
Abstract
Significance:
Hematopoietic stem cells (HSCs) can sustain the production of blood throughout one's lifetime. However, for proper self-renewal of its own population and differentiation to blood, the HSC requires a specialized microenvironment called the “niche.”
Recent Advances:
Recent studies using novel mouse models have shed new light on the cellular architecture and function of the HSC niche. Here, we review the different cells that constitute the HSC niche and the molecular mechanisms that underlie HSC and niche interaction. We discuss the evidence and potential features that distinguish the HSC niche from other microenvironments in the bone marrow. The relevance of the niche in malignant transformation of the HSCs and harboring cancer metastasis to the bone is also outlined. In addition, we address how the niche may regulate reactive oxygen species levels surrounding the HSCs.
Critical Issues and Future Directions:
We propose future directions and remaining challenges in investigating the niche of HSCs. We discuss how a better understanding of the HSC niche may help in restoring an aged hematopoietic system, fighting against malignancies, and transplanting purified HSCs safely and effectively into patients. Antioxid. Redox Signal. 29, 191–204.
Concept of the Stem Cell Niche
T
Many of these mechanisms are intrinsic to the HSCs. However, as early as the 1970s, scientists have proposed that the local bone marrow (BM) microenvironment, referred to as the “niche,” also provides crucial and indispensable factors for HSC self-renewal and differentiation. The formal hypothesis was proposed by Ray Schofield in 1978, who posited that the niche controls the quiescence and cell cycle entry of stem cells, provides the information about tissue and organismal state to stem cells, regulates the fate of stem cells' daughter cells, and curbs the mutation rate in stem cells (76, 78).
The first evidence for the niche concept was presented by Dexter et al., who demonstrated in vitro that BM-derived stromal cells can maintain granulopoiesis (23). Later, Whitlock and Witte isolated stromal BM cultures that initiated and maintained B lymphopoiesis (60, 104). Whitlock and Weissman cloned several stromal lines from these cultures and found several that supported normal populations of BM cells that formed “cobblestones” in culture, with each colony giving rise to both myeloid cells (early) and B-lineage cells (late) (103). The line AC6.2.1 derived from them was used in the screening of fluorescence-activated cell sorting selected mouse and human BM subsets for the initial 2000-fold enrichment of cells containing HSCs (103). Finally, the OP9 stroma cell lines, which are deficient in macrophage colony-stimulating factor, were shown to trigger the differentiation of embryonic stem cells to blood cells (61).
Although these initial experiments proved the importance of the niche for HSC activity, the mechanisms underlying the HSC-niche interactions remained unknown. With the development of new mice models and imaging techniques, we can now more precisely identify the niche components and prospectively investigate the role of the HSC niche. However, many questions still need further clarification: What kinds of cells are essential in the HSC niche? Is the HSC niche unique only to HSCs? What are the mechanisms of competition within the niche? How is the leukemic niche different? How does the niche protect HSCs from exhaustion? In this review, we will summarize recent discoveries addressing these questions and outline the next emerging challenges.
Changing Candidate Cells Making Up the HSC Niche
BM is a complex tissue with a high cell density. Hematopoietic cells constitute a major fraction of the BM, but there are also several other kinds of cells, including mesenchymal stromal cells (MSCs), osteoblasts, endothelial cells, and neuronal cells. A fundamental question was: What are the cells that constitute the HSC niche (94, 102)?
Using the early cell surface markers that could enrich for HSCs, but not isolate pure HSCs, immunohistological evidence implied that the HSCs were in an endosteal region of the BM (109). In engineered mouse strains, in which osteoblasts and osteo-lineage cells were targeted by overexpression of parathyroid hormone (PTH) (11) or by deletion of the BMPr1α gene (109), increased numbers of osteoblasts correlate with a higher number of these phenotypic cells that contain HSCs. Moreover, conditional ablation of osteoblast-lineage cells by ganciclovir treatment of mice that have Col1α1 gene promoter driving thymidine kinase (Col2.3ΔTK) markedly decreased the number of HSCs (95, 113). This suggested that it is the osteoblastic lineage in the bone endosteum that governs the stem cell potential of the HSCs.
However, these observations were only correlative and it has not yet been shown as to whether the observed effect is due to a direct interaction between osteoblasts and HSCs or an indirect mechanism may be involved. It also cannot be ruled out that there exist other cells besides osteoblasts in the BM that express PTH- or PTH-related protein receptor, BMPR1α, and COLLα1. The most definitive way to demonstrate that a cell directly neighbors the HSCs would be by imaging. However, the imaging of the HSC niche has for a long time been impaired by the rarity of HSCs and the necessity to use several markers to identify them. Progress came with the discovery of more specific markers and HSC-reporter mice.
First, Kiel et al. demonstrated that functional HSCs could be enriched by using SLAM family markers: HSCs are negative for CD48 and positive for CD150 (45). They showed that the CD150+CD48− fraction of HSCs resides near perivascular sinusoidal cells (20 out of 35 cells) rather than in the endosteal osteoblastic niche (5 out of 35 cells).
