
Abstract
Significance:
Hydrogen peroxide (H2O2) is a powerful effector of redox signaling. It is able to oxidize cysteine residues, metal ion centers, and lipids. Understanding H2O2-mediated signaling requires, to some extent, measurement of H2O2 level.
Recent Advances:
Chemically and genetically encoded fluorescent probes for the detection of H2O2 are currently the most sensitive and popular. Novel probes are constantly being developed, with the latest progress particular with boronates and genetically encoded probes.
Critical Issues:
All currently available probes display limitations in terms of sensitivity, local and temporal resolution, and specificity in the detection of low H2O2 concentrations. In this review, we discuss the power of fluorescent probes and the systems in which they have been successfully employed. Moreover, we recommend approaches for overcoming probe limitations and for the avoidance of artifacts.
Future Directions:
Constant improvements will lead to the generation of probes that are not only more sensitive but also specifically tailored to individual cellular compartments. Antioxid. Redox Signal. 29, 585–602.
Introduction
H
The chemistry of H2O2 is unique among the class of reactive oxygen species (ROS), which makes it a powerful redox signaling molecule. H2O2 is not a radical and is fairly stable in aqueous solution (30). Its uncharged nature facilitates its diffusion and transport across the membrane or via aquaporins (AQPs) (87, 135), allowing it to signal at sites remote to its production (144). Oxidation of target proteins by H2O2 means that redox signaling requires relatively high concentrations of this oxidant. Therefore, it is likely that H2O2 acts locally at the site of production where concentrations are high enough (90) to trigger compartmentalized signaling.
Molecular Targets of H2O2 and Redox Signaling
Numerous molecules can be oxidized by H2O2 with transition metals and cysteine residues being the most common and well described. Moreover, the oxidative modification is molecule specific, which might enhance or inhibit the activity of the redox target.
Transition metals are oxidized by H2O2, which results in Fenton chemistry. The hydroxyl radical that is subsequently produced could then react with any biomolecule within diffusion distance (144). H2O2 can oxidize metals such as those in the center of the serine–threonine protein phosphatases. One example is protein phosphate 1 that, when inactivated upon oxidation, increases the phosphorylation of eIF2α (translation initiation factor 2α) and its dissociation from the translation initiation complex (120). Consequently, protein synthesis is attenuated and translation of activating transcription factor 4 is enhanced, making this a crucial adaptive pathway to cope with cellular stress. Physiologically, the H2O2-mediated oxidation of enzyme Fe2+ to Fe3+ is rapidly reverted by cytochrome reductases, including the methemoglobin reductase and vitamin C. Thus, the physiological importance of this process is still being discussed.
H2O2-dependent redox signaling is a consequence of intracellular thiol chemistry in the majority of cases. (Fig. 1). By far the most common targets are cysteine residues within peptide chains. There are examples of methionine-dependent redox signaling (63, 113), but this appears less commonly (30). H2O2 is also a substrate for peroxidases such as myeloperoxidase or lactoperoxidase to produce highly reactive molecules for host defense (62, 98). Specialized enzymes such as catalase and glutathione peroxidases react with and detoxify H2O2 (5). Importantly, the reaction speed of any thiol with H2O2 is drastically increased by deprotonation. This can either be achieved in test tube chemistry with a low pH or in proteins wherein surrounding amino acids lower the pKa of the cysteine or methionine of interest (104). This, for example, explains why thiol-based glutathione peroxidase-like enzymes are far more reactive with H2O2 than glutathione itself.

The lowest local pKa is that of the peroxiredoxins (Prxs): professional H2O2 detoxifying enzymes that also exhibit the highest reactivity toward H2O2 in cells. These enzymes can potentially act as redox relays and transfer the oxidation equivalent to other molecules (10). Genetically encoded redox sensors (see below) utilize low pKa cysteines as sensors for H2O2.
Oxidation of cysteines can alter the function of the target protein, leading to distinct biological outcomes. For example, some protein tyrosine phosphatases carry a cysteine in their active center and its oxidation leads to enzyme inactivation (80, 119). The consequence is redox signaling and a lack of dephosphorylation, which results in increased phosphorylation of target proteins (for example, in the endothelial growth factor [EGF] signal transduction pathway) (33, 72).
Another example is the oxidation of cysteines in Kelch-like erythroid cell-derived protein with cap ‘n’ collar homology-associated protein 1 (KEAP-1) (129). Under basal conditions, nuclear factor erythroid-derived 2-like 2 (Nrf2) is polyubiquitylated by KEAP1–cullin 3 (CUL3) complex and targeted to proteasome degradation (64). Upon oxidative or electrophile insult (65), an intramolecular disulfide is formed, which prevents the interaction of KEAP1 with CUL3 (45). Therefore, Nrf2 is no longer degraded and translocates to the nucleus, where it activates a set of genes of the so-called antioxidant response.
Importantly, thiol oxidation, however, is not specific for H2O2 as exemplified by KEAP1. Other electrophiles such as nitro lipids or oxilipids can also oxidize thiols and some enzymes such as endoplasmic reticulum oxidoreductase and protein disulfide isomerase are direct thiol oxidases (127, 154). Finally, slow thiol exchange reactions according to the redox equilibrium also occur in living cells and should not be neglected (96, 110).
Cellular Environment and Its Implication for H2O2 Measurements
Given the highly important role of redox signaling in cellular function, measuring H2O2 should be a common practice in redox biology (108).
H2O2 is produced in a compartmentalized manner and shows different half-lives depending on the cell compartment. Moreover, different cellular redox sinks rapidly react with H2O2, outcompeting reaction of H2O2 with the sensors, which have a far lower constant rate. Therefore, for quantitative detection of H2O2, the sensor has to be present at the local site of its production and at very high concentrations. This, however, is either not feasible or has a detrimental impact on cellular health (59). Many sensors have a preference for accumulation in the mitochondria (116) and can even avoid entry into the cell (146). Already these aspects illustrate that measuring even local and steady state concentrations of H2O2 in biological systems is not trivial. The problem is further complicated by the lack of specificity of the probes. H2O2 measurements in general are based on the direct or indirect oxidation of a probe by H2O2. The nature of such reactions means that oxidants stronger than H2O2, such as peroxynitrite (152), are also capable of oxidizing the H2O2 probes. Several tricks can be applied to demonstrate specificity of the measured signal for H2O2 (108), but these approaches can also display a lack of specificity due to indirect effects. As these considerations are a direct consequence of the reactive nature of H2O2, they are inherent to all H2O2 probes.
