial cells may be important to understand locus of effects and potential targets for intervention (106).
(iii) Role of pulmonary oxidative stress in mediation of systemic responses: Many elements within the PM could directly elicit oxidative damage to the airway and lung tissues. The inflammation and ROS in the lung likely help to remove the injurious stimuli and initiate tissue repair. However, the persistence of pulmonary inflammation and imbalance in ROS and antioxidant response may cause systemic effects. The role of pulmonary oxidative stress and various damage-associated molecular patterns (DAMPs), including oxidatively modified lipoproteins, oxDNA, ssRNA, dsRNA, HMGB1, and mitochondrial protein, and their impact by binding to various receptors (Toll-like receptors [TLRs] and Receptor for Advanced Glycation End Products, [RAGE]) in triggering systemic cytokine and chemokines are important areas of research (35, 36, 59, 106).
PM exposure methodological considerations
Mechanisms obtained from simple cell culture studies and in vitro models although useful to define specific pathways, are of limited utility in predicting systemic responses outside of the lung, especially given the fact that the cells in consideration likely never see these particles (51, 139, 166). Intratracheally administered doses rely on numerous assumptions and often result in uneven intrapulmonary distribution of the particles bypassing the upper airway tracts. Small rodent models, almost exclusively used in research, differ in their breathing patterns, nasal anatomy, and filtering mechanisms from humans, making extrapolation of results difficult. Concentrator systems provide the best compromise as they allow physiologic exposure to higher concentrations over prolonged periods and mimic the physiologic route of entry. Both a strength and limitation, however, are that both concentrations and composition can vary considerably from day-to-day. In addition, only certain particle size ranges are typically concentrated, whereas ambient air contains a mixture of particle sizes and gases. Potential interactions between PM and gaseous copollutants are therefore excluded, unless the latter is reintroduced (22, 93). Other methods of controlled inhalation exposures include diesel engine exhaust (diluted and aged mixtures of high numbers of fresh combustion UFP with vapor-phase components), roadside aerosols, and wood burning sources and are useful in examining effects of source-specific emissions (94, 98, 100). Human studies involving direct exposure although valuable, occur over a few hours and these responses may not be representative of prolonged exposure. Moreover, exposure health risk of experimental subjects, specifically those with underlying heart and lung diseases, and high expense are difficult issues. Panel studies in humans and analysis of surrogate endpoints also provide windows into potential pathways, but are associative. In this review, any reference to PM2.5 exposure is equivalent to exposures to concentrated ambient PM2.5 (CAP) in whole body chambers, unless otherwise specified.
Evidence for Systemic Oxidative Stress with Air Pollution Exposure: Insights from Animal Studies into Sources
Evidence of oxidative stress involvement in the lungs has been noted extensively previously (81). In addition, evidence of oxidative stress has been noted systemically in many organs (30, 54, 180, 184, 186, 192) (Table 1 and Fig. 2). Acute exposure studies have shown a relationship between the vascular dysfunction in systemic microvessels and the release of myeloperoxidase from leukocytes into the vasculature within only hours after the pulmonary instillation of PM (120). In a seminal investigation involving ApoE−/− fed high-fat diet, chronic exposure to CAP exacerbated vascular oxidant stress and promoted atherosclerosis progression (155). The proatherogenic effects of ambient UFP versus PM2.5 in genetically susceptible ApoE−/− mice in a mobile whole-body exposure facility close to a Los Angeles freeway have also been compared (4). Exposure to UFP resulted in a more pronounced atherosclerotic effect (compared to PM2.5), inhibition of anti-inflammatory capacity of high-density lipoprotein (HDL), and greater systemic oxidative stress as evidenced by increased hepatic malondialdehyde (MDA) and upregulation of Nrf2-regulated antioxidant genes (4).

ABTS, 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid); Akt, protein kinase B; BAL, bronchoalveolar lavage; BAT, brown adipose tissue; BC, black carbon; CAPs, concentrated ambient PM2.5; CAT, catalase; DHE, dihydroethidium; eNOS, endothelial nitric oxide synthase; FA, filtered air; Gclm, glutamate-cysteine ligase modifier subunit; GST, glutathione S-transferase; IDL, intermediate-density lipoprotein; IkBa, inhibitor of kappa B; iNOS, inducible nitric oxide synthase; LDL, low-density lipoprotein; MDA, malondialdehyde; mRNA, messenger RNA; NO, nitric oxide; Nqo1, NAD(P)H dehydrogenase (quinone 1); Nrf2, nuclear factor, erythroid derived 2, like 2; oxPAPC, oxidized 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine; PAPC, 1-palmitoyl-2-arachidonyl-sn-glycero-3-phosphorylcholine; PEG-SOD, polyethylene glycol-SOD; PGPC, 1-palmitoyl-2-glutaroyl-sn-glycero-3-phosphorylcholine; PM, particulate matter; PM2.5, particulate matter <2.5 μM; POVPC, 1-palmitoyl-2-(5-oxovaleroyl)-sn-glycero-3-phosphorylcholine; ROS, reactive oxygen species; SOD, superoxide dismutase; TBA-RS, thiobarbituric acid reactive substances; TLR4, Toll-like receptor 4; UCP-1, uncoupling protein-1; WAT, white adipose tissue; WT, wild type.
