
Abstract
Aims:
Mitochondrial fragmentation is a crucial mechanism contributing to tubular cell apoptosis during acute kidney injury (AKI). However, the mechanism of modulating mitochondrial dynamics during AKI remains unclear. Numb is a multifunction adaptor protein that is expressed in renal tubules. The aim of the present study was to evaluate the role of Numb in mitochondrial dysfunction during AKI.
Results:
The expression of Numb was upregulated in both ischemia–reperfusion- and cisplatin-induced AKI. Depletion of Numb from proximal tubules (PT-Nb-KO) exacerbated AKI shown as more severe renal tubular damage and higher serum creatinine than wild-type mice. Numb depletion alone significantly increased mitochondrial fragmentation without altering mitochondrial mass and function, including adenosine triphosphate production, mitochondrial membrane potential, oxygen consumption, and reactive oxygen species production. However, mitochondrial fragmentation and dysfunction were significantly aggravated after cisplatin exposure in PT-Nb-KO mice. Mechanistically, Numb depletion triggered dynamin-related protein 1 (Drp1) recruitment to mitochondria by increasing the phosphorylation of Drp1 at serine 656 residue (human Drp1 ser637). Inhibiting the activity of Rho-associated coiled-coil containing protein kinase (ROCK) by Y-27632 attenuated phosphorylation of Drp1 ser656 and mitochondrial fragmentation in Numb-deficient cells. Administration of mdivi-1, a pharmacological inhibitor of Drp1, restored mitochondrial morphology, attenuated cisplatin-induced tubular injury, and renal dysfunction in PT-Nb-KO mice.
Innovation and Conclusion:
Our data suggest that Numb depletion promotes mitochondrial fragmentation by promoting the phosphorylation of Drp1 Ser637 and thus exacerbates cisplatin-induced mitochondrial dysfunction and tubular cell apoptosis. These findings add a novel insight into modulating mechanism of mitochondrial dynamics during AKI.
Introduction
Acute kidney injury (AKI), often resulting from ischemic, nephrotoxic, and septic insults, is a devastating clinical condition that can result in short-term and long-term complications, including chronic kidney disease, end-stage renal disease, and death (6, 24, 25, 32, 71). Despite the growing incidence of AKI, there is no effective therapy for this condition. In this context, understanding the cellular mechanisms of AKI remains an unmet daunting task.
Renal tubule epithelia, especially the proximal tubular cells, are primary targets for acute injury (11). Depending on the nature and extent of the injurious stimuli, tubular cells lose functional integrity transiently or die by necrosis or apoptosis (23, 52). Central to tubular injury is mitochondrial dysregulation, manifested in a reduction in cell respiration and adenosine triphosphate (ATP) production (4, 35, 69).
In the current study, for the first time, we characterized a novel role of Numb in mitochondrial dynamics. Numb deficiency caused a profound increase of mitochondrial fission process by promoting Rho-associated coiled-coil containing protein kinase (ROCK)1-mediated phosphorylation of Drp1 Ser656 (Ser637 in human dynamin-related protein 1 [Drp1]) and subsequently promoted cisplatin-induced mitochondrial fragmentation, dysfunction, and apoptosis of tubular cells. These findings add a novel insight into modulating mechanism of mitochondrial dynamics during acute kidney injury (AKI) and may offer new opportunities for developing therapeutic strategies to combat AKI.
Mitochondria are dynamic organelles that maintain their morphology by a delicate balance between two opposing processes, fusion and fission (2, 21, 27, 69). Mitochondrial fusion is the lengthening of mitochondria by tethering and joining adjacent mitochondria. Mitofusin-1 (Mfn1) and mitofusin-2 (Mfn2) are mainly responsible for outer membrane fusion, whereas optic atrophy 1 (OPA1) is thought to mediate inner membrane fusion (26, 49, 61). Mitochondrial fission involves the constriction and cleavage of mitochondria by fission proteins, such as dynamin-related protein 1 (Drp1) and Fission 1 (Fis1) (29, 59). The master regulator of mitochondrial fission is Drp1 in mammals. During mitochondrial fission, cytoplasm-localized Drp1 is recruited to the mitochondria, and through GTP, hydrolysis constricts the mitochondrion leading to membrane scission (8, 34, 56). Drp1 is regulated by a variety of post-translational modifications including phosphorylation, which changes the localization, dynamics, and activity of Drp1 (9, 10, 12, 14, 22, 30, 59).
Thus far, mitochondrial fragmentation has been demonstrated as an early event in apoptosis of a variety of mammalian cells, including early renal cell injury (15, 35, 37). Genetic depletion and pharmacological inhibition of Drp1 could block mitochondrial fragmentation and tubular cell apoptosis during AKI, indicating that mitochondrial fragmentation is a crucial mechanism contributing to tubular damage during AKI (4, 5). Therefore, novel therapeutic strategies for AKI may derive from studies targeting regulators of mitochondrial dynamics.
Post-translational modifications of Drp1, especially phosphorylation at two main serine sites (Ser637 and Ser616 in human Drp1) by a series of kinases, are critical in modulating its translocation to the mitochondria (9, 10, 28, 55). Rho-associated coiled-coil containing protein kinases (ROCK), including two isoforms ROCK1 and ROCK2, are protein serine/threonine kinases involved in diverse intracellular signal transduction pathways and regulate a wide range of fundamental cellular functions, such as cell proliferation, apoptosis, contraction, adhesion, and migration by phosphorylating a large cohort of substrates (47, 51). Recent study indicates that ROCK1 mediates hyperglycemia-induced mitochondrial fission by phosphorylating Drp1 at Ser637 (human Drp1; Drp1 at serine 637 residue) in podocytes and endothelial cells (58).
Numb was originally identified as an intrinsic cell fate determinant during peripheral and central nervous system development in Drosophila. Drosophila Numb (d-Numb) is asymmetrically localized in dividing progenitor cells of these lineages, segregating preferentially to one of the two daughter cells, thereby directing the two daughter cells along their separate developmental paths (48). In mammals, Numb has been shown to mediate asymmetric division, endocytosis and recycling of specific proteins, and cell migration (39, 50, 53). In addition, Numb has been shown to behave as a tumor suppressor through stabilization of p53 and promoting Notch and Gli1 degradation (13, 16, 40). However, the role of Numb in kidney and renal injury remains largely unknown.
We previously reported that Numb expression was upregulated in fibrotic kidney. Knocking down Numb in proximal tubules significantly attenuates interstitial fibrosis by reducing cell cycle arrest at G2/M phase (70). The aim of the present study was to evaluate the role of Numb in AKI. We provided evidence that the depletion of Numb from proximal tubules leads to mitochondrial fragmentation by promoting the translocation of Drp1 to the mitochondria and subsequently exacerbates cisplatin-induced mitochondrial dysfunction and tubular cell apoptosis. This finding points the way toward a novel therapeutic target for the treatment of AKI.
Result
Tubular-specific loss of Numb aggravates cisplatin-induced AKI
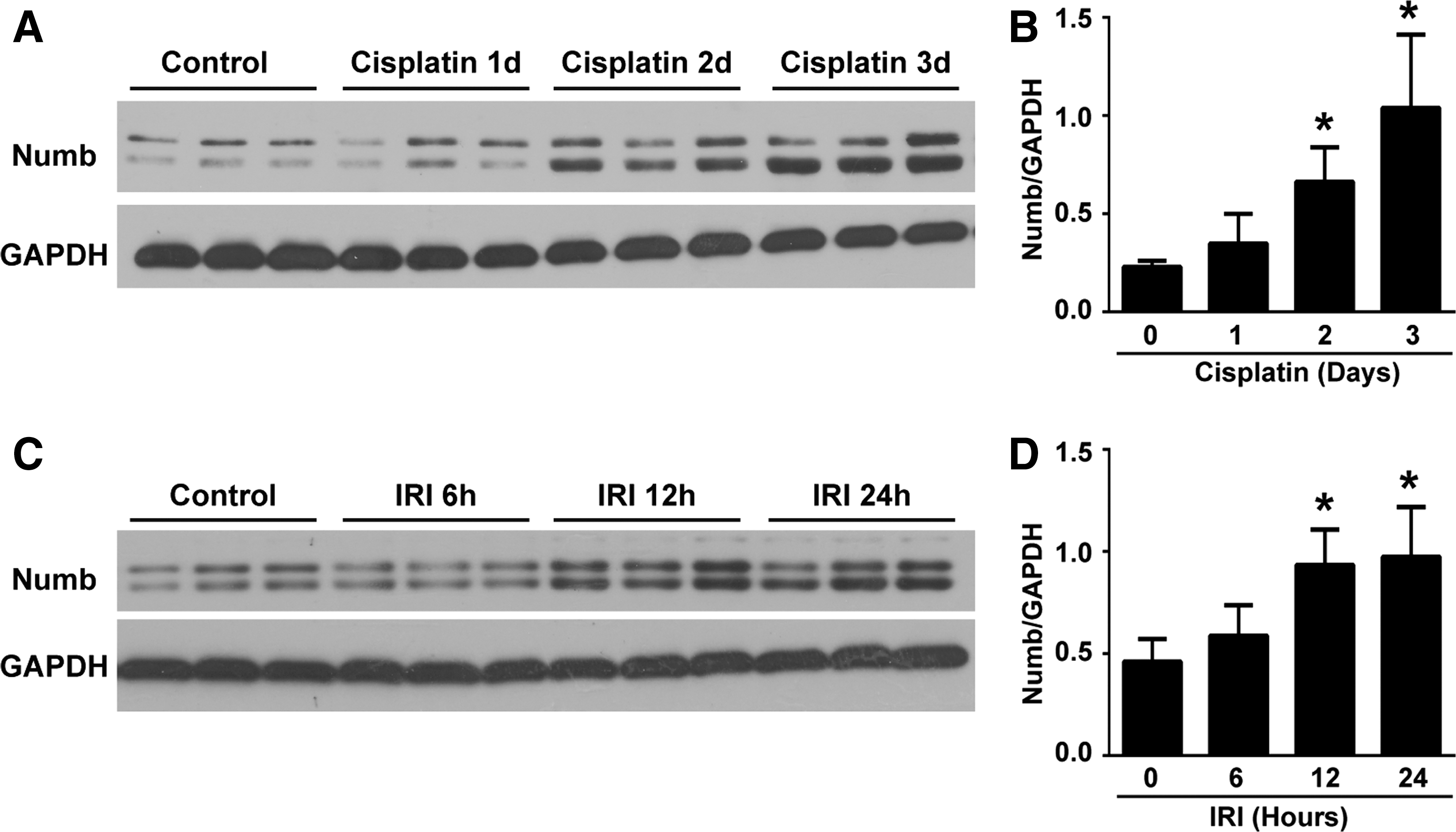
We previously showed that Numb is expressed in proximal tubular cells (70). Since proximal tubular cells are the primary targets for AKI, we wonder whether Numb plays a role in AKI. To address this issue, we first examined Numb expression in the injured kidney induced by cisplatin and ischemia–reperfusion (IR). Western blotting showed that Numb protein was significantly increased at day 2 after cisplatin injection (Fig. 1A, B; Supplementary Fig. S6A, B). Similarly, the upregulation of renal Numb was detected at 12 h after ischemia–reperfusion injury (IRI) (Fig. 1C, D). These data suggest that Numb might play a role in AKI.
