
Abstract
Significance:
Since the discovery of the superoxide dismutase enzyme, the generation and fate of short-lived oxidizing, nitrosating, nitrating, and halogenating species in biological systems has been of great interest. Despite the significance of reactive oxygen species (ROS) and reactive nitrogen species (RNS) in numerous diseases and intracellular signaling, the rigorous detection of ROS and RNS has remained a challenge.
Recent Advances:
Chemical characterization of the reactions of selected ROS and RNS with electron paramagnetic resonance (EPR) spin traps and fluorescent probes led to the establishment of species-specific products, which can be used for specific detection of several forms of ROS and RNS in cell-free systems and in cultured cells in vitro and in animals in vivo. Profiling oxidation products from the ROS and RNS probes provides a rigorous method for detection of those species in biological systems.
Critical Issues:
Formation and detection of species-specific products from the probes enables accurate characterization of the oxidative environment in cells. Measurement of the total signal (fluorescence, chemiluminescence, etc.) intensity does not allow for identification of the ROS/RNS formed. It is critical to identify the products formed by using chromatographic or other rigorous techniques. Product analyses should be accompanied by monitoring of the intracellular probe level, another factor controlling the yield of the product(s) formed.
Future Directions:
More work is required to characterize the chemical reactivity of the ROS/RNS probes, and to develop new probes/detection approaches enabling real-time, selective monitoring of the specific products formed from the probes. Antioxid. Redox Signal. 28, 1416–1432.
Introduction
R
The term ROS refers to a family of oxygen-containing molecules including the superoxide radical anion (O2 •−) (Fig. 1), hydrogen peroxide (H2O2), hydroxyl radical (•OH), and singlet oxygen (1Δg O2). The term RNS includes nitric oxide (•NO), peroxynitrite (ONOO−), nitrogen dioxide (•NO2), nitrosating species (N2O3), and other nitrogen-derived species. All these species have unique reactivity and thermodynamic properties that need to be considered when choosing the method of detection. Further, the medium, or cellular locus where ROS/RNS are being detected, introduces additional challenges in terms of the sensitivity and specificity of the assay.
The strategies to detect O2 •− and •NO are significantly different if studying their generation in cells or the biochemistry of purified enzymes. For example, spin-trapping methodologies are ideal for in vitro detection of radicals such as O2 •−, but, in cells, spin-trapping methodologies are complicated by the intracellular reduction of the radical adducts generated from the reaction of O2 •– with the spin trap. In this case, detection of 2-hydroethidium formed from the reaction between hydroethidine (HE) and O2 •– has been shown to be a robust methodology (145). Moreover, studying mitochondrial O2 •– is also possible with triphenylphosphonium-targeted HE (90, 91).
H2O2 and ONOO− have attracted much attention in biology for their oxidative potential. This property has also served as the basis for their detection by various fluorescein-based probes and cysteine-based molecular probes (9, 30, 61). The advantages and disadvantages of these methodologies are discussed in terms of specificity and sensitivity. Detection of organic peroxides and hypochlorous acid, which represent an important group of reactive species, is clearly essential from the viewpoint of targets and species propagating oxidant signaling.
Over the past two decades, an impressive number of probes have been reported as involving in vitro characterization. After all these years of research, it has become clear that researchers need to understand the chemical and thermodynamic basis of the reaction between probes and reactive species (49, 72, 121, 146). The careful choice of suitable probes, combined with an effort to corroborate the results using additional methods, is, in most cases, necessary to better sustain the conclusions (49, 51, 121).
In this review, we focus on the description and discussion of probes and techniques to detect particular ROS, including the O2 •–, H2O2 and other hydroperoxides, hypochlorous acid (HOCl), •NO, and peroxynitrite (ONOO−), accurately in biological systems. We hope this review assists in clarifying some of the current controversies and discussions about the detection of ROS/RSN in biological systems.
Detection of O2 •–
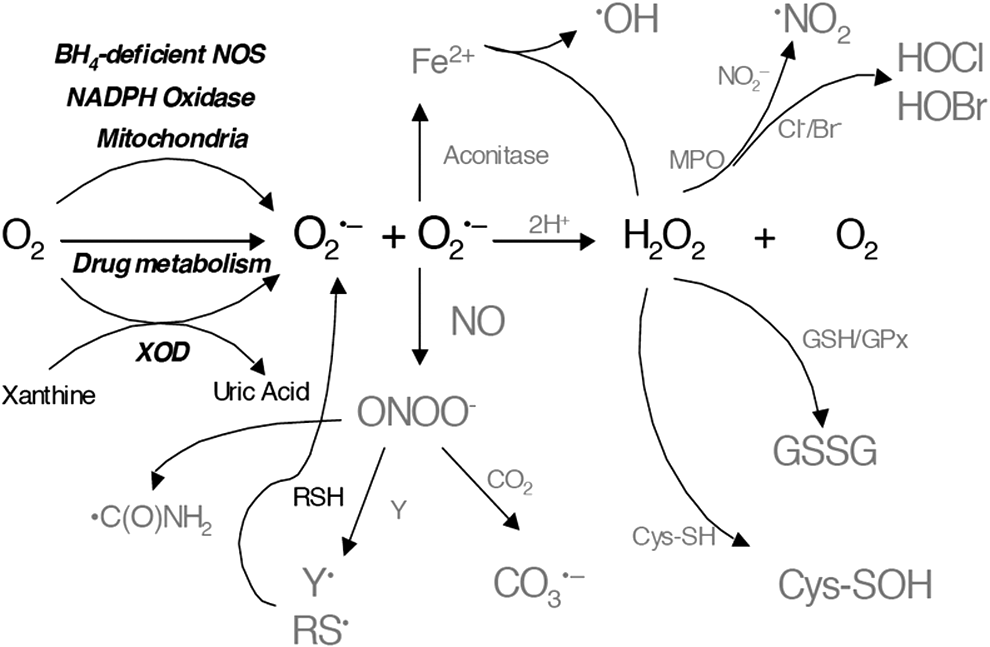
O2 •–, produced by the one-electron reduction of oxygen (O2), plays a central role by initiating the main chains of production of other ROS/RNS (Fig. 1). Its role is essential in pathological, signaling, and other physiological processes such as phagocytosis. The major cellular sources of O2 •– are believed to be mitochondrial complexes I and III and nicotinamide adenine dinucleotide phosphate (NADPH) oxidases. The reactivity of O2 •– with other molecules typically is relatively low, except with •NO and selected metalloproteins (e.g., superoxide dismutases, aconitases), which, in turn, makes it relatively selective. It should be emphasized that O2 •– is not stable in an aqueous (protic) environment and undergoes dismutation to H2O2 and O2 even in the absence of an SOD enzyme. The rate of spontaneous dismutation of O2 •– to O2 and H2O2 is typically high (k ∼ 105 M −1s−1 at pH 7) and becomes diffusion controlled in the presence of SOD (k ∼ 109 M −1s−1 at pH 7).
Detection of O2 •– by the spin-trapping technique
Among the different methods available to detect and characterize O2 •–, the spin-trapping technique combined with electron paramagnetic resonance (EPR) spectroscopy has been shown to be one of the most accurate and valuable methods for in vitro experiments (1, 14, 46, 60). Due to its short lifetime, at physiological conditions, O2 •– cannot be detected directly by EPR, and, thus, a series of spin traps for O2 •– have been developed. The principle of spin trapping (Fig. 2) relies on the addition of a diamagnetic spin trap probe to the sample, which reacts with short-lived free radicals to generate paramagnetic spin adducts exhibiting characteristic EPR signals that are specific to the trapped radicals and a lifetime ranging from a few seconds to hours, depending on the spin trap used and the radical species detected.

Nitrone spin traps have been shown to be efficient probes for O2 •– detection, allowing the detection of superoxide fluxes as low as a few μM .min−1 and generating superoxide adducts with a half-lifetime of up to 1 h. Over the past decade, significant improvement has been achieved in the design of nitrone spin traps for superoxide detection, with improved O2 •– trapping rate constants and longer lifetime of the corresponding adducts, as well as subcellular targeting properties (21, 26, 33 –35, 52, 75 –77, 85a, 118).
