
Abstract
Significance:
Obesity and type 2 diabetes mellitus are increasing globally. There is also increasing associated complications, such as non-alcoholic fatty liver disease (NAFLD) and vascular complications of diabetes. There is currently no licensed treatment for NAFLD and no recent treatments for diabetic complications. New approaches are required, particularly those addressing mechanism-based risk factors for health decline and disease progression.
Recent Advances:
Dicarbonyl stress is the abnormal accumulation of reactive dicarbonyl metabolites such as methylglyoxal (MG) leading to cell and tissue dysfunction. It is a potential driver of obesity, diabetes, and related complications that are unaddressed by current treatments. Increased formation of MG is linked to increased glyceroneogenesis and hyperglycemia in obesity and diabetes and also down-regulation of glyoxalase 1 (Glo1)—which provides the main enzymatic detoxification of MG. Glo1 functional genomics studies suggest that increasing Glo1 expression and activity alleviates dicarbonyl stress; slows development of obesity, related insulin resistance; and prevents development of diabetic nephropathy and other microvascular complications of diabetes. A new therapeutic approach constitutes small-molecule inducers of Glo1 expression—Glo1 inducers—exploiting a regulatory antioxidant response element in the GLO1 gene. A prototype Glo1 inducer, trans-resveratrol (tRES)-hesperetin (HESP) combination, in corrected insulin resistance, improved glycemic control and vascular inflammation in healthy overweight and obese subjects in clinical trial.
Critical Issues:
tRES and HESP synergize pharmacologically, and HESP likely overcomes the low bioavailability of tRES by inhibition of intestinal glucuronosyltransferases.
Future Directions:
Glo1 inducers may now be evaluated in Phase 2 clinical trials for treatment of NAFLD and vascular complications of diabetes.
Health Impact of Obesity and Diabetes and the Related Hepatic and Vascular Complications: Time for Improved, Safe, and Effective Treatment
T
In 2010, being overweight and obese was estimated to cause 3.4 million deaths worldwide. Most deaths are due to cardiovascular disease (CVD)—mostly manifested as coronary heart disease (CHD) and stroke. The increased mortality risk of CHD and stroke is attributed to hypertension and dyslipidemia (36% and 59%) and hyperglycemia (14% and 25%), with residual risk (50% and 16%) unexplained (100). A further complication in overweight and obese subjects is non-alcoholic fatty liver disease (NAFLD) leading to hepatic steatosis, cirrhosis, liver failure, and cancer (81). NAFLD affects 20–40% of the population in Westernized countries and will likely increase direct and indirect medical costs by 25% in the next 5 years. A common metabolic complication in overweight and obese subjects is insulin resistance. This drives the development of T2DM and also NAFLD. Common complications of diabetes are: increased risk of CVD—CHD, peripheral artery disease, and stroke; and microvascular complications—nephropathy, retinopathy, and neuropathy. Diabetes increases mortality risk of CVD by two- to threefold, increasing further to three- to sixfold in advanced diabetic nephropathy (63, 89). There is an urgent requirement for an improved understanding of the risk factors of obesity, insulin resistance, and NAFLD to guide interventions to decrease incidence and health impact (199). There is also an urgent need to improve therapy for diabetic nephropathy and other diabetic complications. It is 15 years since clinical trials produced new drugs licensed for diabetic nephropathy, and the current survival of patients with diabetic nephropathy is similar to that of the average survival of patients with cancer (40).
The Glyoxalase System and Dicarbonyl Stress: Introduction
The glyoxalase system is a cytoplasmic enzyme system present in all mammalian cells. It consists of two enzymes that catalyze successive enzymatic reactions, glyoxalase 1 (Glo1) and glyoxalase 2 (Glo2), and a catalytic amount of reduced glutathione (GSH). Glo1 catalyzes the isomerization of the hemithioacetal formed non-enzymatically from reactive dicarbonyl metabolite methylglyoxal (MG) and GSH to S-

The primary function of the glyoxalase system is to metabolize MG and/or other reactive acyclic α-oxoaldehyde metabolites, thereby suppressing them to low steady-state concentrations. MG is the most reactive and of the highest endogenous flux and so is often the primary concern (145). Typical concentrations of MG are 100–200 nM in plasma and 1–4 μM in tissue (147). MG is formed by trace level, spontaneous degradation of triosephosphates, and other minor pathways [reviewed in Rabbani et al. (152)]. MG is a potent glycating agent, reacting non-oxidatively mainly with arginine residues of proteins to form the hydroimidazolone adduct, MG-H1, and reacting with DNA to form mainly imidazopurinone adduct, MGdG (6, 181). Protein and DNA contents are 0.1–0.8 mmol/mol arg and ∼9 adducts per 106 nucleotides, respectively (Fig. 2). Abnormally high concentrations of MG, and/or other reactive α-oxoaldehyde metabolites, constitute a dysfunctional metabolic state called “dicarbonyl stress” where dicarbonyl glycation is increased (148). In dicarbonyl stress, increased formation of MG and/or decreased activity of Glo1 produces increased cellular and extracellular concentrations of MG driving cell and tissue dysfunction. The terminal product of the glyoxalase system metabolism of MG is
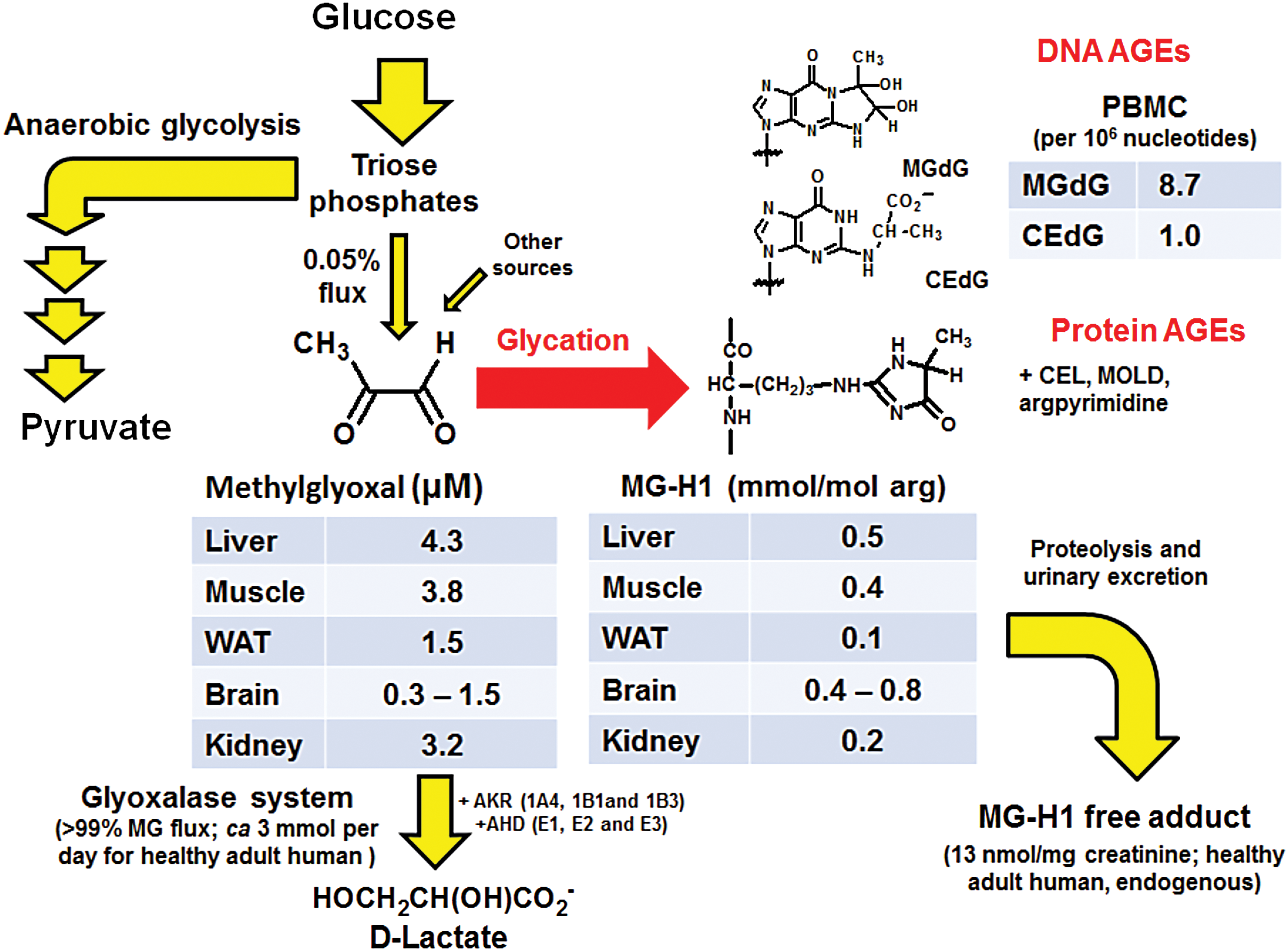
The protection provided by the glyoxalase system against dicarbonyl stress is important both developmentally and functionally. Knockout of the GLO1 gene is embryonically lethal unless induction of compensatory metabolism by aldoketo reductases occurs (11, 116). Silencing of Glo1 expression decreases lifespan and increases susceptibility to glucose intolerance and oxidative stress in the nematode Caenorhabditis elegans model of aging and glucose toxicity (115, 160). In endothelial cells, silencing of Glo1 changed the expression of more than 485 genes linked to the development of CHD (106). The related increase of MG-H1 residue content of cell protein changed the levels of MG-modified and -unmodified cellular and extracellular matrix (ECM) proteins (167). MG-H1 is a quantitatively the major protein-derived advanced glycation endproduct (AGE). MG glycation of proteins is frequently directed to functional sites of proteins. There is a loss of positive charge of modified arginine residues, resulting in protein inactivation and dysfunction (139, 145). The MG nucleotide adduct, MGdG, is a quantitatively major nucleotide AGE. MG-derived nucleotide adducts are mutagenic and may be linked to malignant transformation. Supporting this, Glo1 was a tumor suppressor protein in a mouse model of hepatocellular carcinogenesis (181, 201).