Detection of H2O2 with Fluorescent Dyes
In general, three classes of H2O2-sensitive probes can be discriminated: (i) horseradish peroxide (HRP)-dependent probes convert H2O2 into a reactive peroxyl radical, which oxidizes the probe, (ii) boronates that belong to the few compounds with direct sensitivity to low level of H2O2, and (iii) protein-based redox probes that utilize redox-sensitive cysteines in combination with conformational changes of fluorescent proteins, thereby changing emission wavelength or excitability upon oxidation. In addition to these main classes, some individual probes and assays with specific properties have been generated. One of these is the ferric xylenol orange assay that measures hydroperoxides based on their reduction by Fe2+ under acidic condition to form Fe3+ xylenol orange complex (46). The products can be easily measured at 560 nm, making the assay very simple and user friendly. However, its sensitivity is limited and an understanding of the hydroperoxides, ascorbic acid levels, and iron chelators in the sample is necessary to properly interpret the results (47).
An additional consideration for H2O2 measurements is the limited temporal resolution of fluorescent probes. As a consequence of H2O2-dependent oxidation, a nonfluorescent compound usually turns into a fluorescent dye. This dye accumulates over time and thus provides an integrative signal of H2O2 production, the obvious advantage of such an approach being its high sensitivity. Transient and short lasting changes in H2O2 production are, however, difficult to detect. To obtain this information from fluorescence measurements, the first derivate of the signal over time has to be calculated. This approach is not feasible for some end point techniques and requires the constant or repetitive excitation of the probe. Probe excitation, however, has two side effects: bleaching of the fluorophore and photo oxidation of the nonoxidized probe (81, 146). These considerations are important as, despite clear redox-mediated biological effects, it is often not possible to detect rapid or transient changes in H2O2 cells (81, 108). Thus, transient changes of H2O2 production in cellular systems still remain the major technical challenge.
The number of assay compounds to measure H2O2 is surprisingly large (35, 37). For the purpose of this article, we focus on some common probes that have widely been used and for which reliable results can be expected. For these, we provide hints on good practice and point out caveats of their use.
HRP-Dependent Assays
Overview
The most common chemical assays to detect H2O2 with high sensitivity rely on HRP activity. This enzyme is of great application due to its catalytic activity toward H2O2 favoring the coupled oxidation of fluorogenic or chemiluminogenic substrates (105). The disadvantages of HRP-dependent assays are the formation of high oxidation state intermediates of the enzyme, which are greatly modulated by the cellular environment and the restriction of the assay compartment to the extracellular space (12).
Three main fluorescent/photometric HRP-dependent probes gained popularity during the 1960s–1980s: scopoletin (19, 79, 97), homovanillic acid (51, 118a), and phenol red (phenolsulfonphthalein) (107). In that time, the probes were considered very sensitive as the models studied were often leukocytes that can produce H2O2 (from dismutation of •O2 −) in concentrations far beyond what is currently considered for redox signaling. These probes certainly added important insights into H2O2 detection, but with the emergence of redox signaling concepts, new and more sensitive assays were required to cope with the low H2O2 concentrations involved in signaling.
Amplex® Red
Amplex Red (10-acetyl-3,7-dihydroxyphenoxazine) is a colorless nonfluorescent compound developed in 1997 (148), which can be oxidized to resorufin in a H2O2/HRP-catalyzed reaction (Fig. 2). Resurofin is a highly absorbing and fluorescent pink compound, which can be easily detected by photometry and fluorescence. The Amplex Red assay is overall very sensitive (detecting as little as 5 pmol/L H2O2) (148) and simple to perform. However, as it depends on HRP, detection is limited to extracellularly available H2O2. Quantitative data can be obtained by means of a standard curve of H2O2 or resorufin itself.

Amplex Red is highly susceptible to photooxidation, and light-dependent artifactual formation of resorufin is further enhanced by SOD or HRP (146). The reaction mechanism for the H2O2-dependent formation of resorufin has only recently been described and novel sources of artifacts have been uncovered. As expected, disproportionation of the Amplex Red-derived radical species is not only exclusive to H2O2 but also mediated by HRP-dependent peroxynitrite-derived radicals (32). In addition, mitochondrial carboxylesterase produces resorufin even in absence of oxygen, H2O2, or peroxidase (91). Therefore, one has to be critical, not only of the complex chemistry of the probe and artifactual sources of the assay, but also of how to apply the correct controls.
Examples of the use of Amplex Red
The Amplex Red assay has been applied to a broad variety of samples, such as plant and intact animal tissue, dissected vessels, cell culture system, and isolated mitochondria. Moreover, it has been proven effective in the detection of even small differences in H2O2 formation as illustrated hereunder.
Plants produce and release H2O2, making it easily accessible for HRP-dependent reaction (20, 24). However, in highly vascularized animal tissue, H2O2 is less stable and produced to a lower extent (30), limiting the formation of resorufin. The consequences for quantification were already mentioned as general concerns for all methods of ROS determination. Ex vivo, Amplex Red has been successfully employed in renal tissue to detect Nox4-derived H2O2. The enzyme is highly expressed in the kidney (48) and basal H2O2 is reduced in an Nox4−/− (7) and a triple Nox knockout (Nox1/2/4−/−) mouse (114) as compared with wild type (WT) mice. An exemplary measurement is shown in Figure 2B and C.
In vascular smooth muscle cells of dissected mouse aortic tissue (10–100 μmol/L Amplex Red and 1–2 U/mL HRP), H2O2 level was increased upon targeted overexpression of p22phox [accessory subunit essential for Nox1, Nox2, and Nox4 activity (112)]. Likewise, in vivo treatment with angiotensin II (AngII) further increased H2O2 production, which was again significantly enhanced in p22phox overexpressing tissue as compared with WT AngII-treated tissue (141). In contrast, depletion of Nox4 in mice is accompanied by a reduction of basal levels of H2O2 in thoracic and abdominal aortic rings. Importantly, the resorufin formation in the latter system was largely polyethylene glycol (PEG)–catalase (250 U/mL) sensitive (122). Differential H2O2 production by distinct vascular beds has also been assayed with Amplex Red. Vena cava was shown to produce twice as much H2O2 as the aorta of rats. Such differences were attributed to the xanthine/XO system (131), which is expressed at higher levels in vena cava.
The increased ROS production in well-established in vivo models such as diabetes, atherosclerosis, and hypertension has been recapitulated in cell culture systems using Amplex Red. For instance, H2O2 production in response to high glucose (25 mmol/L as compared with 5 mmol/L) treatment is increased in human aortic endothelial cells (49). Nox1 siRNA largely abrogated the signal, suggesting the enzyme is a mediator of hyperglycemic oxidative stress. In a model of shear stress, human umbilical vein endothelial cells produce fourfold more H2O2 under laminar flow than under static conditions, as judged by the sensitivity of the signal to diphenylene iodonium (DPI, inhibitor of flavine proteins) and apocynin (antioxidant molecule) (77). Furthermore, oscillatory flow (oscillatory shear stress, a condition occurring in sites of the vasculature vulnerable to atherosclerosis, ±15 dyn/cm2) doubled H2O2 production as compared with laminar flow in bovine aortic endothelial cells (BAECs) (21, 84).
