
Abstract
Aims:
Oxidative stress and neuroinflammation play important roles in the pathology of Alzheimer's disease (AD). Thioredoxin-interacting protein (TXNIP), an endogenous inhibitor of antioxidant thioredoxin, is suspected to be an important modulator of oxidative stress and inflammation. However, the underlying mechanism involved in the abnormal homeostasis of TXNIP-thioredoxin (TrX) in AD pathogenesis remains unclear.
Results:
Using the Swedish mutant form of APP (APPswe)/PSEN1dE9 transgenic mouse (APP/PS1) and human-derived neuronal cells as model systems, we disclosed the impairment of the nuclear factor erythroid 2-related factor 2 (Nrf2)-TXNIP-TrX signaling in Alzheimer's-like pathology. We observed that the immune staining of TXNIP was increased in postmortem AD brain. The chronic accumulation of inflammatory mediator in neuronal cells facilitates interactions of TXNIP-nucleotide binding oligomerization domain-like receptor family, pyrin domain containing 3 (NLRP3) and NLRP3-ASC, which increases β-amyloid (Aβ) secretion. The antioxidant Dl-3-n-butylphthalide (Dl-NBP) is commonly used for cerebral ischemia treatment. In our study, we elucidated for new mechanisms by which Dl-NBP enhanced TrX activity, suppressed TXNIP, and ameliorated neuronal apoptosis in the APP/PS1 mouse brains. In human glioblastoma A172 cells and neuroblastoma SH-SY5Y cells, we delineated the Dl-NBP-mediated signaling pathways by which Dl-NBP-dependent upregulation of Nrf2 mediated the reciprocal regulation of reducing proinflammatory cytokine and inhibiting Aβ production in the glial and neuronal cells overexpressing APPswe.
Innovation:
Our data provide a novel insight into the molecular mechanism that impairments of Nrf2-TXNIP-TrX system may be involved in the imbalance of cellular redox homeostasis and inflammatory damage in the AD brain.
Conclusion:
Dl-NBP treatment could suppress TXNIP-NLRP3 interaction and inhibit NLRP3 inflammasome activation via upregulating Nrf2. These findings may provide an instrumental therapeutic approach for AD. Antioxid. Redox Signal. 00, 000–000.
Introduction
I
The underlying mechanism involved in the abnormal homeostasis of thioredoxin-interacting protein-thioredoxin (TXNIP-TrX) in the Alzheimer's disease (AD) remains unclear. Using the APP/PS1 double transgenic mouse and human-derived neuronal cells, we disclosed a novel insight into the molecular mechanism that the impairment of nuclear factor erythroid 2-related factor 2 (Nrf2)-TXNIP-TrX system may be involved in the imbalance of cellular redox homeostasis and inflammatory damage in Alzheimer's-like pathology. Dl-3-n-butylphthalide suppresses TXNIP-nucleotide binding oligomerization domain-like receptor family, pyrin domain containing 3 (NLRP3) interaction and inhibits NLRP3 inflammasome via the upregulation of Nrf2. Our findings provide a potential therapeutic approach for AD.
Nucleotide binding oligomerization domain-like receptor family, pyrin domain containing 3 (NLRP3) inflammasome can be detected in inflammatory cells. NLRP3 inflammasome is activated in AD and contributes to the pathology in the brain of an Alzheimer's transgenic (Tg) mouse (21). Indeed, inflammatory stimuli can trigger β-amyloid (Aβ) deposition in the wild type (WT) mouse brain and are also known to aggravate Aβ pathology in the AD mouse model (34). It is postulated that chronic inflammation in the brain triggers oxidative stress and inhibits synaptic transmission, leading to synaptic deficits (55). Although the temporal profile and precise contribution of oxidative stress and inflammation remain unclear in the pathogenesis of AD, early intervention may be effective, especially at asymptomatic stages.
Oxidative stress and inflammation are modified by the redox status. Nuclear factor erythroid 2-related factor 2 (Nrf2) is a master endogenous regulator in the protection against oxidative stress. In response to oxidative stress or other inducers, such as misfolded proteins or inflammation, Nrf2 localizes to the nucleus and drives the transcription of antioxidant enzymes, which restores proper oxidative balance, inactivates toxic chemicals, and facilitates protein degradation (31). However, dysregulation of Nrf2-regulated genes exacerbates the cytotoxicity of oxidative stress and results in cell dysfunction (25).
Nrf2 protein expression is substantially reduced in the nuclear lysates of the hippocampus and frontal cortex tissues of AD patients (54). Moreover, lower levels of Nrf2 mRNA and protein expression are shown in the brains of AD Tg mice (30), suggesting inactivation or impairment of the Nrf2 system in the AD brain. Importantly, Nrf2 is a key regulator for thioredoxin-interacting protein (TXNIP) transcription, maintaining the basal expression of TXNIP at a low level (20). TXNIP is an endogenous inhibitor of the antioxidant thioredoxin (TrX). TrX is a major intracellular thiol-reducing and reactive oxygen species (ROS)-scavenging protein. The binding of TXNIP to TrX inhibits TrX activation and promotes oxidative stress. During oxidative conditions, ROS accumulation contributes to TXNIP-TrX dissociation, which facilitates the interaction between NLRP3 and TXNIP, and NLRP3-TXNIP is required for the formation and activation of inflammasomes (17, 41). Thus, TXNIP is postulated to be a critical switch for linking oxidative stress to inflammation. Interestingly, Nrf2 has been shown to alleviate oxidative damage of cardiomyocytes in diabetic mice by repressing the expression of TXNIP (20). Nrf2-mediated TrX expression represses the activation of inflammasomes and protects macrophages against inflammatory damage (36). However, the interplay between TXNIP, TrX, and Nrf2 in regulating redox reactions and oxidative stress in AD has not been described.
Dl-3-n-butylphthalide (Dl-NBP) is favorably safe (11) in clinical use for anticerebral ischemia and was approved by the State Food and Drug Administration of China in 2002. It was observed that Dl-NBP could provide a neuroprotective effect on patients with radiation-induced brain injury (73). There are no available data on the usage of Dl-NBP for AD treatment. Dl-NBP has been reported to protect endothelial cells from oxygen glucose deprivation-induced oxidative damage in vitro (37). We previously reported that Dl-NBP administration increased the recruitment of CREB-binding protein to the promoters of Nrf2 downstream genes in the APP/PS1 mouse brain (67). In the present study, we observed that the downregulation of Nrf2 was accompanied by deficits in TXNIP inhibition in the APP/PS1 mouse brain. We evaluated the effects of Dl-NBP on TXNIP/TrX and NLRP3 activity in the brains of APP/PS1 Tg mice. Dl-NBP treatment upregulates Nrf2 and enhances the inhibition to TXNIP in the APP/PS1 mouse brain. Furthermore, the neuroprotective effects of Dl-NBP on the inhibition of TXNIP and the activation of TrX via Nrf2 in the neuron were confirmed using human neuroblastoma SH-SY5Y cells stably transfected with the Swedish mutant form of APP (APPswe) in vitro. These data indicate a multitarget neuroprotective function for dl-NBP. Our results suggest that impairment of the Nrf2/TXNIP/TrX system may be a novel molecular mechanism involved in the imbalance of cellular redox homeostasis in the AD brain, and our results provide a potential therapeutic approach involving the inhibition of TXNIP through the enhancement of Nrf2 signaling.