The historical development of the glyoxalase research, molecular characteristics of glyoxalase enzymes, molecular physiology of glyoxalase metabolites, and techniques available to study the glyoxalase system—the glyoxalase researchers “toolkit”—have been reviewed elsewhere recently (146, 150 –152, 193).
In situ activity of Glo1 is directly proportional to the concentration of GSH; MG metabolism by Glo1 is impaired by GSH decline in oxidative stress (1). Mitochondrial proteins are targets of MG glycation, which drives increased formation of reactive oxygen species (ROS) (115). So dicarbonyl stress may be both a consequence and a cause of oxidative stress. Dicarbonyl stress may be also initiated and sustained without oxidative stress: through increased formation of MG from increased triosephosphate flux driven by increased glucose metabolism in hyperglycemia and glyceroneogenesis in triglyceride synthesis in obesity; and downregulation of Glo1 linked to hypoxia (161), inflammatory signaling conflicting with Nrf2 (192), and hyperglycemia-induced increased proteolysis (196). It is not surprising, therefore, that dicarbonyl stress is beginning to feature strongly as a driver of pathogenesis in obesity and diabetes—particularly vascular complications (62, 106, 110, 183). A new type of experimental therapeutic agent has emerged to counter dicarbonyl stress: an inducer of Glo1 expression or “Glo1 inducer” (194). We review the background in sections Dicarbonyl Stress and the Glyoxalase System in Obesity, Diabetes and Diabetic Vascular Complications, and Development of Therapeutic Agents to Counter Dicarbonyl Stress.
Beneficial Effects of MG Formation for Physiological Function
There is no known direct benefit of MG formation for physiological function in mammals. The major source of MG formation is spontaneous formation from triosephosphates, dihydroxyacetonephosphate (DHAP), and glyceraldehyde-3-phosphate (GA3P). This occurs as a consequence of the use of high-energy triosephosphate intermediates in glycolytic metabolism (135). MG modification of alpha-crystallin improved chaperone function and protein refolding activity but this effect was limited to higher extents of protein modification by MG than found physiologically (59). However, accumulation of MG is implicated in host defense mechanisms—particularly anti-microbial activity of phagocytic cells. The cellular concentration of GSH falls >90% during the activation of the phagocytic respiratory burst as part of the biocidal activity against pathogenic micro-organisms (176). In situ activity of Glo1 will decrease proportionately, and the local concentration of MG will, therefore, increase (1). The pentosephosphate pathway is also activated to produce increased flux of NADPH to sustain NADPH oxidase activity during the respiratory burst (200) and becomes a net producer of triosephosphates and, hence, also indirectly increases MG formation (132). MG is expected to accumulate within activated phagocytic cells and be released into the phagosome and therein modify proteins of engulfed micro-organisms. This suggests that MG has a role in the anti-microbial activity of phagocytes—such as neutrophils, monocytes, and macrophages. Evidence for this has been reported recently (204). It may also play a role in tissue damage in inflammation. Increased synovial and plasma levels of MG-derived AGEs were characteristics of advanced stages of arthritis (7).
In microbial metabolism, there is enzymatic formation of MG from DHAP by MG synthase (117). This provides a bypass for the lower half of the Embden-Meyerhof pathway. Embden-Meyerhof glycolysis converts DHAP to pyruvate with formation of two equivalents of adenosine triphosphate (ATP) and one equivalent of NADH; whereas the MG synthase/glyoxalase/
Dicarbonyl Stress and the Glyoxalase System in Obesity
The plasma concentration of MG in lean, healthy human subjects was 132 ± 63 nM (147). This was increased to 181 ± 61 nM in overweight subjects and increased further to 245 ± 123 nM in obese subjects (110). The increases were sustained on an isocaloric diet. Plasma
A genetic association of GLO1 with obesity was found in genetic linkage analysis and anthropometric measurements (188). In mice, meta-analysis of 34 mouse cross-breeding experiments linked GLO1 to body weight (189). Glo1 expression and activity is decreased in WAT in HFD-fed mice (102). Glo1 protein was decreased 80% in the liver of the mouse overeating model of obesity, leptin mutant (ob/ob) mice (159). Overexpression of Glo1 in HFD-fed mice suppressed gain in body weight and adiposity with similar food consumption as wild-type HFD-fed controls (102). Relatedly, mice with a preference for high energy-rich diets without marked weight gain and health impairment have a relatively high expression of Glo1 (143). This suggests that downregulation of Glo1 as well as increased formation of MG contribute to dicarbonyl stress in obesity and overexpression of Glo1 can suppress health impairment of the obese phenotype.
The origin of increased MG concentration in obesity is likely not an effect of increased food consumption directly as plasma MG was increased in obese subjects on an isocaloric diet (110). There is a relatively low intake of MG in food and beverages compared with endogenous formation (152). Dietary MG also has limited bioavailability and is metabolized and/or reacts with protein in the intestinal lumen (42). Usually, increased glucose metabolism would be implicated in increased formation of MG but insulin resistance in obesity suggests that the usually dominant source of MG formation from increased flux through anaerobic glycolysis may not be the culprit. Rather, increased triosephosphate flux in glyceroneogenesis associated with adipocyte expansion in obesity supporting increased fatty acid esterification for triglyceride deposition is the likely source (53). Most triglyceride synthesis involves glyceroneogenesis via triosephosphate intermediates (124) and glyceroneogenesis is not limited because it uses pyruvate as the carbon source with unrestricted entry and metabolism into major cell types experiencing insulin resistance—such as adipocytes, hepatocytes, and skeletal muscle cells (Fig. 3).