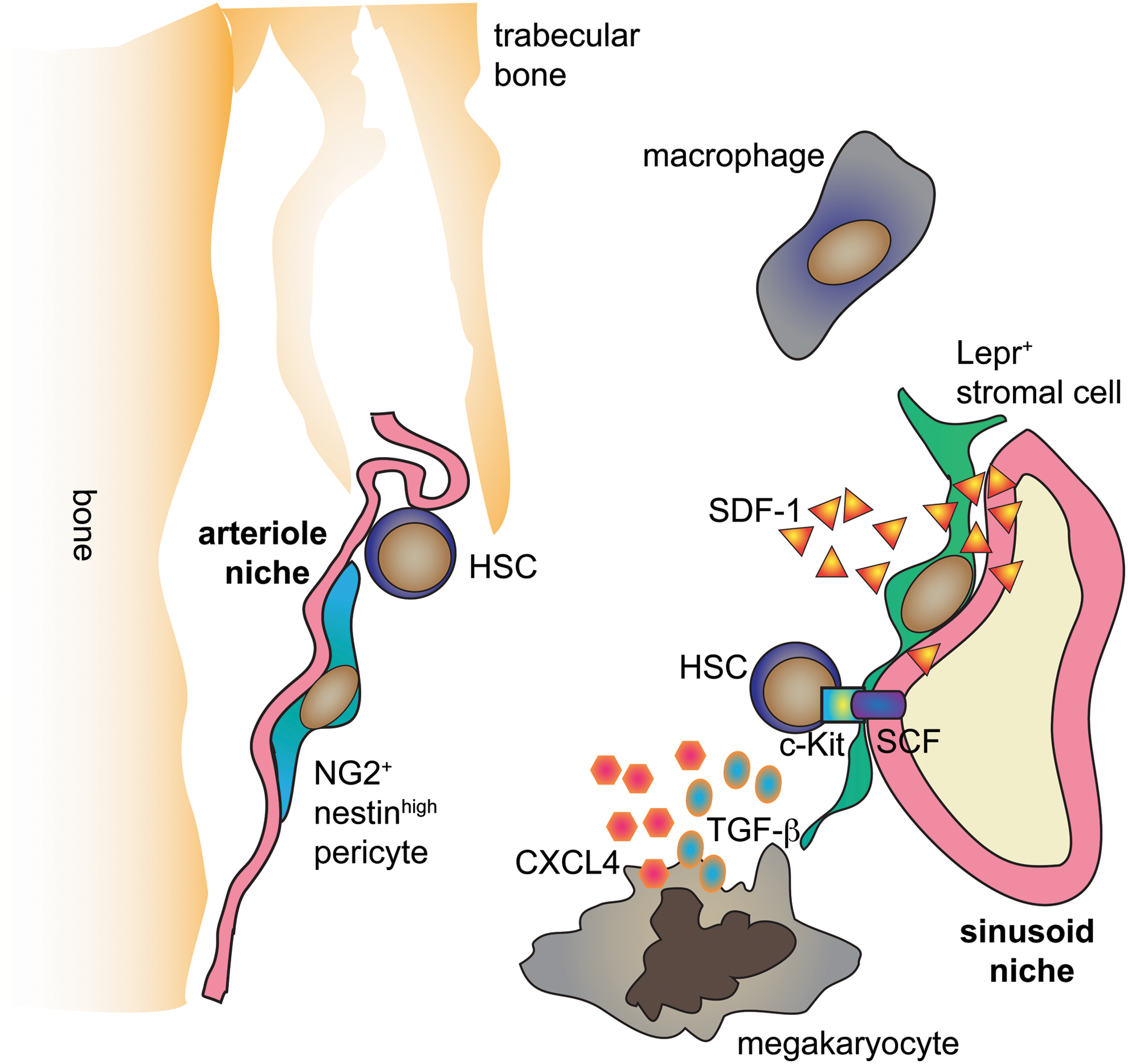
Although now more studies confirm the perivascular niche of HSCs, there are inconsistencies regarding the type of endothelial cells that HSCs attach to and their role in regulating HSC biology. Kunisaki et al. underline the importance of Sca-1+ small arterioles for the maintenance of quiescent HSCs (49). These Sca-1+ arterioles express also VEGFR2, VEGFR3, and Tie2 antigens (49). The whole-mount imaging of BM revealed that cells expressing markers of quiescent HSC preferentially localize to the Sca-1+ arterioles (49), whereas the cycling Ki67+ HSC localize more closely to sinusoids.
Next, Itkin et al. proposed that arterioles and sinusoids differentially regulate the metabolism and reactive oxygen production in neighboring HSCs (35). On an intraperitoneal injection of mice with hydroethidine—a marker of reactive oxygen species (ROS) production—only the HSCs located near the sinusoids (<20 μm) contained a fraction of ROShigh cells (15 out of 41) whereas all HSCs associated with arterioles (22 out of 22) were negative for ROS (35).
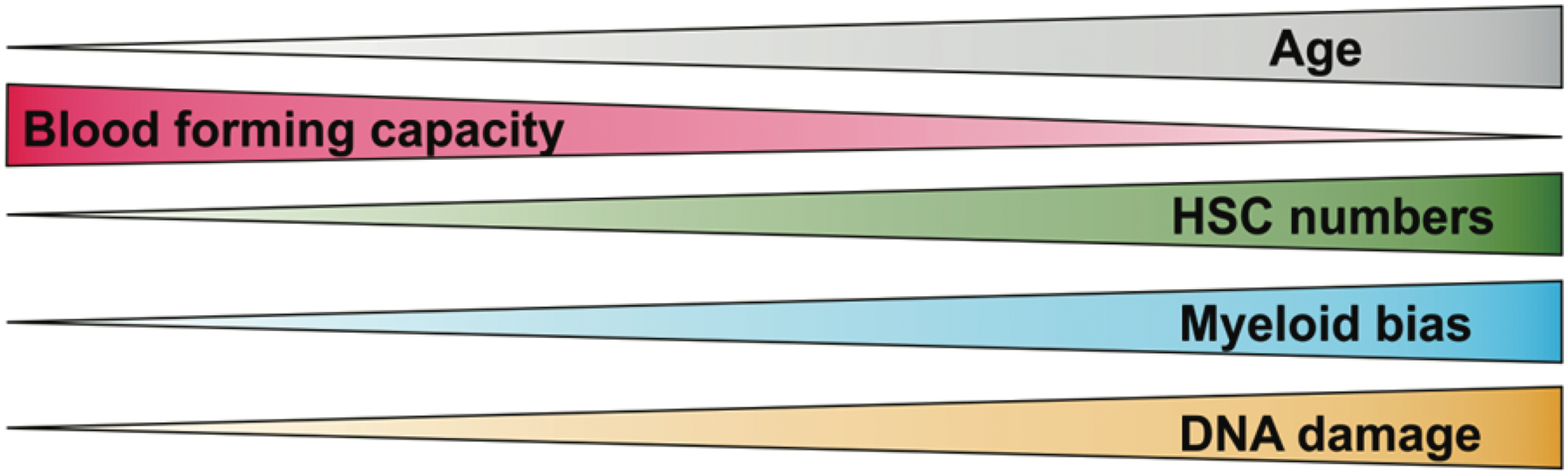
The vascular niche was also proposed to regulate aging of HSCs. During aging, the pool of HSC expands, but their regeneration potential is reduced, with pronounced myeloid differentiation bias (Fig. 1). Kusumbe et al. showed that arteries and capillaries, called H-type endothelium, directly neighbor cells expressing markers found on HSCs, and produce the highest amount of SCF (stem cell factor) among all BM endothelium (50). This fraction of endothelial cells expands in Fbxw7iΔEC mice that have endothelial-specific overactivation of Notch1 signaling, which correlates with a higher number of CD48−CD150+ cells (50). However, arteries and H-type endothelial cells decrease in number with age (50), whereas the number of bona fide HSCs increases (59, 66). The number of functional, transplantable HSCs also does not differ between the aged BM Fbxw7iΔEC and control BM, indicating that restoring the numbers of arteries and H-type endothelial cells in aged animals cannot reverse the effects of HSC aging.