Results
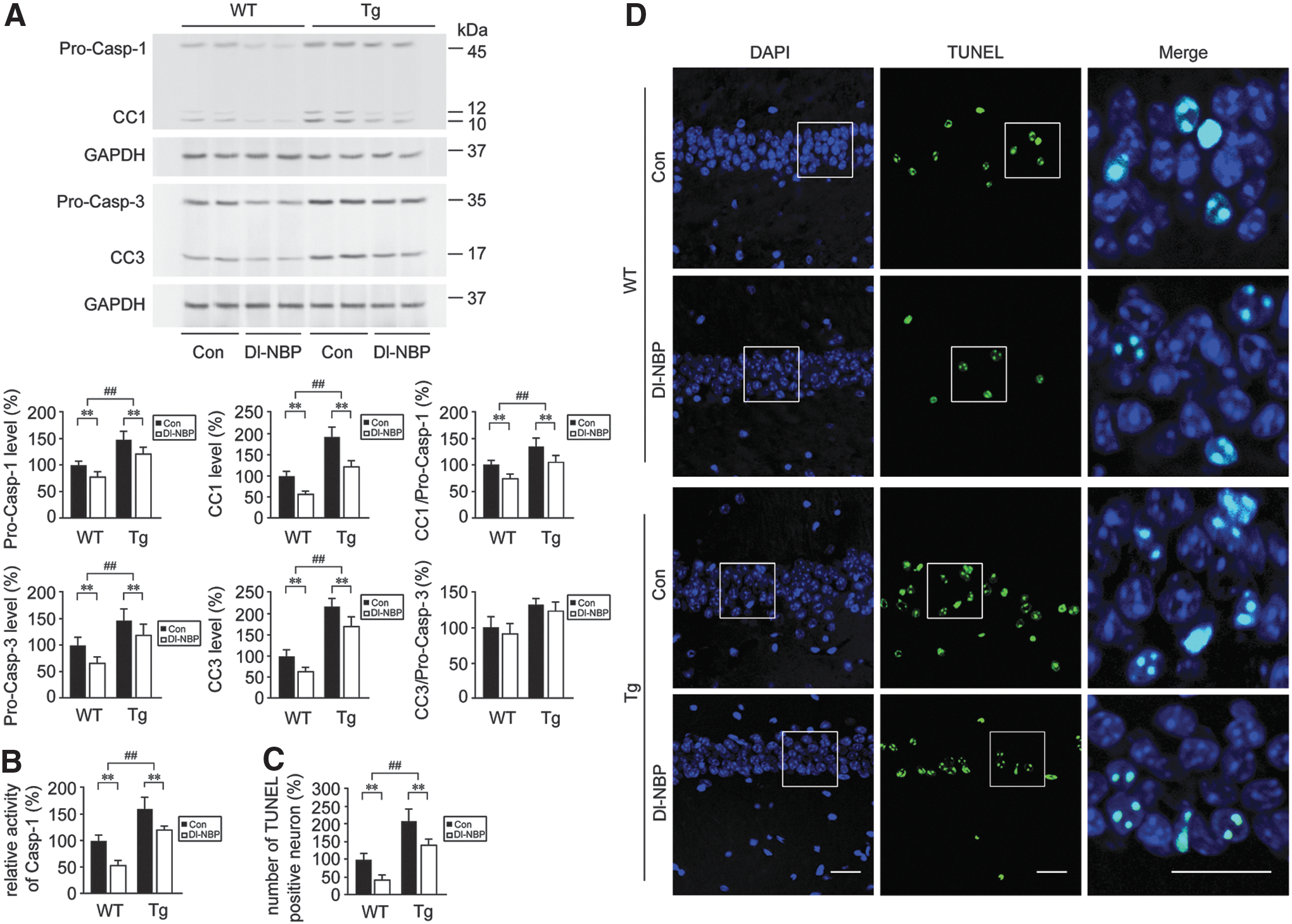
Dl-NBP treatment mitigates neuronal apoptosis in the hippocampus of the APP/PS1 mouse brain
It is considered and reviewed that neuronal cells are vulnerable to apoptosis in the AD brain (42, 57). Caspases are critical mediators of apoptosis and inflammation (24). Caspase-1 activation induces the maturation of IL-1β and subsequently potentiates inflammatory responses (35). Chronic activation of caspase-3 is involved in mediating the degenerative process in the aging brain (60). Increases of caspase-1 and caspase-3 immunoreactivity have been observed in the postmortem brains with AD (21, 63). To determine the level of genotype-related neurodegeneration and whether Dl-NBP could be neuroprotective in the APP/PS1 mouse brain, apoptotic markers were measured. As shown in Figure 1A and Supplementary Figure S1, Western blot analysis for cleaved caspase-1 (CC1) in the hippocampus revealed significant effects of genotype [F(1,20) = 14.293, p < 0.01] and Dl-NBP treatment [F(1,20) = 10.857, p < 0.01] in the absence of genotype × Dl-NBP interaction [F(1,20) = 0.305, p > 0.05]. Dramatic increases in the levels of CC1 were observed in APP/PS1 mice with respect to WT mice (p < 0.01). The effects of genotype [F(1,20) = 20.376, p < 0.01] and Dl-NBP [F(1,20) = 13.814, p < 0.01] or genotype × Dl-NBP interaction [F(1,20) = 0.409, p > 0.05] on the protein expression of procaspase-1 (Pro-Casp-1) were observed as well. We then assess the ratios of CC1 to Pro-Casp-1. Two-way analysis of variance (ANOVA) for CC1/Pro-Casp-1 ratios showed evident effects of genotype [F(1,20) = 86.171, p < 0.01] and Dl-NBP [F(1,20) = 124.117, p < 0.01], but not genotype × Dl-NBP interactions [F(1,20) = 1.237, p > 0.05]. CC1/Pro-Casp-1 ratios were higher in the hippocampus of the Tg groups in comparison with the age-matched WT groups (p < 0.01, Fig. 1A), which indicated increases in the CC-1 from Pro-Casp-1 in the APP/PS1 mouse brain. Western blot analysis for cleaved caspase-3 (CC3) and procaspase-3 (Pro-Casp-3) revealed significant main effects of genotype (p < 0.01) and Dl-NBP (p < 0.01), but not a genotype × Dl-NBP interaction (p > 0.05). The CC3/Pro-Casp-1 ratios were also increased in Tg mouse brains compared with those in the age-matched WT controls (p < 0.01, Fig. 1A). The caspase-1 activities were higher in the Tg group than those in the WT group (p < 0.01, Fig. 1B). Dl-NBP treatment reduced the ratios of CC1 to Pro-Casp-1 in both WT and Tg mice compared with those in the vehicle-treated controls (p < 0.01, Fig. 1A). The ratio of CC3 to Pro-Casp-3 in Dl-NBP-treated group was comparable with respect to that in the vehicle-treated controls in the hippocampus of WT and Tg mice (p > 0.05, Fig. 1A), although a trend toward a decrease. To confirm the level of Dl-NBP-induced mitigation of neuronal apoptosis in the hippocampus, we performed immunostaining analysis using TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling) which reveals DNA fragmentation associated with apoptotic signaling cascades. As shown in Figure 1C and D, the number of TUNEL-positive cells was markedly higher in the hippocampus of the brain from the Tg mice than in the WT mice. Two-way ANOVA for TUNEL-positive cells revealed evident main effects of genotype [F(1,20) = 18.443, p < 0.01] and Dl-NBP [F(1,20) = 16.539, p < 0.01], but not a genotype × Dl-NBP interaction [F(1,20) = 2.062, p > 0.05, Fig. 1C, D]. The number of TUNEL staining cells was significantly lower in the hippocampus of Dl-NBP-treated group in both mouse genotypes than in the vehicle-treated controls (p < 0.01, Fig. 1C, D). Dl-NBP administration did not significantly affect the body weight and motility of the mice (data not shown).
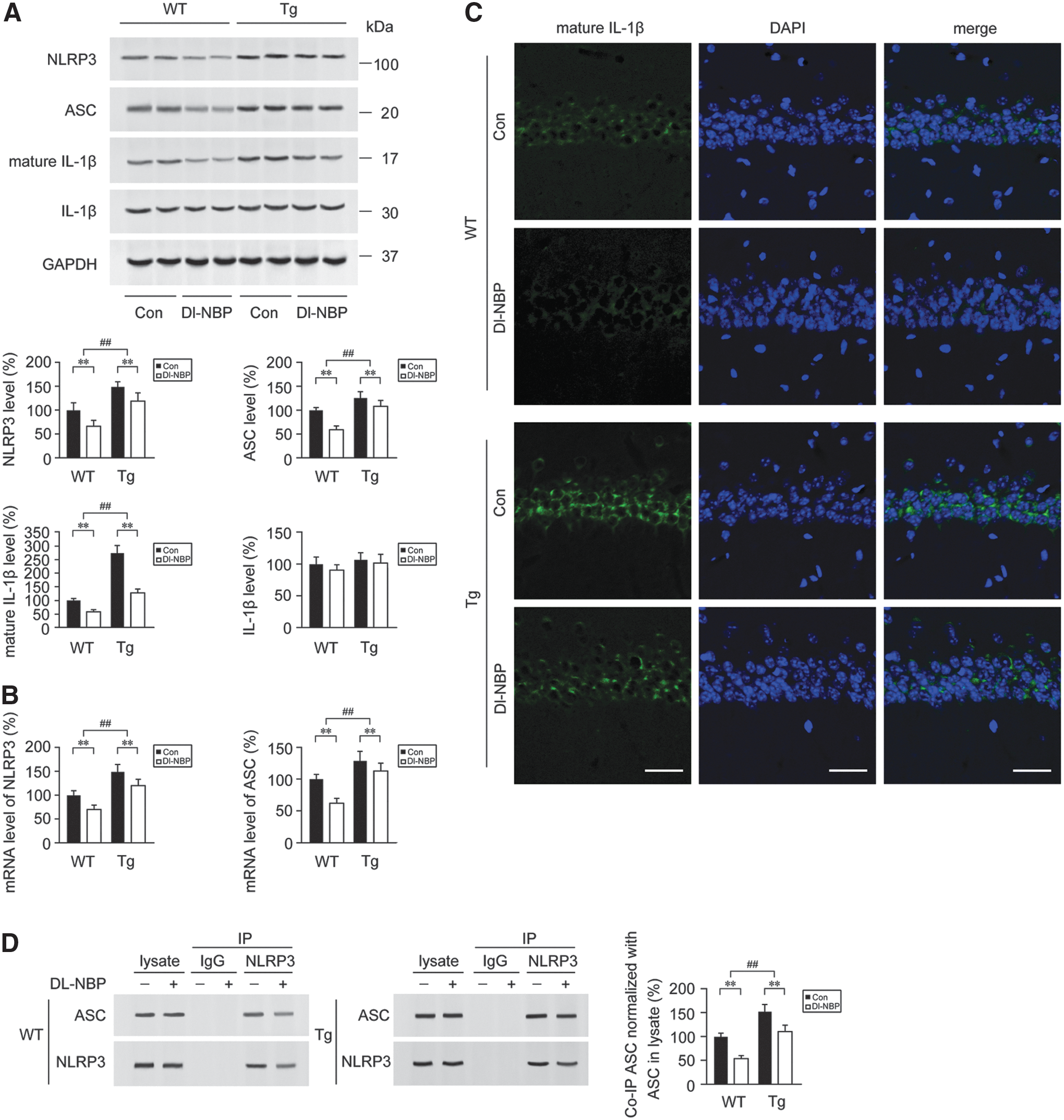
Dl-NBP treatment inhibits the activation of NLRP3 inflammasomes in the APP/PS1 mouse brain
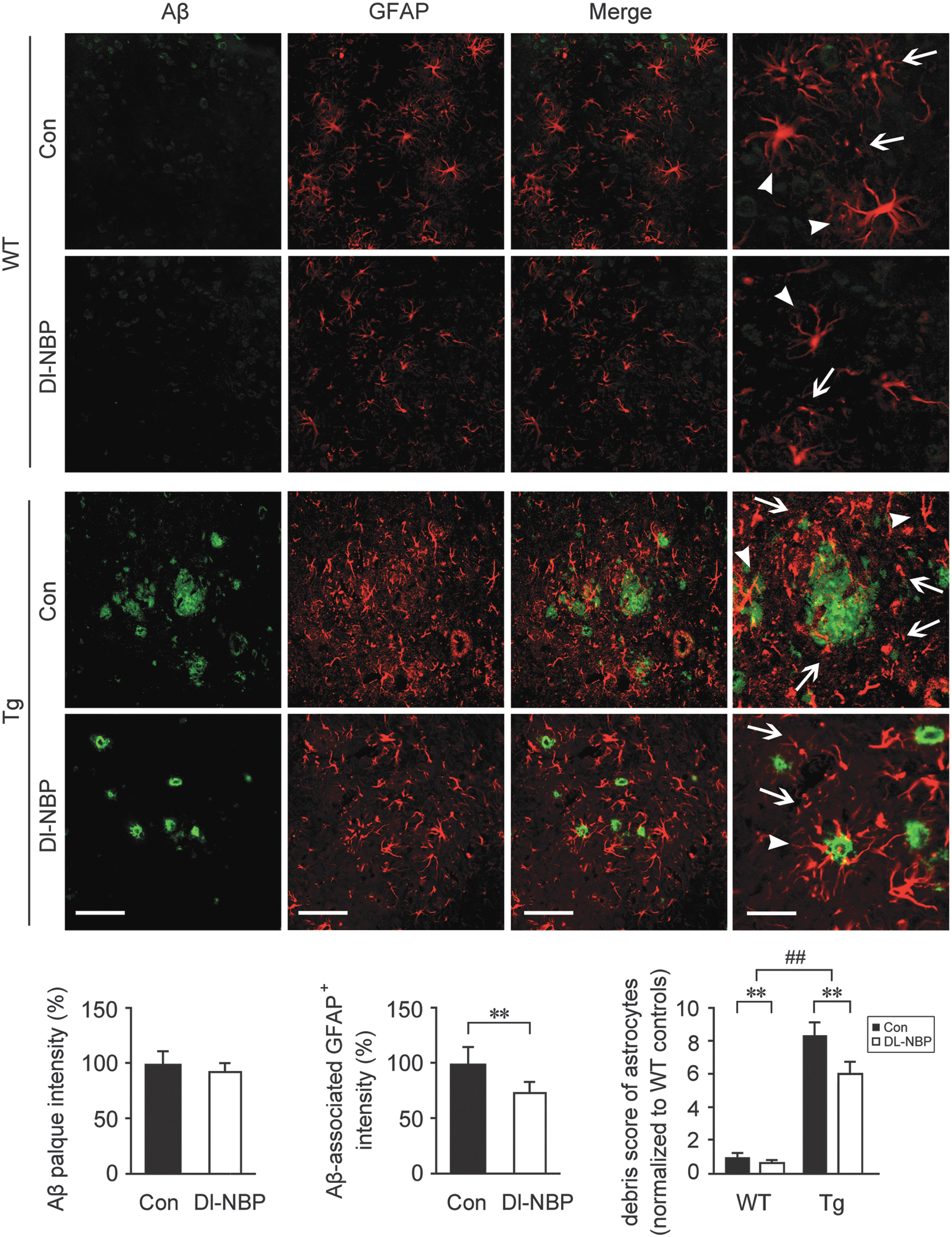
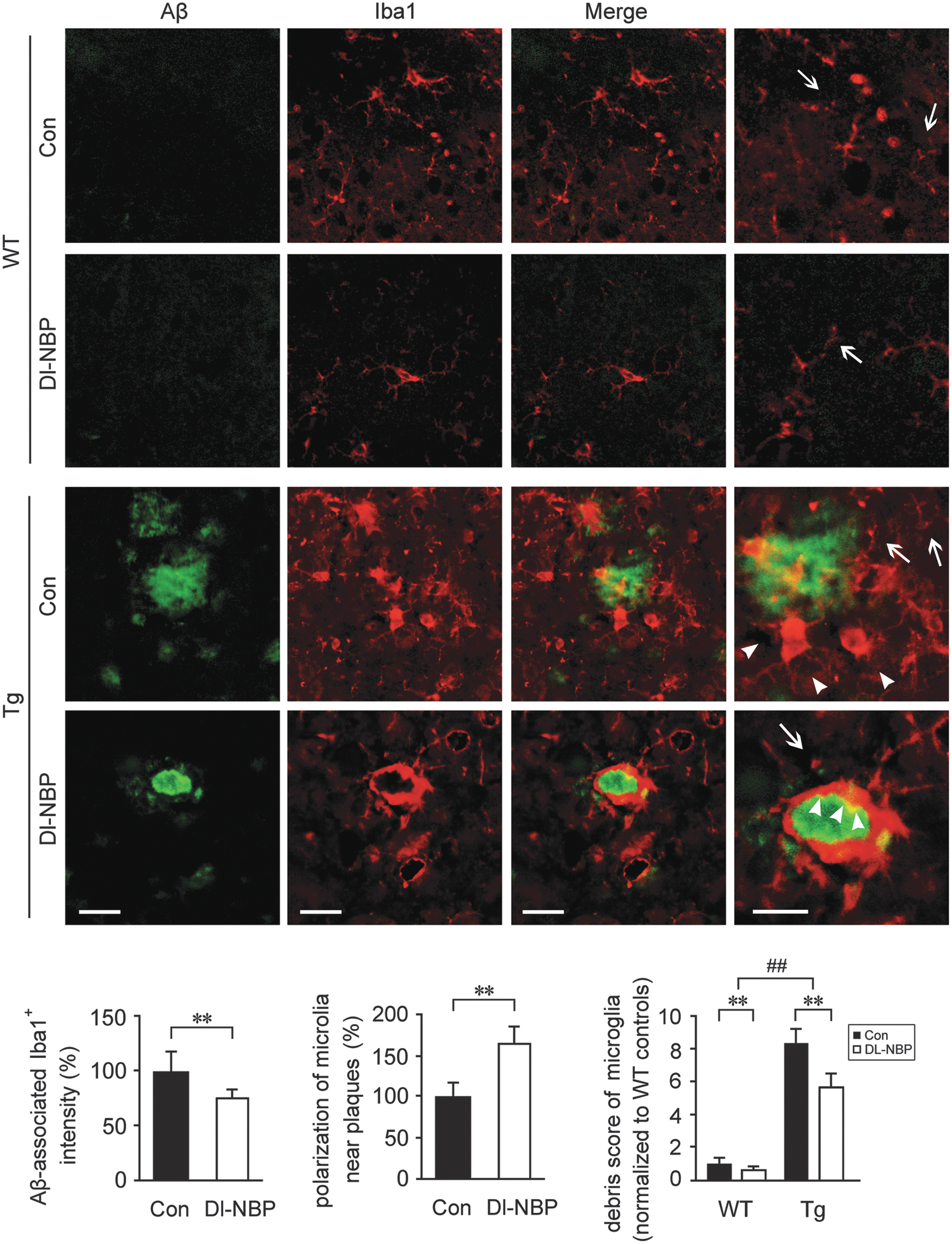