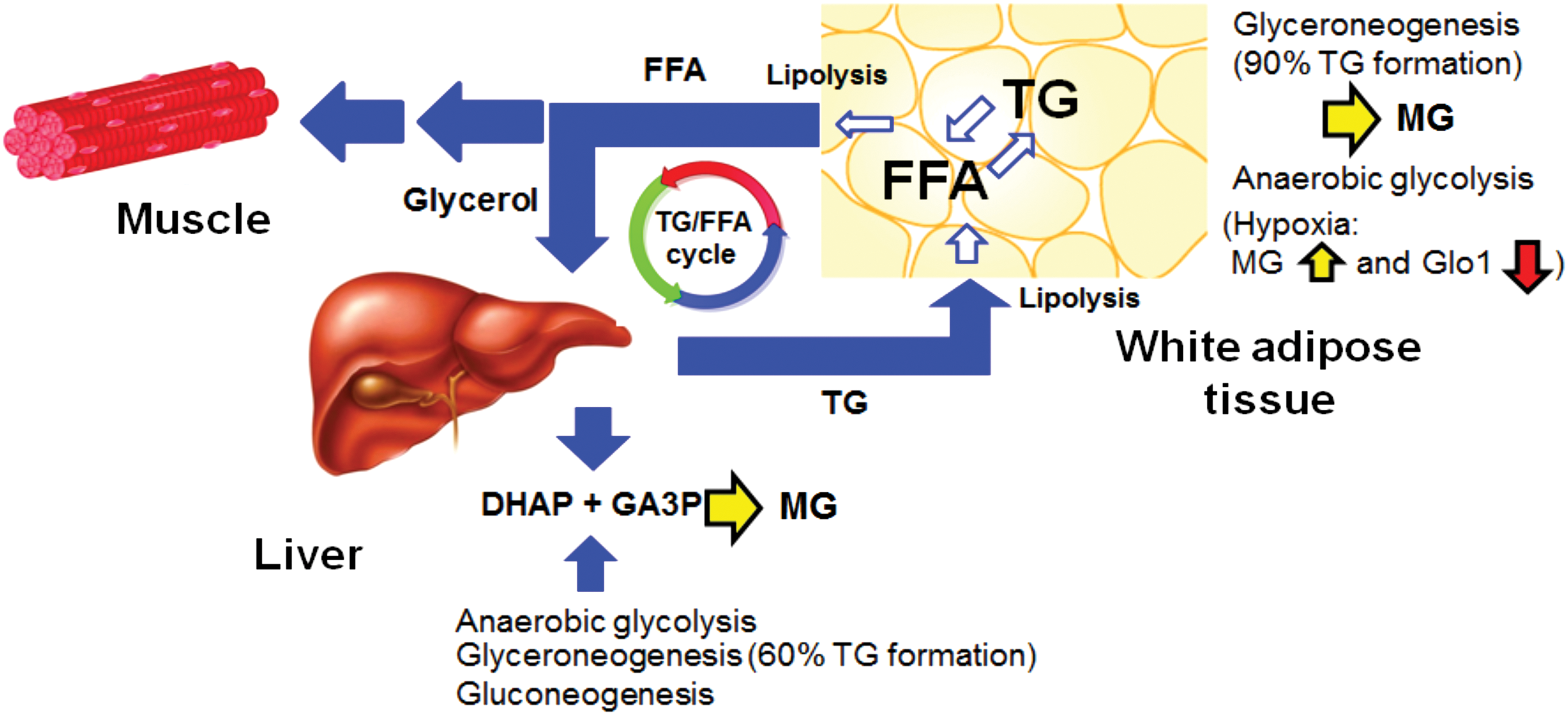
The drivers for downregulation of Glo1 in WAT in obesity are unknown. Glo1 may undergo post-translational modifications: C139 may form a mixed disulfide with GSH, inhibiting Glo1 activity in vitro. Glo1 may be S-nitrosylated on C139 and is a substrate for calcium, calmodulin-dependent protein kinase II, with phosphorylation at T107. There may also be acetylation at K148 (101) and likely de-acetylation by cytosolic sirtuin-2 (153). Glo1 downregulation in obesity may be driven through hypoxia signaling by hypoxia-inducible factor-1α (HIF1α) that downregulates Glo1 expression (161, 203). During adipose tissue expansion, interstitial oxygen tension decreases and adipocytes have high HIF1α protein content driving increased adipose tissue vascularization. Visceral adipose tissue from human obese subjects had high levels of HIF1α. In mice, HIF1α activation in visceral white adipocytes drives glucose intolerance, insulin resistance, and cardiomyopathy (87). Adipocyte-specific deletion of HIF1α decreased HFD-induced adipose tissue inflammation and insulin resistance (92). There are also low levels of SIRT2 in adipose tissue in obesity. Decreased SIRT2 leaves PPARγ coactivator 1α (PGC1α) acetylated, driving decreased expression of β-oxidation and mitochondrial genes and leading to decreased fatty acid catabolism and thermogenesis.
The biochemical consequences of dicarbonyl stress in WAT are unknown but they may contribute to insulin resistance, decreased adipokine expression, and inflammation (102). Recent research has also suggested that β-Klotho may be a target, leading to its downregulation and resistance to fibroblast growth factor-21 (FGF21), which is a major contributor to insulin resistance (58, 194, 205) (see Benefit of a Synergistic Combination of Nrf2 Activators section).
Several studies have attempted to model dicarbonyl stress in obesity by administration of exogenous MG. Difficulties performing such studies are lack of commercial availability of suitable high-purity MG and an appropriate dose to administer. Commercial 40% MG contains 9–17 mol % formaldehyde and many other compounds that potentially interfere in studies of dicarbonyl stress (136). The flux of endogenous formation of MG is ∼3 to 6 mg/kg (∼0.05% glycolytic rate, which is found to be similar in many cell types) (145). Experimental studies have often used 10- to 20-fold higher than this—which likely approaches and exceeds the upper limit of more severe dicarbonyl stress found in poorly controlled clinical diabetes and end-stage renal disease (112, 144). Infusion of MG (60 mg/kg/day) into healthy rats induced impaired glucose tolerance, decreased glucose transporter GLUT-4, phosphoinositide-3-kinase activity, and insulin-stimulated glucose uptake in adipose tissue (44). Exogenous MG (50–75 mg/kg, daily, intraperitoneal) induced insulin resistance in mice (122), inhibited insulin-stimulated phosphorylation of protein kinase B and extracellular-regulated kinase, contributing to insulin resistance in muscle cells (155). It also inhibited insulin-induced insulin receptor substrate tyrosine phosphorylation and phosphatidylinositol 3-kinase/protein kinase B pathway activation in pancreatic β-cells (51), increased free fatty acids, hypoadiponectinemia, hypoxia, and macrophage recruitment of adipose tissue (111). These MG administration models are likely exploring features of MG intoxication but some of the features produced may be similar to those developing in obesity—although less severe.
Obesity has been implicated as a driver for chronic kidney disease (CKD) in the general population. This is believed to occur through vasoconstriction of efferent renal blood vessels and consequent glomerular hyperfiltration driving renal podocyte, mesangial cell, and tubular epithelial cell dysfunction. A link to increased leptin and decreased adiponectin has been suggested (168). Dicarbonyl stress is implicated in renal cell dysfunction in the development of diabetic nephropathy [as reviewed in Rabbani et al. (152)], and may also contribute to the development of obesity-linked CKD in the non-diabetic state.
Dicarbonyl stress may be a contributory mediator of obesity and insulin resistance and thereby a risk factor for the development of T2DM and complications of obesity such as NAFLD, CHD, hepatocellular cancer, and CKD (102, 106, 201).