Although these initial studies address the identity of the perivascular niche of the HSCs, they are still limited by the choice of markers used to detect HSCs. The use of multiple markers to identify HSCs is incompatible with imaging studies as modern microscopy is still limited by the number of detection channels. Therefore, a more feasible identification of long-term (LT)-HSCs with fewer selective markers for imaging studies was warranted. This became possible after the generation of HSC-specific reporter mice. Two groups proposed single markers for distinguishing HSCs from other hematopoietic cells and created reporter mice with these genes: α-catulinGFP/+ (2) and Hoxb5-mCherry (16). Both markers are highly enriched for functional HSCs. The Hoxb5 is more restricted to LT-HSC (Fig. 2) on the transcriptional level [data based on Gene Expression Common platform (79)]; they represent ∼30% of α-catulinGFP/+ cells. However, Acar et al. showed that expression of green fluorescent protein (GFP) in a-catulinGFP/+ mice is also restricted to LT-HSC. The α-catulinGFP/+ mice revealed that 84% of HSCs reside within 10 μm of BM sinusoids and are farther away from arterioles and transition zone capillaries (2). The majority of α-catulin+c-Kit+ HSCs are also localized within the central diaphysis part of the marrow rather than metaphysis and epiphysis and there are no differences between the endothelial compartments of Ki67+ cycling and Ki67- non-cycling HSCs (2). In Hoxb5-mCherry mice, 94% of Hoxb5+ HSCs were found to be in close proximity to BM endothelial cells, which was significantly higher than random spots (16).
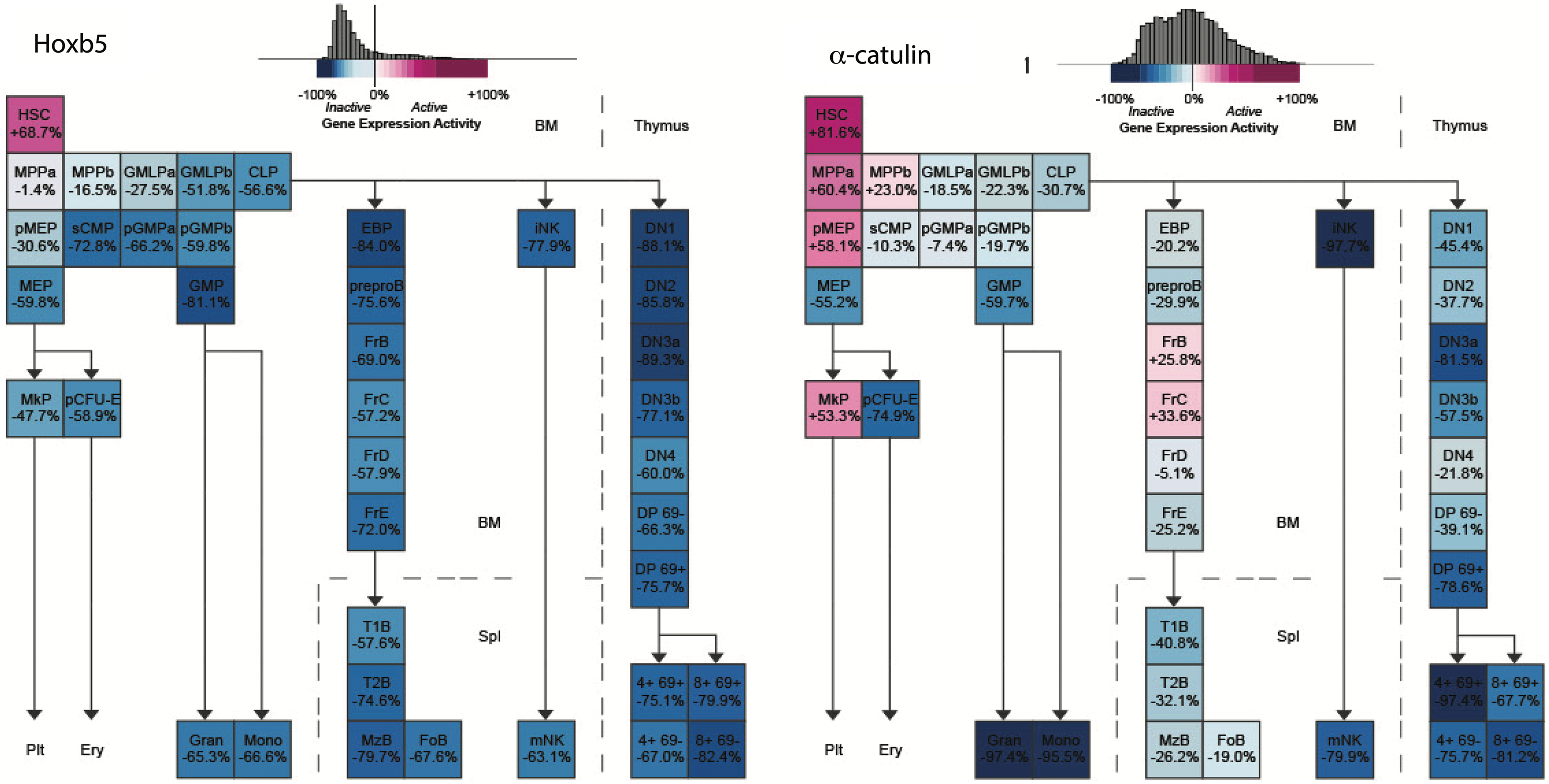
Taken together, direct imaging of the BM indicates the existence of a perivascular niche of HSCs (Fig. 3). Although there are contradictory conclusions regarding the role of arteriole and sinusoid niches for HSCs, recent studies using the HSC-reporter mice indicate that most LT-HSCs occupy the sinusoid niche.