It was observed that caspase-1-induced release of mature IL-1β can be mediated by the activation of NLRP3 inflammasome (21). To determine whether Dl-NBP-mediated inhibition of caspase-1 processing in the APP/PS1 mouse brain was related to the inhibition of NLRP3 inflammasome formation, we detected the inflammasome components, including NLRP3, adaptor molecule apoptosis-associated speck-like protein containing a CARD (ASC), and IL-1β in the APP/PS1 mouse brain. Two-way ANOVA for the protein expression of NLRP3 showed a significant genotype effect [F(1,20) = 8.137, p < 0.01] together with a significant main effect of Dl-NBP [F(1,20) = 49.853, p < 0.01, Fig. 2A and Supplementary Fig. S1] in the absence of genotype × Dl-NBP interaction [F(1,20) = 0.173, p > 0.05]. We observed that the protein expression of NLRP3 was significantly higher in the brains from Tg mice than in the age-matched WT group (p < 0.01, Fig. 2A and Supplementary Fig. S1). The upregulations of inflammasome-related components indicate the activation of the NLRP3 inflammasome (65). Once the NLRP3 is activated, it recruits ASC and procaspase-1. The assembled NLRP3 inflammasome induces the transformation of procaspase-1 to CC1, cleaving IL-1β into mature IL-1β (33). In the present study, Dl-NBP administration led to a decrease in NLRP3 protein expression (p < 0.01, Fig. 2A) and NLRP3 mRNA levels (p < 0.01, Fig. 2B) in both genotypes compared with the vehicle-treated controls, and the effects of Dl-NBP on ASC were similar (p < 0.01, Fig. 2A, B). As demonstrated by Western blot analysis for mature, 17 kDa IL-1β, evident main effects of genotype [F(1,20) = 55.691, p < 0.01, Fig. 2A] and Dl-NBP treatment [F(1,20) = 28.027, p < 0.01, Fig. 2A, B] were observed, but not significant genotype × Dl-NBP interactions [F(1,20) = 1.279, p > 0.05, Fig. 2A]. Dl-NBP treatment repressed the increase of 17 kDa IL-1β in the Tg and WT mouse brains (p < 0.01, Fig. 2A, B). The results of 17 kDa IL-1β immunofluorescence staining confirmed the effects of Dl-NBP on the expression of the inflammatory molecule (Fig. 2C): the number of IL-1β+ cells in the hippocampus of APP/PS1 mice was markedly higher than that of the WT mice (p < 0.01). Under Dl-NBP administration, the number of IL-1β immune-stained cells was significantly reduced in both mouse genotypes compared with controls (p < 0.01, Fig. 2C). Furthermore, coimmunoprecipitation (Co-IP) analyses showed that significant increases in ASC-associated NLRP3 protein were detected in the APP/PS1 mouse brain relative to those in the age-matched WT group. Two-way ANOVA for ASC-associated NLRP3 revealed significant main effects of genotype [F(1,20) = 26.331, p < 0.01] and Dl-NBP [F(1,20) = 13.281, p < 0.01, Fig. 2A and Supplementary Fig. S1], but not a genotype × Dl-NBP interaction [F(1,20) = 0.161, p > 0.05] (Fig. 2D and Supplementary Fig. S2). NLRP3 protein was discovered to be complexed together with ASC, whereas the process of NLRP3 inflammasome formation was interrupted by Dl-NBP administration. Dl-NBP treatment triggered a decrease in ASC-associated NLRP3 protein expression in the brains of WT and Tg mice (p < 0.01, Fig. 2D). We also evaluated the inflammatory response by detecting the distribution and expression of GFAP+ (an astrocytic marker) and Iba1+ (a microglial marker) cells. We have previously reported that Dl-NBP treatment reduced the levels of soluble Aβ and Aβ oligomer in the APP/PS1 mouse brain, but did not alter the fibrillar Aβ levels (67). In the present study, double immunofluorescence staining with anti-Aβ and glial fibrillary acidic protein (GFAP) antibodies showed that there was more severe astrocytosis in Tg mouse brain (Fig. 3). The diffuse plaques were surrounded by astrocyte clusters, which displayed dystrophic characteristics with large somas, hypertrophic primary processes, and intense GFAP immunoreactivity (56). The intensity of Aβ-associated GFAP+ cells in Dl-NBP-treated APP/PS1 mice brain was less than that in the vehicle-treated APP/PS1 mice (p < 0.01), although there was no statistical difference in the fluorescence intensity of Aβ plaque between these two groups (p > 0.05). Two-way ANOVA for debris score of astrocytes revealed significant main effects of genotype [F(1,20) = 142.962, p < 0.01] and Dl-NBP [F(1,20) = 94.308, p < 0.01] in the absence of genotype × Dl-NBP interaction [F(1,20) = 1.724, p > 0.05]. As the GFAP-immunoreactive astrocytes, Iba1+ microglia formed significant inflammatory foci surrounding the Aβ plaque in the Tg mice (Fig. 4). The cell bodies of ionized calcium binding adaptor