Diabetes and Diabetic Vascular Complications
The initial major clinical investigation of dicarbonyl stress in diabetes was a study of whole-blood MG concentrations and red blood cell glyoxalase-linked variables in patients with T1DM, patients with T2DM, and healthy controls (112). Blood samples were collected under casual conditions—without specific control for fasting or postprandial states. Whole-blood MG concentration was increased five- to sixfold in patients with T1DM, and there was a three- to fourfold increase of whole-blood MG in patients with T2DM (Fig. 4). These were disproportionately high increases compared with increases in blood glucose and
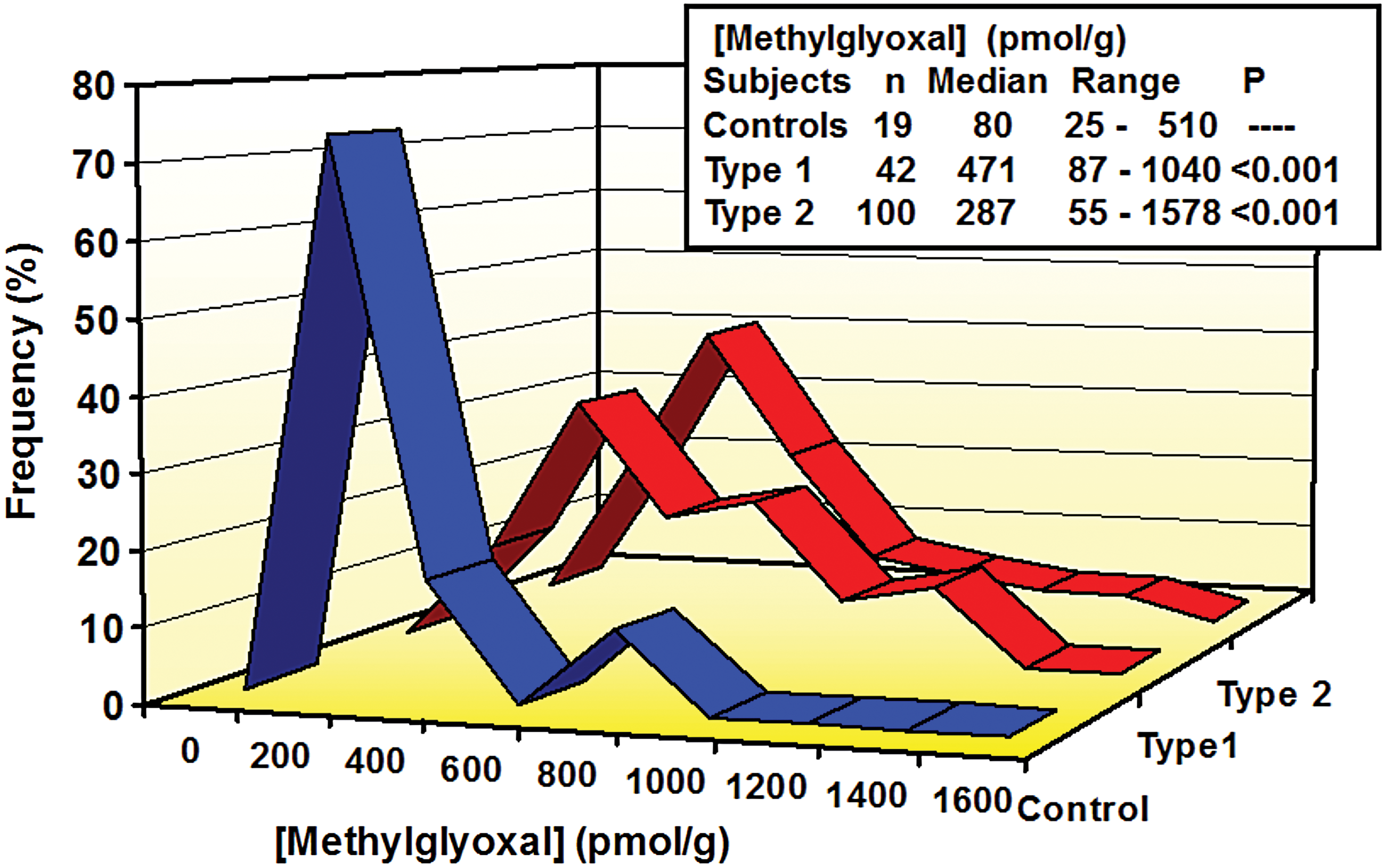
Increased plasma MG and
Studies of rodent models of diabetes have found evidence of increased MG in plasma and tissues suffering vascular complications—kidney and peripheral nerve (13, 23, 29, 30, 60). Evidence of increased MG in the diabetic retina is difficult to obtain as MG leaks out of retinal tissue during isolation. There was increased MG-H1 and other MG-derived glycation adducts in protein of plasma, kidney, retina, and peripheral nerve in experimental diabetes (13, 80). The functional significance of dicarbonyl stress and increased MG-driven protein glycation in these tissues was indicated by prevention of diabetic nephropathy, retinopathy, and neuropathy by overexpression of Glo1 (22, 23, 30) and by therapeutic intervention to decrease MG formation (13, 70). Also, a mouse model of Glo1 deficiency developed a diabetic nephropathy-like phenotype in normoglycemia and accelerated diabetic nephropathy when diabetes was induced. Overexpression of Glo1 limited to vascular endothelial and renal proximal tubular cells also prevented the development of diabetic nephropathy (62). Dosing of atherosclerosis-susceptible apolipoprotein E-deficient mice with the cell-permeable Glo1 inhibitor, S-p-bromobenzylglutathione cyclopentyl diester (177), increased endothelial inflammation and atherogenesis similar to that observed in diabetes (183). These studies suggest that dicarbonyl stress is a driver for diabetic microvascular and macrovascular complications.
Dicarbonyl stress is exacerbated in diabetes by downregulation of Glo1 in the kidney, retina, and peripheral nerve, and likely also vascular endothelial cells (23, 113, 196). This may reflect decreased Glo1 expression in response to inflammatory signaling induced by the receptor for advanced glycation endproducts (RAGEs) and other mechanisms (154), NF-κB conflicting with Nrf2 that regulates basal and inducible expression of Glo1 (97, 192), and also hyperglycemia-induced increased proteolysis of Glo1 (196).
Dicarbonyl stress may promote the development of vascular complications through protein glycation linked to: ECM protein modification leading to cell detachment and dysfunction (45, 137) and changes in ECM synthesis and processing (167), mitochondrial protein modification driving increased ROS formation and oxidative stress (157), modification of p300 and p65 of HIF1α and NF-κB signaling pathways driving abnormal responses to ischemia and persistent vascular inflammation (172, 196), modification of the 20S proteasome and slowing of cellular proteolysis (138), modification of lipoproteins driving dyslipidemia and atherosclerosis (64, 140), β-Klotho depletion driving inflammation, insulin resistance, and nephropathy (205), and other processes.
Development of Therapeutic Agents to Counter Dicarbonyl Stress
Strategies to prevent dicarbonyl stress for therapeutic application are: (i) compounds that prevent dicarbonyl formation—such as high-dose thiamine therapy for MG-linked dicarbonyl stress in diabetes; (ii) compounds that react rapidly with dicarbonyls to outcompete with endogenous proteins in situ—dicarbonyl scavengers; and (iii) compounds that correct Glo1 deficiency in obesity and diabetes by inducing its expression, “Glo1 inducers.”