In a loss of function model using cultured dermal fibroblasts from Nox1/2/4−/−, a lower basal production of H2O2 was observed than that in WT cells. Treatment with transforming growth factor-β led to a 50% increase in H2O2 in WT cells but not in knockout cells (114). Likewise, in lung endothelial cells isolated from Nox4−/− mice, PEG–catalase-sensitive levels of basal H2O2 were reduced by 80% (121). A knockout of Nox4 using CRISPR/cas9 technology in Hela cells resulted in more than a 40% reduction of basal H2O2 level (58).
Amplex Red assays in isolated mitochondria have added invaluable insights into their H2O2 production in a tissue-, substrate-, and site-specific manner. Moreover, characteristics of physiological H2O2 release from Complexes I and III have been determined with this probe (132). AngII-induced mitochondrial dysfunction was demonstrated by using isolated mitochondria of bovine aortic endothelial cells pretreated with AngII and fueled with malate/glutamate or succinate. The AngII-dependent mitochondrial H2O2 release was confirmed with electron spin resonance. Interestingly, genetic deletion or depletion of the p22phox subunit of the NADPH oxidase reduced mitochondrial H2O2 release, attributing a role to Nox enzymes in mitochondrial oxidative damage and for Nox–mitochondrial crosstalk (39).
Amplex Red derivatives
An attractive—but yet to be further explored—technique is the combination of Amplex Ultrared (a more stable and potentially more sensitive probe) and micro dialysis to measure interstitial H2O2 level in vivo in rats and in the organs (kidney, liver, and penis) or tissue (adipose and skeletal muscle) of mice (70). In brief, a microdialysis probe containing a semipermeable membrane is inserted into the tissue or organ of an anesthetized animal. The probe is then perfused with Amplex Red solution, and H2O2, which diffuses from the tissue into the probe, is subsequently detected in the outflowing dialysate. By this technique, AngII was shown to increase renal ROS production by 35%. In addition, the signal obtained in liver was largely catalase sensitive.
The examples already mentioned demonstrate the power of Amplex Red in terms of its versatility and sensitivity. It is important to emphasize that numerous findings with Amplex Red were also confirmed with other probes such as L-012 and dihydroethidium (DHE). Furthermore, this probe has been employed in orthogonal assays together with DHE/high-performance liquid chromatography (HPLC) system to screen for Nox inhibitors (149).
How to perform Amplex Red assay
Amplex Red (50 μmol/L) and HRP (1 U/mL) are well suited for tissue and cell culture experiments. Higher HRP concentrations increase the background oxidation of the assay without increasing sensitivity. At concentrations <0.2 U/mL, HRP becomes rate limiting. Amplex Red (Invitrogen; #A-12222) can be reconstituted in dimethyl sulfoxide (10 mM/L) and aliquots can be stored at −20°C for longer periods. Likewise, HRP (Sigma; #P-6782) can be stored as a frozen stock solution in a concentration of 1 U/μL stocks in Hepes-Tyrode (HT) buffer (in mmol/L: 137 NaCl, 2.7 KCl, 0.5 MgCl2, 1.8 CaCl2, 5 glucose, 0.36 NaH2PO4, and 10 HEPES) for longer periods.
Shortly before the measurement, stocks should be diluted in medium without phenol red or in HT buffer. The reaction solution should be prewarmed at 37°C and well protected from light. A volume of 300 μL is well applicable for most samples.
Samples should be incubated for 30 min at 37°C and detection of resorufin was carried out after transferring 200 μL of the supernatant to a 96-well plate by measuring fluorescence at 530/590 nm excitation/emission. As resorufin is highly absorbing in the visible range, a pink color of the solution might be observed. For cell culture experiments, adherent cells are recommended over freshly detached cells. Cells should be grown on 12-well plates and processed as already described. A standard curve of H2O2 can be used to estimate its concentration in the plate. A range of 25 μmol/L–20 nmol/L (1:2 serial dilutions) is within the detection limit of the probe under the given concentrations. It is very important to add controls such as catalase (PEG–catalase at 250 U/mL) and SOD (at 50 U/mL). Appropriate blanks (no samples but test compound) and blank samples without HRP are indispensable for reliable measurements. In our hands, determination of the part of the reaction sensitive to DPI (10 μmol/L), a broad spectrum flavine inhibitor, which blocks many enzymes such as NADPH oxidases, NO synthase, and the mitochondrial respiratory chain, was superior over catalase in the determination of cellular H2O2 formation. Non-PEGylated catalase preparation has a tendency to increase the Amplex Red signal (possibly due to contamination), whereas NADH and reduced glutathione can potentiate artifactual resorufin formation (138). H2O2 production can be normalized to protein amount or tissue weight and time, yielding μmol/L/sample (mg protein or tissue)/min.
Boronate-based probes
The report of a chemical reaction of H2O2 with boronates dates from 1930 (1) and inspired the first probes in 2003 (76, 78). There are >20 boronate-linked probes to date, with this class of probes undergoing the most development in the past 10 years (2, 3, 34, 75, 76, 86, 88, 130). Boronate-based probes are organic compounds harboring a boron electrophilic center linked to functional groups of different classes: aliphatic, phenolic/aromatic, fluorogenic, or coupled to firefly luciferin. The structure of representative boronate compounds is depicted in Figure 3. Under mild alkaline conditions, H2O2 reacts with boronate groups, converting the probes to their corresponding hydroxyl derivatives or phenolic products (in aromatic-containing probes) (68, 151). This often releases the functional groups that are easy to monitor by absorbance, fluorescence, or fluorescence resonance energy transfer (FRET). In addition, detection can be coupled to HPLC to improve resolution (126, 149). The overall simple mechanism of the H2O2/boronate reaction makes the probes attractive candidates for routine bench assays although they require relatively high probe concentrations to achieve sufficient intracellular retention.