molecule 1 (Iba1)-immunoreactive microglial cells were swollen in the Tg mouse brain. The fluorescence intensity of Aβ plaque-associated Iba1+ cells was reduced in the Dl-NBP-treated Tg mice than in the vehicle-treated Tg controls (p < 0.01). Two-way ANOVA for the fragmented microglia revealed significant main effects of genotype [F(1,20) = 89.206, p < 0.01] and Dl-NBP [F(1,20) = 51.847, p < 0.01], but not genotype × Dl-NBP interactions [F(1,20) = 1.422, p > 0.05]. Dl-NBP treatment reduced microglial debris in both WT and Tg mice with respect to those in their vehicle controls (p < 0.01). Interestingly, the microglial cells in the brain of Dl-NBP-administered Tg mice were closely tracked to the boundaries of the compact Aβ plaques and formed a barrier-like structure with respect to vehicle-treated Tg mice (p < 0.01).
Dl-NBP treatment reduces TXNIP expression and suppresses TXNIP-NLRP3 interaction
TXNIP is the endogenous negative regulator of TrX, which is a major cellular antioxidant and antiapoptotic protein (47). Studies have demonstrated the potential role of TXNIP to serve as a binding partner for NLRP3 and for activating the NLRP3-caspase-1 inflammasome in retinal capillary endothelial cells (51) and in retinal Muller glia (15). In the present study, we assessed the expression of TXNIP in human postmortem brains of AD patients and normal controls. As shown in Figure 5A, the immunoreactivity of TXNIP was markedly induced in the cerebral cortex of patients with AD. The cortex of AD samples demonstrated significant increases in the number of cells with high levels of TXNIP expression (> 40% of total cell) (p < 0.01). The intensity of TXNIP staining was increased in the AD group relative to those in the control subjects (p < 0.01). The high-power micrographs of bottom panels in Figure 5A illustrate that the immunoreactive TXNIP is tightly associated with the neuron and glia morphology. To investigate whether TXNIP is an effective inducer for the activation of the NLRP3 inflammasome in AD-like pathology and to explore whether treatment with Dl-NBP exerts anti-inflammatory effects, the distribution and expression of TXNIP in the APPP/PS1 and age-matched WT mouse brains were analyzed. Western blot assays for TXNIP protein expression revealed the significant main effects of genotype [F(1,20) = 63.112, p < 0.01, Fig. 5B] and Dl-NBP treatment [F(1,20) = 43.699, p < 0.01, Fig. 5B], but not significant genotype × Dl-NBP interactions [F(1,20) = 3.325, p > 0.05, Fig. 5B]. TXNIP expression was significantly increased in the brains of APP/PS1 mice relative to that in the age-matched WT group (p < 0.01, Fig. 5B and Supplementary Fig. S2). Quantitative real-time polymerase chain reaction (PCR) assays showed that the TXNIP mRNA levels were significantly elevated in the Tg mouse brain compared with those in the WT group (p < 0.01, Fig. 5C). In contrast, the protein expression and mRNA levels of TXNIP were markedly lower under Dl-NBP administration than those with the vehicle treatment in both mouse genotypes (p < 0.01, Fig. 5B, C). Co-IP analysis demonstrated the evident binding of TXNIP to NLRP3 in the APP/PS1 mouse brain. Two-way ANOVA for TXNIP-associated NLRP3 showed significant main effects of genotype [F(1,20) = 26.331, p < 0.01] and Dl-NBP [F(1,20) = 13.281, p < 0.01], but not genotype × Dl-NBP interaction [F(1,20) = 0.616, p > 0.05] (Fig. 5D and Supplementary Fig. S2). On the contrary, Dl-NBP-mediated inhibition of TXNIP was accompanied by the prevention of TXNIP-NLRP3 interaction in the APP/PS1 mouse brain (p < 0.01, Fig. 5D). Double immunofluorescence labeling showed the morphological distribution and expression of TXNIP. TXNIP staining in the neuronal cells and neuroglia in the brains of mice was observed (Fig. 5E–H). TXNIP-positive astrocytes and microglia were significantly identifiable in the Tg mice, as exemplified by the cortical and hippocampal section shown, whereas with the decreases of immunoreactive astrocytes and microglia in the brains of Dl-NBP-treated Tg mice (as described in Figs. 3 and 4), the weaker expression of TXNIP immunoproducts was observed in the cytoplasm of granulosa neurons, in the GFAP+ cells of the hippocampus, and in the cortex of Dl-NBP-administered Tg mouse brain (Fig. 5E–H).