Although the spin trapping of O2 •– is considered one of the gold standard methods in in vitro cell-free experiments, current limitations make its use challenging for intracellular and in vivo O2 •– detection (2, 3). The main bottlenecks of the application of the EPR spin-trapping technique for O2 •– detection are the low trapping rate constant relative to the SOD-catalyzed dismutation and the reduction of the EPR-active spin adduct to EPR silent product(s) by endogenous antioxidants/reducing systems. Conversely, spin trapping generates species-specific products, with signature EPR spectral properties, and complementary simple control experiments using SOD or other competitors may be used for easy and accurate assignment of the obtained signal to O2 •–, making the method reliable and robust.
Examples of superoxide detection using the spin-trapping technique in the presence of cells have been reported in the past decades, mainly with stimulated macrophages. In these experiments, the choice of the spin trap has shown to be a key parameter for the successful detection of superoxide radicals, and usually, the EPR spectra exhibited the signals of the superoxide and hydroxyl (or carbon-centered radical) adducts as a mixture (1 –3, 94, 103).
Superoxide spin trapping of O2 •– generated from nitric oxide synthase
EPR spin-trapping detection of O2 •– has been significantly improved with the use of novel substituted DMPO-nitrones (31, 53, 128). These new spin traps produce more persistent O2 •– adducts that increase the sensitivity and reproducibility of O2 •–quantitative analysis. As an example of the significance of this methodological development, later we review the application of EPR spin trapping to clarify the role of the (6R) 5,6,7,8-tetrahydrobiopterin (BH4) cofactor in the regulation of O2 •–release from nitric oxide synthase (NOS), a reaction known as NOS uncoupling.
One possible consequence of O2 •– generation from NOS is that NOS itself could act as a peroxynitrite synthase. Thus, understanding the mechanisms and regulation of this activity has been the focus of several biophysical and biochemical studies. Neuronal NOS (nNOS) and endothelial NOS (eNOS) are homodimeric proteins, and each subunit contains a reductase domain with tightly bound flavin mono and dinucleotide (FMN and FAD, respectively) and a binding site for NADPH. Calcium/calmodulin activates the electron transfer from NADPH to flavins in the reductase domain, which, subsequently, results in the reduction of heme-Fe(III) to heme-Fe(II) in the oxygenase domain (106). Using DEPMPO and EMPO spin traps, we established that recombinant dimeric nNOS and eNOS, produced in Escherichia coli and purified in the absence of BH4, generate O2 •– from the heme-iron oxygenase domain on activation with the calcium/calmodulin complex (113, 115, 128). L-arginine increases O2 •– release from eNOS, whereas it decreases it from nNOS. This indicates that endothelial oxidant stress is likely enhanced on L-arginine supplementation in conditions of limited BH4 availability. Reconstitution of the BH4-free recombinant enzyme with BH4 dose dependently decreased O2 •– generation by nNOS and eNOS. The redox activity of BH4 in NOS catalysis was suggested by results from several experiments, including the fact that the oxidized metabolite 7,8-dihydrobiopterin (BH2) was unable to support •NO formation. Because BH4 but not BH2 inhibited O2 •–release from the oxygenase domain of NOS, it was proposed that BH4 directly reduced the heme-iron-dioxygen to a heme-iron peroxyl species (116). The breakdown of the heme-dioxygen intermediate produces O2 •–, whereas heme-iron-peroxyl generates H2O2. The redox activity of BH4 is also consistent with the requirement of BH4 to support the •NO-producing activity of NOS enzymes (Fig. 3). Low-temperature EPR analysis of the enzyme in single-turnover conditions demonstrated the formation of the BH4 radical, which is reduced back to BH4 in situ (44, 87).
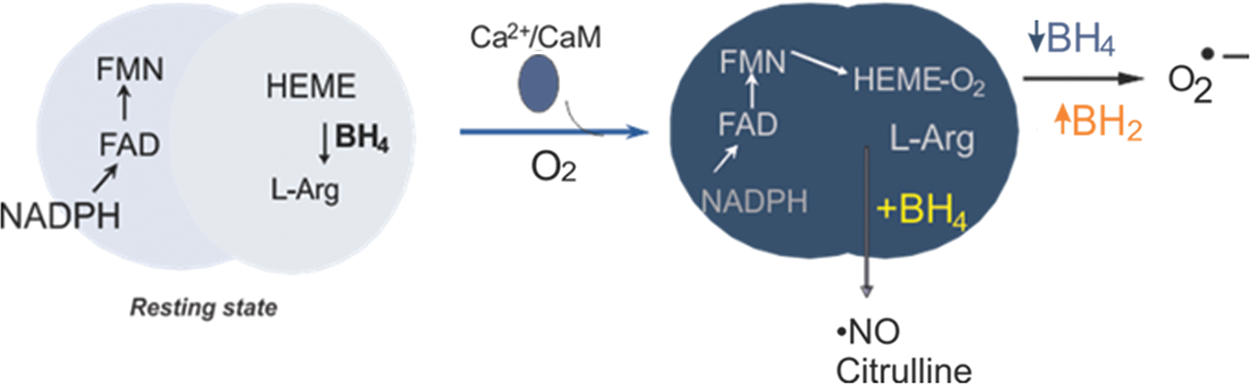
Evidence that the BH4 binding site is readily accessible to other BH4-analogs suggests that competition kinetics is possible. Using DEPMPO as a spin trap, we showed that the addition of BH2 to fully coupled eNOS (i.e., enzyme-producing •NO) but not O2 •– inhibited •NO production but increased O2 •– release, an effect known as NOS uncoupling and referring to the dissociation of NADPH oxidation to arginine oxidation in favor of O2 •– release (116). This mechanism has been implicated in the increased production of ROS and the cellular oxidative injury affecting cardiovascular functions.
Detection of O2 •– with luminescent probes
Luminescence (chemiluminescence and fluorescence)-based assays for O2 •– are widely used when measurements are performed in cells, tissues, and live animals. Among the chemiluminescent probes, luminol, L-012, and lucigenin probes are the most widely used (74). However, luminol is not a species-specific probe and the reliability of those probes for O2 •– measurements has been repeatedly questioned due to the redox properties of the probe-derived intermediates. Since these probes can react with multiple radical species (HO•, RO•, ROO•) and oxidants (ONOOH), it must be noted that these probes can be seen as specific to O2 •– only in purified in vitro cell-free system. Further, it was demonstrated that all those probes are capable of producing O2 •– on one-electron oxidation (luminol and L-012) or reduction (lucigenin) (23, 62, 114). For example, it was shown that lucigenin redox cycles in the presence of NOS and that the L-012 probe produces an SOD-inhibitable signal when incubated with H2O2 and peroxidase (114, 140). A most recent example of the confusion linked to lucigenin usage is the demonstration that lucigenin yields a luminescence signal in NADPH oxidase assays, when tested in tissue homogenates obtained from NADPH oxidase-lacking mice (triple Nox1/Nox2/Nox4 knockout model) (88).
Reduced cyanines (hydrocyanines) have been proposed as probes for O2 •– (56, 133). Although their advantage is the formation of a fluorescence signal in the near-infrared region, compatible with in vivo detection, the selectivity of those probes, as well as the product identity of their reaction with O2 •–, remains to be established. Similarly, other fluorogenic probes developed for O2 •– measurements have been reported recently; however, they still require comprehensive chemical characterization as well as a detailed validation in biological systems (40, 126).
HE (also known as dihydroethidium, DHE, Fig. 4) is the fluorescent probe of choice for the detection of O2 •– in biological systems (139). The major advantage of this probe is the formation of an O2 •–-specific product, 2-hydroxyethidium (2-OH-E+, Fig. 5), which can be detected by using high-performance liquid chromatography (HPLC) or liquid chromatography–mass spectrometry (LC-MS) techniques (50, 135, 145). Also, HE is one of the few probes whose chemistry has been studied in detail, including the reaction kinetics, stoichiometry, and reactivity toward different oxidants (67, 139). Therefore, the HE probe, combined with HPLC-based detection of 2-hydroxyethidium, should be used as the first-line approach to study cellular production of O2 •–.