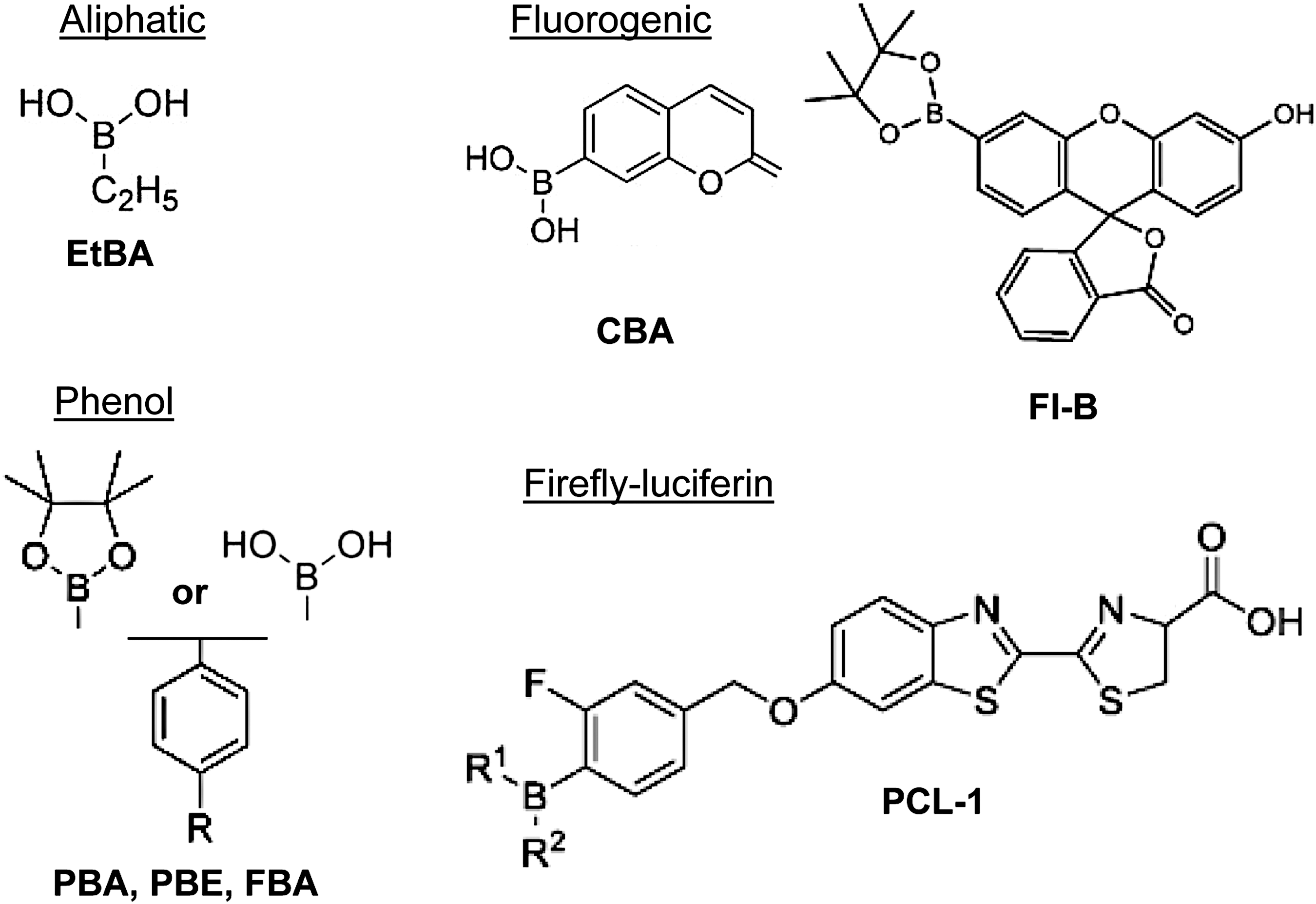
The development of boronate-based probes has been a focus of synthetic chemistry but not of redox biology. Hence, the biological properties and the suitability of boronates for H2O2 detection in vivo have not been completely characterized. Moreover, most boronates have not been extensively employed in cellular studies. Nevertheless, boronates exhibit properties of an almost ideal H2O2 probe: sufficient intracellular retention, ratiometric detection, targeting distinct cellular compartments, detection with multiple laser lines, and in vivo imaging (76). Table 1 summarizes the main probes with their respective characteristics and systems where they have been tested. In our hands, however, the sensitivity of boronates has proven insufficient for the detection of physiological H2O2 concentrations in most cellular systems.
Has the probe been tested with peroxynitrite?
BAEC, bovine aortic endothelial cell; EGF, epidermal growth factor; Fl-B, fluorescein boronate; H2O2, hydrogen peroxide; PC1, peroxy crimson 1; PCL-1, peroxy-caged luciferin 1; PF1, peroxyfluor; PG1, peroxy green 1; PY1-ME, peroxy yellow 1 methyl ester; RPF1, ratio peroxyfluor 1.
Examples for the use of boronates
Two probes within this class were employed in recent studies: peroxy yellow 1 methyl ester (PY1-ME) and peroxy-caged luciferin 1 (PCL-1). PY1-ME was designed to study the diffusion of H2O2 through AQPs. To increase its intracellular retention, the probe was linked to a methyl ester group, which is cleaved in the cell by esterases. The validation of the system was performed in HEK cells overexpressing AQPs (forms 1, 3, and 8) treated with 50 μM H2O2. Only AQP3-expressing cells showed an increased PY1-ME fluorescence upon H2O2 treatment. To further explore this model, HT29 cells that endogenously express AQP3 were treated with EGF, which lead to a ≥10% increase in PY1-ME fluorescent signal. This setup has thus demonstrated cellular H2O2 fluxes through AQPs (87).
The second probe, PCL-1, is a small molecule carrying a boron reactive group linked to a firefly luciferin. Upon reaction with H2O2, luciferin is released and then free to react with luciferase to produce a photon (137). This strategy converted the probe into a bioluminescent reporter with in vivo application in mice under basal and disease conditions.
The insights into H2O2 dynamics gleaned from the boronate-based probes were initially very impressive. However, the characterization of the probes was largely based on overexpressing system or cells treated with exogenous H2O2, conditions that do not reflect redox signaling. Moreover, in vitro validation was carried out separately for H2O2, NO, and •O2 −. The boronate compounds ethylboronic acid, phenylboronic acid, phenyl boronic acid ethylene glycol ester, and phenylalanine boronic acid (EtBA, PBA, PBE, FBA, respectively) and coumarin boronate exhibit higher reactivity with peroxynitrite (k = 1.6 × 106 M −1 s−1) than with H2O2 (k = 2.2 M −1 s−1) (126, 152). The probe PCL-1 was no exception to this rule, reflecting its great potential to detect radical nitrogen species rather than H2O2 in vivo (125). PCL-1 reacts with peroxynitrite to form not only luciferin but also a second nitrated luciferin as recently characterized by HPLC, liquid chromatography-mass spectrometry (LC-MS), and spin trapping (150).