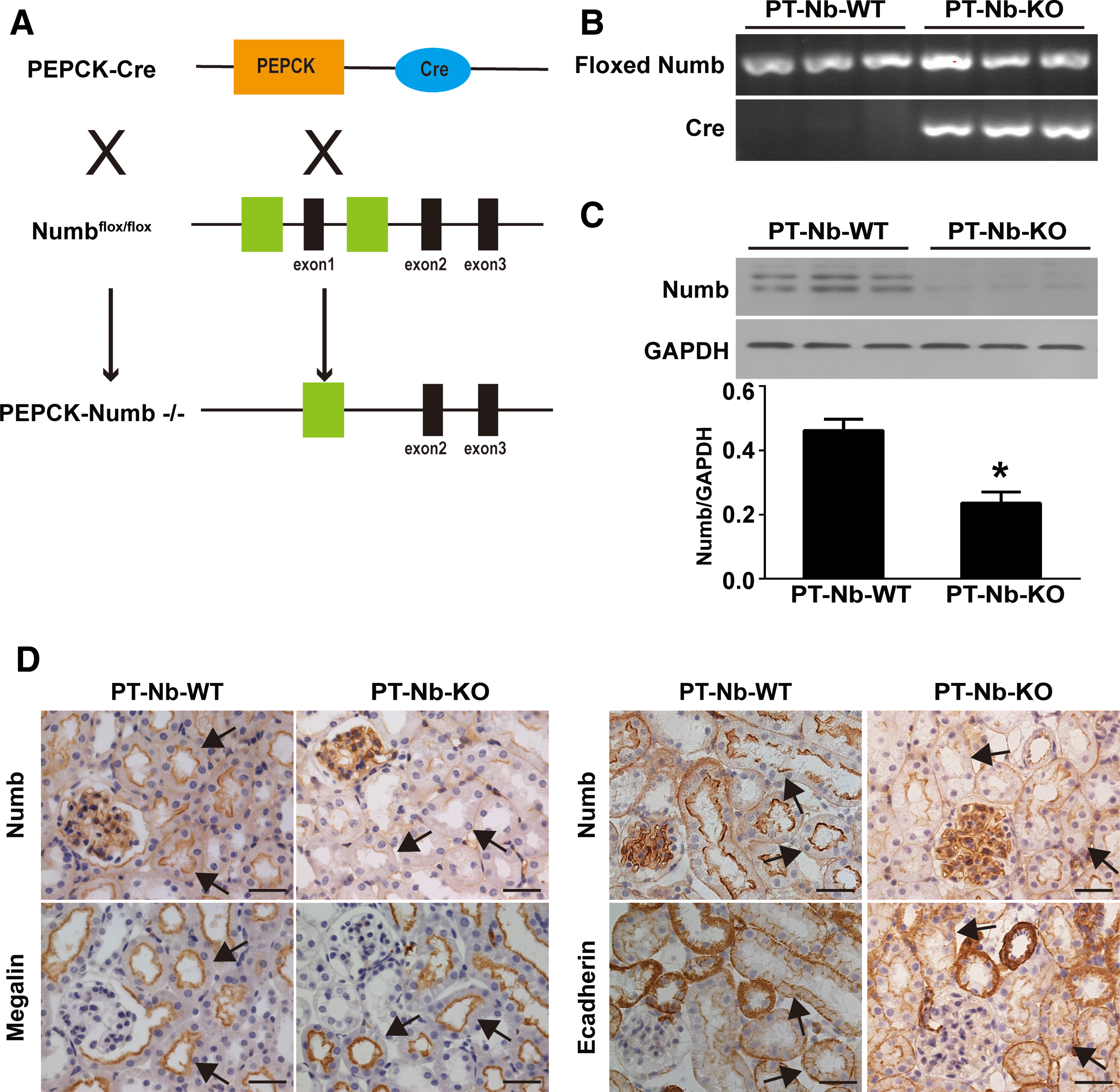
To further define the role of Numb in AKI, Numb gene was specifically disrupted in proximal tubules by intercrossing PEPCK-Cre mice with Numbflox/floxNumblike+/+ mice to generate Numbflox/floxNumblike+/+XcreY mice (depletion of Numb from proximal tubules [PT-Nb-KO]) (Fig. 2A–C; Supplementary Fig. S6C) as described previously (70). Immunohistochemistry staining showed proper localization of megalin at the apical side and E-cadherin at cell–cell contact and basal membrane of proximal tubular cell, indicating intact cell polarity of proximal tubular cells in PT-Nb-KO mice (Fig. 2D, E). There is no appreciable abnormality in kidney function in PT-Nb-KO mice at the age of 10 weeks (70). However, after cisplatin injection, PT-Nb-KO mice exhibited a significant increase in serum creatinine (Scr) and blood urea nitrogen (BUN) compared with their wild-type littermates (PT-Nb-WT) (Fig. 3A, B). Corroborating with the functional analysis, renal histology by hematoxylin and eosin (H&E) staining revealed that cisplatin-treated PT-Nb-KO mice showed more severe renal tubular damage, including loss of brush border, tubular dilation, cast formation in the lumen, and tubular cell necrosis loss (Fig. 3C, D, asterisks).
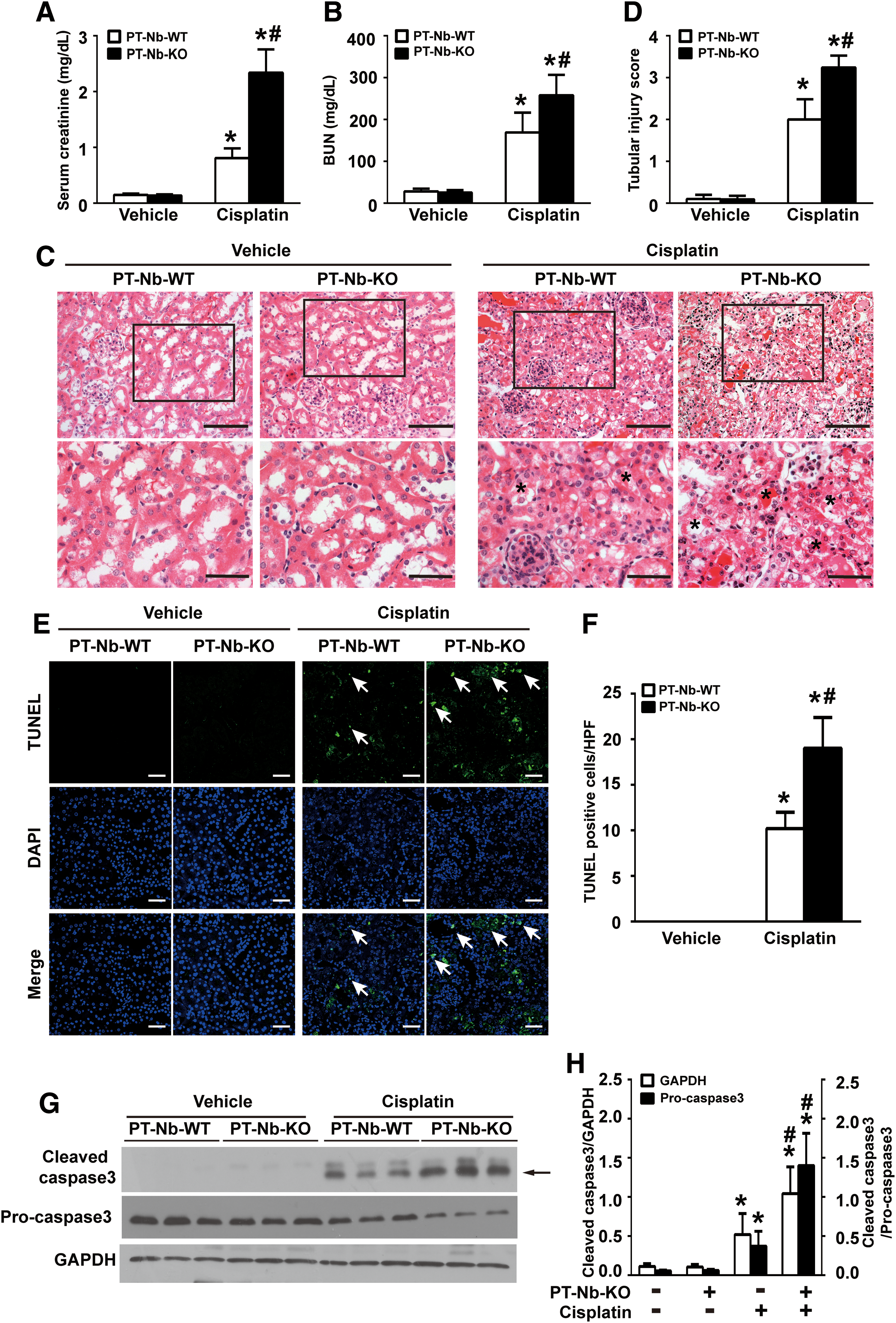
We next assessed tubular cell apoptosis by the TUNEL (TdT-mediated dUTP Nick-End Labeling) assay. As shown in Figure 3E, F, no apoptosis was detected in kidney tissues of both PT-Nb-WT and PT-Nb-KO mice. However, after cisplatin injection, PT-Nb-KO mice exhibited a significant increase in the number of TUNEL-positive cells compared with their wild-type littermates. Consistently, determination of cleaved caspase 3 by Western blotting confirmed the more prevalent tubular cell apoptosis in cisplatin-treated PT-Nb-KO mice (Fig. 3G, H; Supplementary Fig. S6D). These data suggest that tubule-specific loss of Numb exacerbates cisplatin-induced AKI.