MSCs in Niche Architecture
The perivascular niche of HSCs is not based solely on endothelial cells. MSCs on the abluminal surface of sinusoids give rise to multiple diverse progeny that in concert create the niche that supports and nourishes the HSCs (7). The earliest evidence for MSCs comes from work by Friedenstein et al. in 1970, where they showed that single BM cells form discrete colonies in culture, which, when transplanted in vivo, generate multilineage skeletal tissue (bone, cartilage, adipocytes, and fibroblasts) (27). Later, prospective isolation of non-hematopoietic (CD45−), nonvascular (Tie2−) αV+CD105+Thy1.1− progenitor cells from fetal BM and transplantation into the renal capsule of immunodeficient mice demonstrated sufficiency of these cells to give rise to a heterotopic bone capable of reconstitution with host HSCs and vessels (13). In humans, nonhematopoietic, nonvascular CD146+ BM cells similarly generate heterotopic ossicles with host HSCs and vessels. CD146+ cells in these ossicles are positioned around the sinusoidal walls close to where HSCs reside. These xenografted ossicles in mice have also served as valuable models of the human BM niche for the study of normal and leukemic human HSCs (1, 72).
Further characterization of the HSC niche came from studies of cells expressing niche factors that are capable of maintaining and influencing the LT-HSCs. Although many factors and cells have been implicated in supporting LT-HSCs, few have passed the trials and tribulations of cell type-specific conditional deletion experiments in mice. Early models of the osteoblastic niche of the HSC postulated the importance of adhesion molecules, particularly N-Cadherin, in maintaining HSC self-renewal in the BM. However, not only have studies shown that LT-HSCs do not localize to the endosteum, but also conditional deletion of N-Cadherin in hematopoietic cells (Mx1-Cre) and osteolineage cells (Osx-Cre) does not affect HSC maintenance in the BM (30, 44). Another controversial factor is Jagged-1, a ligand for receptors involved in Notch signaling. Conditional deletion of Jagged-1 in hematopoietic cells (Mx1-Cre) was shown to have no effect on HSC self-renewal (52). However, recently, Jagged-1 deletion in endothelial cells (VE-cadherin-Cre) (69) was shown to compromise self-renewal and maintenance of HSCs. Still, the use of a constitutive rather than an inducible Cre recombinase opens the possibility that the impaired HSC maintenance in this model could be a reflection of a developmental defect and may not produce the same results in adults. Angiopoietin-1 (ANGPT1), ligand for the receptor tyrosine kinase Tie2, is another factor that was associated with the suspected osteoblastic niche of the HSC and was shown initially on ex vivo manipulation to affect HSC engraftment and quiescence (3). However, when Angpt1 was tested in vivo by conditional deletion in osteoblasts (Col1a1-Cre), osteoprogenitors (Osx-Cre), LEPR+ stromal cells (Lepr-cre), hematopoietic cells (Mx1-Cre), and globally (UBC-CreER), there was no effect on BM cellularity, HSC frequency, or HSCs reconstitution capacity (111).
The factors reproducibly shown to play a role in HSC maintenance are CXCL12, the niche ligand for CXCR4 on HSCs, and SCF (KITL), the niche ligand for the KIT receptor on HSCs. CXCL12 is important in directing HSC migration to the BM and maintaining HSC quiescence, and SCF (KITL) is required for the maintenance of quiescent HSCs (92, 93). In mice with Cxcl12 promoter driving diphtheria toxin (DT) receptor and GFP, depletion of CXCL12+ cells results in a 50% decrease in HSCs within 2 days of DT administration (29, 62). Consistent with the phenotype of MSCs, these CXCL12-abundant reticular cells reside in the perivascular niche and are capable of both adipogenic and osteogenic differentiation (62, 86). Additional support for MSCs as a crucial part of the HSC niche came after studies done on the Nestin-GFP transgenic mouse model, in which GFP was inserted under the control of the nestin gene promoter. Nestin-GFP+ cells, which are capable of multilineage differentiation toward osteochondral lineages and self-renewal on serial transplantation, localized around blood vessels near HSCs and highly expressed CXCL12 and SCF (55). This population was divided into two distinct subpopulations: Nestin-GFPlow cells that localized perisinusoidally were marked by Leptin receptor (LEPR), and they expressed high levels of CXCL12 and SCF; Nestin-GFPhigh cells that localized periarteriolarly were marked by neural/glial antigen 2 (NG2), and they highly expressed CXCL12 (4, 49, 112).
Conditional deletion of Cxcl12 in Tie2-Cre (vascular) and 2-week-induced NG2-CreERTM (pericyte) mice depleted HSCs, but in Lepr-Cre (mesenchymal) mice only mobilized HSCs, and in Nestin-Cre and 6-week-induced NG2-CreERTM mice had no effect on HSCs (4, 26, 29). The difference in HSC depletion between 2- and 6-week Cxcl12 depletion in NG2-CreERTM mice suggests that NG2-CreERTM-expressing cells are important for HSC maintenance in early development and/or early induced NG2-CreERTM marks a subset of CXCL12+ cells that no longer express NG2-CreERTM in adults. This would be consistent with numerous studies that have shown that stromal reporter lines followed at different developmental timepoints mark distinct waves of MSCs (26, 33, 57, 63). On the other hand, conditional deletion of Scf with Lepr-Cre (mesenchymal) and Tie2-Cre (vascular) cells resulted in HSC depletion, but did not affect HSC frequency in Vav1-Cre (hematopoietic), Col2.3-Cre (osteoblast), 2-week-induced NG2-CreERTM (pericyte), and Nestin-Cre cells (4, 26). Notably, when Scf was conditionally deleted in LepR-expressing cells by crossing Scffl/fl and LepR-Cre mice, HSCs were not depleted in the fetal liver, but they were depleted in the BM with age, suggesting that Scf expression by LEPR+ cells is a critical component of the adult HSC niche. On the other hand, HSC depletion is observed in the fetal liver of Tie2-cre;Scffl/− mice and the magnitude of depletion increases with age, suggesting that SCF production by vessels is persistently important in early and late HSC maintenance.
It may seem puzzling that Scf and Cxcl12 deletions in Nestin-Cre mice did not affect HSCs, especially given the strong evidence for Nestin-GFP and overlap of Nestin-GFPlo cells with LEPR+ cells. However, it has been shown that different Nestin transgenes exhibit different expression patterns such that Nestin-GFP and Nestin-Cre mark separate populations; Nestin-GFP cells actually have low endogenous Nestin, whereas Nestin-Cre expression is more consistently correlated with endogenous transcription (112). Together, it is evident that CSC112 and SCF are differentially expressed by various niche cells and that their effect is dependent on the type and location of the expressing cell (Fig. 4).