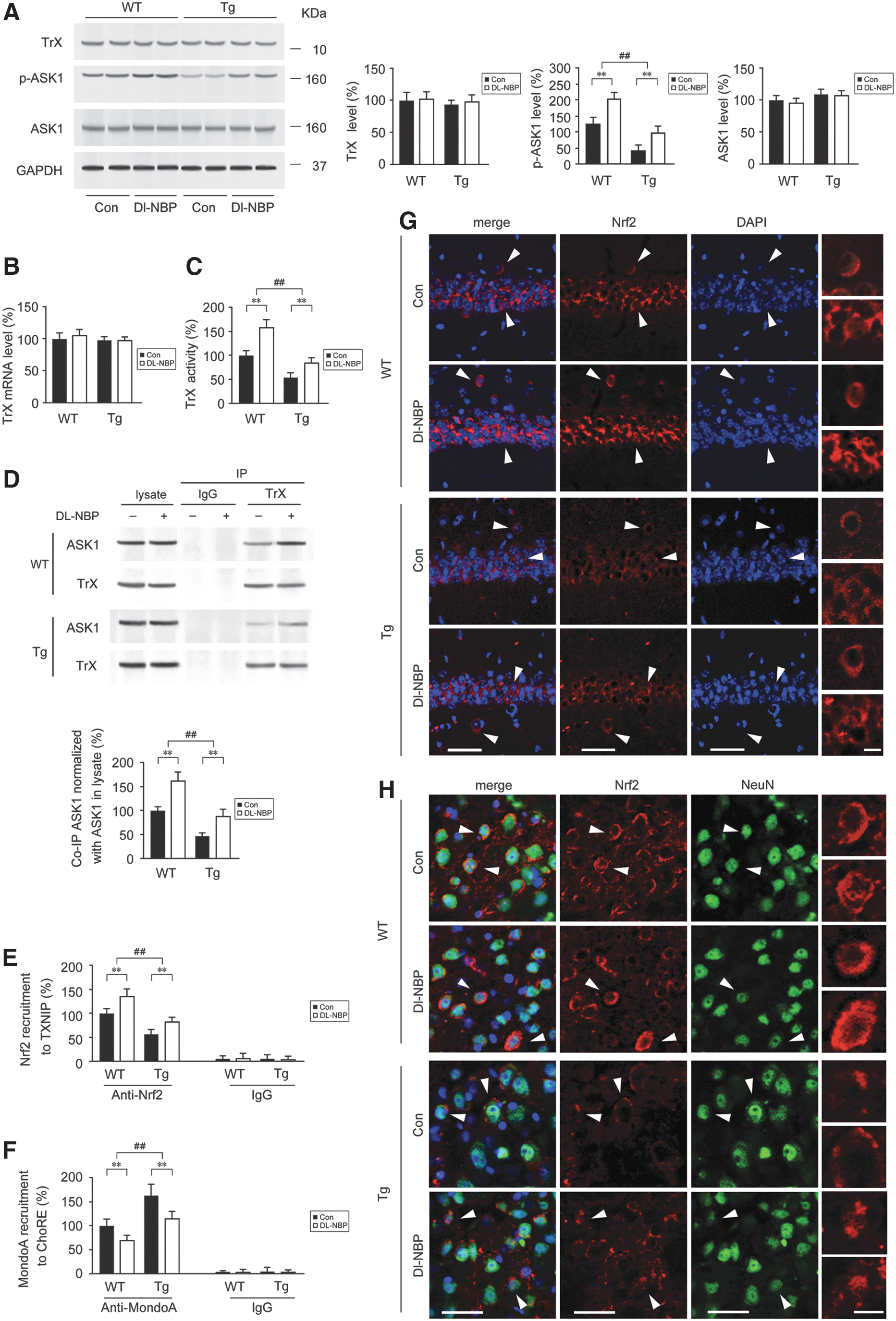
Dl-NBP-mediated upregulation of Nrf2 inhibits TXNIP transcription and enhances TrX activity
We previously reported that Dl-NBP treatment upregulated Nrf2 signaling in the APP/PS1 mouse brain (67). It has been reported that Nrf2 boosted TrX activity by suppressing TXNIP in the streptozotocin-induced diabetes mouse model (20). To explore whether the Dl-NBP-mediated inhibition of TXNIP is involved in the upregulation of Nrf2 signaling, the protein expression, activity, and mRNA level of TrX were detected. As shown in Figure 6A and Supplementary Figure S3, Western blot analysis showed that the protein expression of TrX in the APP/PS1 mouse brain was comparable with that in the WT group: neither significant genotype effect [F(1,20) = 1.213, p > 0.05] nor Dl-NBP effect [F(1,20) = 0.298, p > 0.05] or genotype × Dl-NBP interaction [F(1,20) = 0.016, p > 0.05] was observed, and similar results were obtained for the mRNA levels (p > 0.05, Fig. 6B). Attenuation of TrX activity was observed in the APP/PS1 mouse brain compared with that in the WT mouse brain (p < 0.01, Fig. 6C). Under Dl-NBP treatment, the activity of TrX was significantly higher in both mouse genotypes than that in the vehicle controls (p < 0.01, Fig. 6C). The binding of TXNIP to reduced-TrX inhibits TrX activity and impairs TrX-apoptosis signal-regulating kinase 1 (ASK1) interaction, leading to the release of ASK1, which mediates stress-induced cellular apoptosis (70). Considering the negative regulation of TrX to ASK1 (61), we evaluated the protein expression of ASK1 and in the mouse brain. As shown in Figure 6A, two-way ANOVA for ASK1 protein expression indicated that neither significant genotype effect [F(1,20) = 0.030, p > 0.05] nor Dl-NBP effect [F(1,20) = 2.949, p > 0.05] or genotype × Dl-NBP interaction [F(1,20) = 0.107, p > 0.05] was observed. Phosphorylates of ASK1 at Ser83 indicate the inhibition of ASK1 activity, promoting cell survival (32). In the present study, the levels of phosphorylated Ser 83 of ASK1 (p-ASK1) were markedly lower in the Tg mouse brain compared with those in the WT mouse brain (p < 0.01, Fig. 6A). Dl-NBP treatment led to an increase of p-ASK1 level in both mouse genotypes compared with those in the controls (p < 0.01, Fig. 6A). Furthermore, co-IP assays for TrX-associated ASK1 showed significant main effects of genotype [F(1,20) = 28.743, p < 0.01] and Dl-NBP [F(1,20) = 22.264, p < 0.01], but not significant genotype × Dl-NBP interactions [F(1,20) = 2.420, p > 0.05] (Fig. 6D and Supplementary Fig. S3). The TrX-ASK1 interaction in the Tg mouse brain was less than of the WT group (p < 0.01, Fig. 6D). Thus, according to previous work reported by Zhou and colleagues (75) and our results, the dissociation of TXNIP from oxidized TrX likely facilitates TXNIP activation and interactions with the NLRP3 inflammasome in AD. We then performed chromatin immunoprecipitation (ChIP) to analyze the presence of Nrf2 at ARE sites on the promoters of TXNIP genes in the brains of Tg and WT mice. As shown in Figure 6E, the binding of Nrf2 to the TXNIP ARE was lower in the APP/PS1 mouse brain than WT mice (p < 0.01). Dl-NBP treatment significantly increased the binding of Nrf2 to the TXNIP ARE in the brains of both mouse genotypes compared with the vehicle controls (Fig. 6E, p < 0.01). MondoA, a transcription factor of the basic helix-loop-helix leucine zipper family, is required for TXNIP induction (43, 59). The binding of MondoA to carbohydrate response element (ChoRE) is involved in the positive regulation for TXNIP transcription (20, 71). It is reported that Nrf2 may suppress MondoA-ChoRE interaction to keep TXNIP expression at a low level, preventing diabetes (20). To investigate whether the effects of Nrf2 on TXNIP were related to Nrf2-mediated blocks on MondoA-ChoRE interaction, we assessed the binding of MondoA to TXNIP ChoRE-a by ChIP. The increases of MondoA-ChoRE were observed in the APP/PS1 mouse brain relative to that in the age-matched WT mice (Fig. 6F, p < 0.01), whereas Dl-NBP management significantly reduced the recruitment of MondoA to ChoRE in comparison with those of controls in both mouse genotypes (Fig. 6F, p < 0.01). We previously reported that the nuclear levels of Nrf2 were reduced in the APP/PS1 mouse brain compared with those of the age-matched WT mouse brain (67). In the present study, we detected the location and expression of Nrf2 in the CA1 region of hippocampus and the frontal cortex from WT and Tg mouse brain. Immunofluorescence labeling showed that Nrf2 reactivity was observed in the cytoplasm or in addition within the nucleus of neuronal granulosa cells (Fig. 6G) and in the neurons of the cortex (Fig. 6H). The immunofluorescence staining for nuclear Nrf2 in the hippocampus showed significant main effects of genotype [F(1,20) = 17.640, p < 0.01] and Dl-NBP [F(1,20) = 15.089, p < 0.01], but not genotype × Dl-NBP interaction [F(1,20) = 2.073, p > 0.05] (Fig. 6G and Supplementary Fig. S4). The quantifications of cytosolic Nrf2 staining in the hippocampus were observed as well. Similar results of Nrf2 staining in the frontal cortex were observed (Fig. 6H and Supplementary Fig. S4). The intensity of nuclear and cytosolic Nrf2 staining was weaker in the Tg group than in the WT mice (p < 0.01). In contrast, Dl-NBP management caused significant increases in the intensity of nuclear and cytosolic Nrf2 in both mouse genotypes relative to their vehicle controls (p < 0.01).
Dl-NBP treatment inhibits IL-1β secretion and caspase-1 activation, mitigating amyloidosis in neuronal cells
Considering the hypothesis that Nrf2 is the “master regulator” of the antioxidant response and modulates immunological signaling (25), we verified the effect of Dl-NBP on inflammatory mediator-triggered cell damage in human-derived glial A172 cells in vitro. As a major component of all gram-negative bacterial cell walls, lipopolysaccharides (LPS) are considered to be an inducer to trigger proinflammatory responses. It has been reported that suppression of the LPS-triggered inflammatory system was Nrf2 pathway relevant in macrophages in vitro (44). In the present study, LPS was used to induce neuroinflammation in A172 cells. Western blot analysis revealed that LPS (1 μg/mL) stimulated caspase-1 activation, as confirmed by the appearance of processed caspase-1 (p < 0.01, Fig. 7A and Supplementary Fig. S5). Elevated secretion of mature IL-1β was also observed in the culture medium of LPS-treated A172 cells relative to vehicle controls (p < 0.01, Fig. 7B). However, treatment with Dl-NBP reduced LPS-mediated protein expression of processed caspase-1 (p < 0.01, Fig. 7A) in A172 cells. A significant decrease in IL-1β levels was observed in the culture medium supernatants of Dl-NBP-treated A172 cells compared with those in the LPS-treated group (p < 0.01, Fig. 7B). In addition to caspase-1, we also assessed cell apoptosis by analyzing annexin V-FITC/propidium iodide (PI) staining. As shown in Figure 7C, flow cytometry assays showed that the apoptosis rate was elevated following LPS stimulation compared with controls (p < 0.01), whereas treatment with Dl-NBP significantly reduced the percentage of apoptotic cells (p < 0.01; Fig. 7C).