Tubular-specific deletion of Numb aggravates IR-induced AKI
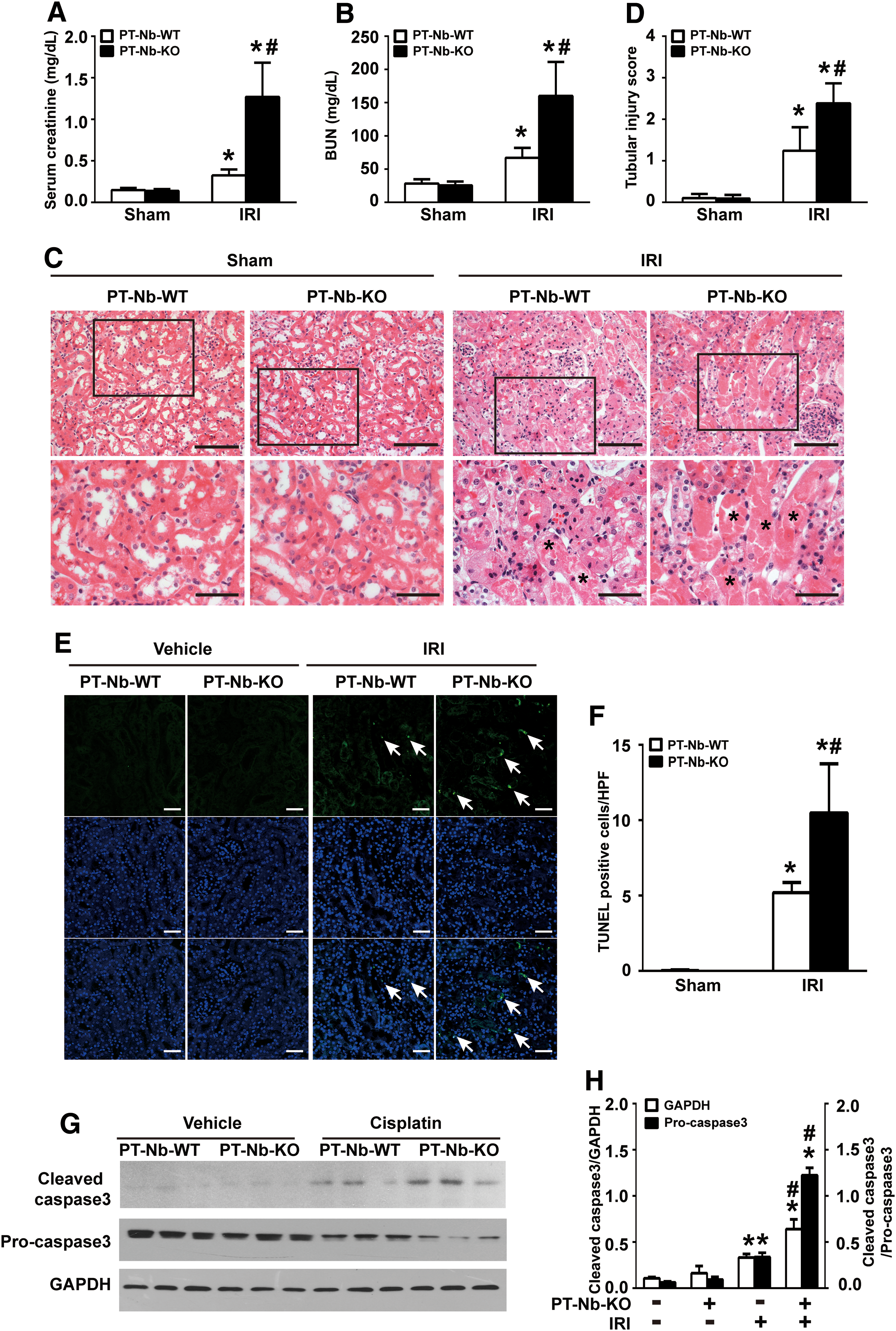
To demonstrate that the sensitivity of PT-Nb-KO mice to AKI is not specific to cisplatin treatment, we next induced AKI by IR. As shown in Figure 4A, B, 24 h after reperfusion, higher levels of Scr (Fig. 4A) and BUN (Fig. 4B) were presented in PT-Nb-KO mice. Renal histology analysis by H&E staining showed that more tubules displayed tubular cell necrosis loss and cast formation in PT-Nb-KO mice compared with PT-Nb-WT mice (Fig. 4C, D, asterisks). Consistently, IR also led to a more severe tubular cell apoptosis in PT-Nb-KO mice compared with their wild-type littermates (Fig. 4E–H; Supplementary Fig. S7A). Collectively, these data indicate that Numb is required to prevent tubular cell apoptosis in AKI.
Knockdown of Numb sensitizes cisplatin-induced tubular cell apoptosis in vitro
To provide direct evidence that links loss of Numb to tubular cell apoptosis, we transfected normal rat kidney epithelial (NRK-52E) cells with Numb siRNA. Figure 5A shows that Numb siRNA decreased Numb protein as well as mRNA expression (Supplementary Fig. S1), about 70% compared with scramble siRNA. We then treated NRK-52E cells with 20 μM cisplatin for 24 h. Flow cytometry analysis revealed that silencing Numb did not cause obvious tubular cell apoptosis (Fig. 5C, D). However, after cisplatin administration, the percentage of apoptosis cells was significantly increased in Numb siRNA-transfected cells compared with scramble siRNA-transfected cells (20.5% vs. 10%) (Fig. 5C). Consistently, Western blotting showed that silencing endogenous Numb significantly exacerbated cisplatin-induced cleavage of poly (ADP-ribose) polymerase (PARP) (Fig. 5A, B; Supplementary Fig. S7B). Additionally, flow cytometry analysis revealed that overexpressing Numb by infecting with a Numb adenovirus (Ad-Numb) significantly attenuated cisplatin-induced apoptosis (Supplementary Figs. S2A, D, and S9D).
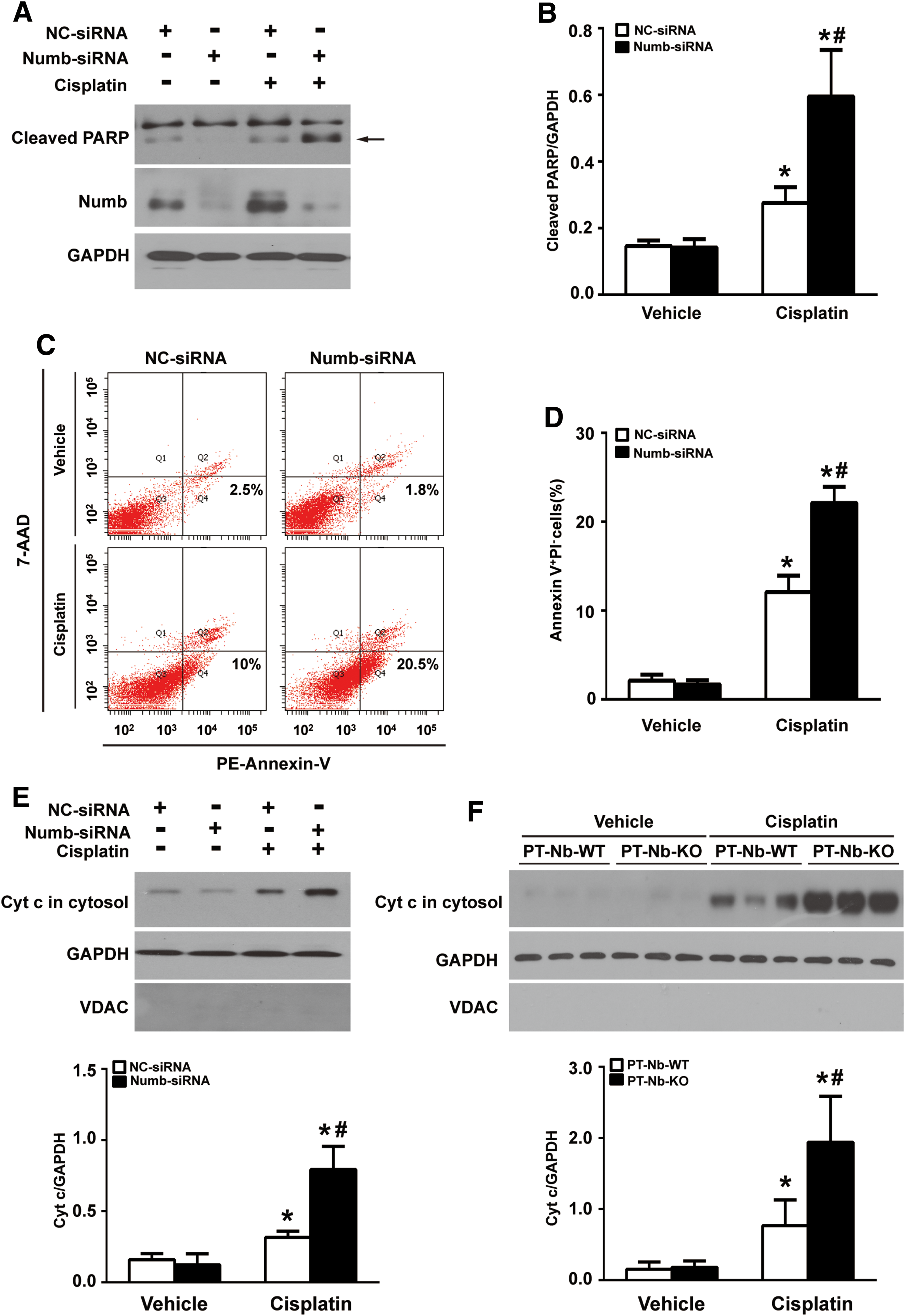
We further examined the effect of Numb on necrotic cell death by measuring propidium iodide (PI) uptake and lactate dehydrogenase (LDH) release. As shown in Supplementary Figure S3A, silencing endogenous Numb by siRNA did not cause obvious tubular cell necrosis but exacerbated cisplatin-induced necrosis measured by PI uptake. Consistently, the LDH release assay revealed that silencing Numb did not cause LDH release but led to higher levels of LDH release after cisplatin exposure compared with scramble siRNA-transfected cells (Supplementary Fig. S3B). These data indicate that loss of Numb promotes cisplatin-induced necrosis.
Cytochrome c (Cyt c) release from mitochondria to cytosol is a hallmark of the intrinsic pathway of apoptosis (21). As shown in Figure 5E; Supplementary Figure S7C, no Cyt c release from mitochondria was detected in Numb siRNA-transfected cells. However, after cisplatin administration, the release of Cyt c to the cytosol was significantly increased in Numb siRNA-transfected cells than in scramble siRNA-transfected cells. Moreover, the amount of Cyt c in cytosol of kidney tissues from PT-Nb-KO mice was significantly higher than that of control mice after cisplatin injection (Fig. 5F; Supplementary Fig. S7D).