In Sprague-Dawley rats exposed to PM2.5 for 10 weeks, before angiotensin II infusion, exposure increased superoxide (O2
•−) production in the aorta as measured by in situ dihydroethidium (DHE) staining, as well as enhancing the vasoconstriction to phenylephrine compared to the filtered air (FA) group (158). The O2
•− production was abolished by NAD(P)H oxidase inhibitor apocyanin and NOS inhibitor N-omega-nitro-
Unfolded Protein Response and Endoplasmic Reticulum Stress
Oxidative stress, endoplasmic reticulum (ER) stress, and inflammation usually coexist in the pathogenesis of multiple cardiometabolic diseases such as diabetes and atherosclerosis (57, 74). Unfolded protein response (UPR)/ER stress is an evolutionarily conserved and sophisticated cellular response to alleviate protein misfolding (171). Various environmental stressors can disrupt ER protein-folding resulting in the accumulation of “unfolded” proteins and ER stress (53). The activation of UPR involves three ER transmembrane stress sensors: inositol-requiring enzyme 1α (IRE1α), PKR-like ER kinase (PERK), and activating transcription factor 6α (ATF6α) (194). These ER stress sensors are normally bound to immunoglobulin protein (BiP) and are inactive, but in response to unfolded/misfolded proteins, BiP dissociates to bind unfolded/misfolded proteins initiating downstream signaling (49).
Recent studies have suggested that UPR/ER stress is involved in PM2.5 exposure-induced adverse effects (69, 101, 133). After 10 weeks of whole-body exposure (mean PM2.5 concentration 74.6 μg/m3), both oxidative stress and ER stress were detected in the lung and liver tissues of C57BL/6 mice (69). To test the hypothesis that ROS are required for PM2.5-induced UPR signaling in macrophages, cells overexpressing manganese superoxide dismutase (SOD) or dominant negative Rac 1 (N17Rac1) were exposed to PM2.5 particles in the media. PM2.5 exposure increased levels of phosphorylated eIF2α, CHOP, and GADD34 in the control RAW264.7 cells but not in cells expressing Mn-SOD or dominant negative N17Rac1, suggesting that PM2.5-induced ER stress depends on the production of ROS (69). A subsequent study confirmed the upregulation of a number of genes associated with ER stress along with enhanced infiltration of macrophage in the white adipose tissue from PM2.5-exposed animals, suggesting that activation of ER stress via ROS pathways may occur in systemic tissues (101). Phosphorylation of eIF2α was increased in the liver along with induction of CHOP/GADD153, a C/EBP homologous transcription factor, associated with apoptosis in the lung and liver (69). This study also demonstrated a critical role for NOX-dependent oxidant stress in the activation of PM2.5-induced ER stress, as p47−/− mice were protected against ER stress. Mendez et al. also reported that inhalational exposure to PM2.5 chronically (10 months) induces UPR/ER stress, lipid deposition, and adipocyte differentiation changes in adipose tissue (101). There was an increase of expression of ER stress-associated genes (such as BiP/GRP78, Xbp-1, and Edem1) in white adipose tissue of PM2.5-exposed mice, along with an increased size of adipocytes. ER stress may also regulate adipocyte lipid metabolism and inflammation (33, 65). Indeed, expression of the genes involved in lipogenesis, lipid transport (cluster of differentiation 36 [CD36]), TG synthesis, and adipocyte differentiation/lipid droplet formation in white adipose were affected by exposure to PM2.5 (101). In a subsequent study, exposure to PM2.5 led to activation of c-Jun N-terminal kinase (JNK), nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), and TLR4, and suppression of the insulin receptor substrate 1 (IRS1)-mediated signaling in the liver, with fat deposition and glycogen depletion consistent with a nonalcoholic steatohepatitis picture (69). PM2.5 exposure repressed expression of the peroxisome proliferator-activated receptor PPARγ and PPARα in the liver, suggesting that inflammatory activation, perhaps through ER stress, may modulate an inflammatory response that then leads to downregulation of PPARα and PPARγ leading to inflammation and reduction of fatty acid oxidation and hepatic glycogen production (69).
Xu et al. observed that in response to PM2.5 exposure, ApoE−/− mice (96.89 μg/m3, 2 months) reduced the expression of brown adipocyte-specific genes, while white adipocyte-specific genes were differentially upregulated in brown adipose tissue (BAT) (186). Thus, exposure to air pollutants and likely PM2.5-mediated ROS and ER stress may adversely affect adipose tissue lipid metabolism and brown to white adipose transition. Indeed, in additional careful experiments performed in metabolic cages, PM2.5 exposure resulted in reduced VO2 and VCO2 levels, consistent with a reduction in metabolism. These effects were associated with reduction in uncoupled protein-1 (UCP-1) expression in BAT consistent with an impact of CAP on thermogenesis (87). Whether mitochondrial ROS sources are additionally involved remains to be determined, but parenthetically, chronic inhalational PM2.5 exposure results in reduction in BAT mitochondria and downregulation of BAT genes, including UCP-1 (181, 186).
Antioxidant Mechanisms and Redox Balance with Air Pollution Exposure
In contrast to the rather robust in vivo evidence base supporting the upregulation of ROS pathways in response to air pollution exposure, the regulation and expression of antioxidant defenses with in vivo are relatively sparse. There are a multitude of endogenous pathways that scavenge a range of ROS, including enzymatic systems (such as SOD, catalase [CAT], glutathione peroxide, thioredoxin reductases, NADPH quinone oxidoreductase 1 [NQO1], and methionine sulfoxide reductases) and nonenzymatic entities (such as glutathione [GSH], vitamins A, C, and E, and flavonoids) (97). Many of these defense mechanisms are regulated at the transcriptional and post-transcriptional levels. For example, SOD, glutathione S-transferase A2 (GSTA2), and NQO1, the major detoxication enzymes, contain antioxidant response elements in their promoter region. In response to oxidative stress, Keap1 dissociates from Nrf2, releasing it to bind to the antioxidant response element of target antioxidant genes (61, 143). The magnitude of antioxidative response may depend on the duration, concentration, and toxicity of exposure, and susceptibility of the subjects to air pollutants. For instance, antioxidants may increase in the early phase to limit oxidative damage, while long-term exposure or high-level exposure or highly toxic air pollutants may cause exhaustion of endogenous antioxidant responses. In this regard, there is a paucity of understanding of temporal regulation of antioxidant responses at the transcriptional and post-transcriptional level.