Coumarin boronate has been used for measurements of hydroperoxides of free amino acids and amino acid residues (tyrosine, cysteine, tryptophan, and histidine) in proteins during oxidative modification induced by ROS (85). Fluorescein boronate (Fl-B) detects peroxynitrite (115) with higher sensitivity than coumarin boronate. Endogenous peroxynitrite formation is detected with boronates after endothelial NO synthase activation: in response to ionomycin (4 μmol/L) in BAECs (incubated with 50 μM/L of the boronate Fl-B), the assay signal doubled and this increase in Fl-B fluorescence was abolished by nitroarginine methyl ester (
Limitations of boronates
The reaction of boronates with peroxynitrite raises concerns about their overall specificity and suitability as H2O2 probes in the presence of even low concentrations of peroxynitrite. Addition of the NO synthase inhibitor
ROS-Glo™
ROS-Glo is a luciferase-based boronate assay from Promega. Its major advantage over other boronates is its great sensitivity. This is achieved through the detection of an accumulated chemiluminescent reaction product, which signals an end point. This assay is several orders of magnitude more sensitive than fluorescence-based assays or chemiluminescence-based assays at detecting the generation of a reaction product. The assay does not require extensive sample preparation and offers easy multiplexing application. However, H2O2 detection is limited to the extracellular compartment, as is the case with HRP-dependent probes. There are only a few studies utilizing this probe and none have a major focus on redox signaling (17, 57, 61, 66). The probe is purchased as a kit and reaction of the sensor with H2O2 releases luciferin, which accumulates in the sample (first phase). At the end of the reaction, luciferase is added, and the light produced reflects the amount of luciferin released during the reaction with H2O2. The two-step assay condition makes the ROS-Glo more susceptible to artifacts, but its sensitivity is still superior over all other boronates. Nevertheless, even with this technique, basal H2O2 release from cultured cells is hardly detected and measurements can only be performed as an end point determination, that is, with very limited temporal resolution.
Genetically encoded probes
To overcome the complex and often undefined chemistry of chemical probes and to cope with the growing need for enhanced intracellular availability in defined compartments, genetically encoded fluorescent indicators have been developed. The first redox-sensitivity probes were variants of the green fluorescent protein (GFP) termed roGFP1 and roGFP2 (38, 54). These were generated by inserting redox-active cysteines, which change the excitability of GFP depending on their oxidation state. The use of these redox sensors was, however, limited by their low reaction speed and low sensitivity toward H2O2. In essence, these probes were slow indicators of the cellular redox potential according to the Nernst pair of –SH and –SS—but not detectors of H2O2. To render the fluorescent probes sensitive to H2O2, they were coupled to redox-sensitive transcription factors. The overall advantage of genetically encoded probes is the visualization of compartmentalized redox signaling with real-time dynamics and subcellular resolution.
The two main probes are HyPer (from hydrogen peroxide) and roGFP2-Orp1 (Fig. 4). Additional probes are the FRET-based detectors OxyFRET, PerFRET (41), and Redoxfluor (145), but still very little is known about the mechanistic behavior and practical applicability of these sensors. The most recently developed probe, a fusion of a Prx-based redox sensor to roGFP2 (roGFP2-Tsa2ΔCR), was claimed to exhibit superior sensitivity over the other probes. In this section we discuss the rationale of HyPer and roGFP2-Orp1 and general protocols: the application of the probes has been extensively reviewed recently (13, 15, 123). Table 2 summarizes the genetically encoded probes.

Dynamic range is often determined with the enriched probe incubated with exogenous H2O2.
GFP, green fluorescent protein; PDGF, platelet derived growth factor; pHi, intracellular pH; roGFP, redox-sensitive GFP. All Hyper probes are sensitive to pHi.
HyPer family
The first HyPer construct was developed in 2006 (10). Of all genetically encoded probes, HyPer gained the most popularity as it is easy to use and is sensitive. Reported limits of detection are 5 μmol/L and 250 nmol/L H2O2 in vitro and in mammalian cells, respectively (10). Currently, four HyPer constructs have been published (HyPer 1–3 and HyPer-Red) (10, 16, 42, 82) with the recent constructs having advantages of sensitivity over the initial HyPer 1. In general, HyPer probes take advantage of the redox sensitivity of the prokaryote transcription factor OxyR. Its low pKa cysteine (Cys 199) forms a disulfide bridge with Cys 208 upon oxidation with H2O2, leading to conformational changes and DNA binding properties (10). Through this mechanism, bacterial OxyR senses H2O2 and activates antioxidant genes to protect the cells against oxidative damage (27).
In HyPer probes, the C-terminal domain of OxyR is fused to the fluorescent protein cpYFP for HyPer 1–3 and to mApple for HyPer-Red. Oxidation by H2O2 triggers conformational changes in OxyR, which alters the excitation features of those fluorescent proteins. HyPer 1–3 have two excitation peaks (420 and 500 nm) and only one emission peak at 516 nm. Under basal and low H2O2 concentrations, respectively, the ratio F420/500 nm is high. With increased oxidation and higher H2O2 levels, the ratio F500/420 nm is proportionally increased. This property classifies HyPer 1–3 as ratiometric probes. These are favorable characteristics for comparative studies as ratio values are independent of the expression level of the protein and measurements can be corrected for bleaching.
HyPer 1 had a narrow dynamic range upon H2O2 exposure. HyPer2 (a A406 V mutation of HyPer 1) increased the dynamic range from 1.5–3 in HyPer1 to 6–7. Unlike HyPer1, HyPer2 is a dimer with a greater dynamic range and, therefore, reduced sensitivity. (82). HyPer 3, a H34Y mutation of HyPer2 (16), exhibits increased sensitivity despite maintained dynamics. HyPer-Red is a red variant obtained by replacing cpYFP with mApple. HyPer-Red is not a ratiometric but an intensiometric probe, with one excitation peak at 575 nm and one emission peak at 605 nm (42). The advantage is a second probe with an excitation/emission behavior distinct from that of HyPer1–3. Therefore, measurements in two cellular compartments can be performed simultaneously. For this, variants of the plasmid retaining the protein in the endoplasmic reticulum (ER), in mitochondria, the cytosol, and at the plasma membrane, have been developed (84a).
Limitations of the HyPer probes
All HyPer probes share one limitation apart from the narrow dynamic range: they are all fused to fluorescent proteins with a pKa of 8.6, which are highly sensitive even to low pH changes within the physiological range (123). In an alkalinized environment, there is an increase in the F500 nm and, conversely, under acidic conditions the F420 nm is proportionally increased (142). Therefore, it is essential to control measurements with HyPer C199S, SypHer, or SypHer-2. These probes exhibit no redox sensitivity but maintained pH sensitivity (53). Studies lacking these important controls have to be interpreted with caution and should be re-evaluated.
Owing to its pH sensitivity, SypHer even became a useful tool to measure intracellular pH, which can often be challenging to monitor (83, 109, 124). The next improvement in the HyPer family might potentially address the pH limitation using circular permutations of red fluorescent proteins with low pKa, such as mKate (pKa 5.4) or FusionRed (pKa 4.6) (42).
Being a cysteine redox sensor, specificity of HyPer for H2O2 is limited and electrophiles and strong oxidants can also oxidize the probe. Moreover, the sensor might also consume H2O2, potentially interfering with the biological outcome. Another limitation of the protein-based sensors is their high rates of oxidation but low rates of rereduction by the endogenous antioxidant enzymes such as thioredoxins and glutathione reductases (15). Therefore, the reducing capacity of the cell and glutathione pool may affect the results (101).