Numb deficiency promotes cisplatin-induced mitochondrial fragmentation
Since mitochondrial fragmentation is an early event of tubular apoptosis during AKI (35), we monitored mitochondrial morphology by transfecting MitoTracker Red probe into NRK-52E cells to label mitochondria. As shown in Figure 6A, upper panel, mitochondria in scramble siRNA-transfected cells were filamentous with a tubular or thread-like appearance, whereas mitochondria in Numb siRNA-transfected cells appeared smaller and punctate pattern. To objectively quantify mitochondrial morphology, morphometric analyses were performed by calculating the form factor (FF) and the aspect ratio (AR) as described previously (66 –68). Both parameters have a minimal value of 1, which represents a perfect circle, and the values increase as mitochondria elongate. Plotting AR against FF revealed that most mitochondria in Numb siRNA-transfected cells had lower values of FF and AR under both physiological and cisplatin-stimulated condition (Fig. 6A, bottom panel). Compared with that of scramble siRNA-transfected cells, both the average values of FF and AR were significantly decreased in Numb siRNA-transfected cells (AR 2.02 vs. 2.36; FF 1.82 vs. 2.99) (Fig. 6B, C). After cisplatin administration, the average values of FF and AR were further decreased to 1.58 and 1.80, respectively, in Numb siRNA-transfected cells, indicating more severed mitochondrial fragmentation (Fig. 6B, C).
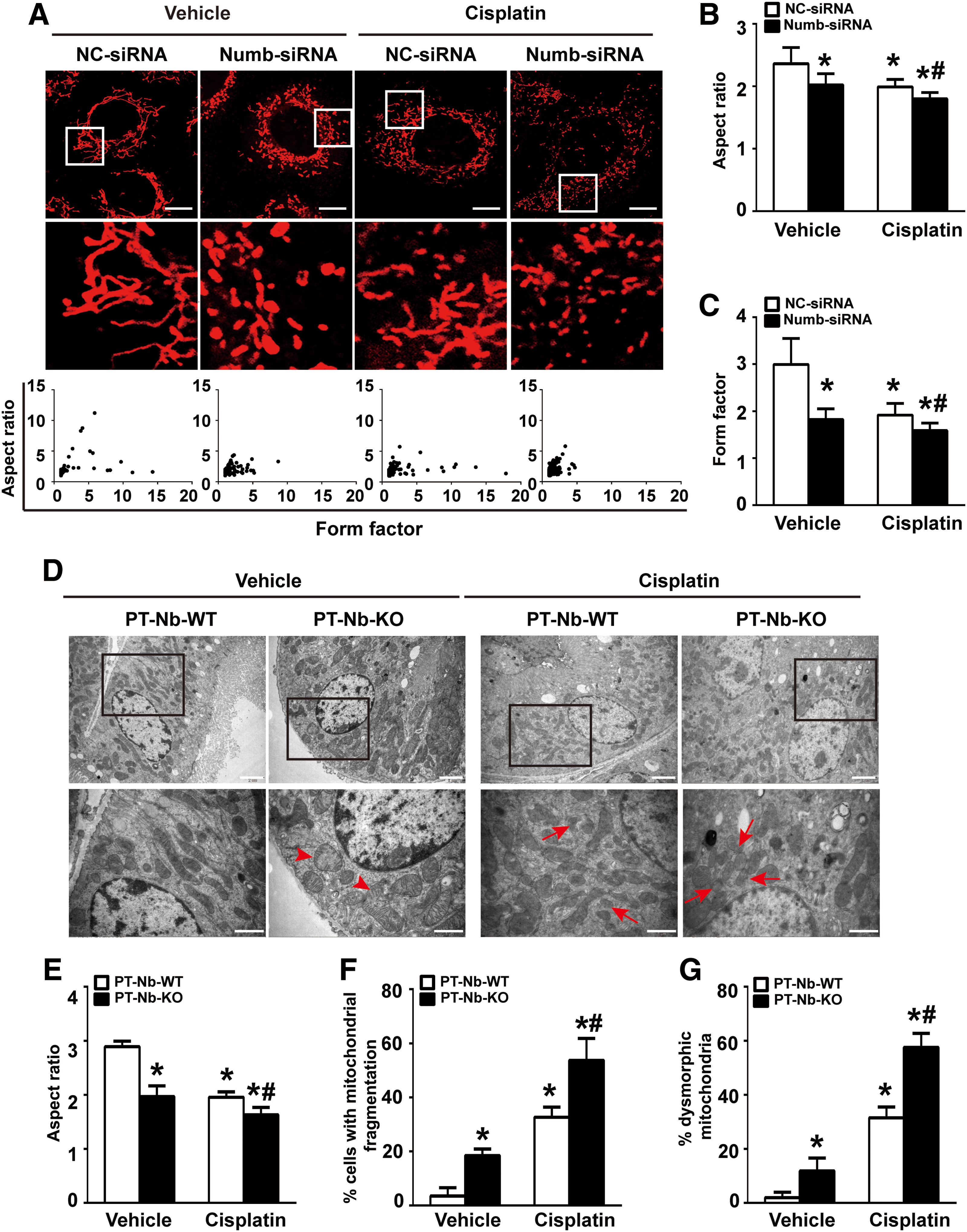
Electron microscopy revealed that mitochondria in tubular cells of PT-Nb-WT mice showed a typical filamentous morphology, whereas mitochondria in tubular cells of PT-Nb-KO mice were shortened and globular (Fig. 6D). To quantitatively evaluate mitochondrial fragmentation, we determined the AR in each group as shown in Figure 6E. The average value of AR decreased from 2.88 in PT-Nb-WT mice to 1.96 in PT-Nb-KO mice. After cisplatin exposure, the value of AR in PT-Nb-KO mice further decreased to 1.63 compared with that of 1.95 in PT-Nb-WT mice, indicating that the loss of Numb significantly triggers mitochondrial fragmentation in both physiological and AKI conditions. Consistently, the percentage of cells with fragmented mitochondria, which is <2 μm, was also significantly increased in renal tubular cells of PT-Nb-KO mice with or without cisplatin treatment (Fig. 6F). Moreover, the percentage of dysmorphic mitochondria, which is <2 μm with partial or completely loss of cristae, in renal tubular cells of PT-Nb-KO mice was also significantly increased (11.8% vs. 1.9%). Cisplatin exposure further increased it to 57.6%, which is significantly higher than that in PT-Nb-WT mice (31.5%) (Fig. 6G).
Given that mitochondrial fission facilitates mitophagy, especially phosphatase and tension homolog (PTEN)-induced putative kinase 1 (Pink1)-Parkin signaling-mediated mitophagy (57, 60, 65), we evaluated mitophagy in Numb-deficient cells. Both pMXs-YFP-Parkin and pMXs-mtDsRed (mitochondrial targeted-DsRed) recombinant lentivirus were delivered into scramble siRNA- or Numb siRNA-transfected NRK-52E cells to mark Parkin and mitochondria, respectively. Compared with scramble siRNA-transfected cells, no obvious increase of the colocalization of YFP-Parkin and mtDsRed was detected in Numb siRNA-transfected cells (Supplementary Fig. S4A). Moreover, Western blotting showed that the amount of Parkin and Pink1 in mitochondrial fraction in Numb siRNA-transfected cells was comparable with that in scramble siRNA-transfected cells (Supplementary Figs. S4B and S9E). These data indicate that Numb depletion does not initiate mitophagy in renal tubular cells.
Numb deficiency promotes cisplatin-induced mitochondrial dysfunction
We further evaluated the effect of Numb on mitochondrial function. First of all, mitochondrial DNA (mtDNA) content was measured. As shown in Figure 7A, the ratio of mtDNA/nuclear DNA (mtDNA/nDNA) was significantly increased in Numb siRNA-transfected cells than in scramble siRNA-transfected cells. This increase was more pronounced after cisplatin exposure, suggesting that Numb depletion promotes mitochondrial biogenesis, whereas Numb depletion did not cause obvious change on mitochondrial mass with or without cisplatin treatment (Fig. 7E). Moreover, Numb depletion did not alter mitochondrial membrane potential (Δψm) (Supplementary Fig. S5), which is the driving force for energy production. Cisplatin significantly decreased Δψm in both control and Numb-deficient cells (Fig. 7B). Next, compared with control cells, Numb depletion did not obviously alter intracellular ATP content but exacerbated the reduction of intracellular ATP content after cisplatin exposure (Fig. 7C). Consistently, Numb depletion did not alter either total reactive oxygen species (ROS) or mitochondrial reactive oxygen species (mitoROS) production but dramatically exacerbated cisplatin-induced total ROS and mitoROS production (Fig. 7D, E).
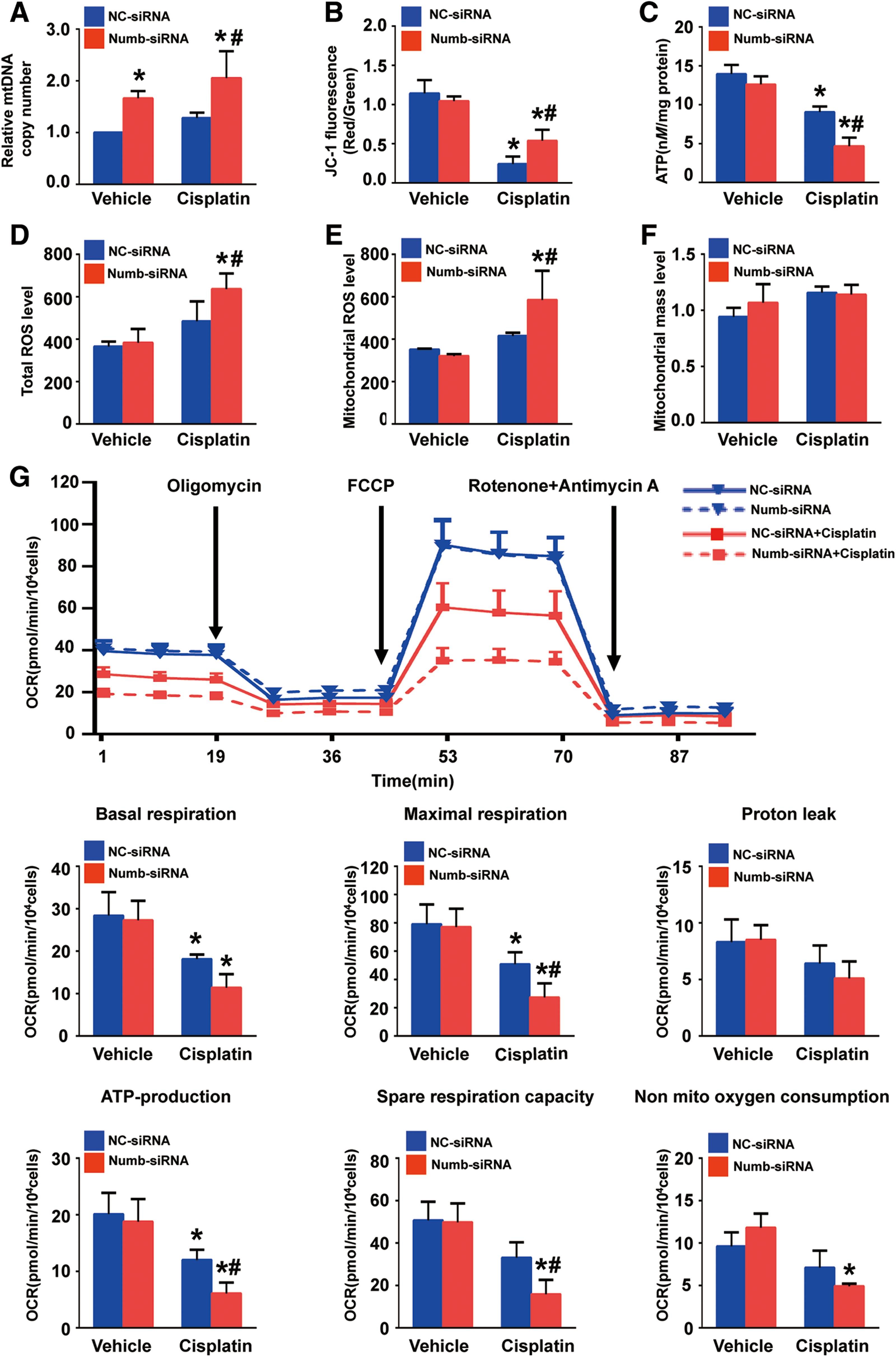
To better characterize the metabolic and mitochondrial consequences of Numb inhibition, we further evaluated oxygen consumption rate (OCR) in NRK-52E cells. As shown in Figure 7F, under physiological condition, basal respiration, ATP-production-dependent respiration, maximal respiration, and spare respiratory capacity were similar between scramble siRNA- and Numb siRNA-transfected cells, whereas nonmitochondrial respiration was slightly increased in Numb siRNA-transfected cells. However, cisplatin-induced reduction of mitochondrial respiration was more prominent in Numb siRNA-transfected cells compared with scramble siRNA-transfected cells. Collectively, these data suggest that Numb depletion alone does not alter mitochondrial function but significantly aggravates cisplatin-induced mitochondrial dysfunction.