Prevention of MG formation by high-dose thiamine therapy
The major source of formation of MG in physiological systems is degradation of triosephosphates (135). Interventions to prevent the abnormal accumulation of triosephosphates in diabetes are expected to decrease MG formation. A strategy explored to achieve this was to increase activity of transketolase, thereby stimulating the reductive pentosephosphate pathway that operates with net consumption of GA3P and fructose-6-phosphate with conversion to ribose-5-phosphate. Initial studies in cultured cells showed that this intervention was achievable, although at high, supraphysiological concentrations (179). This was later translated to the pre-clinical model of streptozotocin (STZ)-induced diabetic rats where 7 and 70 mg/kg thiamine decreased plasma MG, tissue, and protein glycation by MG in plasma and tissues at sites of microvascular complications development (13, 14, 80). Thiamine therapy reversed increases of plasma and urinary
Dicarbonyl Scavengers
Scavengers of dicarbonyl compounds may potentially decrease plasma and tissue concentrations of all α-oxoaldehyde metabolites—although they tend to be more effective against reactive acyclic dicarbonyls such as glyoxal and MG and less effective against α-oxoaldehydes of lower reactivity with major cyclic hemiacetal solution species, such as 3-deoxyglucosone (182). A disadvantage is that removal of dicarbonyls is stoichiometric: cf. scavenging of ROS by α-tocopherol, ascorbic acid, and GSH, which has reductive regeneration and cycling of the antioxidant in cells and body fluids under normal physiological function (39, 94, 96). Several approaches have been explored (Fig. 5).

Aminoguanidine (Pimagedine)
The initial experimental therapeutic developed for treatment of vascular complications of diabetes that has dicarbonyl scavenging activity is aminoguanidine (Pimagedine). Aminoguanidine was initially believed to react with fructosamine residues and prevent formation of AGEs (31), but lack of effect on fructosamine levels in glycated proteins in vivo suggested that other mechanisms of intervening in glycation processes may be involved (43). Aminoguanidine rather reacts with MG and other dicarbonyls to form 3-amino-1,2,4-triazine derivatives (3, 182). It was ∼100-fold more effective than arginine in preventing glycation of plasma protein in an ex vivo system mimicking physiological glycation—human plasma without significant dilution at 37°C with 1 μM [14C]MG (98). The reaction kinetics of MG with aminoguanidine are complex because there are two parallel reaction pathways: (i) reaction of aminoguanidine with the unhydrated form of MG forming 5-methyl-3-amino-1,2,4-triazine, and (ii) reaction of aminoguanidine with the MG monohydrate forming 6-methyl-3-amino-1,2,4-triazine (182). Under physiological conditions of <10 μM MG and peak plasma aminoguanidine of ∼50 to 60 μM, the major reaction is via the former reaction pathway (182). In healthy human subjects, an oral dose of 300 mg per day aminoguanidine achieved a peak plasma concentration of ∼50 μM with a half-life of ∼1.4 h (125); and in patients with end-stage renal disease receiving hemodialysis renal replacement therapy, the peak plasma concentration was 61 μM with a half-life of 37.9 h between dialysis and ∼1.5 h while on hemodialysis (52). Plasma concentrations of up to 400 μM have been achieved experimentally in rats after intraperitoneal injection of 100 mg/kg aminoguanidine (43). The estimated rate of formation of MG-derived glycation adducts of plasma protein in human plasma in situ is 0.58 μM/day (147, 175), and the estimated rate of reaction of 50 μM aminoguanidine with physiological concentrations of MG (100 nM) is 0.86 μM/day (182). So, at its peak plasma concentration, aminoguanidine is an effective MG scavenger. In contrast, inside cells, aminoguanidine is predicted to compete very ineffectively with Glo1 for MG scavenging. Even at high experimental doses up to ∼400 mg/kg, aminoguanidine achieving tissue concentrations of 200 μM–1 mM aminoguanidine, in situ Glo1 activity is predicted to scavenge MG >1000-fold faster than aminoguanidine (125, 174).
Problems associated with aminoguanidine development in clinical trial were adverse effects (gastrointestinal disturbance, abnormalities in liver function tests, flu-like symptoms, and a rare vasculitis) (54) and failure to meet the clinical endpoint in evaluation for treatment of diabetic nephropathy (25). Clinical therapeutic development was questioned further when renal tumors were found in aminoguanidine-treated diabetic rats (129). The rapid clearance of aminoguanidine impaired its clinical efficacy and, with toxicity and adverse effects, further clinical development is unlikely.
Dicarbonyl scavenging likely contributed to the experimental prevention of vascular complications of diabetes by aminoguanidine [reviewed in Thornalley (174)] and obesity (57, 162). At pharmacologically relevant concentrations in vivo, aminoguanidine is also an inhibitor of inducible nitric oxide synthase, Ki = 31 μM; although it is probably not an inhibitor of neuronal nitric oxide synthase, Ki = 170 μM, and endothelial nitric oxide synthase, Ki = 330 μM in vivo (9). At very high supra-physiological concentrations of aminoguanidine, there are also significant reactions with pyruvate and glucose for which effective median inhibitory concentration IC50 values were estimated to be 7 and 59 mM, respectively (174). In vitro studies of cell systems using aminoguanidine concentrations of >50–100 μM likely compromise physiological relevance for dicarbonyl scavenging effects.
Metformin
Metformin is a therapeutic agent that is widely used for the treatment of patients with T2DM. It has long been considered to work by decreasing hepatic glucose production, improving glucose uptake and utilization (65). Recent studies suggest that decreased hepatic glucose production may occur through activation of the duodenal AMP-activated protein kinase (AMPK)-dependent pathway (46). It has also found to be of benefit but is not in widespread use for treatment of prediabetes in obesity (130). It was associated with decreased plasma MG concentration in patients with T2DM (20), and also decreased plasma protein glycation by MG—including MG glycation of low-density lipoprotein (149). It reacts with MG to form mainly hydroimidazolone and also triazepinone adducts (158). These adducts are detected in vivo but they are quantitatively minor compared with the flux of MG formation (18, 85). The peak plasma concentration of metformin is 23 μM at the 1.5 g dose but may be ∼4 mM in the jejunum after ingestion (66). The kinetics of the reaction of MG with metformin are relatively slow (15). It is likely, therefore, that metformin decreases MG formation exposure by improving glycemic control in both diabetes and prediabetes and also has a weak MG scavenging activity.
Phenacylthiazolium bromide
Phenacylthiazolium bromide (PTB) was developed as a prototype therapeutic agent for the cleavage of putative α,β-carbonyl AGE protein crosslinks (187). Subsequent studies revealed instability of PTB and misinterpretation of some of the effects observed (180). PTB is an efficient scavenger of MG; we found that it was approximately sixfold more effective than aminoguanidine under physiological conditions, corroborated by others (84, 182). PTB is, however, unstable under physiological conditions and degrades by hydrolysis with a half-life of 44 min (180). The pharmacokinetics of PTB have not been published but those of a similar compound, TRC4186, found that the peak plasma concentration was ∼3 μM at a dose of 2500 mg with a half-life of 2–3 h (32). The short half-life and instability of this type of compound impairs the MG scavenging activity, although this may have contributed to the beneficial effects found (38).
Arginine-rich peptides
Arginine and arginine-rich peptides are potential scavengers of MG and other dicarbonyls. However, they have to achieve and maintain high steady-state concentrations in vivo to outcompete with endogenous cellular and extracellular arginine residue concentration: ∼20 and 80 mM, respectively (128, 134). Arginine itself has poor scavenger activity with an IC50 of 23 mM for plasma protein glycation by MG in the ex vivo model (99). Clinical pharmacokinetics of arginine indicate that oral high-dose arginine achieves a peak plasma concentration of ∼300 μM and is an ineffective dicarbonyl scavenger (170). Arginine residues may be made more reactive for scavenging of MG by decrease of the side chain pKa with a proximate positivity charged amino acid residue. Improved MG scavenging has been found with arg-arg and arg-lys residue containing peptides and with a large 40-amino-acid-long peptide (GERP10) (23, 28).
Histidine glyoxalase mimetics
In an attempt to produce catalytic scavengers of MG, we explored the possibility of histidine-containing peptides, building on the prior observation that imidazole has a Glo1 mimetic activity by a reaction with MG and isomerization to N-lactoylimidazole (69). We hypothesized that the imidazole moiety of histidine and histidyl peptides may react with MG to form N-lactoylimidazole derivatives and this would be hydrolyzed by endogenous esterase activity in plasma and tissues. The esterase site of albumin, for example, has N-acylimidazole hydrolase activity (127). Histidine and β-alanylhistidine (carnosine) had Glo1 mimetic activity, but the kinetics of MG scavenging is low such that at physiological concentrations of histidine in plasma (270 μM), carnosine in skeletal muscle (10 mM), and typical Glo1 and GSH content of mammalian cells, the reaction of MG is 260,000- and 20,000-fold faster with Glo1 than with histidine and carnosine in situ (16).