These observations with conditional deletion niche models have drawn focus to the multiple niche hypothesis in which HSCs occupy anatomically distinct regions in the BM that tightly control their quiescence, proliferation, and differentiation. From recent reports, it is evident that the perisinusoidal (LepR+/Nestin-GFPlow) and periarteriolar (NG2+/Nestin-GFPhi) niches represent separate microenvironments that influence HSC proliferation and quiescence, respectively. However, the heterogeneity of MSCs within any particular perivascular niche, much less between the niches, has yet to be understood. Recently, characterization of the putative mouse skeletal stem cell (mSSC) and its downstream progeny unveiled the presence of diverse niche stroma distinguished by their differential surface marker expression for Thy, 6C3, CD105, and CD200. Different subsets of the mSSC lineage tree produce different HSC-supportive cytokines, with the Thy+ cells most closely resembling the LEPR+ and CXCL12hi cells previously identified (14, 15). Remarkably, LEPR+ cells are highly specific for CXCL12 and SCF, marking virtually all CXCL12 and SCF-expressing cells in the BM.
The advent of high-throughput single-cell RNA-sequencing platforms will enable the capture of interniche diversity in the BM and the identification of rare MSCs, pericytes, and endothelial cells that maintain HSCs in the niche. Combining transcriptional data with spatial information resolved by state-of-the-art clearing and microscopy tools can then lead to the determination of the direct neighbors of the HSC and the binding interactions that influence HSC localization in one niche versus another.
Apart from endothelial and stromal populations, other non-hematopoietic cells have been shown to contribute to the HSC niche. The sympathetic nerves interact with perivascular cells and control CXCL12 expression (43). This regulation is responsible for circadian oscillations of CXCL12 levels and subsequent daily fluctuations of HSC numbers that are mobilized into peripheral circulation (54). Other regulation of the HSC niche governed by the neuronal system is the activation of transforming growth factor beta (TGF-β) by nonmyelinating Schwann cells (106). However, the study using α-catulinGFP/+ reporter mice did not find much overlap between the HSC niche and GFAP+ Schwann cells.
Hematopoietic Neighborhood of HSCs
Although the endothelial and MSCs play crucial roles in the HSC niche, the majority of the cells in BM are hematopoietic cells. Thus, several studies have interrogated the role of the hematopoietic cells in the HSC niche (Fig. 2).
BM macrophages emerge as an essential population that shapes the HSC niche. The fraction of CD169+ macrophages is essential for the retention of HSCs in the niche (19). The depletion of CD169+ macrophages mobilizes HSCs into a blood stream in steady-state conditions and further augments the pharmacological mobilization by granulocyte colony stimulating factor (G-CSF) or CXCR4 antagonists (19). The osteoclasts responsible for the bone resorption derive from macrophages and caused egress of HSCs into circulation (48). However, the role of macrophages is not clear, as another study identified a macrophage population called “osteomacs” that exhibited opposite activity—their depletion increased HSC egress (105). Nevertheless, these studies indicate that macrophages may be important cellular elements of the HSC niche.
Another population of hematopoietic cells, megakaryocytes, also controls the HSCs niche (9, 110). When the megakaryocytes were depleted from the BM, the number of HSCs increased (9, 110). However, this increase in the number of HSCs is not direct evidence for megakaryocyte role in the niche, as we showed that even bleeding mice three times leads to compensatory HSC entry into the cell cycle, and this is blocked by reinfusing the RBC from the taken blood (17). It was proposed that megakaryocytes restrict the proliferation of HSCs possibly by two mechanisms. First, they produce the CXCL4 chemokine that inhibits HSC proliferation (9). Second, they are a source of TGF-β, which also governs the HSC quiescence (110).
The BM niche provides an immune-suppressive environment that protects HSCs from immune attack. The immune privilege of the HSC niche is provided by T regulatory cells (Tregs) (28). In vivo imaging reveals colocalization of FoxP3-positive Tregs with HSC (28). The presence of Tregs enables HSC survival from allogenic donors in an interleukin (IL)-10-dependent fashion (28).
No Guests Invited: Specificity of the HSC Niche
The next big question addressed within the past few years concerns the specificity of the HSC niche. Is the niche where HSCs reside unique or is the same niche occupied by HSCs also occupied by other downstream progenitor cells (Fig. 5)? The answer for this question has important clinical implications for developing new strategies for BM transplantation and HSC mobilization. If there is, indeed, a unique niche for HSCs, then we could specifically empty the HSCs from their niche without affecting other hematopoietic cells. Such a targeted strategy would be preferential to currently used recipient conditioning by irradiation and chemotherapy for multiple reasons, with the most important being the reduced side effects and toxicity to the patient.

The first evidence for a specific HSC niche came from experiments involving transplantation of purified HSCs to unconditioned RAG2−/−γc−/− mice (22). Czechowicz et al. transplanted 1000 purified HSCs or 1000 purified HSCs together with 100,000 CD34+ progenitors and observed the same level of chimerism among granulocytes after 16 weeks (22). Osawa et al. showed that CD34+ progenitors do not provide any chimerism 16 weeks after transplantation (64); thus, the chimerism observed after 16 weeks had to be derived from CD34- HSC. If CD34+ progenitors had competed for the niche with CD34− HSC, the transplantation of 100,000 CD34+ cells together with 1000 HSC would have blocked the homing of HSC and no/reduced chimerism would have been observed. Thus, the fact that transplantation of the prevailing number of CD34+ progenitors did not affect the chimerism levels indicates that CD34+ progenitors occupy distinct niches than CD34− progenitors (22). This interpretation is valid under the assumption that the number of HSC niches is limited. The existence of a limited number of HSC niches was supported by experiments when transplanting increasing number of HSCs does not lead to increasing chimerism, but leads to saturation of HSC engraftment. However, recent work by Shimoto et al. indicates that transplantation of a high number of HSCs may overcome the niche saturation limit (80).