Numb deficiency promotes Drp1 translocation to mitochondria by activating ROCK1
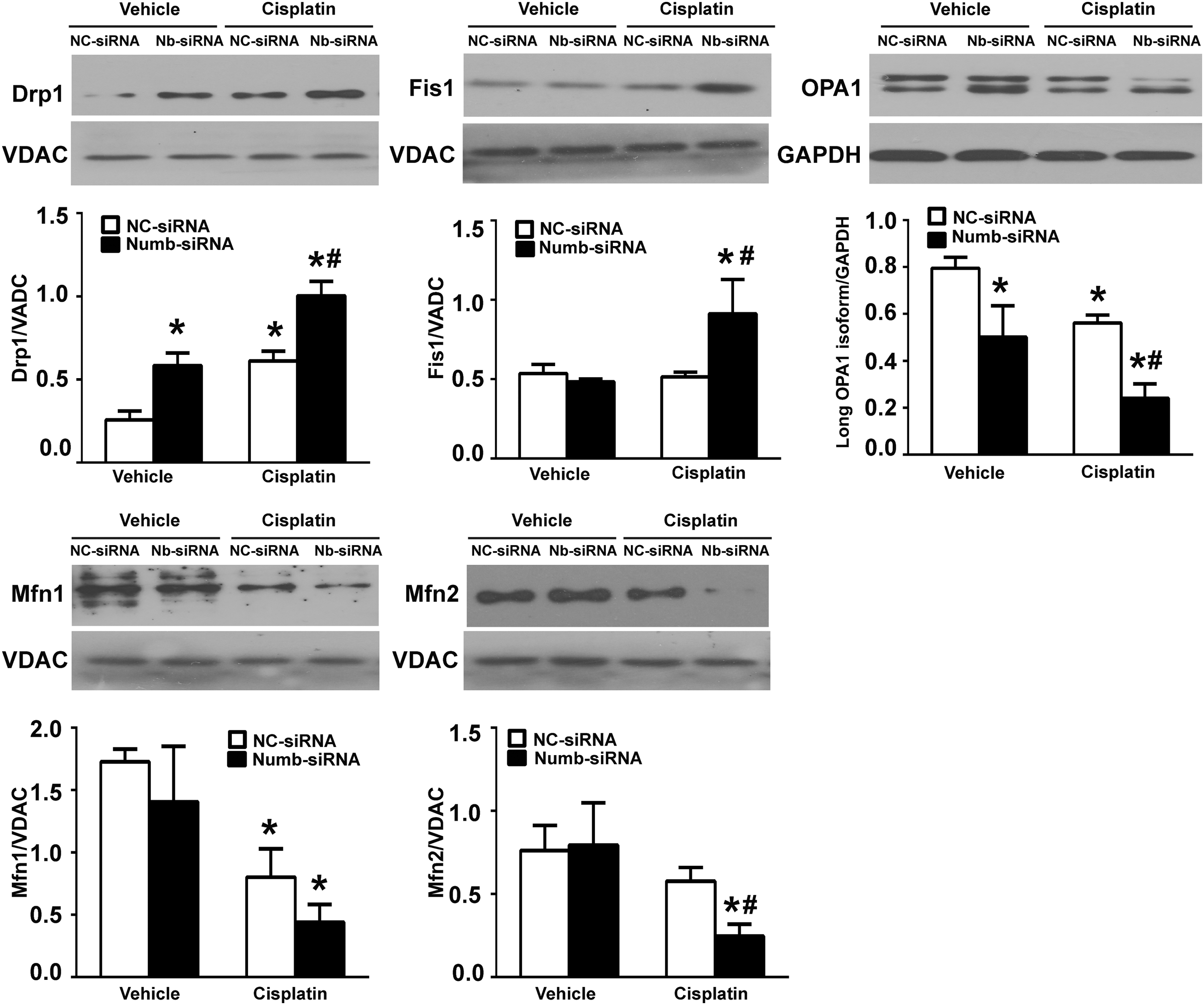
Mitochondrial fragmentation can be a result of increased fission, suppressed fusion, or a combination of both (29, 33, 61, 64). To further explore the role of Numb in mitochondrial fragmentation, we examined the expression of several dynamin-related GTPases. As shown in Figure 8 and Supplementary Figure S8A, a significant increase of Drp1 in mitochondrial fraction was detected in Numb-deficient cells, and this increase was further enhanced after cisplatin exposure. The amount of Fis1 in mitochondrial fraction was not altered in Numb-deficient cells but was significantly increased after cisplatin exposure. The abundance of Mfn1 and Mfn2, which are responsible for outer membrane fusion, in mitochondrial fraction was not altered in Numb-deficient cells but was significantly decreased after cisplatin exposure. In addition, the short isoforms of OPA1 were slightly increased in Numb-deficient cells, and this increase was enhanced after cisplatin exposure accompanied with the reduction of the long isoforms of OPA1. Collectively, these data suggest that Numb depletion promotes mitochondrial fission and subsequently exacerbates cisplatin-induced mitochondrial fragmentation at least partially by promoting the recruitment of Drp1 to mitochondria.
Given the critical role of Drp1 phosphorylation on its translocation, we next examined the amount of phosphorylated Drp1 in mitochondrial fraction. As shown in Figure 9D and Supplementary Figure S8B, compared with scramble siRNA-transfected cells, the amount of phosphorylated Drp1 at serine 656 residue (Drp1 ser656, ser637 in human Drp1), but not Drp1 ser585 (Ser616 in human Drp1), was significantly increased in Numb siRNA-transfected cells, suggesting that Numb modulates the phosphorylation of Drp1 ser656.
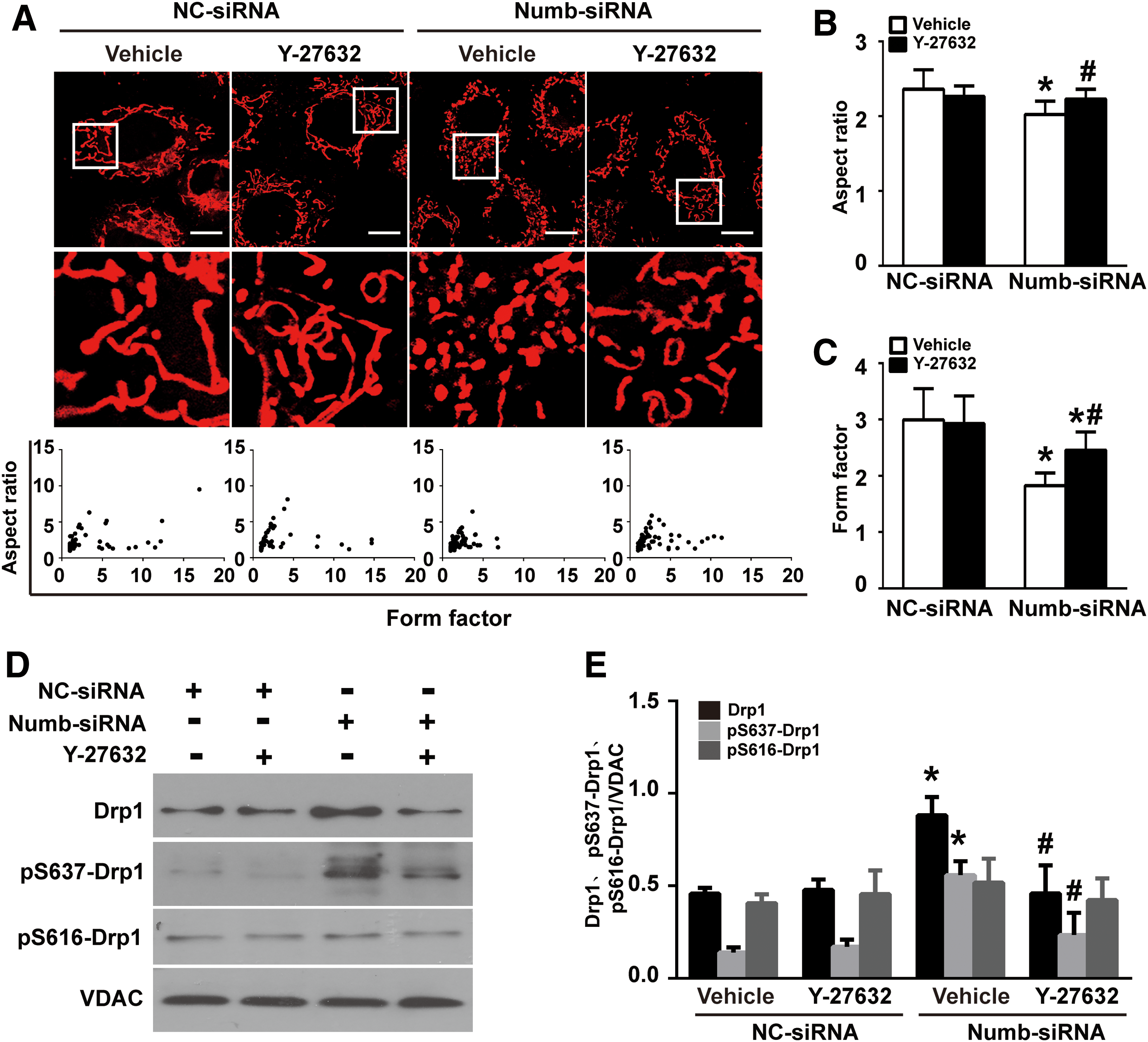
Since a previous study showed that ROCK1 triggers the phosphorylation of Drp1 ser637 in response to hyperglycemic stimulation in podocyte, we thus treated NRK-52E cells with 10 μM of ROCK inhibitor Y-27632 for 24 h. Western blotting showed that Numb depletion-induced phosphorylation of Drp ser656 (ser637 in human Drp1) was significantly attenuated by Y-27632 (Fig. 9D, compare lanes 3 and 4). Moreover, plotting AR against FF revealed that Numb depletion-induced mitochondrial fragmentation was significantly blocked by Y-27632 (Fig. 9A–C). These data suggest that ROCK1 mediates Numb depletion-promoted mitochondrial fragmentation.