From the observations and kinetic considerations cited earlier, we conclude that it is challenging to design scavengers of MG that compete effectively with Glo1. Therefore, optimum therapeutic intervention is likely with Glo1 inducers—particularly reversing downregulation of Glo1 expression in obesity and diabetes.
Glo1 Inducers
When surveying the regulatory elements of the mammalian GLO1 gene to exploit for induction of increased Glo1 expression, the functional ARE was a prime choice. Basal and inducible expression of Glo1 is thereby regulated by Nrf2 (192). A Glo1 inducer directed to Nrf2 would also likely have beneficial off-target effects on other ARE-linked genes. As the regulatory mechanism of Nrf2 remains unclear, we developed and applied a functional screen for Glo1 inducer activity by using a luciferase reporter linked to the functional GLO1-ARE motif. Small-molecule activators of Nrf2 were required for screening. We sought to develop Glo1 inducer therapeutic agents for treatment of obesity and diabetes and their complications. Regulatory agencies require prospective novel drugs to have very low frequency of adverse effects for these therapeutics applications and so, we restricted our Glo1 inducer search to dietary bioactive compounds that often have known high tolerability and low risk of adverse effects. Dietary bioactive compound selection criteria were: ability to activate Nrf2 at concentrations achieved or likely achievable at tolerable doses clinically and/or decrease glycation and/or toxicity by MG or similar compounds. GLO1-ARE and functionally inactive mutated GLO1-ARE luciferase stable transfectant cell lines were incubated with and without bioactive compounds (0.625–20.0 μM) for 6 h. Early in our studies, we found that 10 μM trans-resveratrol (tRES) gave a strong Glo1 inducer response and so, we normalized Glo1 inducer activity in the screening assay to this as the positive control (192). Nrf2-dependent Glo1 transcriptional response was verified by siRNA silencing of Nrf2. Screening hit criteria were: increased transcriptional response at ≤5 μM without significant cytotoxicity to human cells in primary culture. Human aortal endothelial cells (HAECs) and BJ fibroblasts were used.
For the Glo1 inducer activity screening studies in vitro, the bioactive compound giving the lowest median effective concentration EC50 for GLO1-ARE transcriptional activity was hesperetin (HESP) for which EC50 = 0.59 μM and Emax 24.4%. The bioactive compound giving the highest Emax was tRES for EC50 = 2.52 μM and Emax 100% (194). The Glo1 inducer response was validated by assessment of Glo1 messenger RNA (mRNA) and Glo1 protein in human cell lines and primary cultures. To enhance the pharmacological efficacy, we studied the synergism of tRES and HESP together. A study of the GLO1-ARE transcriptional response of 5 μM HESP with 0.625–10 μM tRES showed that HESP combined synergistically with tRES, decreasing the EC50 of tRES to 1.46 μM while maintaining the Emax, lifting the Glo1 induction response to high levels with a clinically achievable concentration range for both compounds. The predicted increase of GLO1-ARE transcriptional response of 0.1–1.0 tRES in the presence of 5 μM HESP was 3- to 79-fold, including up to 79% increased GLO1-ARE transcriptional response over additive effects. This suggested that improved pharmacological efficacy and decreased risk of toxicity may be achieved in clinical translation from use of a tRES-HESP co-formulation through pharmacological additive and synergistic effects (194).
Other compounds with Glo1 inducer activity are: allyl isothiocyanate (AITC) and sulforaphane (SFN), butylated hydroxyanisole (BHA) and n-3 polyunsaturated fatty acids (PUFA) (Table 1). SFN and AITC have a relatively narrow therapeutic window, and BHA and PUFA have relatively low potency.
Evidence excludes studies where Glo1 is only increased in disease state or concurrent with treatment with supraphysiological concentrations of exogenous MG.
AITC, allyl isothiocyanate; BHA, butylated hydroxyanisole; Glo1, glyoxalase 1; HAEC, human aortal endothelial cell; HESP, hesperetin; MG, methylglyoxal; mRNA, messenger RNA; PBMC, peripheral blood mononuclear cell; PUFA, polyunsaturated fatty acids; SFN, sulforaphane; tRES, trans-resveratrol.
Clinical Evaluation of Glo1 Inducer, tRES-HESP
The Glo1 inducer, tRES-HESP, was evaluated in a recent clinical study of healthy overweight and obese subjects. The study was a randomized, double-blind, placebo-controlled crossover study in 29 subjects. Dosing was by oral capsule, once daily, containing 90 mg tRES-120 mg HESP or placebo. Dosing periods were 8 weeks with 6 weeks washout between in the crossover period. There was high compliance to tRES-HESP treatment and urinary excretion of tRES and HESP metabolites was increased >2000-fold and >100-fold, respectively, compared with placebo. Food consumption was similar throughout the study. Clinical safety indicators were normal at study entry and remained unchanged throughout the placebo and tRES-HESP treatment periods.
tRES-HESP treatment produced a 22% increase in Glo1 activity of peripheral blood mononuclear cells (PBMCs) of all subjects, compared with placebo in all subjects. The increase was 27% in highly overweight subjects, defined as subjects with BMI >27.5 kg/m2, and 30% in obese subjects (with BMI ≥30 kg/m2). Most changes were found in highly overweight subjects, and outcomes are presented for this subgroup. Concomitant with increased Glo1 activity, there was a 37% decrease in plasma MG post-supplementation with tRES-HESP but not with placebo. The whole-body flux of MG glycation of proteins was assessed by measuring the urinary excretion of MG-H1 free adduct, corrected for MG-H1 absorbed from food. The flux of endogenously generated MG-H1 adducts was decreased by 14% with tRES-HESP treatment but not with placebo. MG-H1 is also absorbed from food. The mean proportion of total MG-H1 produced endogenously was 67%, indicating that in overweight and obese subjects MG-H1 is mainly of endogenous origin—although this is highly variable (194).
Physiological effects of tRES-HESP treatment were profound. Insulin resistance, assessed by the Oral Glucose Insulin Sensitivity (OGIS) index, was corrected to levels typical of lean subjects with normal insulin sensitivity. The increase in OGIS of obese subjects was +58 mL/min/m2, which is a change similar to that found with pharmaceutical treatment of patients with T2DM (e.g., 1.7 g metformin per day, ΔOGIS = +54 mL/min/m2) (82) and extreme weight loss with gastric band surgery in morbid obesity (ΔOGIS = +62 mL/min/m2) (72). The main contributory factors to this effect were decreased fasting plasma glucose (FPG) and decreased postprandial plasma glucose. There was a 5% decrease in FPG post-supplementation with tRES-HESP that exceeds that found in similar trials with metformin and matches that found with Olristat—a lipase inhibitor anti-obesity drug (107, 130). A decrease in FPG in the normal range is associated with reduced risk of developing T2DM (184). Therefore, tRES-HESP treatment would likely decrease exposure to increased glucose concentration in the fasting and postprandial states in the highly overweight and obese populations and decrease risk of developing T2DM (194).
Concomitant with increased metabolic health there were small decreases in BMI and body weight in the obese subjects with tRES-HESP. There were also suggestions of improved vascular function, indicated by increased brachial artery flow-mediated dilatation response, decreased plasma vascular inflammation marker sICAM1, and improved renal function—indicated by both increased estimated glomerular filtration rate and decreased plasma urea (194).