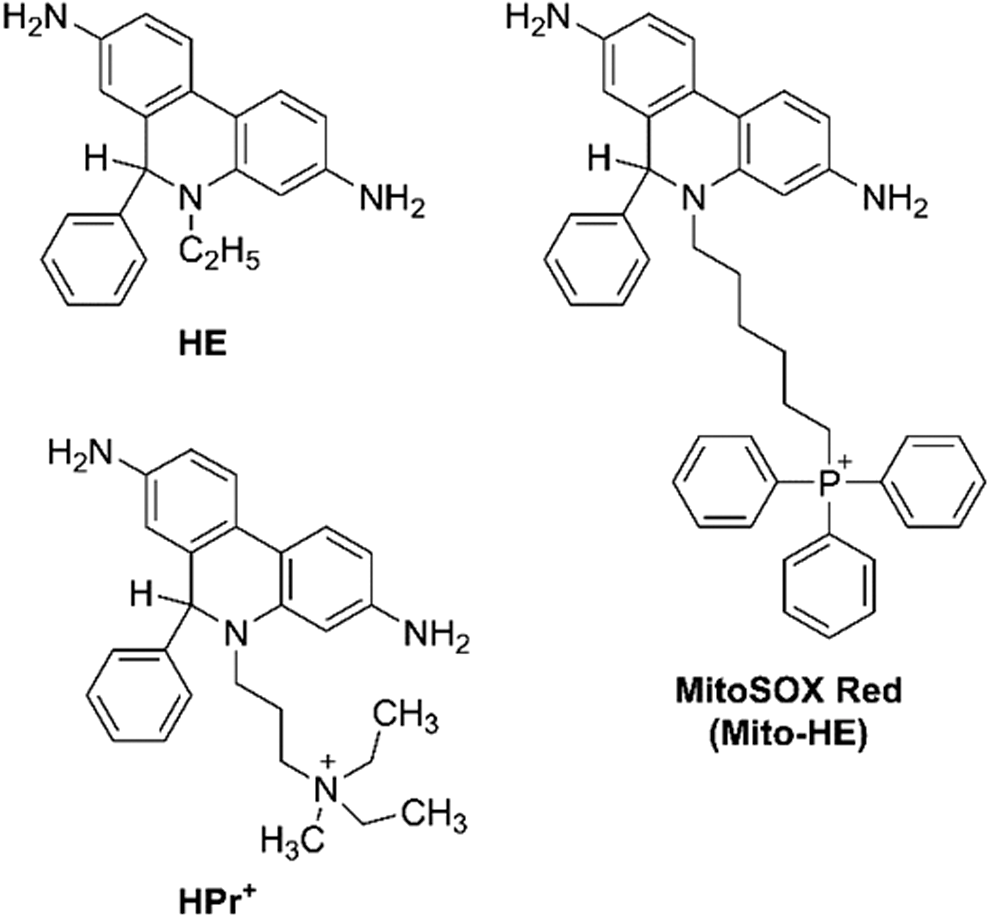

Although HE was initially believed to be selective toward O2 •– and produce red fluorescent ethidium in a reaction with O2 •–, both assumptions were shown to be untrue (93, 139). HE reacts with several biologically relevant oxidants, including peroxynitrite-derived radicals, iron-derived oxidants, and heme proteins (142, 144, 146). In case of all one-electron oxidants, the first intermediate formed is the HE radical cation (HE•+, Fig. 5), which can be converted into different products depending on the reaction conditions. In the presence of superoxide, HE•+ produces 2-OH-E+; in the absence of O2 •–, ethidium (E+, Fig. 5) and dimeric products (HE-HE, HE-E+, and E+-E+) are formed. The factors controlling the distribution of the oxidation products in the absence of O2 •– are not fully understood and are currently under investigation.
As indicated earlier, the product of the reaction of HE by superoxide is 2-OH-E+ and not E+ (67, 132). It is important to note that E+ is typically formed in cells in excess of 2-OH-E+, and for specific monitoring of 2-OH-E+, HPLC-based techniques are necessary (139). One-electron oxidation of HE by heme proteins and specific one-electron oxidants lead to the formation of E+ and several dimeric products, including diethidium (144, 146). Thus, HPLC-based profiling of HE oxidation products can be used to monitor the production of O2 •– as well as other oxidants. As described later, the reaction of HE with HOCl produces the 2-chloroethidium cation (2-Cl-E+, Fig. 5), which was proposed to be used as a specific marker of HOCl (or of myeloperoxidase [MPO] activity) both in in vitro and in vivo studies.
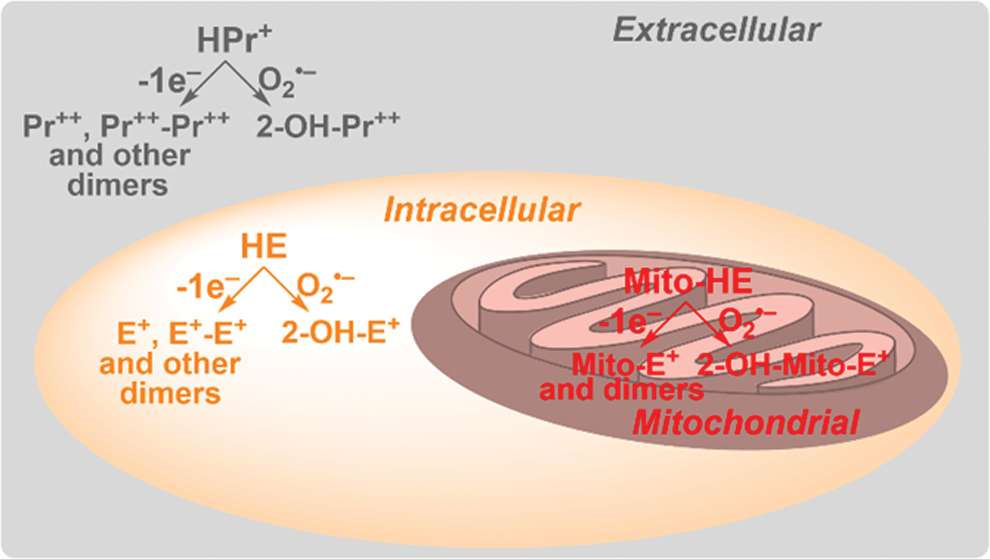
As a small lipophilic molecule with a neutral charge, HE is assumed to passively diffuse through cellular membranes, as it is distributed over most subcellular compartments. To detect O2 •– in mitochondria, the HE molecule has been linked to the triphenylphosphonium cation (TPP+), which targets the probe to energized mitochondria (90, 91, 137, 144). The probe, known as MitoSOX Red (or Mito-HE, Fig. 4), has been widely used to detect mitochondrial O2 •– in intact cells. However, in most reports, the identity of the oxidation product(s) was not determined. Another derivative of HE, called hydropropidine (HPr+, Fig. 4), has been designed to trap extracellular O2 •–, as the probe does not easily enter the cells due to the localized positive charge attached to the molecule (69). Although HE, MitoSOX Red, and HPr+ differ in their physicochemical properties and subcellular distribution (Fig. 6) and can be used for site-specific superoxide detection, they share their chemical reactivity and, in all cases, a 2-hydroxylated cationic product is formed in reaction with O2 •–. In all cases, heme proteins were shown to rapidly oxidize the probes, leading to the nonhydroxylated cation and dimeric products (69, 144). Therefore, regardless of whether HE, MitoSOX Red, or HPr+ is used, the identity of the product(s) formed from the probe needs to be established for the determination of the oxidant(s) being detected (50).