Pharmacological inhibition of Drp1 ameliorates Numb depletion-promoted cisplatin nephrotoxicity
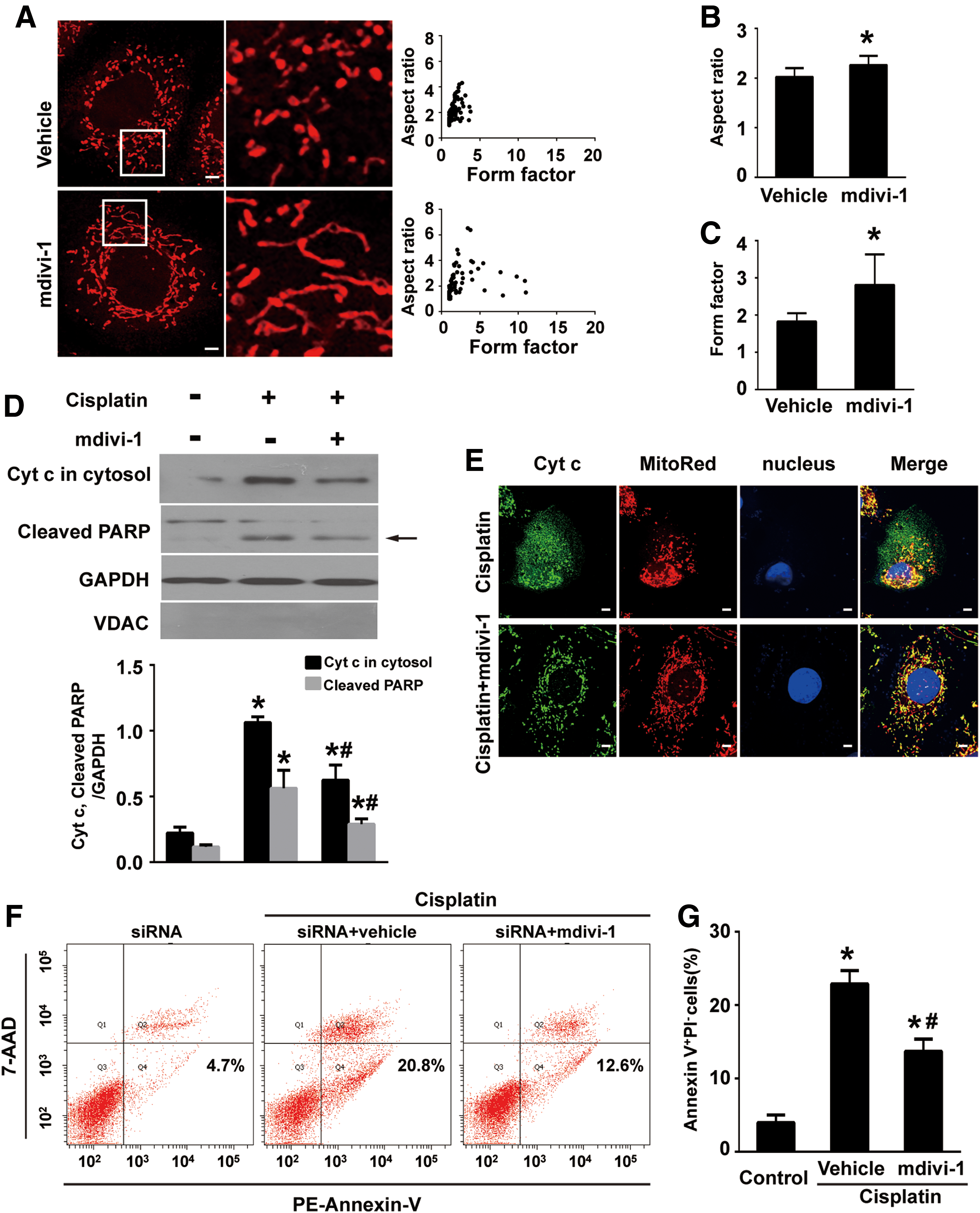
Since Numb depletion promoted Drp1 translocation to mitochondria, we pretreated Numb siRNA-transfected cells with mdivi-1, a pharmacological inhibitor of Drp1, before cisplatin administration. Immunofluorescence staining revealed that mdivi-1 restored the tubular pattern of mitochondria in Numb siRNA-transfected cells, shown as increased average values of AR and FF (Fig. 10A–C). Western blot and immunofluorescence staining demonstrated that mdivi-1 decreased cisplatin-induced Cyt c release to the cytosol (Fig. 10D, E). Moreover, flow cytometry analysis demonstrated that mdivi-1 significantly inhibited cisplatin-induced apoptosis from 20.8% to 12.6% (Fig. 10F, G). Consistently, the amount of cleaved PARP was significantly reduced in mdivi-1-treated cells (Fig. 10D; Supplementary Fig. S9A).
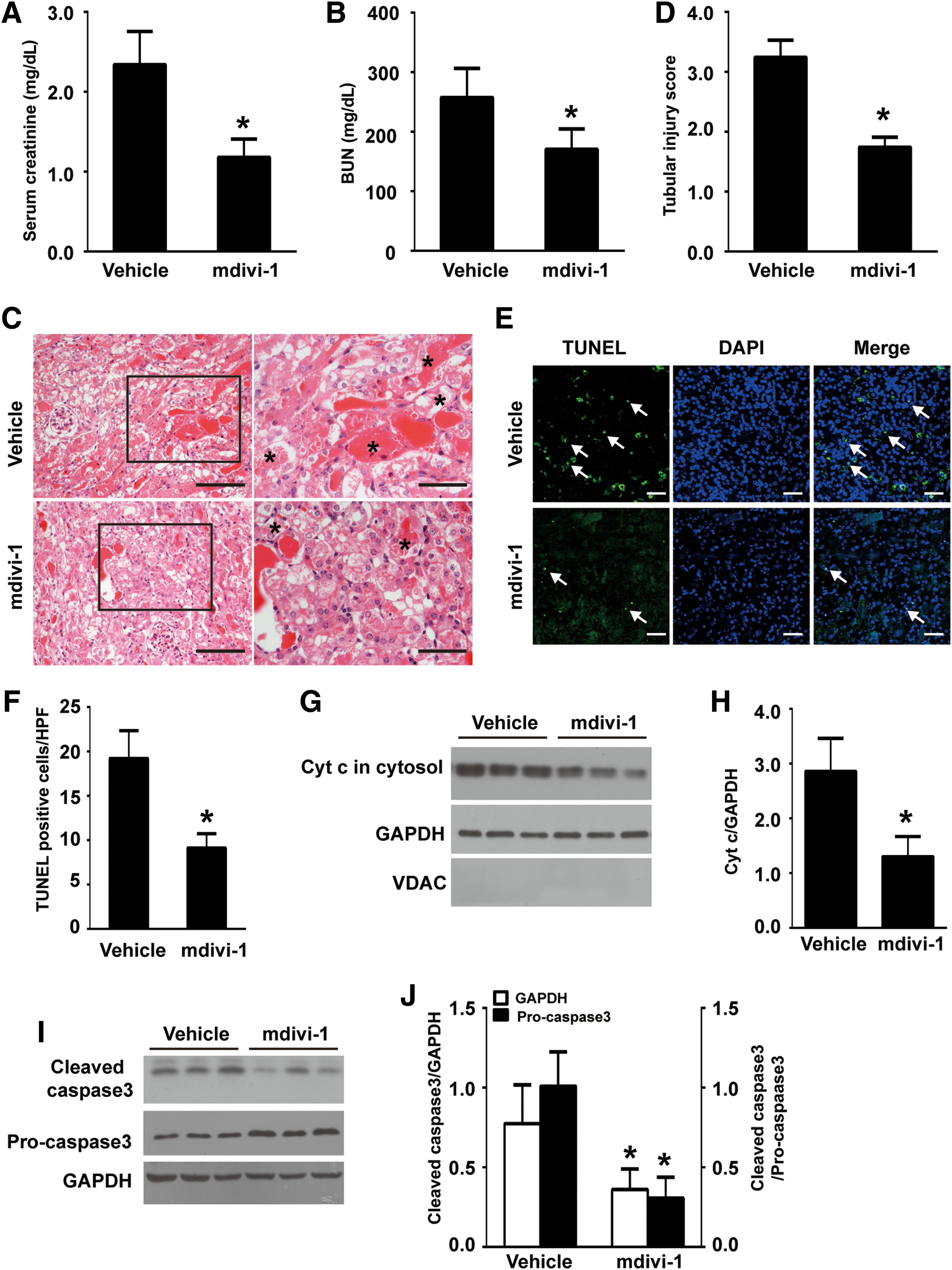
Next, mdivi-1 was given to PT-Nb-KO mice by intraperitoneal injection before cisplatin injection. Cisplatin-induced decline of renal function, as indicated by increases in Scr (Fig. 11A) and BUN (Fig. 11B), was significantly attenuated by mdivi-1. Moreover, H&E staining showed that mdivi-1 significantly ameliorated renal tubular damage characterized by loss of brush border, tubular dilation, cast formation, and tubular cell necrosis loss (Fig. 11C, D, asterisks). Moreover, the TUNEL assay demonstrated that mdivi-1 attenuated cisplatin-induced apoptosis (Fig. 11E, F). Western blotting showed that mdivi-1 prevented Cyt c release and cleavage of caspase 3 (Fig. 11G–J; Supplementary Fig. S9B, C). Collectively, these in vitro and in vivo data demonstrate that Numb protects cisplatin-induced AKI at least partially through preventing Drp1-mediated mitochondrial fragmentation.