Examples for the use of HyPer probes
Numerous studies ranging from subcellular compartments over single cells up to living organisms such as zebrafish, Caenorhabditis elegans, and Xenopus have been performed with HyPer (13). Cells can be transiently transfected with standard reagents and stable clones can be selected. In zebrafish models, one-cell-stage embryos were injected with HyPer mRNA to express the sensor (99). In addition, the sources of H2O2 in several systems have been addressed with this probe. Finally, comparative studies with HyPer 1/3 and HyPer 3/HyPer-Red have been carried out to experimentally demonstrate the advances during development of the probes (16, 42).
Expression vectors encoding HyPer are commercially available (Evrogen) and insertion of targeting sequences directs HyPer to defined cellular compartments such as mitochondria, cytosol, or the nucleus. ER-targeted HyPer has a fairly low dynamic range as the compartment is highly oxidizing and most of the HyPer is already oxidized under basal conditions.
The plasmid coding for HyPer (pHyPer-cyto) contains a Kozak sequence upstream of HyPer and can be subcloned into other vectors to generate a fusion protein of HyPer with other target proteins (90). Such fusion constructs could be of use to determine redox modifications of the respective target protein. Obviously, validation of the construct is required as such a fusion can change the function or localization of the construct.
HyPer fluorescence can be visualized by wide field fluorescence microscopy, confocal systems, or fluorescence lifetime imaging microscopy (13, 90). HyPer 1–3 oxidation should be determined from the differential emission at 516 nm after excitation with 420 and 500 nm (90). To control the experiments, the following interventions are required: exogenous H2O2 and dithiothreitol have to be added as positive controls to assay the functionality of the probe by driving it to its fully oxidized or reduced states. Second, as already pointed out, it is necessary to control the pH. This can be done by the use of SypHer-2 or C199S-HyPer-Red. In the case of cpYFP imaging, a 488 nm laser line may be performed to control cellular distribution of the probe.
roGFP-Orp1 probe
roGFPs were engineered by inserting cysteines at positions 147 and 204 in adjacent β-strands of the GFP surface (54). These residues are close to the chromophore, and upon formation of an intramolecular disulfide bond, conformational changes alter its fluorescent properties (54, 123). Two roGFPs have been developed: roGFP1 with mutations at C48S, S147C, and Q204C and roGFP2 with an additional S65T (38). The probes were initially employed for redox status measurements, as response times of roGFPs to H2O2 were in a range of minutes, a resolution too low for signaling studies. Moreover, the midpoint potential E′0 is −291 and −280 mV for roGFP1 and roGFP2, respectively. This implies that the sensors require highly reducing conditions to maintain responsiveness to changes in redox status. To overcome these limitations, roGFP2 was fused to the peroxidase Orp1 (Gpx3) protein from Saccharomyces cerevisiae, a homologue of mammalian glutathione peroxidases 4 and 5 (52). The conceptual idea behind this genetic fusion is inspired by the redox relay model, where a professional peroxidase-active site cysteine (Cys36) is specifically oxidized by H2O2 to a sulfenic acid; the latter transduces redox equivalents to target proteins. This mechanism underlies the Orp1-Yap activation in yeast (136) and Prx 2-STAT3 in mammalian cells (128).
The probe roGFP-Orp1, developed in 2009, consists of Orp1 fused to the C-terminus of roGFP2 by a 32 amino acids linker to guarantee flexibility to the molecule (Fig. 4B). It is a ratiometric probe with 405 and 488 nm excitation and 510–530 nm emission (52). A ratiometric increase in 405/488 nm occurs with increased H2O2 availability or decreased rate of rereduction by thioredoxins, glutathione reductases, and glutathione pool.
The sensor can be transiently expressed in defined cell compartments such as cytosol and mitochondria. Alternatively, roGFP-Orp1 can also be integrated into the chromosome. Studies characterizing the probe showed its function in vitro as well as in cells treated with exogenous H2O2 and primary cells. However, as for HyPer, the dynamic range of roGFP-Orp1 (i.e., the fold increase) to H2O2 is relatively low. The highest fold increase (≥7-fold) was obtained by exposing the probe to H2O2 at 750 nM. HeLa cells expressing roGFP-Orp1 and treated with 50 μM H2O2 show a more than threefold change in fluorescence. In T cells, receptor activation by crosslinking (anti-CD3 antibody) resulted in an increase of the detected H2O2 by twofold. Although the fold increase is narrow, the probe was successfully employed for tracing in vivo H2O2 levels in Drosophila with resolution to the cellular level during development and aging (4) and in the cardiovascular tissue of the living zebrafish (102). More recent characterization of roGFP-Orp1 revealed that it can also react with peroxynitrite and hypochlorous acid under nonphysiological high concentrations (93).
Compared with HyPer, roGFP-Orp1 showed a slower reaction response with a similar sensitivity. This is likely because of the different reaction mechanisms of both probes. Although the signal in roGFP-Orp1 depends on redox transfer and disulfide exchange between two proteins, HyPer signal depends on formation of a single intramolecular disulfide. However, a critical advantage of roGFP-Orp1 over HyPer is that the ratiometric signal is not pH sensitive.
How to use roGFP-Orp1 probe
Despite similarities with HyPer and advantages including pH insensitivity, roGFP-Orp1 has not yet gained comparable popularity. Nevertheless, the probe has been used in human cell models (HEK293, HeLa, and T cells), whole organism (Drosophila), and yeast (S. cerevisiae) [recently reviewed in Schwarzlander et al. (123)].
The plasmids for targeted roGFP-Orp1 expression in mammalian and insect systems are available through the nonprofit repository Addgene (
The oxidation of roGFP-Orp1 by H2O2 is reversible in vivo. As with HyPer sensors, redox couples including thioredoxin and glutathione reductase can potentially reduce the signal. Therefore, the reducing capacity of the system must be taken into consideration. Also, the highly oxidizing environment of the ER may already bring the sensor to its maximum oxidized state. Low glutathione pools change the probe redox dynamics, and resident protein disulfide isomerases may catalyze roGFP oxidation and make the sensor irresponsive to changes in H2O2 levels. Therefore, appropriated controls such as roGFP alone, inhibition of thioredoxin reductase with auranofin, and H2O2 scavengers are helpful if the probe is targeted to the ER.
Prx-based probes—roGFP2-Tsa2ΔCR
Of all proteins, Prxs exhibit the highest reaction speed with H2O2 [rate constant of 107 M −1 s−1 (106)]. This outcompetes the redox sensors OxyR and Orp1 by two orders of magnitude (105 M −1 s−1) (6). Therefore, sensors utilizing Prxs promise increased sensitivity. On this basis, a 2-Cys Prx (one peroxidatic Cys and one catalytic) from S. cerevisiae (Tsa2) was genetically fused to roGFP2 (92) inspired by the redox relay model. A mutation of the resolving cysteine to alanine (roGFP2-Tsa2ΔCR) rendered the sensor resistant to the endogenous glutaredoxins, whereas an additional mutation of the peroxidatic cysteines produces a catalytically inactive probe. roGFP2-Tsa2ΔCR is thus ahead of all previously known genetically coded probes: it is more sensitive (by 20-fold), not affected by rereduction, and does not interfere with H2O2 homeostasis. The probe is ratiometric and detects basal H2O2 in nanomolar and potentially picomolar concentrations with subcellular resolution and real-time dynamics. Moreover, it responds to increasing but also decreasing concentrations with subcellular resolution. So far, the characterization of the probe has only been performed in yeast.