HE and its analogs have been used to monitor O2 •– and/or general cellular oxidants in numerous studies in vitro in cell culture, as well as in animal studies in vivo. As the scientific community begins to realize the requirement and advantages of HPLC-based product profiling, an increasing number of in vitro and in vivo studies will involve the detection of the hydroxylated product as a specific marker of O2 •– (20, 24, 25, 65, 97, 109). However, currently, the in vivo experiments are mostly limited to local administration of the probe, as HE is rapidly oxidized by heme proteins in blood, confusing data analyses. It should also be noted that the pharmacokinetic properties of HE have not yet been studied in detail. However, local administration of HE, in combination with HPLC-based analyses, was successfully applied to study O2 •– and production of other oxidants (e.g., in carotids and in the brain) in vivo (97, 109).
The limitations of HE-based analyses are linked to HE's reactivity toward various oxidants and the formation of several products in cells. In fact, the pathways of E+ formation in cells remain to be established, although both one-electron oxidation and enzymatic two-electron oxidation have been proposed (139, 144). Although HPLC-based analyses convert this limitation into an advantage, the assay requires termination of the experiment and sample preparation for HPLC-based profiling of the oxidation products, limiting the ability for real-time analyses of the dynamics of O2 •– production. New derivatives of HE, with complete spectral separation between E+ and 2-OH-E+, are needed to overcome this limitation.
Detection of H2O2
H2O2 is an important biological oxidant produced by cells under physiological and pathophysiological conditions, and it is believed that under physiological conditions H2O2 plays an important role in cell signaling as a redox messenger (123).
H2O2 can be generated by direct two-electron reduction of molecular oxygen, or by spontaneous or SOD-catalyzed dismutation of O2 •–. For that reason, all cellular sources producing O2 •− (e.g., mitochondrial respiratory chain and NADPH oxidases) generate a flux of H2O2. H2O2 is also generated as an end product of some metabolic reactions, especially peroxisomal oxidations.
It is important to keep in mind that estimated basal cytosolic steady-state H2O2 concentrations are in the nanomolar range, due to both the low basal production and the efficient cellular antioxidant defense system against that oxidant, which include peroxiredoxins, glutathione peroxidases, and catalase. The chemical reactivity and biological chemistry of H2O2 is well recognized (123), but the mechanism of H2O2-dependent redox signaling is still elusive, although great progress in that field has been made in recent years (98, 104).
Intracellular detection of H2O2
Despite the progress in ROS probes' design and characterization, detection of intracellular H2O2 remains a challenge. In general, two different approaches can be used: one based on the use redox-sensitive genetically encoded fluorescent proteins and the other based on the usage of the low-molecular fluorogenic probe. In both cases, the selectivity of the sensor and/or specificity of the oxidation product for H2O2 seems to be a serious limitation.
Redox-sensitive fluorescent proteins
In the case of redox-sensitive fluorescent proteins, those from the HyPer family or those based on roGFP, the mechanism of the probe fluorescence enhancement is based on the oxidation of redox-sensitive cysteines leading to the disulfide bond formation (5, 66). This mechanism, however, does not guarantee selectivity toward H2O2 as any agent (e.g., ONOO−, HNO) reacting with those cysteines with disulfide formation will result in an increased fluorescence signal. The other shortcoming of this approach is that the HyPer probes are extremely sensitive to changes in physiological pH, and, therefore, precise pH control is required (for example, SypHer probe can be used to monitor the changes in intracellular pH).
Fluorogenic probes
In the past, several small-molecule probes producing fluorescent products on oxidation were used to monitor H2O2 production in cells (16, 121). Among those, reduced fluorescent dyes—dichlorodihydrofluorescein (DCFH) and dihydrorhodamine (DHR)—were the most popular for H2O2 detection. It should be emphasized that neither DCFH nor DHR reacts with H2O2 directly, and that the oxidation of those probes requires involvement of a catalyst (transition metal ions, peroxidases, etc.). Moreover, both probes are converted into fluorescent dyes via radical intermediates that can react with molecular oxygen, producing O2 •– and, after its dismutation, H2O2, which is what results in artificial enhancement of the signal intensity. (It even has been pointed out that the oxidation of DCFH is a “self-fulfilling prophesy” (7).) It has also been demonstrated that many other biological oxidants, for example, •NO2, carbonate radical anion (CO3 •−), glutathionyl (GS•), or cysteinyl (CysS•) radicals, are able to oxidize DCFH and DHR (121). With no information about what is detected, the use of those probes gives only qualitative information on the production of cellular oxidants, or activity of cellular peroxidases/heme proteins. It is important to note that cytochrome c can serve as a catalyst for oxidation of DCFH (8, 59). Because cytochrome c is released from mitochondria during the apoptotic events, the use of the probe under the conditions leading to cell apoptosis should be avoided.
In the first decade of the 21st century, boronate derivatives of fluorescent dyes were proposed as a novel class of sensors for the selective detection of H2O2, and now there exists a large pool of fluorogenic boronate probes that have been designed and claimed as selective toward this cellular oxidant (63, 143). As mentioned earlier, the main problem here is the selectivity. Boronates react with H2O2, but the oxidation is rather slow with typical rate constants in the range of 1–10 M −1s−1, and the probe cannot compete effectively for H2O2 with other cellular H2O2 scavengers. In addition, boronates also react with hypohalous acids (e.g., HOCl) and other peroxides (ROOH, O2COO−, ONOO−, O2NOO−) with the formation of the same phenolic products. Among listed oxidants, only ONOO− and HOCl produce additional, oxidant-specific products, as discussed in a subsequent section.
Extracellular detection of H2O2
The measurement of extracellular H2O2 in cell cultures is definitely less problematic than intracellular detection. Either boronate probes or an Amplex Red/HRP assay can be used (134, 147). For kinetic reasons, the detection of extracellular H2O2 with the usage of boronates is effective in the cellular systems generating high quantities of that oxidant (e.g., activated neutrophils). Recently, we have proposed the use of coumarin-based boronate probe CBA in the high-throughput assay for the measurement of the NADPH oxidases activity (134, 136, 147).
Much more sensitive than boronate-based extracellular H2O2 detection is an assay using 10-acetyl-3,7-dihydroxyphenoxazine (Amplex Red) probe. The method is based on the enzymatic oxidation of the probe by H2O2 in the presence of horseradish peroxidase (HRP), resulting in the formation of highly fluorescent resorufin. Although the Amplex Red assay is assumed to be selective toward H2O2, it is important to keep in mind that peroxynitrite is also able to oxidize Amplex Red to resorufin, and the yield of resorufin is increased in the presence of HRP (18). Thus, catalase should be used to confirm the identity of detected species. H2O2 quantitation may also be affected by NADH, GSH, and light, which should be taken into account when designing the experiment and interpreting the data (107, 119, 130).
Organic hydroperoxides
While describing the techniques and methods of the detection of H2O2, we must mention the detection of other organic hydroperoxides, especially when parts of the techniques and methods are common to both species.
Organic hydroperoxides are produced during the reactions of free radicals and excited-state species with biological molecules in the presence of molecular oxygen (15, 73). These species are particularly readily formed not only on lipids and proteins but also on the small molecules present in biological systems. Organic hydroperoxides are associated with the pathogenesis of various diseases, but their actual biological significance and sources are still being investigated. Organic hydroperoxides can propagate oxidative injury to the proteins or other surrounding biomolecules, especially low-molecular-weight hydroperoxides, such as tyrosyl hydroperoxide or urate hydroperoxide, that can diffuse far from the site of their formation (80, 124).
Organic hydroperoxides are not stable in biological matrixes; thus, the methods of their detection need to be simple and relatively fast. This excludes the chromatographic and spectroscopic detection methods based on complicated sample preparation procedures. The available hydroperoxides detection techniques can be divided into titration and colorimetric methods based on iodide or iron oxidation (e.g., the FOX assay) and methods based on the oxidative transformation of the profluorescent probes into the highly fluorescent products. The use of profluorescent probes seems to be a more convenient method for hydroperoxides determination because it enables one-step direct detection of hydroperoxides, in contrast to the FOX assay and iodometric method, where organic hydroperoxides need to be isolated from biological samples before analysis.