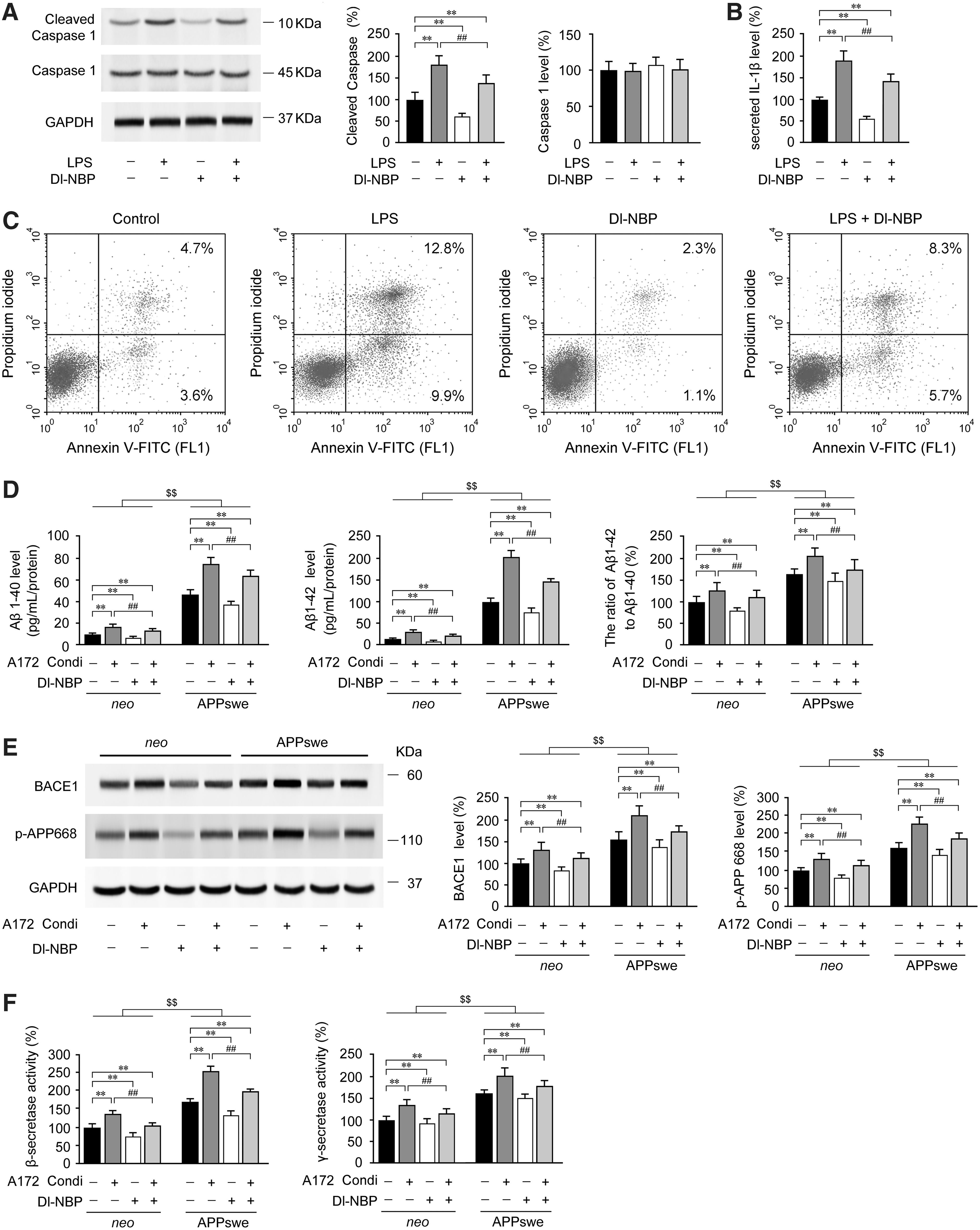
It has been reported that elevated IL-1β levels contribute to an increased risk of AD (45). We previously observed that Dl-NBP treatment led to decreases in soluble Aβ1–40, soluble Aβ1–42, and Aβ oligomers in the APP/PS1 mouse brain (67). It is speculated that sustained activation or impairment of glia results in a chronic inflammatory process, giving rise to neuronal dysfunction in AD-like pathology (7). To investigate whether the increased secretion of IL-1β in A172 cells facilitates the production of Aβ in human neuronal cells and to confirm the effects of Dl-NBP-mediated inhibition of neuroinflammation on Aβ accumulation, experiments were performed to treat SH-SY5Y cells overexpressing the APPswe or empty vector (neo) with conditioned medium from A172 cells with or without Dl-NBP treatment. IL-1β antibody (1 μg/mL) was added to confirm that IL-1β secretion from LPS-primed A172 cells was essential for inducing amyloidosis in APPswe or neo cells. We discovered that Dl-NBP (p < 0.01, Fig. 7D) or IL-1β antibody (data not shown) incubation abrogated the effects of LPS-treated conditioned medium from A172 cells, and the conditioned medium facilitated the increases of Aβ1–40 and Aβ1–42 secretion in both APPswe and neo cells (p < 0.01, Fig. 7D). Compared with the neo cells, the protein expression of β-site of APP-cleaving enzyme (BACE-1) and phospho-APP (p-APP668) was significantly higher in the APPswe cells cultured with the conditioned medium from LPS-primed A172 cells (p < 0.01; Fig. 7E and Supplementary Fig. S5), and the alterations of the activity in the β- and γ-secretase were as well (p < 0.01, Fig. 7F), however, Dl-NBP treatment partly inhibited the protein expression of BACE-1 and p-APP668 (p < 0.01, Fig. 7E), and repressed the upregulation of β- and γ-secretase activity (p < 0.01, Fig. 7F). Our results highlight an inflammatory-mediated reciprocal regulation between glial and neuronal cells that may contribute AD-like pathology, and suggest that Dl-NBP-mediated inhibition of IL-1β secretion facilitates the non-amyloidogenic processing in neuronal cells.
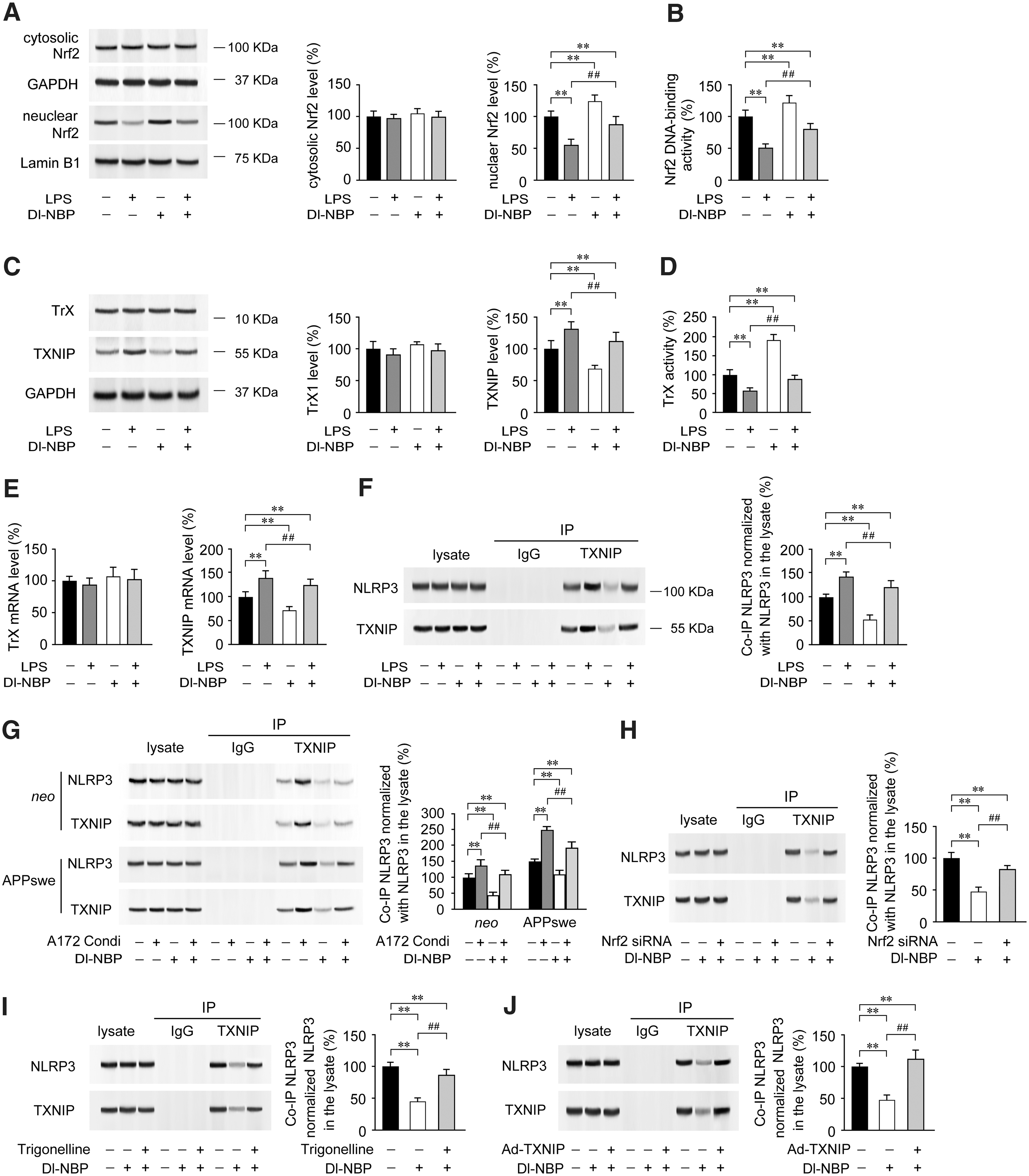
Dl-NBP-mediated upregulation of Nrf2 induces TXNIP inhibition and reduces TXNIP-NLRP3 binding
Previous study has demonstrated that TXNIP is inhibited by Nrf2 in the diabetic mouse heart (20). Considering our present findings that Dl-NBP treatment upregulates Nrf2 and inhibits TXNIP transcription in the APP/PS1 mouse brain, we examined the effects of Dl-NBP on the inflammation-induced signaling cascade of TXNIP in A172 cells in vitro. Our data revealed that LPS chronic treatment reduced nuclear protein expression (p < 0.01, Fig. 8A and Supplementary Fig. S5) and DNA binding activity of Nrf2, when compared with A172 cells on vehicle incubation (p < 0.01, Fig. 8B). However, the protein expression of cytosolic Nrf2 was not significantly altered (p > 0.05, Fig. 8A). Marked increases in protein expression (p < 0.01, Fig. 8C and Supplementary Fig. S6) and mRNA levels (p < 0.01, Fig. 8D) of TXNIP were observed in LPS-primed A172 cells compared with those of vehicle controls. There were no significant differences in the protein expression (p > 0.05, Fig. 8C) and mRNA levels (p > 0.05, Fig. 8D) of TrX among groups under indicated treatment. The activity of TrX in LPS-primed A172 cells was lower than in the controls (p < 0.01, Fig. 8E). Interestingly, the incubation of A172 cells with Dl-NBP (10 μM) significantly increased the levels of nuclear Nrf2 (p < 0.01, Fig. 8A) and Nrf2 DNA binding activity (p < 0.01, Fig. 8B). Dl-NBP treatment also inhibited the LPS-induced upregulation of TXNIP (p < 0.01, Fig. 8C). Accordingly, in the LPS-primed A172 cells, the TrX activity was higher under Dl-NBP administration than that without Dl-NBP addition (p < 0.01, Fig. 8D). IP assays showed a differential increase of TXNIP-associated NLRP3 protein expression in the LPS-primed A172 cells, when compared with A172 cells on vehicle incubation (p < 0.01, Fig. 8F and Supplementary Fig. S6). In contrast, Dl-NBP treatment caused a significant reduction in the interaction between TXNIP and NLRP3 relative to those of controls or LPS-primed A172 cells (p < 0.01, Fig. 8F). Interestingly, under treatment with the A172 condition medium, the binding of TXNIP to NLRP3 in the APPswe cells was higher than that in the neo cells (p < 0.01, Fig. 8G and Supplementary Fig. S6). Importantly, the TXNIP-associated NLRP3 protein was reduced under Dl-NBP treatment in both neo cells and APPswe cells, when compared with those treated by conditioned medium without Dl-NBP addition (p < 0.01, Fig. 8G). To verify the specificity of the pharmacological effects of Dl-NBP, A172 cells were transfected with small interfering RNAs (siRNAs) targeting Nrf2, treated with adenoviral overexpression of TXNIP (Ad-TXNIP) or incubated with 10 μM trigonelline (C7H7NO2, Nrf2 inhibitor) (1) before being incubated with Dl-NBP (10 μM). Co-IP analysis showed that Nrf2 siRNA (Fig. 8H and Supplementary Fig. S7), trigonelline treatment (Fig. 8I and Supplementary Fig. S7), or Ad-TXNIP (Fig. 8J and Supplementary Fig. S7) partly abrogated Dl-NBP-mediated inhibition of NLRP3-TXNIP interaction (p < 0.01) in A172 cells. These data further indicate that Dl-NBP-induced upregulation of Nrf2 inhibits TXNIP transcription and expression, blocks the TXNIP-NLRP3 interaction, and enhances TrX-mediated redox homeostasis.