Probes that should not be used to measure H2O2
Dichlorodihydrofluorescein diacetate
Dichlorodihydrofluorescein diacetate (H2DCF-DA) is an extensively used probe to assess cellular H2O2 despite serious limitations (59). The assay is overall very easy to perform and a high fluorescent signal can be detected by routine techniques such as confocal microscopy, flow cytometry, and multiwell plate fluorimetry (8, 59, 139). This, and the large number of publications utilizing this probe, frequently misleads “newcomers” to choose H2DCF-DA, its derivatives, or dihydrorhodamine as their tool of choice.
The H2DCF-DA probe is colorless and cell permeable. Inside the cell, the compound is readily cleaved by esterases, which leads to H2O2 production (117) (Fig. 5). The resulting H2DCF is retained within the cell and has a tendency to accumulate in mitochondria. H2DCF does not react with H2O2 directly but with several other one-electron oxidants to form •DCF−. This intermediate radical is further oxidized to 2′-7′-dichlorofluorescein (DCF), which exhibits high fluorescence at λex/em 495/529 nm.

Several publications aimed to uncover the complex in vivo chemistry of H2DCF/•DCF−/DCF and reveal artifactual sources (18, 29, 67, 95, 100, 111, 117, 134). Essentially, hydroxyl radical (•OH), reaction of H2O2 with HRP/myeloperoxidase, iron, and heme at high concentrations will form DCF. Likewise, reactive nitrogen species such as •NO2 and products from peroxynitrite decomposition also increase DCF formation. Furthermore, the •DCF− radical reacts with oxygen to form •O2 − (81), which subsequently dismutates to H2O2 and fuels the system by redox cycling. Last but not least, the dichlorodihydrofluorescein (DCFH) system is highly photosensitive. Light exposure can artificially increase fluorescence, leading to false positive results (81). Once oxidized, DCF leaves the cell to some degree by diffusion, resulting in a loss of fluorescence with time.
Although measurements obtained with H2DCF-DA have added important insights into cellular oxidative stress responses, it is nowadays unacceptable for scientists working on redox biology to exclusively rest their conclusions on H2DCF-DA. As reviewers are aware of the limitations of the dye, they tend not to accept H2DCF-DA measurements at all. Readers are, therefore, advised against using this compound so as to avoid ambiguous or artifactual results.
Dihydroethidium
DHE is currently among the “gold standard” probes (35) to measure •O2 −. DHE is membrane permeable and was first believed to react only with •O2 −. The reaction product ethidium is a highly fluorescent compound that intercalates DNA, further enhancing its fluorescence and retaining the dye inside the cell (11, 118). The first studies using DHE utilized fluorescence detection and reported that the detected signals were decreased by SOD and by knockout of members of NADPH oxidases. On this basis, the assay signal was attributed to •O2 − (23, 56, 89). Later studies indeed showed that H2O2 does not react with DHE. The situation changes in the presence of heme proteins wherein DHE forms nonhomogenous mixtures of various oxidation products with an overall increase in fluorescence. These observations raised serious doubts on the reliability of the DHE fluorescence assay (103). To overcome this problem, the oxidation products of DHE were separated by HPLC to distinctly detect specific products of •O2 −. This predominantly includes 2-dihydroxyethidium (2EOH), whereas unspecific one-electron oxidation reactions yield ethidium (43, 71, 147, 153). Figure 6 shows exemplary HPLC traces of in vitro reactions of •O2 − generated by the XO/xanthine system and H2O2 with DHE. The reaction of DHE can be monitored by absorbance at 350–400 nm, whereas its oxidation products are detected by fluorescence λex/em: 510/595 nm.
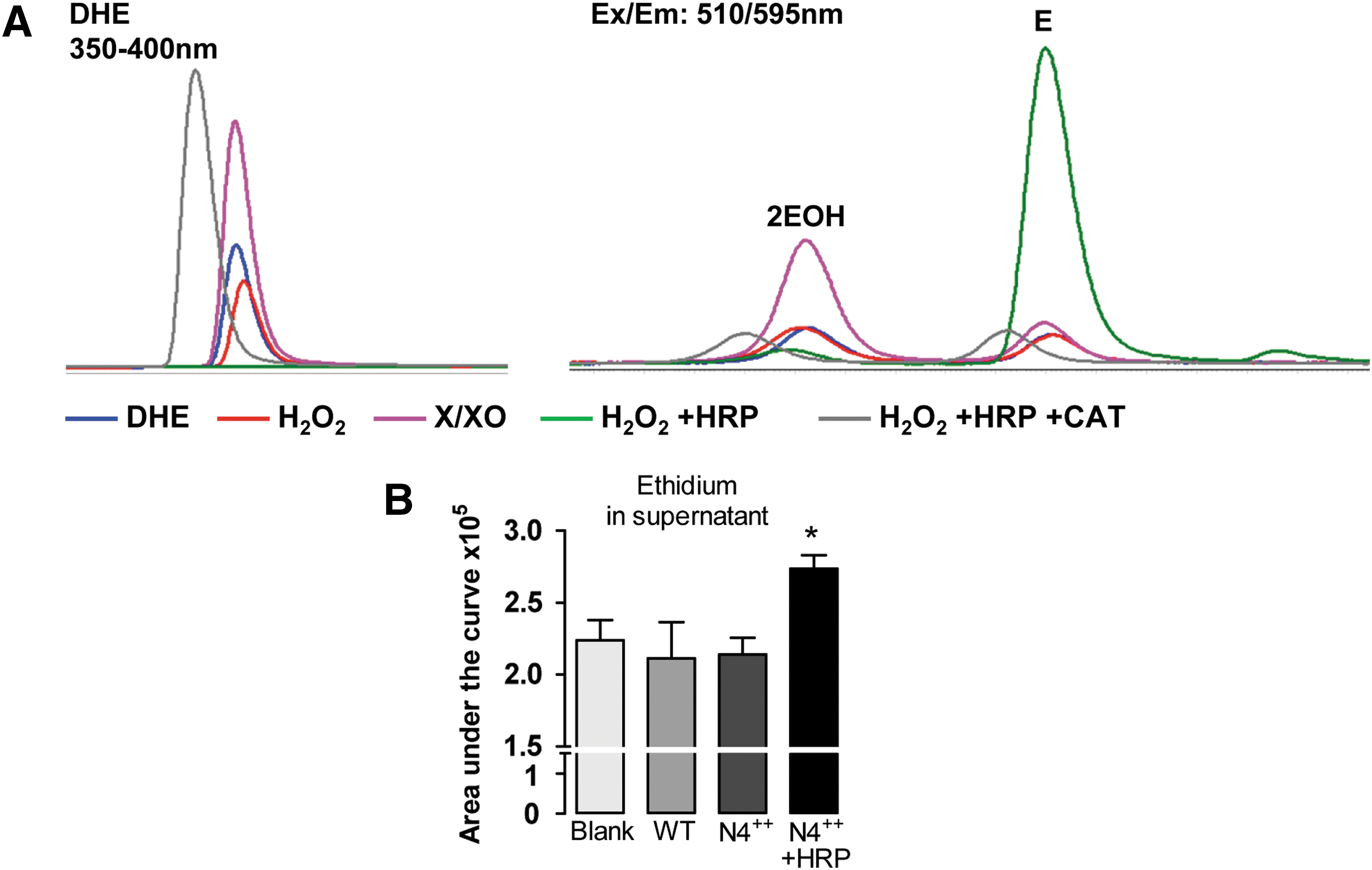
2EOH contains a hydroxyl group, making it more hydrophilic than ethidium, so that the compounds can be separated by reverse phase chromatography. H2O2 alone does not react with DHE but, in the presence of HRP, massively increases the formation of ethidium (E) in a catalase-sensitive manner. The overall formation of the products (independent of 2EOH and E ratios) is proportional to the consumption of DHE. Based on this, a simple normalization of 2EOH/DHE and E/DHE has been commonly applied. However, these reactions are not linear and might over-represent small differences (153).