Gene expression changes of PBMCs with tRES-HESP suggested a profound anti-inflammatory response: 30% decrease of prostaglandin-endoperoxide synthase 2 (PTGS2 or COX2) and 39% decrease of interleukin-8 (IL-8). This was maintained in highly overweight subjects with also a 49% decrease in expression of chemokine (C-C motif) ligand 2 (CCL2) or monocyte chemoattractant protein-1 (MCP-1) and a 37% decrease in expression of the RAGE. In the obesity subgroup, there was also a decrease in Kelch-like ECH-associated protein 1 (KEAP1) and tumor necrosis factor-α (TNFα). Treatment with placebo produced no change in gene expression of PBMCs (194).
The Glo1 inducer appears to be particularly appropriate and timely for treatment of high prevalence complications of obesity such as NAFLD. Decreases in expression of KEAP1 and TNFα will likely also be beneficial in facilitating activation of Nrf2 and decreasing insulin resistance, respectively (114, 186). Countering insulin resistance and inflammation are targets for treatment of NAFLD (118), and, therefore, prospective Glo1 inducer therapy would address multiple treatment targets. These responses are also likely to be beneficial in clinical diabetes: IL-8 drives renal inflammation (4, 171) and PTGS2 increases angiotensin production, renal cell dysfunction, and albuminuria (35). Downregulation of both MCP-1 and RAGE would likely contribute to treatment of diabetic kidney disease (36, 166). Inhibition of MCP-1 and RAGE is an alternative strategy for development of new treatments for diabetic kidney disease that have yet to be proved tractable by direct inhibitor drug therapy.
Benefit of a Synergistic Combination of Nrf2 Activators
Before the HATFF study, tRES and HESP were evaluated for health benefits individually but were without similar effect. Synergistic combination of Nrf2 activators was crucial to successful clinical translation of Nrf2 activators.
There have been many studies on the potential health benefits of tRES—including the pioneering studies of Sinclair and colleagues (17) and many others, reviewed elsewhere in this journal (133). Disappointing outcomes have been in clinical translation. From meta-analysis, it was concluded that tRES alone does not affect glycemic status in overweight and obese human subjects (97a). Prolonged treatment of patients with T2DM with tRES at doses approaching the maximum tolerable was without beneficial effect (24). Studies showing beneficial effects of tRES in diabetes have often been open trials or studies with baseline difference in treatment and placebo arms, suggesting problems with randomization. This may produce misleading outcomes. Lack of therapeutic effect is at odds with evidence from rodent models (28) and is likely due to interspecies differences in pharmacology, host interactions, and maximum tolerable dose. The maximum tolerable dose of tRES clinically to avoid adverse effects is <14 mg/kg (48). Evidence for health benefits of tRES in rodent models was mainly found at doses that markedly exceeded this and so safe clinical translation of tRES alone is unlikely. HESP has been mostly studied as a metabolite absorbed from clinical dosing with hesperidin—a related dietary glycoside found in citrus fruits (33). HESP alone also did not improve plasma glucose nor insulin resistance (156). Use of HESP rather than hesperidin is likely also crucial since HESP has higher bioavailability than hesperidin and ∼70-fold greater potency in Nrf2 activation (121).
A mechanistic interpretation of the pharmacological synergism of tRES and HESP came from consideration of the regulation of Nrf2 as a constitutive translocation oscillator whereby functional activation of Nrf2 is increased by increasing the frequency of cytoplasmic-nuclear oscillations of Nrf2—through increasing the reactivation or refresh rate of Nrf2, and decreasing the rate of inactivation of Nrf2 in the nucleus (190, 191) (Fig. 6). HESP likely drives increased Nrf2 oscillation frequency through activation of protein kinase A (PKA) and Fyn kinase downstream at ≥1 μM (75, 191, 194), and tRES decreases acetylation-driven nuclear inactivation of Nrf2 by increasing in situ activity through inhibiting cAMP phosphodiesterases (PDE), activating AMPK, and increasing NAD+ and in situ activity of sirtuin-1 (131); with HESP also synergizing for AMPK activation and sirtuin-1 through the PKA pathway (61, 75, 131) (Fig. 7). This is the first time that the oscillatory model of Nrf2 regulation has been exploited pharmacologically to achieve potent transcriptional response at low concentrations of co-agonists. This enables maximal pharmacological effect at agonist concentrations that are 50- to 100-fold lower than the minimum concentration inducing toxicity. Synergistic combinations of small-molecule agonists addressing different features of Nrf2 regulation may provide for successful clinical translation of Nrf2 activator pharmaceutical combination therapeutics.


Although evidence from Glo1 transgenic animal studies suggests that correcting downregulation of Glo1 in obesity and diabetes is likely to provide treatment for NAFLD and vascular complications of diabetes, change in the expression of other protective genes may contribute to the therapeutic mechanism of action. tRES-HESP increased expression of γ-glutamylcysteine ligase, catalytic and modulatory subunits GCLC, GCLM, in HAECs, BJ fibroblasts, and HepG2 cells, for example, and increased cellular GSH concentration—which increases in situ activity of Glo1. Expression of many antioxidant genes was unchanged in PBMCs of the clinical intervention study; however, Glo1 expression and activity were increased. Larger clinical studies in the future may have increased statistical power to detect changes in antioxidant-linked genes.
Mechanism of Action of the tRES-HESP Glo1 Inducer
tRES-HESP corrected insulin resistance, as judged by increases in the OGIS index. The OGIS index correlates strongly with the gold-standard method to assess insulin sensitivity, the hyperinsulinemic-euglycemic clamp technique (41), and is deduced from plasma glucose and insulin levels in the fasting and glucose-challenge states of an oral glucose tolerance test (108). OGIS was initially proposed as a marker of insulin resistance but also improves with increased β-cell sensitivity to glucose and decreased glucose absorption (74). tRES-HESP may intervene to correct insulin resistance by activating Nrf2-induced increase in expression of FGF21 (55). However, obesity is an FGF21-resistant state due to downregulation of the FGF21 receptor cofactor β-Klotho (58). MG-driven protein glycation is implicated in the downregulation of β-Klotho (205). By inducing Glo1 expression and decreasing MG protein glycation, therefore, we likely corrected the functional deficit of β-Klotho and re-engaged FGF21. This is expected to improve insulin sensitivity and stimulate browning of WAT, potentiate fatty acid storage and oxidation in brown adipose tissue (47, 109). A characteristic of increased β-Klotho expression and activity was its blocking of inflammatory signaling to downregulate expression of pro-inflammatory mediators IL-8, MCP-1, ICAM1, and RAGE (205), and via decreasing MCP-1 it also suppresses expression of PTGS2 (56). Remarkably, these were the exact features of the transcriptional signature found in PBMCs with tRES-HESP treatment (194).
For pancreatic β-cell sensitivity to glucose, the pancreatic islet is also a target of FGF21 and displays decreased and reduced β-Klotho expression and resistance to FGF21 in obesity and diabetes, leading to decreased β-cell function and cell mass (165). Increasing β-Klotho expression was associated with conservation of β-cells mass, and increased insulin storage and glucose-stimulated insulin secretion response (95). tRES (7 mg/kg) improved β-cell function in nonhuman primates given a high-fat/high-sugar diet (50). Lower doses of tRES may synergize with HESP to produce similar effect.
The positive correlation of tRES-HESP therapeutic potency with increasing BMI in overweight and obese subjects is consistent with the correction of metabolic dysfunction to regain that of lean controls rather than imposition of a pharmacological response in all subjects. This could relate to the correction of β-Klotho inactivation to normal levels. Lack of effect in healthy subjects of BMI close to the upper limit of the lean range suggests lack of an anorexic adverse effect.
There may be pre-systemic effects of the tRES-HESP treatment. An extremely high dose of HESP, ∼500 mg/kg, impaired digestion of starch and changed the composition of gut microbiota in rats (185), but this may not apply to the markedly lower clinical dose of HESP in the HATFF study (∼1.3 mg/kg HESP). tRES at the clinically maximum tolerable dose (15 mg/kg) did not affect the composition of gut microbiota in high-fat sucrose-fed rats (49). Also of relevance is activation of AMPK and sirtuin-1 by 20–40 μM tRES infused in the duodenum, which initiated a gut–brain–liver neuronal axis that improved hypothalamic insulin sensitivity and decreased hepatic glucose production (37). This may be potentiated by HESP co-administration.