As previously described, the extracellular detection of H2O2 is done by the Amplex Red/HRP system. Although this assay could be useful in detecting other organic hydroperoxides, this method cannot be applied for the detection of most organic hydroperoxides, because bulky organic hydroperoxides are not good substrates for HRP (70).
Another profluorescent probe that can be used for detection of hydroperoxides is diphenyl-1-pyrenylphosphine (96). This probe reacts stoichiometrically with hydroperoxides to produce fluorescent diphenyl-1-pyrenylphosphine oxide and corresponding alcohols. Nevertheless, very little is known about the kinetics of oxidation of this probe and its specificity. Another strategy in the detection of organic hydroperoxides is the use of the earlier mentioned, redox-active fluorescent proteins, such as OHSer (Organic Hydroperoxide Sensor) (131). Unfortunately, this probe has the same limitations as other fluorescent proteins used for detection of H2O2. The enhancement of the intensity of the protein's fluorescent signal is followed by the oxidation of two cysteines and the formation of a disulfide bond between two subunits. Thus, any oxidant able to oxidize these cysteines (e.g., ONOO−) may cause a false positive response.
Recently, it has also been shown that boronate probes are oxidatively transformed into fluorescent derivatives by amino acid and protein hydroperoxides (17, 68). It has been determined that the resulting products of hydroperoxides' reduction by boronate probes are the corresponding alcohols (68). Although the stoichiometry of this reaction has not been confirmed directly because of the lack of appropriate standards, it is reasonable to assume that it is 1:1, the same as that of the boronates/H2O2 reaction (101, 143). Boronates are unreactive toward lipid hydroperoxides, and this property can be used to determine the protein hydroperoxides generated in cells without separating the protein hydroperoxides from lipid hydroperoxides (68). Boronate probes react at least 10 times faster with amino acid hydroperoxides when compared with hydrogen peroxide, but in the presence of a millimolar concentration of cellular reductants, such as glutathione, boronates cannot compete effectively for them (17). Another inconvenience is that boronate probes are not selective for organic hydroperoxides and are oxidized to the phenolic product, similar to H2O2.
The reaction of organic hydroperoxides with profluorescent probes may be an alternative to the existing methods that usually require the onerous procedure of separating hydroperoxides from biological samples. This approach enables high-throughput studies and allows kinetic measurements. Nevertheless, the lack of specificity of existing probes, lack of authentic hydroperoxide standards, and uncertainty as to whether all the peroxides present undergo reaction are the main obstacles for real-time monitoring of organic hydroperoxides in intact cells. However, protein-based hydroperoxides are characterized by their longer lifetime than ONOO−, and this can be taken advantage of for their selective detection by analyzing boronate probes oxidation in cell lysates, when performed in the presence of catalase to remove residual H2O2 (68).
Detection of •NO
•NO is a small hydrophobic molecule containing one unpaired electron exhibiting paramagnetic properties. It is produced in vivo by three isoforms of NOS in the presence of L-arginine as well as several NOS cofactors. In 1998, R.F. Furchgott, L.J. Ignaro, and F. Murad received the Nobel Prize of Medicine for showing that •NO is responsible for the vascular smooth muscle vasorelaxation previously known as EDRF (endothelium-derived relaxing factor) (29, 45). Since then, •NO has raised major interest in many researchers' groups. Among its numerous physiological roles, •NO is a cytotoxic mediator of the immune system (79) and a neurotransmitter in the central nervous system (11).
•NO is stable in an oxygen-free environment and can diffuse across cell membranes, but it reacts with superoxide to yield ONOO− at a nearly diffusion-controlled rate (43). Peroxynitrite is a potent oxidative molecule that is capable of severe cellular damages reacting with thiols, lipids, and DNA (86). •NO has been implicated in the pathology of numerous diseases such as cardiac diseases (myocardial ischemia and reperfusion injury, myocarditis); vascular diseases (atherosclerosis); cancer; and neurodegenerative disorders such as Parkinson's, Alzheimer's, and Huntington's diseases and amyotrophic lateral sclerosis (ALS) (78). •NO also plays an important role in plants and, since the pioneering work of Lamb and colleagues, •NO research is mostly devoted to its role in plant immunity (19, 71). •NO is a simple molecule with the tremendous ability to affect numerous processes in mammalians and plants. •NO reactivity and its low steady-state concentration in biological systems represent a challenge for researchers trying to develop methods to detect and quantify it. However, to understand the physiology and the pathology of •NO, it is imperative that •NO production is detected and quantified in living systems. Direct detection of •NO using EPR is very tedious (due to the reactivity of •NO with molecular oxygen, and its steady-state concentration below the EPR limit), so conventional EPR associated with a spin-trapping technique has been developed. Usual spin traps such as nitrones and nitroso compounds are not suitable because of the instability of the resulting spin adduct (85). Therefore, other systems such as exogenous iron chelates and nitronyl nitroxides (NNO) were developed for •NO detection (38). Each method is discussed, and recent developments are exposed later.
To elaborate, for in vitro experiments involving •NO, exogenous sources of •NO are usually necessary. Among the •NO donors commercially available, NONOates (
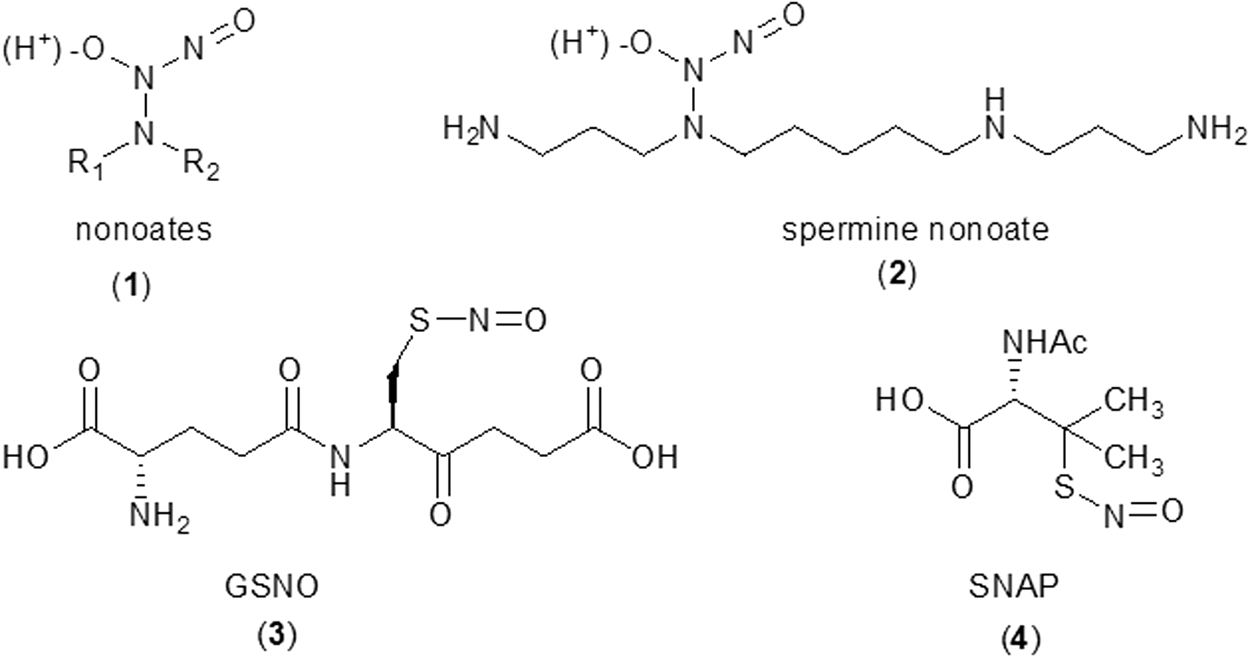
•
Detection of •NO Using Fe(II)-Dithiocarbamates
•NO can bind efficiently to Fe2+ chelates, and the diethyldithiocarbamate ferrous complex, (DETC)2-Fe2+, (
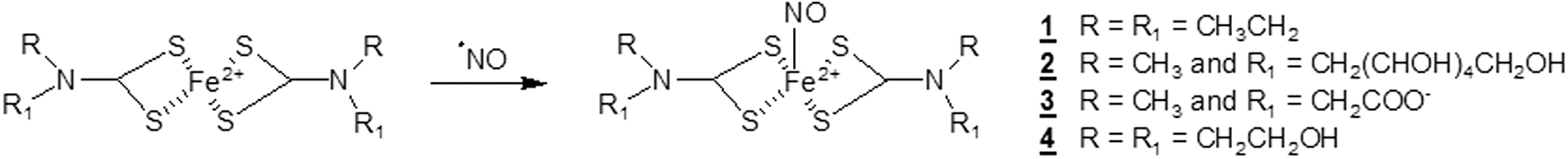
The resulting (DETC)2-Fe2+-•NO complex (
However, the trapping of •NO by (MGD)2-Fe2+ is not selective; Mason and colleagues showed that nitrite, an oxidation product of •NO, can react with (MGD)2-Fe2+ to produce •NO (112). Moreover, Ichikawa's and Rosen's groups have reported that the iron contained in iron-dithiocarbamate complexes is an inhibitor of •NO production by the NOS enzymes, further confusing data interpretations (84, 127).