Two weeks of exposure to PM2.5 resulted in vascular oxidative stress and upregulated the expression of Cu/Zn-SOD and Mn-SOD together with a reduction in endothelial nitric oxide synthase (eNOS) and vasorelaxation in the pulmonary artery (30). Findings by Xu et al. indicate an increase in messenger RNA (mRNA) expression of Nrf2 and downstream genes Nqo1 and glutamate-cysteine ligase modifier subunit (Gclm) in C57BL/6 mice exposed to CAP for 10 months (181). The contribution of locus and tissue-specific Nrf2 in attenuating PM2.5 effects are important questions and would need to be examined. At least in in vitro studies, stimulation of Nrf2−/− dendritic cells with PM, augmented oxidative stress, and cytokine production compared with resting or Nrf2+/+ cells. In contrast to Nrf2+/+ cells, coincubation of Nrf2−/− dendritic cells with PM and the antioxidant N-acetyl cysteine attenuated PM-induced upregulation of CD80 and CD86 (173). These findings suggest that broad transcriptional regulators of antioxidant responses may regulate initiation and maintenance of T cell proliferation in response to antigenic stimulation to components of PM.
Mechanisms by Which Oxidative Stress Mediates Effects of PM in the Lung
The molecular events by which pulmonary oxidative stress triggers inflammation, along with the contributions and interactions between nonimmune and immune cells, the role of protective proteins (e.g., surfactants, proteins, and antioxidants) is highly complex and may differ depending on models (39, 111, 149). In the human condition, the situation is decidedly more complex, with the ultimate effect depending on particle fate (e.g., lung clearance vs. retention), sequestration, intracellular distribution, pathways of potential systemic transmission, and ultimately on multiple host factors, including susceptibility (39, 149). The importance of oxidant stress mechanisms and the contribution of alveolar macrophages to generation of cytokines and chemokines have been previously extensively reviewed and we touch on broad themes (11, 55, 106, 113, 168).
Role of direct ROS generating capacity of particle/particle constituents and the oxidant stress paradigm
It is often difficult to determine if particle-derived ROS formation or secondary endogenous ROS by cells contribute to toxicity. This is important in the correct interpretation of the direct toxic effects of particles, where prior studies have attempted to correlate the oxidative capacity of particles in cell-free systems and their ability to induce inflammatory responses or other effects in cells, animals, or humans (51, 139, 166). The effects of PM in animal models with inhalational exposure may vary depending on the composition and sources of particulates and since UFPs have more reactive components and greater ROS potential, they may be expected to have greater systemic effects (3). For example, Manhattan PM2.5 composed of traffic-related UFP has larger effects on atherosclerosis (89% increase) (190) compared with those in Sterling Forest, New York (58–68% increase) (155, 157). Araujo et al. also reported that UFP-exposed mice developed 25% greater atherosclerotic plaques compared with those exposed to PM2.5 (4). In humans, the evidence suggests an inconsistent correlation between oxidative capacity of particles and their effects in humans (166). In the graded ramp model of oxidative stress, low levels of ROS result in activation of antioxidant and phase II defenses (114). When the antioxidant response is inadequate, pathological oxidative stress can initiate a variety of pulmonary inflammatory responses. For example, ROS in the lungs have been shown to augment the signal transduction of membrane ligands (e.g., epidermal growth factor by disrupting phosphatases) and pattern recognition (e.g., TLRs) (9, 24, 56, 58, 83) that lead to the increased expression of a variety of cytokines and chemokines. With higher levels of ROS, there could be activation of kinase pathways (MAPK) and transcription factors such as NF-κB and AP1 leading to increased synthesis of inflammatory proteins. At extreme levels of ROS, there may be direct alterations in membrane permeability and mitochondrial damage. This rather simplistic model does not account for host defense mechanisms, including antioxidant response that may curb the effects of ROS.
PM interaction with cellular membranes
Carbon nanoparticles have been reported to alter the composition of lipid rafts in airway epithelial cells, by increasing the content of ceramides (sphingolipids) (129). Components such as PAHs may alter the fluidity of cellular membranes and affect lipid raft formation (164). Since lipid rafts play a central role in aggregation of receptor complexes, the alterations in this key membrane component could affect a variety of signaling pathways and cellular functions. Notably, this response seems to be mediated through activation of the cytosolic aryl hydrocarbon receptor (AhR) and inhibition of cholesterol synthesis (163, 164). PAHs have also been found to increase the fluidity of cellular model membranes directly, and benzo[a]pyrene (B[a]P) may interact with carbonyl groups of phospholipids (67, 86). Negatively charged surface groups on the surface of particles may interact with positively charged moieties on the head group of membrane phospholipids resulting in alteration of membrane bilayer and induction of membrane permeability. Crystalline particles such as asbestos and quartz while efficiently taken up by pulmonary macrophages without toxicity (due to coating by protective proteins, including surfactants), lysed in the acidic environment of the lysozyme, where their surface is stripped away resulting in lysosomal membrane damage and rupture. Membrane binding may induce both interleukin-1β (IL-1β) transcription and cleavage of pro-IL-1β to mature IL-1β through combined activation of NF-κB and ROS-mediated stimulation of the nucleotide-binding oligomerization domain (NOD)-like receptor containing pyrin domain 3 (NLRP3) resulting in so called frustrated phagocytosis (66, 72, 119, 159).
Activation of membrane-associated surface receptors
Membrane-associated receptors have been noted to be involved in a variety of in vitro studies in recognizing particles and particle components (148). Transient receptor potential channels (TRP) have been implicated in particle sensing and may be activated by combustion particles or soluble organics (transient receptor potential cation channel, subfamily A, member 1 [TRPA1], and transient receptor potential cation channel, subfamily V, member 1 [TRPV1]) (68, 140). Some of these receptors appear to be activated directly by combustion particles or soluble organics, such as TRPA1 and TRPV1, while others such as TRPV4 have been suggested to be activated more indirectly through transactivation (40, 52, 150). TRP-mediated calcium signaling, at least in the case of TRPV4, seems to regulate inflammatory gene transcription through ERK1/2 cascade (77). The precise role of ROS in regulation of TRP channels is not clear.