Discussion
AD is considered a neurodegenerative disorder involving in Aβ plaque deposition, neurofibrillary tangle formation, and synapse loss in the brain (53). Clinical trials targeting anti-Aβ therapies failed to inhibit the progression of AD patients' cognitive decline (13), leading the field to explore novel targets for therapeutic intervention (9, 10). In AD brains, oxidative damage and decreases in antioxidant enzyme activity are predominantly localized to synapses, and synaptic alterations are associated with the severity of the disease (4) apart from Aβ plaques (6). TrXs, which are antioxidant defense proteins, play an essential role in cell function by participating in redox-dependent processes by regulating apoptosis and interacting with transcription factors to maintain protein folding (28, 49). TrX levels are predominately reduced in AD brain regions (40). Furthermore, TrX can be oxidized by Aβ in a rapid manner (2). These findings suggest that the deregulation of TrX antioxidant systems might contribute to the pathogenesis of AD. In the present study, the activity of TrX is significantly lower in the APP/PS1 mouse brain than that in the age-matched WT mouse brain, which indicates the imbalance of cellular redox homeostasis in AD-like pathology.
TXNIP, an endogenous inhibitor of TrX, interacts with the catalytic center of reduced TrX and inhibits its reducing activity (27). Studies have indicated that TXNIP is necessary for inducing IL-1β expression and promoting neurodegeneration under diabetic conditions (51) or apoptotic stimulus (58). The expression of TXNIP is enhanced in several diseases, such as diabetes mellitus, hypertension (70), and ischemia (5), which are all risk factors for the onset and progression of AD. Early overexpression of TXNIP was observed in the hippocampus of the brain from a 5 × AD mouse model (48). Aβ induced TXNIP expression in brain-derived endothelial cells in vitro (52). Furthermore, Zhou and colleagues identified TXNIP as a critical link between oxidative stress and inflammasome activation using cultured macrophages or TXNIP knockout (KO) mice (75). Under oxidative stress conditions, TXNIP dissociates from TrX and allows the binding of TXNIP to NLRP3, which contributes to the formation and activation of the inflammasome. The assembly of the NLRP3 inflammasome controls the process and production of the proinflammatory, cytokine IL-1β, which has been reported to be significantly increased in the brains of AD patients (19). It is postulated that NLRP3 inflammasome activation may mediate synaptic failure, cognitive deficits, and the observed beneficial microglial clearance dysfunction (21). Conversely, NLRP3-deficient AD Tg mice are protected from memory impairment (21). Suppressing the activation of the NLRP3 inflammasome to alleviate neuroinflammation may be an innovative therapeutic approach for AD (50). In the present study, the protein expression and mRNA levels of TXNIP were increased in the APP/PS1 mouse brains compared with those in the age-matched WT group. Co-IP assays showed increases in TXNIP-NLRP3 binding in the APP/PS1 mouse brain compared with those in age-matched WT mice. Accordingly, ASC-associated NLRP3 protein exhibited an increased expression in APP/PS1 mouse brain relative to the age-matched WT group, which indicates the conformation and the activation of the NLRP3 inflammasome in the APP/PS1 mouse brain. Our results are consistent with the observations of Oakley and colleagues (48). Our findings further support the hypothesis that TXNIP is implicated in Alzheimer's-like pathology.
Nrf2, a transcription factor, plays a pivotal role in the cellular defense against oxidative stress. Jones and colleagues observed that impaired Nrf2 responses during aging result in a delayed recovery of the redox balance following oxidative stress (26). Studies have shown a predominate decline in Nrf2-mediated antioxidant responses with age (12, 16). Previous preclinical studies showed that Nrf2 levels were reduced in AD brains (54). Kanninen and colleagues have reported reduced mRNA and protein levels of Nrf2 in the brains of Tg mice with AD-like pathology (30). The results above indicate that the Nrf2 system is impaired in the brains of AD, while exogenous Nrf2 overexpression improves learning and memory retention in a mouse model of AD (29). Interestingly, Nrf2 enhances TrX redox signaling via inhibiting the transcription of TXNIP in the heart of the diabetic mouse model. In contrast, the Nrf2 KO mouse exhibits increased ROS production and apoptosis. TXNIP is upregulated in the hearts of Nrf2 KO mice compared with WT controls (20). Although few studies have investigated the alterations of the Nrf2-TXNIP-TrX axis in AD, shared characteristics between diabetes mellitus and AD, such as oxidative stress, accumulation of misfolded proteins, and inflammation, imply that there might be a common underlying pathogenic mechanism.