The notion that HSCs occupy a specific and distinct niche was confirmed by functional studies done by Ding and Morrison (25). Given that different cell types, including HSCs and downstream progenitors, depend on CXCL12, they conditionally deleted this chemokine in various niche cells and observed how it differentially affects HSCs and progenitors. As described earlier, conditional deletion of Cxcl12 in endothelial cells by using the Tie-2-cre mouse model decreased the frequency of HSCs in the BM, but it did not affect any of the tested downstream progenitors: multipotent progenitors (MPP), lymphoid-primed multipotent progenitors (LMPP), common myeloid progenitors (CMP), common lymphoid progenitors (CLP), megakaryocyte–erythroid progenitors (MEP), granulocyte–monocyte progenitors (GMP), or B-lineage progenitors (25). The deletion of Cxcl12 in stromal cells by using LepR-Cre mobilized both HSCs and progenitors to the peripheral blood, indicating that LEPR+ stromal cells contribute to both the HSC and progenitor niche. Next, to address the role of osteoblasts, they used the Col2.3-cre;Cxcl12fl/fl model. These mice possess normal HSCs, CMPs, MEPs, and GMP numbers, but have reduced numbers of lymphoid progenitors, which was functionally confirmed by worse lymphoid contribution on whole BM transplantation. Further microscopic analysis confirmed the presence of Lin−Il7R+ in the endosteal niche (25). Therefore, these results confirm that the HSC niche is distinct from the progenitor niche, and that the endothelial and stromal cells are most crucial for providing the niche specificity. These conclusions were consistent with the study by Greenbaum et al. based on similar conditional deletion mice models (29).
Another study suggests that not only is the niche for lymphoid progenitors and HSCs different, but the GMP niche is also distinct from the HSC niche (80). Shimoto et al. transplanted a high number of purified HSCs into wildtype nonirradiated mice. They proposed that there are empty HSC niches distinct from HSC-filled niches that allow HSC engraftment, because after transplantation the total number of cells enriched for HSC increased (80). However, the total number of GMP fraction did not increase and recipient GMP was replaced by donor-derived GMP. Therefore, this suggests that there are empty HSC niches that could not be filled by GMP cells (80).
All the aforementioned evidence indicates that the HSC niche is unique and distinct from the progenitor niche. The next step would be to identify the molecular signals and interactions that define this specific HSC niche. As has already been shown, the mobilization of HSCs from their niche as well as their homing to the niche can be regulated by antibodies against the adhesive antigens, for example, α4β1 (67, 97). Other antibodies against c-Kit and CD47 (18) are currently being tested in clinical trials as nonmyeloablative strategies of patient conditioning. However, these antigens are still not specific to the HSC niche. Therefore, identifying the HSC-specific mechanisms, which are present in the human HSC niche, will result in the development of new clinical strategies for the mobilization and transplantation of HSCs.
Normal and Leukemic Niche
The genetic mouse models revealed that the deletion of even one factor from the niche can result in hematopoietic collapse. Naturally, this raises questions of whether the disturbances in the niche can contribute to malignant transformation and drive leukemic development. One of the first evidences came from the work of Walkley et al. The widespread inactivation of Retinoblastoma (Rb) resulted in the loss of HSCs from BM and pronounced myeloproliferation syndrome (MPS) (99). However, the transplantation of Rb-deficient cells to the wild-type recipients did not recapitulate the MPS (99). The MPS was observed only when the Rb-deficient cells were transplanted to the Rb-deficient recipients, indicating that MPS requires the Rb deficiency in the niche to occur (99). However, the Rb-deficient niche was not able to transform the wild-type cells, so the transformation to MPS in this model is not entirely noncell autonomous (99). In subsequent work, Walkley showed that retinoic acid receptor gamma (RARγ−/−) deficiency in recipient mice can trigger MPS even when transplanted cells are wild-type (98). The authors showed that this was correlated with elevated tumor necrosis factor (TNF)α levels in RARγ−/−-deficient mice (98). Other studies revealed a similar role for the Mbi-1 gene involved in the Notch pathway (46). Transplantation of wild-type BM cells to Mbi-1-deficient animals caused MPS, but this could be reversed by secondary transplantation to wild-type recipients (46). The authors also showed that in this model the MPS-initiating cells were included in hematopoietic stem and progenitor fraction Lin−c-Kit+Sca-1+ (46).
These studies provided evidence for HSC-extrinsic factors that may play a causative role in MPS. However, the HSC-extrinsic role may depend directly on the local HSC niche or indirectly on systemic effects. Several studies tried to address this problem by using more specific genetic mouse models. Wang et al. demonstrated that disruption of the canonical Notch pathway by the deletion of Rbpj in endothelial cells by using the Tie-2-CreER mouse model was enough to trigger MPS (101). However, it should be stated that the Tie2 is also expressed in the HSCs (3); therefore, the MPS may be the result of Rbpj deletion in both the endothelial cells and hematopoietic lineage. Nevertheless, authors showed that defective Notch signaling affects the hematopoiesis by mechanisms that are dependent on nuclear factor kappa B (NF-κB) and miR-155 (101).
Although the existing literature suggests that osteoblasts are not in direct proximity to the HSC niche, genetic models evidenced that they can contribute to malignant transformation. A study by Kode et al. showed that constitutive expression of β-catenin in osteo-lineage (Col2.3-Cre mice model) can drive acute myeloid leukemia (AML) that has the typical mutation signature (47). This inducible AML was also dependent on the Notch pathway as Jagged-1 deficiency and γ-secretase inhibitors prevented AML development (47). Other studies demonstrated that deletion of the enzyme involved in miRNA processing, Dicer-1, in osteoblasts recapitulates myelodysplastic syndrome, a precursor to AML (70). Similar effects were observed on the deletion of the Sbds gene in osteoblasts (70). This correlates with clinical observations as mutations in the Sbds gene are associated with Shwachman-Bodian-Diamond syndrome, a congenital disorder marked by abnormal neutropenia and progression to marrow failure or AML (8).