Multiple families of pattern recognition receptors (PRRs) exist and include C-type lectin receptors, TLRs, and pentraxins that survey the extracellular milieu, as well as the nucleotide-binding domain leucine-rich repeats (NLRs) and RIG-I-like receptors (RLRs), which detect intracellular signals. Biological components of PM2.5 such as endotoxin, DNA, may activate TLRs directly or indirectly through secondary mediators such as ROS (Fig. 3) (10, 60, 148). Scavenger receptors such as CD36 are also involved in sensing PM-generated oxidation products and may cooperate with TLRs for mediation of effects (146). For instance, oxidized lipid derivatives such as 1-palmitoyl-2-arachidonyl-sn-glycero-3-phosphorylcholine (PAPC) and 7-ketocholesterol may be recognized by CD36 and phagocytosed by macrophage and ultimately synergize with TLR4-dependent pathways to amplify inflammation (136). ROS as an activator of TLR and NLRP3 has been suggested by many studies, in which inhibition and/or deficiency of NOX-derived ROS prevented TLR activation or ATP-induced caspase-1 activation and IL-1β production in alveolar macrophages (26, 36). In an important study, innate immune signaling via TLR4-TIR-domain-containing adapter-inducing interferon-β (TRIF)-TNF receptor-associated factor (TRAF)-6 was shown to be a key genetic pathway that determines the susceptibility to acute lung failure in vivo (59). In the same study, oxidized phospholipids generated in the lung in response to acute lung injury were shown to activate the TLR4 signaling cascade. Local lung injury not only triggered activation of the oxidative stress but also induced upregulation of TLR4 and amplified inflammatory response. Deficiency of p47 phox significantly ameliorated the generation of ROS and production of oxidized phospholipids.

Although multiple PRR families in conjunction with uptake receptors such as CD36 converge in the regulation of cytokine and chemokine transcription, the NLR family is more specifically responsible for maturation of proinflammatory cytokines IL-1β or IL-18 especially in response to crystalline or foreign biomaterials (38, 96, 146). The activation of NlrP3 appears to be a two-step mechanism, with the primary signal from the activation of TLRs and transcriptional upregulation of NLRP3 and pro-IL-1β via NF-κB. Secondary signals come from multiple pathways: K+ efflux via P2X7 receptor activation, ER stress, mitochondrial dysfunction, NOX, frustrated phagocytosis, and lysosomal rupture pathways, all of which appear to converge in the production of ROS. The hypothesis that ROS could serve as an NLRP3-activating trigger was initially proposed when NOX-derived ROS prevented ATP-induced caspase-1 activation and IL-1β production in alveolar macrophages (26). Knockdown of the p22 phox subunit of NOX significantly suppressed IL-1β release in THP1 cells in response to asbestos and monosodium urate challenge, both of which are classic triggers for NLRP3 activation (36). The crystal structure of NLRP3 contains a highly conserved disulfide bond connecting the PYD domain and the nucleotide-binding site domain, which is sensitive to redox alterations (5).
Mechanisms by Which Pulmonary Oxidative Stress Is Systemically Transduced
The mechanisms underlying initiation of systemic inflammation in response to air pollution and the involvement of oxidative stress in this transduction are still evolving but almost certainly have to involve the lung in some capacity. Hypothesized mechanisms include the following. (i) Release of inflammatory cytokines and chemokines systemically (46, 63). (ii) Recruitment of a systemic innate immune response (42, 46, 63, 89, 188). (iii) Secondary antigens generated in response to oxidation, by antigen-presenting cells that may then present antigens to the T cells in the draining lymph node and activate adaptive immunity (32). (iv) Leachable components such as transition metals and organic secondary intermediates such as oxidized phospholipids, quinines, semiquinones, and aldehydes generated in the lung that may overflow into the circulation (34, 63, 85). (v) Activation of central nerve system pathways via TRP receptors, C-fibers, taste receptors, or other afferent mechanisms that may facilitate systemic inflammation (91, 133, 150). (vi) Small particles (especially UFPs) entering blood stream and impair endothelial function by direct interaction with the endothelium.
Reactive intermediates generated in response to air pollution exposure and role in systemic responses
PM-induced oxidation may also result in a number of oxidatively modified molecules, which serve as a secondary mediator to induce systemic effects. These mediators include oxidatively modified proteins, lipids, surfactants, and matrix proteins.
To illustrate the role of oxidized lipids as a secondary mediator, we examined the oxidative stress response in the lungs and oxidized lipid generation in mice exposed chronically (>20 weeks) to concentrated PM2.5. Airborne PM2.5 markedly increased oxidized derivatives of PAPC in the bronchoalveolar lavage (BAL) fluid. Both 1-palmitoyl-2-(5-oxovaleroyl)-sn-glycero-3-phosphocholine (POVPC) and 1-palmitoyl-2-glutaryl phosphatidylcholine (PGPC) increased with PM2.5 exposure. Incubation of PM2.5 by itself with BAL had no effect on oxidized phospholipids, arguing against direct particle-mediated ROS generation and suggesting an endogenous mechanism of oxidation of PAPC (63). The production of NOX-derived O2 •− was increased in monocytes, aortic tissue, and perivascular fat from wild-type (WT) mice exposed to concentrated PM2.5, accompanied by impaired vascular function suggesting a systemic oxidative stress response. In another study, an increase of oxidized 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine (oxPAPC) was noted in the brain of PM2.5-exposed mice, as measured by mass spectrometry (88).