Discussion
Numb is a highly conserved and ubiquitously expressed adaptor protein. In the kidney, Numb was mainly expressed in proximal tubule, and a small amount could be detected in distal tubule and collecting duct, but its role in the kidney and renal injury remains largely unknown. We previously reported that upregulation of Numb leads to G2/M arrest of tubular cells and promotes interstitial fibrosis (70). In the current study, we aimed to determine the role of Numb in AKI. First of all, we found that the expression of Numb was upregulated in both IR- and cisplatin-induced AKI. Second, depleting Numb from proximal tubular cells (in PT-Nb-KO mice) resulted in worsened morphological lesions, renal function, and tubular cell apoptosis in cisplatin-induced AKI, indicating a protective role of Numb in the setting of AKI. To further confirm the in vivo finding, endogenous Numb in cultured tubular cells was silenced by siRNA. Flow cytometry analysis detected increased apoptosis in Numb siRNA-transfected cells after cisplatin treatment. These quantitative in vitro data confirmed the result of TUNEL assay in PT-Nb-KO mice. Interestingly, the function of Numb in AKI is in contrast to the setting of chronic kidney diseases. A better understanding of the function of Numb in different pathophysiological settings is needed.
The role of Numb in mitochondrial remodeling or the molecular mechanism by which Numb potentially modulates mitochondrial dynamics is completely unknown. In the current study, for the first time, we demonstrated that Numb deficiency caused a profound increase of Drp1-mediated mitochondrial fission. This conclusion is supported by the following findings: First of all, smaller mitochondria were detected in renal tubular cells of PT-Nb-KO mice as well as in Numb siRNA-transfected NRK-52E cells. Mdivi-1, a pharmacological inhibitor of Drp1, restored the tubular pattern of mitochondria in Numb-deficient cells both in Numb siRNA-transfected cells and in renal tubular cells of PT-Nb-KO mice. Second, Numb depletion increased the amount of Drp1 in mitochondrial fraction but did not alter the abundance of fusion proteins Mfn1/2 in mitochondrial fraction. In line with our finding, a study of the heart indicates that only Drp1 was increased on mitochondrial fraction in renal IR-induced cardiomyocyte apoptosis (54). In addition, Danesh's group demonstrated that conditional deletion of Drp1 improves mitochondrial fitness in podocytes and ameliorates the progression of DN (3). Together with our current finding, these studies highlight a critical role of Drp1-mediated mitochondrial fission in cell apoptosis during renal injury.
The next question is how Numb modulates Drp1 translocation. Previous studies indicate that post-translational modifications of Drp1, such as phosphorylation, ubiquitination, and sumoylation, are critical in controlling Drp1 assembly on mitochondria (9, 10, 22, 30, 55, 59). In the current study, a significant increase in phosphorylation of Drp1 ser656 (ser637 in human Drp1), but not ser585 (Drp1 ser600 in human Drp1), was detected in Numb-deficient cells. Moreover, pharmacologically inhibiting the ROCK activity ameliorated Drp1 ser656 phosphorylation and mitochondrial fragmentation in Numb-deficient cells, as previous study reported that ROCK1-mediated Drp1 ser637 phosphorylation in mitochondrial fragmentation in response to hyperglycemic stimulation in podocytes; thus, we suggested that the ROCK1 activity mediates the role of Numb in modulating mitochondrial morphology in AKI.
Consistently, inhibition of the ROCK1 activity with small-molecule inhibitors could also prevent morphological changes in endothelial cells that accompany ischemic injury (20).
We previously showed that silencing Numb sensitizes proximal tubular cells to puromycin aminonucleoside-induced apoptosis by enhancing the activity of Notch signaling (17). Recent study showed that the activation of Notch1 upregulates nuclear and mitochondrial genes that encode electron transport chain (ETC) components and proteins involved in ETC assembly, mtDNA replication, and mitochondrial protein synthesis (62). Whether Notch signaling is involved in regulating Drp1-related mitochondrial fission needs further investigation.
Collectively, we provided both in vitro and in vivo data demonstrating that Numb depletion enhances mitochondrial fission by promoting ROCK1-mediated phosphorylation of Drp1 Ser656 (Ser637 in human Drp1). Of note, Numb-depletion-promoted mitochondrial fragmentation does not perturb mitochondrial function but significantly aggravates cisplatin-induced mitochondrial dysfunction. These findings add a novel insight into modulating mechanism of mitochondrial dynamics during AKI and may offer new opportunities for developing therapeutic strategies to combat AKI.
Materials and Methods
Mouse strains and genotyping
Numbflox/floxNumblikeflox/flox mice (C57BL/6J background) were purchased from the Jackson Laboratory (Stock No. 005384). PEPCK-Cre mice were kindly provided by Dr. Volker Haase (Vanderbilt University School of Medicine, Nashville, TN). The protocol of mouse intercrossing, breeding, and genotyping was depicted as previously described (70). All animals were housed in the Nanfang Hospital Animal Center and were born normally at the expected Mendelian frequency. All animal experiments were performed according to a protocol approved by Ethics Committee for Animal Experiments of the Southern Medical University.
Murine models of AKI
For cisplatin injury, male mice (body weight 20–25 g) were intraperitoneally injected with a single dose of cisplatin (p4393; Sigma, St. Louis, MO) at 25 mg/kg and sacrificed 3 days after injection. Blood and renal tissues were collected for further analyses. For analyzing the effect of mdivi-1, PT-Nb-KO mice were intraperitoneally injected with 50 mg/kg mdivi-1 (M0199; Sigma) 1 h before cisplatin administration.
For IRI, renal pedicles were exposed by a midline abdominal incision and were clamped for 30 min before release. Sham-operated mice underwent the same procedure except for the renal pedicle clamping. During the ischemic period, body temperature was maintained at 37.5°C using a temperature-controlled heating system. Blood and renal tissues were obtained at 24 h after reperfusion.
Cell culture and treatment
NRK-52E cells (Rattus norvegicus, cell strain) were cultured as previously described (1). For silencing Numb, NRK-52E cells were transfected with scramble siRNA or Numb siRNA (GenePharma, Shanghai, China) using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to the manufacturer's instruction. Eighteen hours after transfection, cells were incubated with 20 μM cisplatin for 24 h. The RNA sequences of RNAi oligonucleotides were as follows: scramble siRNA: GCGACGAUCUGCC UAAGAUdTdT; Numb siRNA: GCACCUGCCCAGUGGAUCCTT.
To determine the role of Numb overexpression in cisplatin-induced apoptosis, NRK-52E cells were infected with the Ad-Ctrl and Ad-Numb (SinoGenoMax, Beijing, China) as previously described (70). Eighteen hours after infection, cells were incubated with 20 μM cisplatin for 24 h.
To evaluate the role of Drp1 in cisplatin-induced apoptosis, Numb siRNA-transfected cells were pretreated with mdivi-1 (5 μM) 1 h followed by incubating with cisplatin for 24 h.
Renal function and histology
Renal function was assessed by measuring Scr and BUN with analytical kits (Beckman Coulter, CA) using automatic biochemistry analyzer (Au480; Beckman Coulter, CA).
For histology, 2 μm paraffin-embedded kidney sections were subjected to H&E staining by standard protocol. Briefly, sections were deparaffinized and rehydrated in ethanol and then processed to stain in hematoxylin solution for 15 min and in eosin solution for 1 min, respectively. Images were taken by the Olympus BX51 microscope (Olympus, Tokyo, Japan). Tubular injury was defined by the loss of brush border, tubular dilation, cast formation, and cell lysis. A pathologist without knowing the experimental protocol evaluated and quantified histological tubular damages by a semiquantitative scoring method as previously described (61). Briefly, score 0 represents no damage. Scores 1, 2, 3, and 4 represent the injury area involving 10–25%, 25–50%, 50–75%, and >75%, respectively. Ten fields were chosen randomly under the microscope (200 × ) from each mouse, five mice in each group were evaluated, and an average score was calculated.
Necrotic cell death determination
Necrotic cell death was evaluated by assessing plasma membrane integrity with PI uptake and LDH release assay as described previously (43, 63). For detecting PI uptake, NRK-52E cells were collected and resuspended in phosphate buffered saline (PBS) containing 5 μg/mL PI (#006990; Invitrogen, Carlsbad, CA) for 10 min at room temperature in the dark, and then, PI incorporation was assessed by using the FACSCanto II flow cytometry (Becton Dickinson, CA). Cells positive for PI was defined as necrotic cells.
The release of the LDH into the culture medium was quantified with LDH cytotoxicity assay kit (#88953; Invitrogen, Carlsbad, CA). Briefly, 50 μL of culture medium was transferred to a fresh 96-well, black flat-bottomed plate, and the colorimetric LDH release assay was performed according to the manufacturer's instruction. Absorbance was read at 490 and 680 nm, respectively. The LDH activity was determined by subtracting the absorbance value at 680 nm from that at 490 nm.
Preparation of the cytoplasmic and mitochondrial fractions
Cytoplasmic and mitochondrial proteins were collected using Qproteome Mitochondrial Isolation Kits (#37612; QIAGEN, Germany) according to the manufacturer's instructions. Briefly, cells and fresh renal tissues were smashed in lysis buffer. After centrifuging, the supernatant was collected as cytoplasmic protein. The pellet was resuspended in disruption buffer, homogenized, and then subjected to centrifugation at 1000 g for 10 min at 4°C to remove nuclei and unbroken cells. The supernatant was further centrifuged at 6000 g for 10 min at 4°C to obtain the mitochondrial fraction.
Intracellular ATP content
ATP levels were determined using the luciferase-based luminescence ATP Assay Kit (A22066; Molecular Probes, Life Technologies) according to the manufacturer's instruction. Cells were homogenized in an ice-cold ATP-releasing buffer and centrifuged at 12,000 g for 5 min. Twenty microliters of supernatant was mixed with 100 μL of ATP detection working dilution in 96-well plates. Luminance (relative luminescence units) was assayed by a multilabel plate reader (Victor-V; Perkin Elmer, USA). Luminescence was then normalized to protein concentration.