Despite those limitations, dithiocarbamate complexes are widely used for in vivo detection of •NO, even if the high quantities of Fe2+ added and dithiocarbamate ligands can initiate unwanted reactions and a cytotoxicity.
Detection of NO by NNO
NNO are stable paramagnetic species bearing a nitronyl and an aminoxyl moiety in the same molecule. NNOs are characterized by a five-line EPR signal with an intensity ratio 1:2:3:2:1 due to the coupling of the single electron with two equivalent nitrogen atoms (Fig. 9). It was suggested that nitronyl nitroxides could be a viable alternative to iron (II)-dithiocarbamate complexes (54). Indeed, nitronyl nitroxides react specifically with •NO at reasonable rates (103–104 M −1s−1) (4), giving rise to an imino nitroxide (INO), which shows a completely different EPR spectrum (seven lines, Fig. 9) (48).
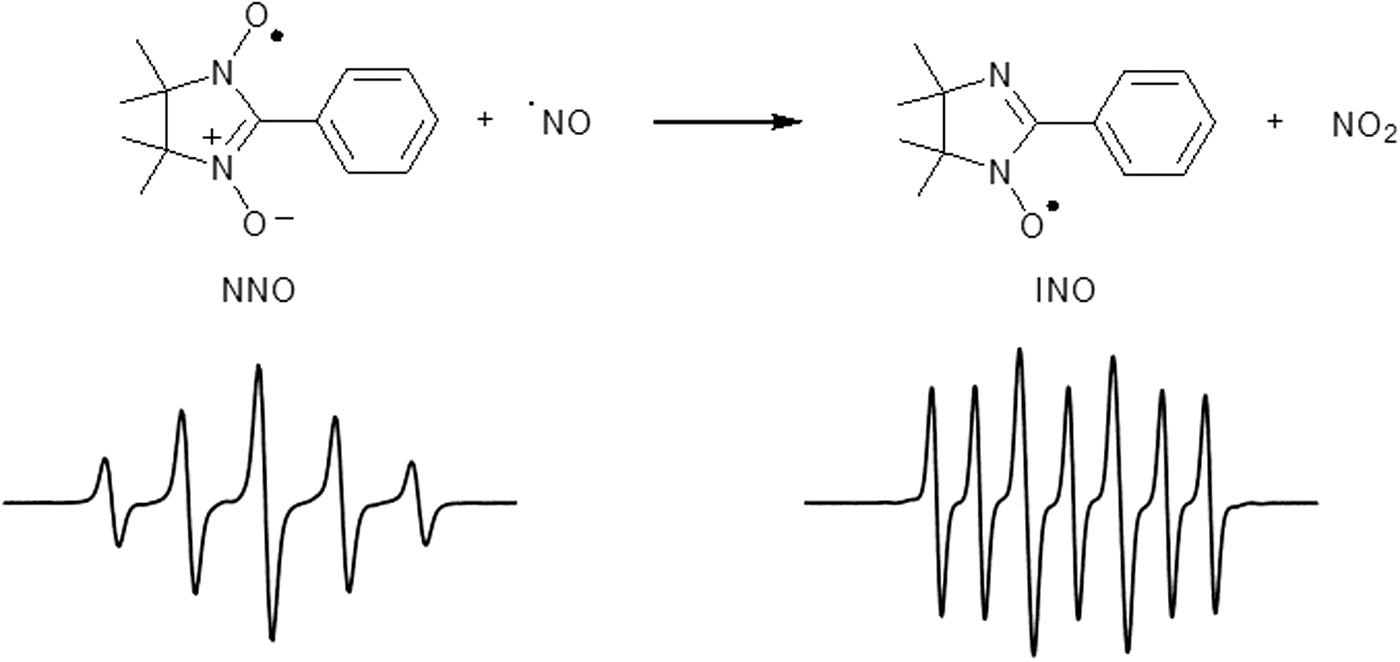
The stoichiometry of the reaction between •NO and the NNO probe is 2 to 1, respectively (Scheme 1) (39).
However, the use of NNO for •NO detection is not without limitation. NNO can react with HNO and O2 •–, affording the reduced hydroxylamine form (6, 95). Indeed, Blasig and colleagues reported that O2 •– can reduce NNO into an EPR-silent diamagnetic product with a rate constant of 8.8 × 105 M −1s−1, competing directly with the trapping of •NO (36).
The possibility of modulating NNO solubility and specificity makes possible the detection of •NO at different tissue sites; an example was published by Peng and colleagues, who report the synthesis of 30 different NNO labeled with amino acid fragments (129).
To improve the sensitivity of the •NO detection method and to circumvent NNO bioreduction, several strategies were used. Rosen et al. reported the first synthesis of dendrimer-linked NNOs (from 2 to 8 U of NNO) (92). Duan and colleagues associated an NNO with a fluorescent metal-organic tetrahedron, which mimics the enzymatic pocket, combining EPR spectroscopy with fluorescence detection for improved sensitivity (120). They report that the trapping reaction of NO by the metal-organic tetrahedron–NNO complex was enhanced and has a limit of detection of 5 nM.
Detection of •NO via nitrosation of diamino-substituted fluorophores
For real-time monitoring of •NO in biological systems, the various fluorescent probes have been used, but diamino-substituted fluorophores (e.g., diaminofluorescein, DAF-2) gained the highest popularity. The advantage of those probes is the formation of the highly fluorescent product, triazole derivative (e.g., DAF-2T), which incorporates a nitrogen atom from •NO. HPLC-based analyses indicate that several fluorescent products can be formed from DAF-2, and, thus, selective detection of DAF-2T using the chromatographic techniques is recommended (110, 146).
It must be noted that •NO does not react directly with DAF-2, and that two pathways have been proposed for DAF-2T formation. One involved direct nitrosation by N2O3, and the other involved one-electron oxidation to the aminyl radical, followed by its reaction with •NO (58). This mechanism would implicate the sensitivity of DAF-2T yield to one-electron oxidants, as demonstrated experimentally (138). It is important to note that the probe nitrosation can also result from the reaction of the probe with nitrite under acidic conditions.
Detection of Peroxynitrite with the Use of Boronate Probes
For many years, intracellular detection of peroxynitrite has been a significant challenge. In the past, the reduced leuco-derivatives of fluorescein and rhodamine, DCFH and DHR, previously discussed, were used (12). Unfortunately, there are some serious disadvantages to their use: the radical mechanism of their oxidation, the production of O2 •– in the reaction of radical intermediates with molecular oxygen, and the lack of selectivity (121).