To delineate the involvement of TLR4 and NOX pathways in the generation of oxidized lipids, and recruitment of a systemic inflammatory response, Nox2−/− , WT mice, and mice deficient in TLR4 (Tlr4Lps-d [TLR4d]) were exposed to air pollution. PM2.5 increased inflammatory monocytes (CD11b+Ly6Chi) in the peripheral circulation in TLR4 wt mice, an effect diminished in TLR4 d mice (16). PM2.5 exposure-associated vascular O2 •− production, vascular dysfunction, and macrophage infiltration were reversed in TLR4 d mice and in Nox2−/− mice (63). TLR4wt mice demonstrated an increase in TNFα, MCP-1, and IL12p70 and a decrease of IL-10 levels in the lung, while TLR4 deficiency attenuated these responses. Increase in plasma levels of TNFα and MCP-1 with PM2.5 exposure was normalized in TLR4d mice. Incubation of bone marrow-derived macrophages to oxidized PAPC recapitulated a stereotypical inflammatory response reminiscent of the in vivo effects in the lung with PM2.5 exposure, including phosphorylation of a cytosolic subunit of NOX p47 and interleukin-1 receptor-associated kinase (IRAK) phosphorylation, abolished by TLR deficiency. Using atherosclerosis-prone ApoE−/− mice, we demonstrated that PM2.5 exposure also increased 7-ketocholesterol, an oxidatively modified form of cholesterol, in plasma LDL and intermediate-density lipoprotein fractions concomitant with progression of atherosclerosis. In addition, CD36, a scavenger receptor recognizing specific oxidized phospholipids and lipoproteins, was upregulated in PM2.5-exposed mice in peripheral monocytes and within plaque macrophages. Increased CD36 expression resulted in foam cell formation and accumulation of 7-ketocholesterol in atherosclerotic plaque, while CD36 deficiency in myeloid cells abolished PM2.5-induced 7-ketocholesterol accumulation, foam cell formation, and plaque progression in LDLR−/− mice (136).
Gaseous air pollutants such as ozone have been demonstrated to have important effects on immune activation (153, 193). Garantziotis reported that O3 exposure increased hyaluronan levels in the lavage fluid and enhanced airway hyperactivity in C57BL/6 mice. Short-fragment hyaluronan (100–400 kDa) but not high-molecular-weight hyaluronan activated macrophages and induced airway hyperactivity in CD44 (a hyaluronan surface receptor)- and TLR4/myeloid differentiation primary response gene 88 (MyD88)-dependent mechanisms (43 –45, 84). Mice deficient in CD44 (43, 44), TLR4, MyD88, or TIRAP (84) attenuated O3-induced airway hyperactivity and production of proinflammatory cytokines, including TNFα, IL-1β, MCP-1, IL-6, and keratinocyte chemoattractant (CXCL1).
Recruitment of a systemic innate immune response in response to PM
A role for ROS
Multiple early studies have demonstrated that acute exposure results in recruitment of an innate bone marrow response, but the identity of these cells and specific populations was unclear (46, 162). Using a model of fluorescently labeled monocytes under control of a c-fms (receptor for macrophage-colony stimulating factor, M-CSF), Sun et al. confirmed that intratracheal PM2.5 enhanced YFP+ migration, adhesion to the mesenteric microcirculation, and accumulation in visceral fat (156). These results were redemonstrated with chronic exposure to ambient PM2.5 where significantly more adherent YFP+ cells were found in the cremasteric endothelial wall in response to chronic PM2.5 (63). Xu et al., while investigating the effects of PM2.5 exposure over 10 weeks in C57Bl/6 mice, detected increased macrophage infiltration in visceral adipose tissue and vascular dysfunction (184). Deficiency of p47phox improved abnormalities in insulin resistance, vascular function, and reduced visceral inflammation (F4/80+ macrophages) in response to PM2.5, suggesting a proximal role for NOX in this process (184). TLR4 and NOX appear to mediate the effect of PM2.5 as deficiency of TLR4 diminished the effect of PM2.5 on Ly6Chigh cells (F4/80+, CD11b+, CD115+). The preponderant majority of PM2.5-mediated circulating Ly6Chigh cells appeared to originate from the bone marrow. Deficiency of TLR4 and Nox2 abolished the effects of PM2.5 on systemic tissues, suggesting a cooperation of these pathways in mediating exposure effects (63).
We examined the role of CCR2 and chemokine (C-X-C motif) receptor 3 (CXCR3) in two separate experiments involving separate groups of WT C57BL/6, CCR2−/− , and CXCR3−/− knockout mice, exposed to variable but chronic periods to PM2.5 or FA. In the first article, PM2.5 exposure resulted in increased CD11b+CD11c+ macrophages and CXCR3+ T cells in the lung, mediastinal lymph nodes, and spleen (32). While circulating CD4+ cell numbers remained unchanged, CD8+ cell numbers increased in WT-PM2.5 mice. The number of activated CD44+CD62L−CD4+ T cells in the lung was also increased in PM2.5-exposed WT mice. CXCR3 deficiency completely prevented the movement of CXCR3+CD4+ and CXCR3+CD8+ T cells into the lung, reduced the numbers of activated T cells (CD44+CD62L− CD4+), in response to PM2.5, and also reduced increases in CD11c+CD11b+ dendritic cells in the lung seen in response to PM2.5. CXCR3 deficiency attenuated the total number of CD4+ and CD8+ T cells exposed to PM2.5 seen in WT and was associated with increased retention of these populations in the spleen, suggesting impairment of homing to the lung. Central memory cell populations (CCR7+ CD44+CD62+) were increased with PM2.5 in both WT and CXCR3−/− mice compared with FA groups, suggesting that CXCR3 is not involved in the trafficking of this subset of cells (32). Consistent with the prior study by Kampfrath et al., PM2.5-exposed mice had significantly higher ratios of oxidized derivatives of PAPC (POVPC and PGPC) in the BAL fluid, with the ratio of PGPC/POVPC to PAPC nearly doubling with exposure to PM2.5 (63). These findings suggest robust innate and adaptive immune activation in response to the air pollution, likely via oxidative stress-mediated transformation of intermediate proteins with recruitment via CXCR3 mechanisms. In a subsequent article, we tested the involvement of CCR2 in mobilization of innate immune cell populations. CCR2−/− or WT C57Bl/6 mice were exposed by inhalation to either FA or PM2.5 (PM) 17 weeks. We noted an increase in circulating CD11b+Gr-1low7/4hi cells, the inflammatory subtype in response to PM2.5 exposure, seen previously (63). The levels of CD11b+Gr-1low7/4hi in circulation following PM2.5 inhalation were significantly reduced in CCR2−/− mice with a corresponding decrease in the spleen. F4/80+ adipose tissue macrophages were increased in visceral adipose of WT mice but not in CCR2−/− exposed to PM2.5. PPARγ, a transcription factor required for alternate macrophage differentiation, was downregulated in visceral adipose tissue (VAT) of WT-PM2.5 mice, but was only partially downregulated in CCR2-PM2.5 mice. Nrf1 levels were significantly lower in the WT-PM group than that in the WT-FA group, and this was partially restored in CCR2−/− PM2.5 mice. CCR2−/− mice demonstrated reduction in whole-body insulin resistance and improvements in hepatic lipid accumulation in the liver. This occurred via SREBP1c-mediated transcriptional reprogramming, decreased fatty acid uptake, and suppression of hepatic p38 MAPK activity likely related to decreased hepatic infiltration of CCR2+ inflammatory cells in the CCR2−/− mice exposed to PM2.5. Abnormal phosphorylation levels of protein kinase B (AKT), AMPK in visceral adipose tissue in response to PM2.5 exposure, and adipose tissue macrophage content in WT mice exposed to PM were not present in CCR2−/− mice. In contrast to the studies by Kampfrath et al., vascular function impaired by PM2.5 was not significantly different between CCR2−/− and WT mice (63, 89).
A recent study by Haberzettl et al. provides definitive evidence that oxidative stress emanating from the lung may modulate systemic responses. In this work, exposure to concentrated PM2.5 using an exposure chamber for 9 or 30 days reduced insulin-stimulated Akt/eNOS activation in the lung and circulation system. Thirty-day PM2.5 exposure also increased adipose tissue inflammation and systemic glucose intolerance in WT mice. Treatment with antioxidant TEMPOL or overexpression of lung-specific extracellular SOD prevented PM2.5-induced suppression of Akt/eNOS activation and inflammation (50).
Activation of central nerve system pathways
The activation of autonomic nervous system has also been suggested to be involved in the systemic transmission of the adverse effect of air pollution and oxidative stress (21, 135). Air pollutants have been shown to permeate into the olfactory bulb through the olfactory nerve and induce inflammation in the central nervous system. In addition, the TRPA1 receptors in airway sensory neurons can also sense the environmental toxicants and aerogenic oxidants, resulting in neurogenic inflammation (150). We observed increased hippocampal inflammation and impaired spatial learning memory in animals exposed to chronic PM2.5 (41). In follow-up studies, we have provided evidence that activation of sympathetic nervous system and hypothalamic inflammation occurs in response to PM2.5 exposure, with inhibition of central IκB kinase β (IKKβ) preventing the adverse effects of air pollution on peripheral inflammation and abnormalities in insulin resistance and changes in whole-body metabolism (88, 191). In addition, PM particles may directly permeate via the olfactory nerve into the central nervous system (13, 112). Indeed, disruption of antioxidative response by deleting Nrf2 enhanced nerve injury and hypothalamus oxidative stress, while nanoceria, an anti-inflammatory and antioxidant stress biomaterial, restrained PM2.5-induced metabolic syndrome and inflammation (180). We also demonstrated that PM2.5 exposure increased the level of oxPAPC, an oxidatively modified lipid that has been shown to activate TLR/NF-κB pathway (36, 63), in the brain (88).
Endothelial dysfunction induced by direct interaction between PM and endothelial cells
Air pollution exposure-associated endothelial dysfunction has been evidenced in both humans and animals and considered to be an instigating factor for CV disease (28). Ultrafine components UFPs are able to penetrate lung epithelium and enter the bloodstream where they could mediate systemic effects (115, 117, 147, 161). Findings in both humans and mice suggest that nanoparticles inhaled into the lung could rapidly cross the alveolar membrane and appear in the circulation (115, 117). Individual toxic components such as heavy metals, hydrocarbons, and other organic chemicals on the surface of circulating UFPs may directly interact with vascular endothelium, causing oxidative stress and endothelium damage (116). Both endothelial vasodilation and endogenous fibrinolysis were found to be impaired in healthy adults exposed to dilute diesel exhaust (300 μg/m3) (104). We have previously demonstrated redox alterations in the vessel wall through NOX pathways, which in turn may increase aortic vascular tone through O2 •−-mediated upregulation of the Rho/ROCK pathway (158). Increased peripheral vasoconstriction due to short-term exposure to PM2.5 may result in elevation in systemic blood pressure and increased cardiac afterload, and over a longer term result in left ventricular hypertrophy, myocardial fibrosis, and alteration in coronary flow reserve (175).