All these studies support the hypothesis that the niche significantly contributes to leukemogenesis [further reviewed in Refs. (20, 77)]. However, it is known that malignant transformation is a multistep process and requires the accumulation of several mutations (31). Given that only stem cells self-renew, they are predisposed to accumulate mutations through time. Indeed, studies where single human HSCs were examined with probes for all of the mutations in that leukemia demonstrate that HSCs accumulate mutations in an established order—the first mutations are found among genes involved in epigenetic regulation, whereas the final mutations govern the self-renewal of cells at the progenitor stage, which finally results in leukemia development (21, 39) (Fig. 6). Importantly, the HSCs that have the initial leukemic mutations appear to expand in the BM at the expense of normal HSCs, providing evidence for stem cell competitions. The final proliferative mutations or epigenetic changes occur as the last mutations in the clone, and they are only apparent as the massive expansion of the leukemia stem cells (LSCs) that are one to four steps of differentiation from HSCs at the MPP (or ST-HSC) level. Therefore, it is of outstanding importance to understand how the pre-leukemic HSCs outcompete normal HSCs in their niches.

Several studies imply that residual disease AML LSCs are endosteal (34, 75), in contrast to recent studies indicating that HSC are perivascular (2, 16, 45). Thus, we propose that LT-HSCs may remain perivascular, whereas MPPs and ST-HSCs, and their LSC counterparts, may be endosteal. This hypothesis will need to be verified by imaging methods that can distinguish the different stem and progenitor subtypes. Similarly, whether the endosteal niches can also support LMPPs and perhaps CLPs (IL7R+ in mice) should be clarified with markers that are restricted to these cells. Understanding these mechanisms will be crucial for developing strategies to stop leukemia development at early stages. Recent studies also demonstrate that in later stages of disease, leukemic cells affect the niche that, in turn, loses its supportive function for non-transformed HSCs [reviewed in Ref. (77)]. Therefore, we can consider that restoring the proper niche function during leukemia progression may also help maintain non-transformed HSC activity and normal hematopoiesis.
Noteworthy, some tumors preferentially metastasize to bones and BM. This leads to the hypothesis that BM metastasizing tumor cells might use a similar mechanism that governs HSC or MPP homing and interaction with BM niches (82). Bone metastasis is a frequent and debilitating complication of cancer. An estimated 70% of patients with prostate and breast cancer and 15–30% of patients with lung, colon, stomach, bladder, uterus, rectum, thyroid, or kidney cancer develop skeletal metastasis (73). The colonization of bone by distant cancer cells depends on two main components: “the seed,” or a cancer cell that migrates and initiates distant tumor expansion and “the soil,” or the bone or BM environment that supports its invasion and growth (65). Although a great deal is understood about the metastatic cancer cell and the genetic and epigenetic changes that underlie its evolution from a primary cancer cell, little is known about the cells and factors that nurture it in the bone (41, 107). An attractive hypothesis is that the metastatic cancer cell hijacks the BM niche of the HSCs or that of the MPP/ST-HSC, and co-opts the factors usually produced for Hematopoietic stem and progenitor cell (HSPC) maintenance to support its own growth. The HSC niche is rich in factors such as CXCL12 and SCF whose cognate receptors are found on cancer cells and is located in a region of the BM that is protected from immune surveillance and ROS. Preliminary evidence for this hypothesis comes from studies that show that human metastatic prostate cancer (MPCa) cells when injected into mice can exploit the same homing mechanisms, such as the CXCR4/CXCL12 pathway, that the HSC uses to localize to its niche (81, 100). MPCa cells have been reported to outcompete HSCs (KLS CD150+CD48−CD41−) for adhesion molecules such as Annexin A2 (81, 100); however, these are on osteoblasts, and by our proposal, are more likely to be MPP or more lymphoid supporting niches. It remains unclear as to whether the MPCa cells specifically target the HSCs and their niche or rapid tumor or osteoblast expansion damages the HSC niche by mass effect. However, in unpublished work by our group, we have observed that MPCa readily migrate to heterotopic ossicles derived from CD105+Thy− cells and knockdown of Osterix and VEGF-C, key regulators of niche formation, abrogates cancer metastasis to the bone. It is important to note that Osterix is not just an osteoid lineage marker, but is a marker of the full set of the skeletal stem cell (SSC) that can give rise to both bone and stromal cells (15). Stronger evidence for the relationship between MPCa and the different marrow niches will require high-resolution microscopy coupled with clearing technologies to visualize the location of micro-metastasized MPCa cells with respect to the HSCs, MPPs, CMPs, and CLPs, as well as the SSC-derived stromal cells that comprise the perivenous versus endosteal niches.
ROS and Oxygenation of the Niche
Given that HSCs constitute the reservoir of blood regeneration potential throughout life, they have acquired throughout evolution several protective mechanisms that minimize the risk of cell damage and accumulation of mutation. Among these mechanisms, the regulation of ROS seems to be especially important. It is well accepted that excess ROS can damage biological molecules, including DNA, but ROS can also act as an important signaling molecule [reviewed in Ref. (71)]. The reduction of ROS below physiological levels impaired HSC differentiation (42), suggesting that ROS levels in the niche must be tightly regulated. It has been demonstrated that HSCs possess several cell-intrinsic mechanisms that maintain the balance of ROS levels [reviewed in Ref. (51)], such as ATM (37) and FOXO3a (56). In comparison, breast and breast cancer stem cells express higher levels of glutathione synthetase than their progeny cells, providing the glutathione sink for ROS at higher levels (24). However, beyond these cell-intrinsic mechanisms, several properties of the HSC niche are also crucial for the regulation of ROS levels.
First, it is believed that HSCs and other quiescent adult stem cells reside in low oxygen conditions (58, 85), and in this way, they reduce their oxidative stress. Indeed, indirect evidence such as the constant activity of the hypoxia-inducible factor 1 alpha (HIF-1α) pathway (88), dependence on glycolytic metabolism instead of aerobic respiration (89), and analysis with pimonidazol hypoxic marker (68) suggests that the HSC occupies a low oxygen niche. However, only recently, the direct oxygen concentration in different BM niches was measured. Interestingly, the highest oxygen tension was found close to the bone near endosteal regions within the reach of arterioles. It was also higher near BM vessels, whereas it quickly decreased with distance from the vessels, toward the center of the marrow (84). Thus, despite being well vascularized, BM is a relatively hypoxic tissue due to its high cellular density and oxygen consumption (84). Recent studies using α-catulin and Hoxb5-mCherry reporter mice revealed that most HSCs reside in the central marrow region and not in the endosteal region (2, 16), which implies that the HSC niche is hypoxic. However, within this hypoxic region, HSCs are in direct proximity to sinusoids, and, therefore, relative to other cells in the central marrow farther away from the sinusoids, the HSC niche may be slightly more oxygenated.