The discovery of a rapid, direct, and stoichiometric reaction between ONOO− and boronate compounds, resulting in the formation of a peroxynitrite-specific mixture of oxidation products, has opened the possibility of specific detection of this elusive oxidant (101). It should be emphasized that the boronate group containing the trivalent, sp2-hybridized electrophilic boron atom in boronate-based molecular probes can be easily oxidized to corresponding phenols by different nucleophilic biological oxidants—hydrogen peroxide and organic hydroperoxides, peroxynitrite, peroxynitrate, and hypohalous acids—but the reaction with peroxynitrite leads to the parallel formation of oxidant-specific products (99 –102, 147). The mechanism of peroxynitrite reaction with boronates has already been described in detail (99, 100), and it has been shown that the formation of peroxynitrite-specific products can be used to identify the oxidant reacting with boronates (102, 147). The first step of the peroxynitrite reaction with boronates is the formation of an anionic oxidant adduct to the boronate group that further decomposes to products via two different pathways: The heterolytic cleavage of the O-O bond leads to the formation of a major, phenolic product (η ∼ 90%), whereas the hemolytic cleavage of that O-O bond results in the formation of an unstable transient radical anion PhB(OH)2O•− (η ∼ 10%) that decomposes with the formation of phenyl radicals (Ph•). Those radicals can easily react with hydrogen atom donors (e.g., GSH) or with the •NO2 radicals that are also produced during hemolytic cleavage, resulting in the formation of peroxynitrite-specific products (16). Recently, we have shown that, in the case of mitochondria-targeted arylboronate o-MitoPhB(OH)2, the transient phenyl radical, o-MitoPh•, undergoes rapid intramolecular cyclization, resulting in the formation of the peroxynitrite-specific product cyclo-o-MitoPh (147). The yield of that product in the reaction of peroxynitrite and o-MitoPhB(OH)2 is equal to 10.5%, so the major product o-MitoPhOH and the minor, peroxynitrite-specific product cyclo-o-MitoPh are formed in the ratio 9:1 (147). The observed yield of these two products can be used to estimate the contribution of peroxynitrite and other oxidants to o-MitoPhB(OH)2 oxidation.
Detection of HOCl
Over the past decade, numerous fluorescent probes—typically containing a sulfur atom as an HOCl-reactive center—have been proposed for the selective detection of HOCl, as reviewed elsewhere (41). Because most of those probes still require detailed characterization of chemical reactivity and biological validation, detecting the product(s) of probe chlorination seems to be the most reliable method for selective monitoring of HOCl generation in biological systems. Chlorination of endogenous phenols (tyrosyl residues of proteins) and exogenous phenolic probes was applied to detect HOCl production in cell-free and cellular systems (Fig. 10) (13, 27, 37, 47, 122). Taking into account the high reactivity of HOCl toward thiols and amines, only a very small fraction of HOCl is expected to react directly with the probe intracellularily, whereas additional chlorination can be accomplished by chloramines, the products of the reaction of HOCl with endogenous amino acid residues (42, 81 –83, 105).
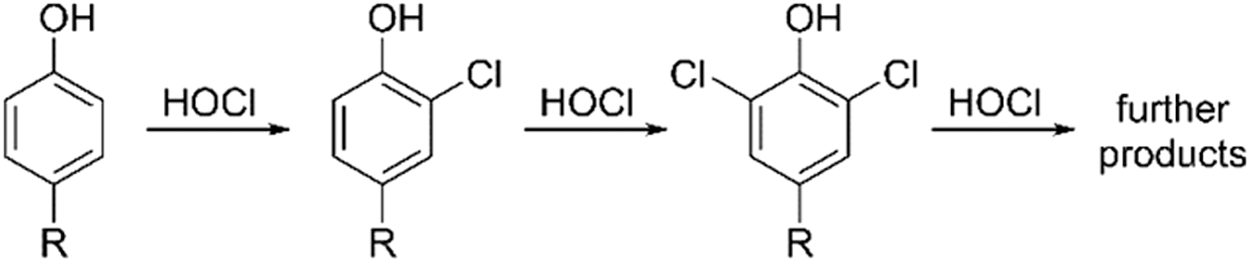
As phenols also undergo nitration by •NO2 radicals, the same probe could be used to gain information about nitrating and chlorinating species. As an example, monitoring fluorescein chlorination and nitration was used to establish the identity of the oxidants produced by MPO under various concentrations of nitrite, in both cell-free and cellular systems (47). More recently, it was established that the HE probe, widely used for O2 •– detection, reacts with HOCl, leading to the formation of 2-Cl-E+ (Fig. 5), which was proposed as a specific marker for HOCl (64, 108). It was shown that stimulation of human neutrophils with PMA leads to the formation of 2-Cl-E+, which correlates with the extent of tyrosine chlorination (64). Similarly, 2-Cl-E+ formation was detected in activated peritoneal mouse phagocytes and in a mouse model of arterial inflammation in vivo. In both in vivo systems, knocking down MPO led to significant attenuation of 2-Cl-E+ formation (64).
As previously mentioned, boronate probes have been used to detect H2O2 and ONOO− (63, 101, 143). It was also demonstrated that boronate probes can be oxidized by HOCl (101, 143). Although the phenolic product is not specific to HOCl, its further reaction with HOCl produces chlorophenol, which can be used as a specific marker for HOCl (101). As an example, it was shown that the luminescent probe PCL-1 undergoes oxidation to luciferin, which in the presence of HOCl is further converted to chloroluciferin (Luc-Cl, Fig. 11) (141).
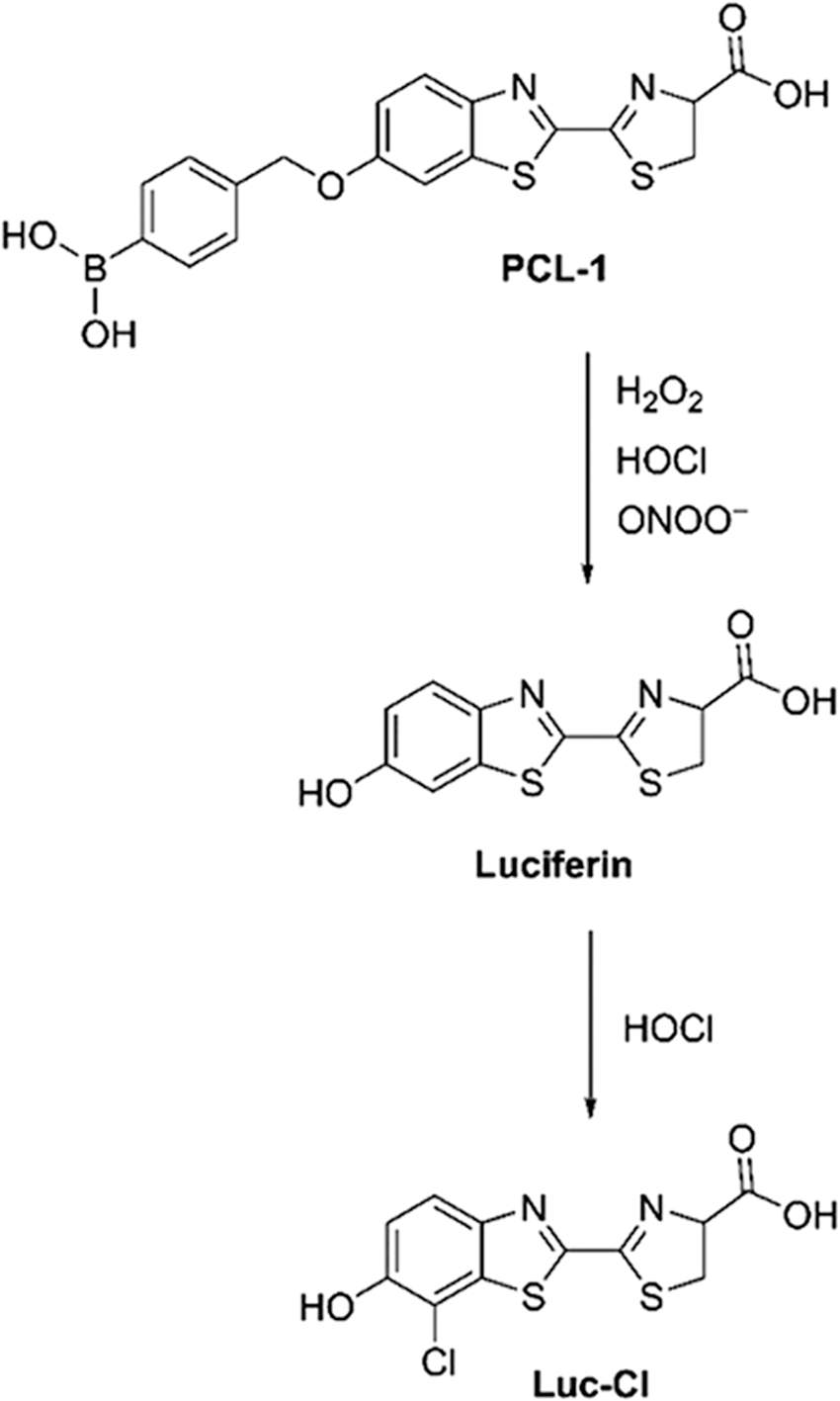
Simultaneous Detection of Various ROS/RNS
As discussed earlier, several probes have been shown to yield various products, depending on the identity of the ROS/RNS with which they react; the HE probe can be used for detection of O2 •–, HOCl, and one-electron oxidants, forming different, specific products.