In a biological system with high levels of H2O2, heme, and peroxidases, the formation of E must not be ignored. Figure 6B shows the formation of E in the supernatant of HEK293 cells overexpressing Nox4 upon treatment with HRP. An increase in E by 22% is detected after separation of products by HPLC. With the recent advances of HPLC being coupled to LC-MS/MS, more and more one-electron oxidation products are being discovered. Dimers of ethidium that have distinct retention times and are formed differentially by Fenton reaction, or in the presence of H2O2 or peroxynitrite and NO donors (Rezende F. and Luck B., unpublished data), can be readily observed. Currently, these numerous products of DHE cannot be attributed to specific sources or reaction mechanisms. Therefore, HPLC separation of DHE products and detection by fluorescence are neither quantitative nor qualitative for H2O2 or •O2 −. This is because several dimers (DHE–DHE, DHE–E, and E–E) display similar spectra, but distinct molar extinction coefficients and different reaction stoichiometry.
Conclusion—Measuring H2O2 Is Difficult
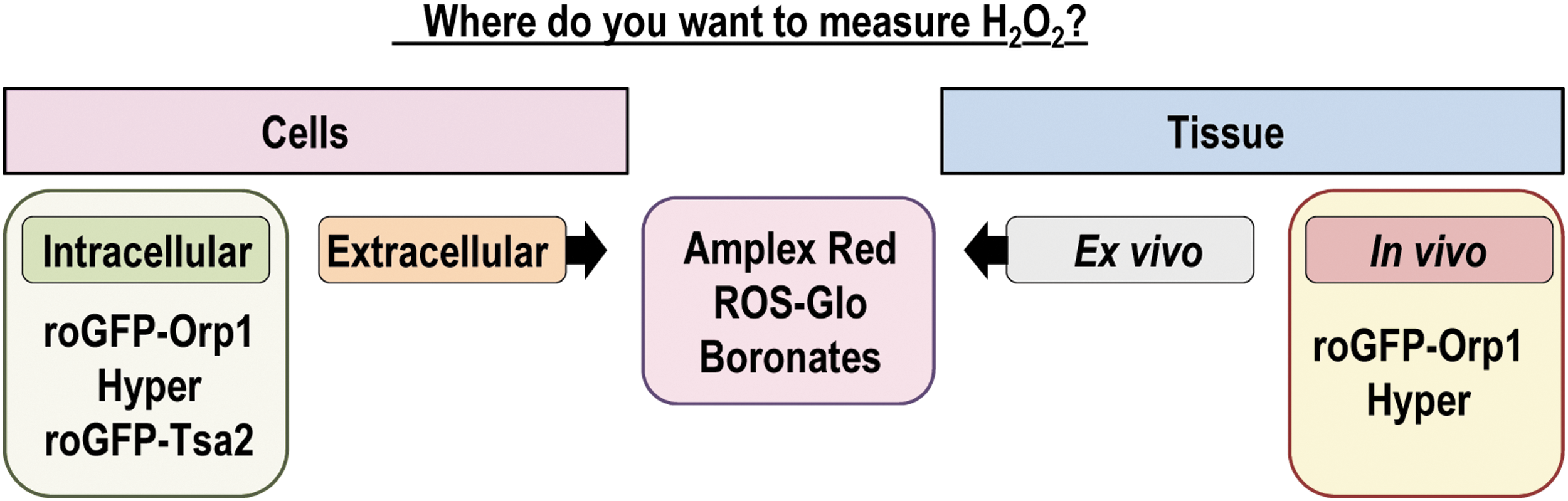
Redox biology has undergone great development concerning the understanding of mitochondrial function and signaling and the highly controlled and powerful signaling elicited by the nonphagocytic Noxes as professional sources of cellular •O2 − and H2O2. However, determination of H2O2 in biological systems still remains a substantial challenge. With the current knowledge on the individual roles of the different types of ROS, the need for specific, yet sensitive, ROS probes has increased. Progress in redox research has been seriously limited by the available tools. Current research aims to define specific redox pathways activated by locally produced and defined doses of H2O2. Detailed understanding of these processes not only depends on progress in redox proteomics but also on development of assays that reliably measure H2O2 in subcellular compartments. With the current toolbox, it is possible to approximate the biologically active H2O2 level if precautions are taken. Table 3 summarizes the main probes, their advantages, disadvantages, and a word of caution on their usage. Figure 7 shows the application of fluorescent probes in distinct samples. To provide a certain level of validity to measurements by these probes, it is generally advisable not to rely on one single assay but rather to combine several detection methods with different reaction mechanisms. In addition, H2O2 footprints such as protein thiol oxidation might illustrate compatible biological consequences (26, 31, 55).
DPI, diphenylene iodonium; GRX, glutaredoxin; HRP, horseradish peroxidase; L-NG, nitroarginine methyl ester; PEG, polyethylene glycol; TRX, thioredoxin.
Even so, one should remember to handle data with utmost care and to acknowledge that measurements differing from the observed biological response are probably artifactual.
Footnotes
Acknowledgments
This work was supported by grants from the German Research Foundation (DFG): SFB 815 TPA1, SFB 834 TPA2, and SFB1039 TPA1 and the Goethe-University: Heinrich und Fritz-Riese-Stiftung to F.R. The authors are grateful to James A. Oo, MRes, MSc, for proofreading the article.