Evidence of the Association Between Particulate Matter and Oxidative Stress in Humans
Evidence from panel studies
The demonstration of systemic oxidative stress is difficult in humans as the techniques are relatively simple and can be applied only on plasma or airway fluid. The assays used to assess the footprint of systemic “oxidative stress” or damage may significantly influence the results. As such, the clinical evidence for increased oxidative stress following exposure to air pollutants is limited, and existing studies somewhat inconsistent (Table 2). Nevertheless, a recently published meta-analysis suggests that there is a robust relationship between exposure to particulate air pollution and increases in oxidative DNA adducts and oxidized lipids in man (107). The studies that have demonstrated a positive relationship include an increase in urinary excretion of free 8-iso-prostaglandin2α among healthy adults following a 4-h exposure to concentrated wood smoke (8), and an increase in plasma antioxidant capacity 24 h after exposure to diesel exhaust in a group of healthy volunteers after a 1-h exposure (167). Other investigators (127) have observed significant differences in expression of genes involved in oxidative stress pathways due to diesel exhaust exposure. In a study that examined the effect of ultrafine traffic particles on oxidative stress-induced damage to DNA in healthy young adults exposed to low concentrations of ambient urban particles (PM2.5 and PM2.5–10 mass of 9.7 and 12.6 μg/m3, respectively) in an exposure chamber above a busy road with high traffic density, increased levels of DNA strand breaks and formamidopyrimidine-DNA glycosylase sites in monocytes were seen without any change in the DNA repair enzyme 7,8-dihydro-8-oxoguanine-DNA glycosylase (14). Similar to their previous findings with ambient exposure (170), the results seemed to suggest that short-term exposure to UFP may result in damage to DNA. Results from the same investigators failed to demonstrate significant biomarker signals for lipid or protein oxidative damage after similar near-roadway exposures (15).
8-epi-PGF2α, 8-epi-prostaglandin F2α; 8-OHdG, 8-hydroxy-2′-deoxyguanosine; 8-oxodG, 8-oxo-7,8-dihydro-2′-deoxyguanosine; 15-F2t-IsoP, 15-f2t-isoprostane; AMI, acute myocardial infarction; BP, blood pressure; CI, confidence interval; FeNO, fractional exhaled nitric oxide; GSS, glutathione synthetase; GSTCD, glutathione S-transferase C-terminal domain containing; GSTM1, glutathione S-transferase M1; GSTP1, glutathione S-transferase P1; GSTT1, glutathione S-transferase theta-1; HBAAS, 2-aminoadipic semialdehyde in hemoglobin; HBGGS, gamma-glutamyl semialdehyde in hemoglobin; HF HRV, high-frequency heart rate variability; HFE, high iron Fe; HO1, heme oxygenase-1; HRV, heart rate variability; LF HRV, low-frequency heart rate variability; NO2 •, nitrogen dioxide; oxLDL, oxidized low-density lipoprotein; PLAAS, 2-aminoadipic semialdehyde in plasma protein; QTc, corrected QT interval; SDNN, standard deviation of all normal RR intervals; sICAM-1, intercellular adhesion molecule 1; sVCAM-1, vascular cell adhesion protein 1.
Although not entirely consistent, the available studies demonstrate that acute exposure to PM, perhaps even at ambient levels, may induce acute systemic oxidative stress in human subjects under certain circumstances that may depend on host susceptibility factors. To investigate the effect of air pollution on oxidative stress markers, the association between plasma oxidized low-density lipoprotein (oxLDL) level and traffic-related air pollution, determined by the distance from the patient's residence to a major road and by airway macrophage carbon load, was studied in a cross-sectional study of nonsmoking adult outpatients with diabetes. Each interquartile range increase (0.25 μm2) in airway macrophage carbon load was associated with a 7.3 U/L (95% confidence interval [CI]: 1.3–13.3 U/L) increase in plasma oxLDL, and each doubling in distance from patient residence to major roads was associated with decrease in plasma oxLDL (62). In a study of 40 healthy university students before and after relocating from a suburban campus to an urban campus with high air pollution levels in Beijing, China, oxLDL was associated with iron and nickel concentration in PM2.5 particles (177). In another study carried out in Copenhagen, the association between personal PM2.5 exposure and MDA, a marker for lipid peroxidation, was examined in 50 students. A 3.7% increase in the concentration of MDA per 10 μg/m3 increase in personal PM2.5 exposure was found in women, although no significance was observed in men (152). The levels of urine 8-isoprostane, MDA in breath condensate, and alveolar NO were increased in humans exposed to wood smoke-derived particulate air pollution, along with the increase of serum amyloid A, a CV risk factor (7, 8). In addition, increased levels of oxidized nucleic acids such as 8-hydroxy-2′-deoxyguanosine (8-OHdG, also known as 8-oxo-7,8-dihydro-2′-deoxyguanosine [8-oxodG]) and 8-oxoguanine (8-oxoguanine) have also been observed in human subjects exposed to particulate air pollution (1, 2, 29, 75, 142, 160). Mitochondrial DNA is also a primary target of oxidative stress in response to environmental stimulation. Byun et al. measured personal PM2.5 exposure and blood mitochondrial DNA methylation in 48 healthy men in Massachusetts. They reported that the level of blood mitochondrial DNA methylation in the D-loop promoter was associated with PM2.5 level and heart rate variability (HRV) (20). Collectively, these data provide evidence that oxidative stress markers are associated with a variety of exposures in humans.
Epidemiological evidence of oxidative stress in particulate air pollution exposure
Larger epidemiologic studies investigating the susceptibility to air pollution have provided some evidence that oxidative stress plays an essential role in air pollution exposure. There are several studies conducted in the same population of elderly Caucasian men (The Normative Aging Study) that have studied the effects of polymorphisms in different antioxidant genes. By examining the association between PM2.5 and the high-frequency component of HRV in 497 participants in the Normative Aging Study, Schwartz et al. noticed an association between decreased high-frequency component and increased PM2.5 exposure during prior 48 h only in subjects without glutathione-S-transferase M1 (GSTM1) allele (144). Similarly, only subject with GT long tandem repeat polymorphism in the heme oxygenase-1 (HO1) promoter, but not those with short repeat variant, showed associations between PM2.5 and HRV measures (normal-to-normal intervals, high frequency, and low frequency) (25). Polymorphisms in other antioxidative genes, including high iron Fe (HFE) C282Y, CAT (rs2300181), glutathione Stransferase P1 (GSTP1), and glutathione S-transferase theta-1 (GSTT1) also modified the effects of PM2.5 (125, 138). However, there were also some studies that showed a negative impact of certain variants in antioxidative genes on certain effects of PM2.5 such as blood pressure (76, 108).