It has been proposed that the activation of HIF-1α and switch to glycolytic metabolism in HSCs is potentially not only a direct result of the low oxygen concentration in the HSC niche but also may be driven by other factors [reviewed in Refs. (38, 85)]. Nevertheless, a low oxygen environment does not necessarily reduce ROS production. Oxygen is the acceptor of electrons in the mitochondrial electron transport chain, and its deficit can lead to electron leak and induce ROS generation (40). In addition, the role of HIF-1α in HSCs is still a matter of debate (87). Previous work by Takubo et al. concluded that HIF-1α is necessary for HSC maintenance (88), whereas recent reports indicate that HIF-1α deficiency does not affect HSC function (96).
It should be pointed out that the previous studies on ROS levels were carried out with the less pure populations of HSPC, not HoxB5+ HSC. The time course of induced DNA strand breaks is slow, but HSCs accumulate them without repair if they are in the cell cycle G0 (6). Amazingly, when these HSCs from old (∼2 years old) mice are placed in culture media that bring them artificially into the cycle, they repair the DNA breaks (measured by both γH2AX foci and DNA migration in the comet assay) and nearly all cells survive, multiply, and differentiate (6). This is accompanied by G1 phase induction of all tested DNA repair pathways (6).
The ROS levels in HSCs might also be regulated more directly by the niche cells. It was shown that FGF-2 could expand the pool of long-term repopulating HSCs in BM without increasing their ROS levels (36). However, in mice that have mutated c-Kit receptor (Wv/Wv strain), FGF-2 expansion elevates ROS levels and reduces HSC long-term repopulating potential (36). This indicates that c-Kit on HSCs and its ligand SCF, which is produced by the niche, constitute essential HSC-niche interaction for maintaining low ROS levels in HSC. Again, c-Kit is expressed on not only HSCs but also most multipotent and oligopotent progenitors (10).
Other extrinsic mechanisms that were shown to be implicated in the regulation of HSC ROS levels are gap junctions mediated by CXN43 channels (12). It has been proposed that these gap junctions transfer ROS produced in HSCs to adjacent stromal cells, and, by this mechanism, protect HSCs from ROS-induced damage (90). However, the proposed mechanism is hard to explain, given the biochemical properties of ROS. It would be more likely that not ROS, but ROS scavengers are transported between the HSCs and stromal cells. Again, these studies must be redone with purer populations of HSCs.
ROS in the HSC microenvironment might also play a crucial role during the mobilization of stem cells from their niches to the peripheral circulation. The classical mobilization factor, G-CSF, increases the ROS levels in BM [reviewed in Ref. (51)]. This increase in ROS levels might be necessary for stem cell mobilization as when G-CSF is administrated along with ROS inhibitors the mobilization is reduced (91). It was proposed that high ROS levels reduce adhesion of stem and progenitor cells to stromal cells and, thus, facilitate their mobilization (83). Others also proposed that ROS promotes the mobilization by activation of proteolytic enzymes (53, 108), such as matrix metalloproteinases, which are crucial to release cells from BM to circulation (32). Altogether, these results indicate that ROS regulation by the extrinsic niche is necessary for steady-state function of HSCs, as well as for their mobilization in emergency hematopoiesis.
Conclusion and Future Directions
Recent studies employed new mouse transgenic models that have enabled the investigation of the niche in a prospective manner. These models combined with new HSC-reporter mice and imaging methods have consistently revealed the perivascular niche of HSC with its two main players: endothelial cells and MSCs mainly derived from skeletal stem cells. Although the perivenous vascular niche is well evidenced, there are still diverse views on the different roles of endosteal sites, arterioles, and marrow venous sinusoids in the regulation of HSCs. These almost certainly reflect the purity of cells tested. There is also limited consensus on the importance of particular subsets of MSCs that express LEPR, Nestin, and/or NG2 markers. One possible reason for these inconsistences is that we still underestimate the heterogeneity of the populations that compose the niche. Although the vascular bed can be divided into arteries, capillaries, sinusoids, and transition zone vessels (sinusoids likely represent the L-type capillaries, and transition zones possibly are related to H-type capillaries), we do not yet know whether the prevailing sinusoids contain distinct subfractions that may differentially contribute to the HSC niche. Therefore, the next direction would be to employ new single-cell transcriptome methods to explore the diversity of the niche and combine this knowledge with already analyzed single-cell transcriptomes of HSCs. Given the evidence for specificity of the HSC niche, the emerging challenge now is to find the mechanism and specific cells that make this HSC niche unique and the new single-cell methods combined with the quickly developing bioinformatics tools will make this possible.
Although several mechanisms have been proposed for how HSCs age, HSC aging is mainly believed to be a cell-intrinsic process. Our own current model proposes that diverse HSC subsets exist, each with their own intrinsic canonical gene expression repertoire, and it is the changes in niches that specify which subset is hosted more efficiently with age or inflammation or other environmental influences (5). Providing a better understanding of how the niches regulate HSCs is needed, and that requires understanding the phenotypes, functions, and developmental origins of all niche-related cells.
Finally, we have to recognize the differences between the mouse and human BM niche. Mouse models have given us a great understanding of the basic biology underlying the HSC-niche interactions. However, particular mechanisms may differ in humans, for example, long bones of humans are filled with yellow marrow or adipose tissue, a feature that is not observed to the same degree in mice (74). Therefore, to translate our understanding of the HSC niche to clinical applications, we need to continue the development of new humanized preclinical models. This is of outstanding importance, especially as we try to better understand the mechanism for niche competition between malignant and nonmalignant HSCs. The far-reaching aim will be to block the expansion of HSCs that have the preleukemic mutations but are still asymptomatic. Moreover, by understanding HSC-niche interactions, we want to develop strategies of noninvasive preconditioning with minimal side effects.
To conclude, the study of the HSC niche gives us the basic knowledge for how the highly dynamic process of blood regeneration is regulated. Gaining a deeper understanding about the mechanisms that are specific to the HSC niche and translation of these findings to the human niche may result in new clinical applications. These challenges are definitely tough, but worth tackling, as they might lead us to solutions for restoring an aged hematopoietic system, fighting against malignancies, and transplanting purified HSCs safely and effectively into patients.
Footnotes
Acknowledgments
This work was supported by the Polish Ministry of Science and Higher Education within Mobility Plus program granted to Krzysztof Szade.