EPR spin traps can be used to detect various radical species, each producing a product with very characteristic spectral features. For example, the 5-tert-butoxycarbonyl-5-methyl-1-pyrroline N-oxide (BMPO) spin trap was used to characterize radical species formed under conditions of cogenerated fluxes of O2 •– and •NO (Fig. 12) (55). In the absence of •NO, an EPR spectrum characteristic for the adduct of O2 •– is observed. With increasing flux of •NO, the superoxide adduct disappears, with concomitant build-up of the •OH adduct. This is consistent with the rapid reaction between O2 •– and •NO producing ONOO−, which decomposes to the •OH radical.

Also, testing the reactivity of o-MitoPhB(OH)2 with various oxidizing, nitrating, and halogenating species revealed a range of products, including a relatively nonspecific phenolic product (o-MitoPhOH); ONOO−-specific products (cyclo-o-MitoPh and o-MitoPhNO2); nitrated phenolic products (o-MitoPh(NO2)OH), indicative of the presence of •NO2 radicals; and halogenated products (o-MitoPh(Cl)OH and o-MitoPh(Br)OH), formed in the presence of HOCl and HOBr, respectively (Fig. 13) (147). Thus, complementation of real-time monitoring of boronate-probe-derived fluorescence or luminescence by HPLC or LC-MS-based profiling of the products formed can provide a rigorous method for identifying HOCl and/or other species that are capable of oxidation of boronates.
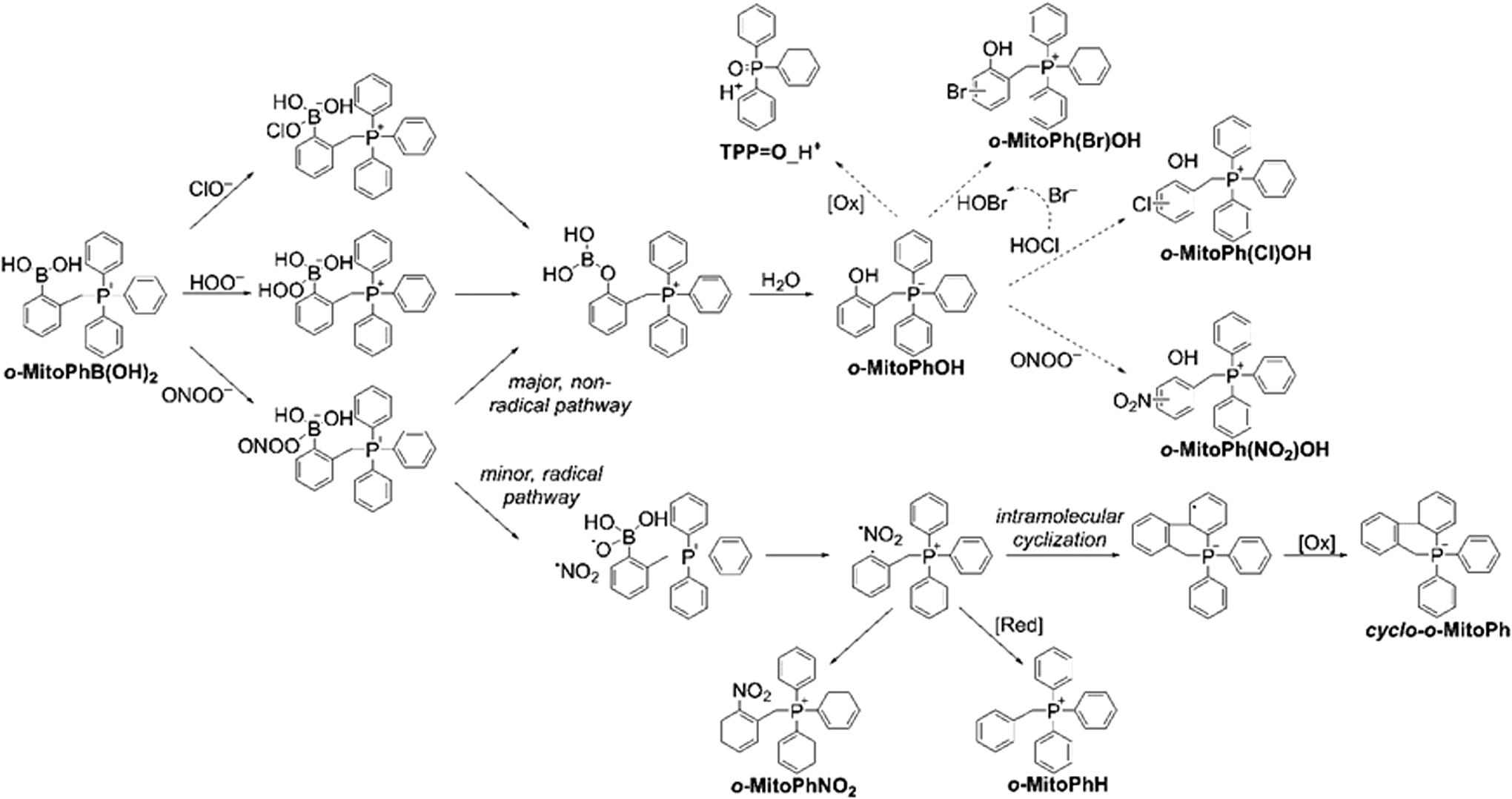
Summary
This review is unique in that it focuses on the detection of specific products formed from the interaction between various ROS/RNS, spin traps, and fluorescent/nonfluorescent probes. Using this approach, we are able to rigorously identify the specific ROS/RNS generated in cell-free and cellular systems.
Nitrone spin traps (e.g., DEPMPO, DMPO, BMPO) react with O2 •– to form nitroxide spin adducts that exhibit highly characteristic EPR spectra. This approach is suitable for detecting O2 •– formed in biochemical systems and in the extracellular milieu. However, this approach is not suitable for detecting intracellular superoxide. HE and its analogs react with intracellular and extracellular superoxide, forming hydroxylated products that can be rapidly separated and identified by using HPLC-based techniques. HE analogs also react with HOCl to form specific chlorinated products.
Boronate-based probes react slowly with H2O2, and amino acid/protein hydroperoxides to form hydroxylated products that can be detected by using fluorescence or chromatographic techniques. Boronate probes react rapidly with ONOO− to form the hydroxylated product (major) and nitrated products (minor). These minor nitrated products are formed only through a reaction with ONOO−. Boronate probes react with HOCl to form specific chlorinated products. The techniques to detect and characterize these products are EPR, HPLC, and/or LC-MS.
Footnotes
Acknowledgments
This work was supported by grants from the NIH National Cancer Institute (R01 CA152810 to B. Kalyanaraman), the French National Research Agency (ANR-09-BLAN-0193-02, ANR-15-CE29-0022-01 to O. Ouari., and ANR-16-CE07-0023-01 to M. Hardy), and a grant from the Polish National Science Centre (NCN) within the SONATA BIS 5 program (Grant no. 2015/18/E/ST4/00235).