Controlled exposure studies
Several controlled exposure studies demonstrate that acute exposure to PM2.5 and dilute diesel exhaust results in rapid vascular dysfunction that manifests as endothelial dysfunction or transient constriction of a peripheral conduit vessel that is reversible at least in some studies (16, 19, 103, 104, 128, 167). In some of these studies, fine CAP exposure diminished conduit artery endothelial-dependent vasodilatation 24 h (but not immediately) postexposure (19). PM2.5 mass and TNF-α level postexposure were associated with the degree of endothelial dysfunction, suggesting that systemic inflammation induced by higher levels of particles was likely responsible (19). However, depending on the location and composition of exposure, concentrated PM2.5 may not always induce endothelial dysfunction, underscoring the importance of composition in determining vascular responses (102).
In studies of UFP, composed of elemental carbon, a 2-h exposure impaired peak forearm blood flow response to ischemia 3.5 h later. There were no other vascular changes or alterations at other time points (145). Several studies have also shown that dilute diesel exhaust can impair peripheral resistance vessel responses to agonists and reactive hyperemia responses (104, 128). The blunted responses to acetylcholine persisted for 24 h in healthy adults (167). In contrast, bradykinin- and sodium nitroprusside (SNP)-mediated vasodilatation and bradykinin-induced acute plasma tissue plasminogen activator (tPA) release were not altered 24 h later. In subsequent studies, patients with stable coronary artery disease exposed to dilute diesel exhaust for 1 h during intermittent exercise demonstrated reduced bradykinin-mediated tPA release; however, microvascular endothelial function was not impaired (103). This may perhaps be related to some degree of pre-existing endothelial dysfunction in these patients that prevented additional impairment. However, exercise-induced ST-segment depression and ischemic burden were significantly greater during diesel compared to FA exposure (103). In some studies, even a 24-h-long exposure to ambient pollution shunted into a chamber next to a busy street did not impair microvascular endothelial function assessed by digital tonometry (15). This exposure to near-roadway air, consisting of ambient UFP and PM2.5, also did not alter biomarkers of inflammation or markers of protein and lipid oxidation.
In a randomized controlled clinical trial, individuals with metabolic syndrome were exposed to diesel exhaust, 200 mg/m3 of fine PM, and FA for 120 min on days separated by ≥2 weeks, systolic blood pressure increased at all of the points measured during and after diesel exhaust exposure; the mean effect peaked between 30 and 60 min after exposure initiation (3.8 mm Hg [95% CI: −0.4 to 8.0 mm Hg] and 5.1 mm Hg [95% CI: 0.7–9.5 mm Hg], respectively). The alterations of endothelial function have been speculated to represent enhanced degradation of NO. This latter concept was tested in a randomized double-blind crossover study; healthy nonsmokers were exposed to diesel exhaust or FA. Bilateral forearm blood flow was measured during intrabrachial infusions of acetylcholine and SNP in the presence of an NO synthase inhibitor NG-monomethyl-
Effects of air pollution prevention on disease
Multiple studies have demonstrated that an improvement in air quality results in a favorable reduction in cardiopulmonary disease. Pope et al. showed in 2009 that reduction in air pollution was associated with as much as 15% of the overall increase in life expectancy (130). Data from personalized intervention studies are also available, demonstrating that use of domestic air-filtration devices, particle-filtration masks, and car air filtration/air conditioning leads to meaningful reduction in CV surrogates such as systolic blood pressure, improvements in microvascular function, autonomic tone, and lower levels of inflammatory biomarkers in adults exposed to PM2.5 (110).
Polymorphisms in several oxidative stress-related genes such as GSTM1, GSTP1, GSTT1, HFE C282Y, and CAT are found to be associated with the vulnerability to PM2.5 (125, 138, 144). There have been a few intervention studies that have demonstrated that the harmful effects of air pollution could be modulated by intake of antioxidant and anti-inflammatory nutrients. Canova et al. showed that serum levels of antioxidants (vitamin C, uric acid, and vitamin E) modified the effect of PM10 on asthma/chronic obstructive pulmonary disease exacerbations (23). Vitamin C and E supplementation for 6 months normalized biomarkers of oxidative stress in individuals exposed to coal electric power plant-derived PM to control levels, suggesting a protective effect of vitamins C and E against PM-associated oxidative insult (132). Omega-3 polyunsaturated fatty acid intake from fish oil has been associated with increased levels of antioxidant proteins such as SOD and GSH. In a randomized controlled trial in Mexico City, the elderly residents of a nursing home supplemented with 2 g/day of fish oil showed a 7% decrease in high-frequency component of HRV/standard deviation increase in indoor PM2.5, compared with a 54% decrease in those individuals supplemented with 2 g/day of soy oil (141). In another randomized, controlled exposure study, 4 weeks of fish oil supplement attenuated concentrated ambient fine/ultrafine PM-induced HRV changes and increases in very low-density lipoprotein/triglyceride (165). These studies appear to suggest that common interventions thought to be protective from a CV standpoint can be beneficial in protecting against air pollution.
Conclusions and Future Direction
Taken together, oxidative stress appears to be a common thread in the effects of air pollution (Fig. 4). While the role of oxidative stress is compelling in the lung, the role of oxidative stress in mediating systemic effects and its role in processes such as inflammation and injury need further work. The role of endogenous antioxidant defenses particularly with chronic exposure will need further exploration. The importance of personal interventions to reduce air pollution and their effects on key oxidative stress pathways, including endogenous antioxidant defense mechanisms, are important emerging areas in future research. Finally, the effects of targeted interventions that disrupt oxidant stress pathways and/or enhance antioxidant defenses in reducing effects of air pollution are also important areas of interest.

Footnotes
Acknowledgments
This work was supported by grants from the NIH (R01ES015146, R01ES019616, K99ES026241, and T32DK098107). J.Z. was supported by K01DK105108 and 17GRNT33670485.