Mitochondrial membrane potential
The Δψm was determined using JC-1 (MP03168; Molecular Probes, Invitrogen) according to the manufacturer's protocol as previously described (42, 45). Briefly, NRK-52E cells were initially stained with 2 μM JC-1 for 30 min at 37°C in the dark, and then, labeled cells were analyzed by a flow cytometer (Becton Dickinson, CA) with a 488-nm excitation. The JC-1 monomer and the J-aggregates were detected separately at emission of 525 nm (green) and 590 nm (red), respectively. Δψm is presented as the ratio of red to green fluorescence intensity. In some experiments, the Δψm was determined by tetramethylrhodamine methyl ester (TMRM) (T668; Molecular Probes, Invitrogen) staining according to the manufacturer's instruction. Briefly, NRK-52E cells were initially stained with 100 nM TMRM for 30 min at 37°C in the dark. Then, cells were washed twice with PBS and resuspended in 300 μL cold PBS. Labeled cells were analyzed by a flow cytometer (Becton Dickinson, CA). Δψm is presented as the fold change of fluorescence intensity relative to scramble siRNA-transfected cells.
mtDNA copy number assay
Relative mtDNA copy number was determined as the ratio of the amount of mtDNA-encoded gene to the amount of nuclear-encoded gene as previously described (41). Total DNA was extracted from NRK-52E cells using DNA isolation kit (#51304; QIAGEN, Germany) according to the manufacturer's protocol. Real-time quantitative polymerase chain reaction (PCR) of the mtDNA-encoded NADH dehydrogenase 3 (ND3) and the nuclear-encoded S18 gene was carried out on the ABI PRISM 7500 Fast sequence detection system (Applied Biosystems, Foster City, CA). Relative mtDNA copy number was determined as the mtDNA-to-nDNA ratio. The primer pairs were listed as follows: ND3 was 5′-TGGCTTACAAGACGCCACAT-3′ and 3′-TGGGCGTCTAT TGTGCTTGT-5′; S18 was 5′-TCTTCCACAGGAGGCCTACAC-3′ and 3′- CCCA CACCCTTAATGGCAGTGAT-5′.
Measurement of mitochondrial biomass
Mitochondrial biomass was measured using MitoTracker Green FM dye (M7514; Molecular Probes, Invitrogen) as previously described (18). Briefly, 1 mL of the cell suspension was incubated with MitoTracker Green FM dye (50 nM, 30 min) at 37°C in the dark. Then, cells were washed twice with PBS and resuspended in 300 μL cold PBS. Labeled cells were then analyzed by a flow cytometer (Becton Dickinson, CA).
Measurement of OCR
The OCR was analyzed by the XF24 Extracellular Flux Analyzer (Seahorse Bioscience, North Billerica, MA) using a cell mitochondrial stress test kit (Seahorse Bioscience, North Billerica, MA) according to the manufacturer's instructions (46). Briefly, NRK-52E cells were seeded at a density of 4000 cells per well in XFp cell culture miniplates (Seahorse Bioscience) and incubated in the 5% CO2 incubator at 37°C for 24 h. After transfected with NC-siRNA or Numb-siRNA for 18 h, cells were treated with 10 μM cisplatin for 24 h, and then, cells were washed twice with XF assay medium (XF base medium supplemented with 4 mM glutamine, 1 mM pyruvate, and 25 mM glucose) and equilibrated in 525 μL assay medium per well in a CO2-free incubator at 37°C for 1 h. Subsequently, the OCR of cells were analyzed by sequential injections of 1 μM oligomycin, 2 μM p-trifluoromethoxy carbonyl cyanide phenylhydrazone (FCCP), and 0.5 μM Rotenone plus antimycin A to assess mitochondrial function (basal consumption rate, ATP-linked respiration, maximal and spare respiration capacity) in the XFp Extracellular Flux Analyzer.
Nonmitochondrial respiration was calculated by the minimum rate measurement after R&A injection. Basal respiration was calculated by the last rate measurement before oligomycin injection minus nonmitochondrial respiration. ATP-production-dependent respiration was calculated by the last rate measurement before oligomycin injection minus the minimum rate measurement after oligomycin injection. Maximal respiration was calculated by the maximum rate measurement after FCCP injection minus nonmitochondrial respiration. Spare mitochondrial capacity was calculated by maximal respiration minus basal respiration. Proton leak was calculated by the minimum rate measurement after oligomycin injection minus nonmitochondrial respiration. OCR was measured at 37°C with 3-min mix, 2-min wait, and 3-min measurement protocol. OCR was measured three times in respective phase and expressed as units of picomoles (pmol) per minute. After respirometric assessment, cell numbers were recounted for normalization of the OCR.
Measurement of ROS
Total intracellular ROS and mitoROS were measured by incubating cells with the 2′,7′-dichlorofluorescein diacetate (H2DCFDA) (C6827; Molecular Probes, Invitrogen) and mitoSOX (M36008; Molecular Probes, Invitrogen), respectively, as previously described (38).
Immunofluorescence staining
Mitochondria in cultured cells were labeled by incubating living cells with 200 nM of MitoTracker Red CMXRos (M7512; Molecular Probes, Invitrogen) for 30 min at 37°C. Cells were then fixed with 3.7% formaldehyde. Mitochondrial images were taken by confocal microscopy (Olympus, Tokyo, Japan).
To assess Cyt c release, cells cultured on coverslips were processed to mitochondrial stain by incubating with MitoTracker Red CMXRos. After fixation and permeabilization with 0.1% Triton X-100, cells were then incubated with anti-cytochrome c (#12963; Cell Signaling Technology, Beverly, MA) followed by incubating with Alexa Fluor 488-conjugated secondary antibody (Invitrogen). Slides were then mounted in Fluoroshield Mounting Medium (Abcam). Images were taken and analyzed by confocal microscopy (Olympus, Tokyo, Japan).
Morphometric analysis of mitochondria in vitro
Morphometric analysis of mitochondria was performed as described previously (7, 66 –68). Briefly, mitochondria were labeled by MitoTracker Red CMXRos. The NIH-developed ImageJ software (Wayne Rasband; NIH) was used for the analysis. Digital images were processed through converting image to eight bite, reducing unspecific noise by using the despeckle function, and followed by a convolve filter to obtain isolated and equalized fluorescent pixels. After thresholding, individual particles (mitochondria) were subjected to the particle analysis to acquire values for circularity (4π area/perimeter2) and lengths of major and minor axes. From these values, FF (the reciprocal of circularity value) and AR (major axis/minor axis) were calculated.
Morphometric analysis of mitochondria was performed by an investigator blinded to the experimental protocol.
Transmission electron microscopy and mitochondrial morphological analysis
Kidney sections were fixed in 2.5% glutaraldehyde overnight at 4°C. Ultrathin renal sections were stained with uranyl acetate and lead citrate, mounted on a copper grid, and photographed under the Hitachi 7700 transmission electron microscope (Tokyo, Japan).
After adding the scale bar on the images of electron microscopies, the length of mitochondria in proximal tubular cells was measured using the NIH ImageJ software (5). Cells that had <1% long filamentous mitochondria (>2 μm) were defined as cells with fragmented mitochondria (5). The percentage of cells with fragmented mitochondria was determined by analyzing mitochondria in 50–150 cells from three mice in each group.
The AR (the ratio between the major and minor axes of the ellipse equivalent to the mitochondrion) was calculated based on the major and minor axes of each mitochondrion measured by the NIH ImageJ software (19, 58). Five hundred mitochondria from three mice in each group were analyzed.
Mitochondria <2 μm in length (fragmented mitochondria) together with partial or complete loss of cristae are defined as dysmorphic mitochondria as described previously (5, 31, 36). The percentage of dysmorphic mitochondria was determined by measuring 500 mitochondria from three mice in each group.
Mitochondrial morphological analysis was performed by an investigator blinded to the experimental protocol.
Flow cytometry analysis
Apoptosis was detected by using the Annexin V-APC/7-ADD Apoptosis Detection Kit (Becton Dickinson) according to the manufacturer's instruction. Stained cells were assessed by the apoptotic activity using the FACSCanto II flow cytometry (Becton Dickinson, CA). Apoptotic cells positive for Annexin V, but negative for PI, were defined as apoptotic cells.
Western blot analysis
Western blot analysis was performed according to a standard procedure as previously described (1). The following primary antibodies were used: anti-Numb (#2756), anti-PARP (#9542), anti-cleaved caspase 3 (#9664), anti-caspase3 (#9662), anti-Mfn2 (#9482), anti-OPA1 (#80471), anti-Cytochrome c (#11940), anti-VDAC (#4866), anti-pS637-Drp1 (#4867), anti-pS616-Drp1 (#3455), anti-Parkin (#2132), anti-GAPDH (#5174) (Cell Signaling Technology, Beverly, MA), anti-Drp1 (ab184247), anti-Mfn1 (#ab104274), anti-Fis1 (ab71498) (Abcam, Cambridge, United Kingdom), anti-Pink1 (sc-517353) (Santa Cruz).
Real-time PCR
Total RNA isolation and real-time reverse transcription-polymerase chain reaction (RT-PCR) were carried out by the routine procedures, as described previously (70). Briefly, the first-strand cDNA synthesis was carried out by using a reverse transcription system kit according to the manufacturer's instructions (Takara, Tokyo, Japan). Real-time RT-PCR was performed on the ABI PRISM 7500 Fast sequence detection system (Applied Biosystems, Foster City, CA). The primer pair for Numb was 5′-GCAGCTTCCCAGTTAAGTACC-3′ and 5′-AAC GGCCTTCACTGCTTTCT-3′. The mRNA level was calculated after normalizing with GAPDH by the comparative CT method (2-DDCT).
Detection of mitophagy
Mitophagy was determined by the translocation of cytosolic Parkin to the mitochondria. In line with this, cells were coinfected with pMXs-YFP-Parkin and pMXs-mtDsRed constructs to mark Parkin and mitochondria, respectively (44). Then, cells were transfected with scramble siRNA and Numb siRNA using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to the manufacturer's instruction; 48 h after transfection, cells were fixed with methanol. Images were taken and analyzed by confocal microscopy (Olympus, Tokyo, Japan). pMXs-YFP-Parkin and pMXs-mtDsRed were gifts from Xingguo Liu (CAS Key Laboratory of Regenerative Biology, Joint School of Life Sciences, Guangzhou Institutes of Biomedicine and Health).
Immunohistochemical staining
Immunohistochemical staining was performed according to a standard protocol, as previously described (70). Briefly, kidney sections were processed to deparaffinization, hydration, and antigen retrieval. After blocked with normal donkey serum, sections were incubated with the primary antibodies and followed by secondary antibody incubation. Images were taken by the Olympus BX51 microscope (Olympus, Tokyo, Japan). The primary antibodies used in this study include anti-Numb (#2756; Cell Signaling Technology, Beverly, MA), anti-megalin (ab184676; Abcam, Cambridge, United Kingdom), and E-cadherin (#610812, BD, Transduction Laboratories).
Statistical analyses
Data were expressed as means ± SD. Comparisons between the two groups were conducted using the two-tailed t-test. Differences among the four groups were compared using two-way analysis, followed by the Student–Newman–Keuls post hoc test. Differences among more than two groups (with one variable) were compared using one-way analysis (IBM SPSS software, version 19.0). p-Value of <0.05 was considered statistically significant.
Footnotes
Acknowledgments
The authors thank Dr. Volker Haase (Vanderbilt University School of Medicine) and Dr. Katalin Susztak (Perelman School of Medicine, University of Pennsylvania) for kindly providing the PEPCK-Cre mice. This work was supported by grants from the Nature and Science Foundation of China (81288001, 81730019, 81521003) and also by grants from the Natural Science Foundation of Guangdong province (S2013020012748) to J.N. and from the Nature and Science Foundation of China (81500514) to J.A.
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.