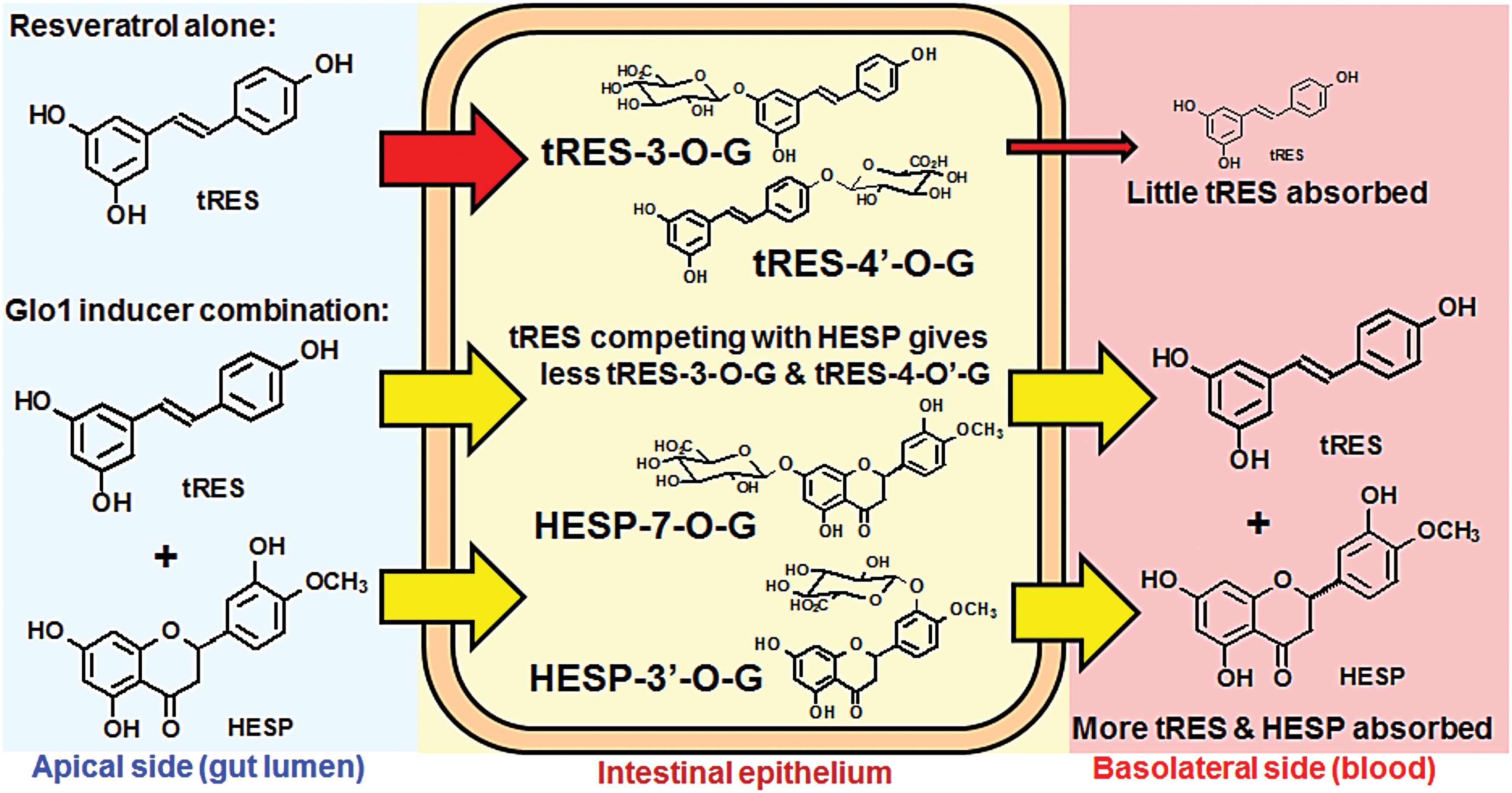
The major site in the body where HESP and tRES are at relatively high concentration is the stomach and, for a short time after administration, the small intestine. At the latter site, tRES and HESP are absorbed and rapidly conjugated to mainly glucuronides and, also to a lesser extent, sulfates (27, 164). Conjugates may be secreted on the apical and basolateral sides into the gut lumen and portal venous circulation, respectively. Studies of HESP infusion into the small intestine have shown that the absorption of free HESP is limited by this conjugation that is overwhelmed by 120 μM HESP, and increased unconjugated HESP is then absorbed (164). As tRES also draws on the capacity of the small intestine epithelium for conjugation, co-presence of ≥120 μM HESP may permit increased absorption of tRES. After an average meal, the mean stomach volume of a healthy human subject is 0.94 L. With 90 mg tRES and 120 mg HESP, the expected mean concentrations in the stomach lumen are 420 μM for both compounds. Gastric emptying occurs ∼40 min after food and/beverage ingestion. When tRES is administered alone, the peak plasma concentration of tRES at doses similar to those used in the HATFF study occurs at ∼50 min post-administration (26). So when tRES and HESP are given individually, the relatively high concentrations in the small intestine are short-lived. Co-administration of HESP with tRES likely exhausts or slows the intestinal conjugation capacity for both agents and facilitates increased absorption of the unconjugated compounds. This remains to be explored in follow-up pharmacokinetic studies but is an intriguing strategy to circumvent the problems of limited bioavailability of tRES (Fig. 8).
Glucose absorption in the porcine jejunum and ileum ex vivo was inhibited ∼50% by 300 μM tRES and so, there is potentially pharmacological competence to slow glucose absorption for a short period after dosing (68).
Next Steps for Development of the tRES-HESP Glo1 Inducer for Treatment of Complications of Obesity and Diabetes
In rodent models, there is evidence that tRES and HESP or hesperidin alone improve metabolic health in obesity and prevent diabetic nephropathy but at doses that exceed the maximum tolerable clinical dose. tRES improved metabolic health in HFD-fed mice at ∼30 to 60 mg/kg (10, 17) and improved renal function in rodent models of diabetic nephropathy at 20–300 mg/kg (83, 86). HESP had moderate anti-obesogenic activity and was evaluated at ∼500 mg/kg (73), and hesperidin decreased urinary albumin excretion in diabetic rats at 100 and 200 mg/kg (77).
The Glo1 inducer combination may also be considered for prevention of T1DM. tRES alone has previously been found to be effective in prevention of diabetes in an experimental mouse model of non-obese of type 1 diabetes (NOD) at doses of tRES higher than tolerated clinically (91).
For further development of tRES-HESP combination therapy, there is a regulatory requirement to show synergistic therapy in an appropriate animal model at clinically translatable doses. The challenge now is to demonstrate synergism of tRES and HESP in rodent models for prevention of weight gain and NAFLD in obesity and treatment of nephropathy and other vascular complications in experimental diabetes. In reverse translation of the outcomes of the HATFF study, the chances of success look good. A clinical phase 2 trial for diabetic would then be viable.
Concluding Remarks
Dicarbonyl stress is a dysfunctional metabolic state that appears to have a key role in the development of obesity, diabetes, and related metabolic and vascular complications. It is intrinsically linked to oxidative stress but may also be established and produce protein and DNA damage non-oxidatively. The recent emergence of a strategy to counter dicarbonyl stress with Glo1 inducers has shown promise in early clinical evaluation and builds on a wealth of knowledge accumulated on dietary bioactive compounds—particularly tRES.
Footnotes
Acknowledgments
The authors thank the Biosciences and Biotechnology Research Council (BBSRC), U.K., the U.K. innovation agency Innovate UK and Unilever for funding in development of Glo1 inducers.