Neuroinflammation is considered to be a prime suspect in the development of age-related cognitive decline (39). Dl-NBP administration has been shown to mitigate neuroinflammatory processes and reduce neuronal damage in a mouse model of amyotrophic lateral sclerosis (18). Furthermore, Wang and colleagues observed that Dl-NBP treatment ameliorated cataract by inhibiting oxidative stress in streptozotocin-triggered diabetic rat (68). It has been reported that NBP attenuates Aβ-induced inflammation in cultured astrocytes in vitro (69). We have previously demonstrated that Dl-NBP treatment upregulated Nrf2 signaling in the APP/PS1 mouse brain (67). To investigate whether Dl-NBP treatment could exert anti-inflammatory effects in AD-like pathology and to explore the mechanism, we determined the inflammatory levels and Nrf2/TXNIP/TrX signaling in the APP/PS1 and age-matched WT mouse brain. In our present study, a significant decrease of TXNIP expression and mRNA levels was observed in Dl-NBP-treated mouse brain. Dl-NBP management enhanced TrX activity without altering the TrX protein expression or mRNA levels. The CC1/Pro-Casp-1 ratios were also suppressed under Dl-NBP administration. As a comparison, Dl-NBP treatment did not affect the ratio of CC3 to procaspase-3. These results indicate the effects of Dl-NBP on the inhibition of NLRP3-mediated caspase-1 cleavage. Dl-NBP treatment reduces Aβ plaque-associated neuritic dystrophy, which is consistent with Dl-NBP-induced increases of p-ASK1 (Ser83). Phosphorylates of ASK1 at Ser83 attenuates ASK1 activity and might promote cell survival (32). Dl-NBP administration increases the TrX-ASK1 interaction, which probably indicates that Dl-NBP inhibits ASK1 activity by blocking the dissociation of TrX from ASK1, contributing to the inhibition of neuronal cell apoptosis. Dl-NBP treatment also markedly reduced the TXNIP-associated NLRP3 protein levels, which indicated that Dl-NBP-induced inhibition of TXNIP represses the interaction of TXNIP with NLRP3. In addition, the increases in ASC protein expression are reversed under Dl-NBP treatment, which implies that Dl-NBP treatment can mitigate the conformation and activation of the NLRP3 inflammasome. Liu and colleagues reported that Dl-NBP could protect cortical neuronal cells against apoptosis in a mouse model of traumatic brain injury via upregulating Nrf2 signaling (38). In our study, Dl-NBP administration leads to Nrf2 upregulation in the hippocampus and frontal cortex in the APP/PS1 mouse brain. Interestingly, ChIP assays showed that Dl-NBP-mediated increases of nuclear Nrf2 led to a decrease in TXNIP transcription, which may inhibit caspase-1 activity, reduce IL-1β secretion, and alleviate inflammation-induced apoptosis in the APP/PS1 mouse brain. The effects of Dl-NBP on regulating Nrf2-TXNIP were confirmed in vitro. Under the treatment with conditioned medium from LPS-primed human-derived A172 glial cells, the secretion of Aβ was significantly increased in the APPswe cells. The ratio of Aβ1–42 to Aβ1–40 was increased, which indicated the increases of amyloidosis. And IL-1β from the conditioned medium above may be the critical factor for facilitating Aβ production. The results have further verified the work of Buxbaum and colleagues (8), showing that the increased inflammatory response of astrocytes or microglia cells might enhance the progression of Alzheimer's-like pathology. In contrast, the Aβ levels were markedly reduced following the Dl-NBP addition. Interestingly, we observed that Nrf2 inhibitor, Nrf2 siRNA or TXNIP, overexpression abrogated the Dl-NBP-triggered decreases of NLRP3-ASC interaction in A172 cells, which suggests that the nature of Dl-NBP-mediated inhibition on neuroinflammation is via Nrf2-TXNIP signaling.
Considering both ex vivo and in vitro results, our study reveals the neuroprotective effects of Dl-NBP against oxidative damage and inflammation. Our observations show that TXNIP links oxidative stress and NLRP3 inflammasome activation in the APP/PS1 mouse brain. Limitations of this study include that the limited accessibility of human AD postmortem brain and the TXNIP immunostaining in the postmortem AD brain do not provide direct effects of TXNIP on the activation of NLRP3 inflammasome in the AD brain. There remain uncertainties as to the temporal pattern of TXNIP and Nrf2 changes in the AD brain. Our current understanding of the mechanisms by which Nrf2/TrX impairment mediates chronic inflammation in AD is limited. Further animal experiences and preclinical trials need to be performed to address a time window of targeting the Nrf2/TXNIP/TrX axis. Although experiments with APP/PS1 mouse and A172 and APPswe cell models certainly have limitations relative to direct comparisons with the brain condition of AD patients, our results indicate that Nrf2 impairment-induced deficiency in TXNIP inhibition facilitates the formation of NLRP3 inflammasome and contributes to the progress of AD-like pathology. Herein, we elucidated the potential mechanism by which Dl-NBP may alleviate AD pathology. Owing to the upregulation of Nrf2 signaling, Dl-NBP enhances TrX activity, reduces oxidative stress, represses TXNIP expression, inhibits the TXNIP-NLRP3 interaction, and attenuates inflammation and neurodegeneration. The present data together with our previous studies (67) suggest that Dl-NBP may serve as an effective therapeutic agent for the treatment of AD.
Materials and Methods
Animals and drug treatments
Male APP/PS1 (B6C3-Tg [APPswe, PSEN1dE9] 85Dbo/Mmjax) double Tg mice were originally generated by and purchased from the Jackson Laboratory (West Grove, PA). The APP/PS1 mouse expresses a chimeric mouse/human Swedish mutation for amyloid precursor protein and a mutant form of human presenilin 1 driven by a central nervous system-specific neuronal promoter. Age-matched C57BL/6 mice (Jackson Laboratory) were used as WT controls. The experimental procedures were performed in compliance with the Chinese regulations involving animal protection and were approved by the Ethics Committee for Animal Use of China Medical University.
The procedures for Dl-NBP administration were performed exactly as previously described (67). Briefly, 4-month-old APP/PS1 mice and age-matched WT mice were randomly divided into two groups: one was treated with Dl-NBP at a dosage of 20 mg/kg body weight by oral gavage once daily for 5 months and the other was administered vehicle (vegetable oil, 50 μL). Dl-NBP (purity, 99.6%; Shijiazhuang Pharma Group NBP Pharmaceutical Co., Ltd, Shijiazhuang, Hebei, China) was dissolved in vegetable oil at 10 mg/mL. The general behavior of all mice was monitored daily, and the body weight of each mouse was recorded weekly.
Tissue preparation
Twenty-four hours after the last oral administration with Dl-NBP or vehicle, mice were intraperitoneally anesthetized with sodium pentobarbital at a dose of 50 mg/kg. After transcardial perfusion with ice-cold saline, the mice were sacrificed by decapitation. The brains were sagittally dissected into two halves on an ice-cold surface. The right hemisphere was prepared for morphological assessment. The left hemisphere was snap-frozen in liquid nitrogen and stored at −80°C for biochemical analyses.
Quantification of apoptosis
For the analysis of TUNEL, a commercial kit was used (In Situ Cell Death Detection Kit, Roche Diagnostics, Indianapolis, IN) according to the manufacturer's instructions. Briefly, coronal frozen mouse brain sections (6 μm) underwent 4% paraformaldehyde fixation, and antigen retrieval was performed by boiling in Tris, EDTA, and glycine buffer (pH 8.0) for 4 min in a microwave oven. After washes with phosphate-buffered saline (pH 7.4), the slides were treated with 0.1% sodium citrate plus 0.1% Triton X-100 for 8 min to permeate the nucleus. After rinsing, the slides were blocked in blocking buffer for 30 min and then were incubated with TUNEL reaction mixture (60 min, 37°C). After several washes, the slides were analyzed under a confocal laser scanning microscope (Model SP2; Leica, Wetzlar, Germany). The images of apoptosis-positive cells were captured using an excitation filter of 488 nm.
After the indicated treatment periods in vitro, apoptotic cells were quantified by flow cytometry using the annexin V-FITC/PI apoptosis detection kit (Biovision, Milpitas, CA). Briefly, after trypsinization, the cultured cells were treated with serum-containing media. The cells were rinsed using ice-cold rinsing buffer (pH 7.4) containing 10 mM HEPES, 140 mM NaCl, and 2.5 mM CaCl2. The cells were then incubated using 500 μL of binding buffer with 5 μL of annexin V-FITC and 5 μL of PI. The samples were then incubated for 15 min at room temperature in the dark. Cell apoptosis was quantified using the FITC (FL1) and PI (FL2) signal detectors.
Determination of TrX activity
TrX activity was measured by colorimetry with the Thioredoxin Reductase Assay Kit (Sigma-Aldrich, St. Louis, MO). Briefly, brain tissues or treated cells were homogenized and lysed in lysis buffer containing 150 mM NaCl, 1% Nonidet P-40, 0.5% deoxycholic acid, 0.1% sodium dodecyl sulfate (SDS), and 50 mM Tris (pH 8.0) plus protease inhibitor cocktail (Sigma-Aldrich). The lysate was centrifuged at 10,000 g for 10 min at 4°C. The total protein concentration was determined using a UV 1700 PharmaSpec ultraviolet spectrophotometer (Shimadzu, Tokyo, Japan). The substrate in the kit is 5,5-dithiobis (2-nitrobenzoic) acid (DTNB). The TrX reductase reduction reactions occur as follows: two moles of 5-thio-2-nitrobenzoic acid (TNB) are formed for every 1 mol of NADPH oxidized. The reduction of DTNB with NADPH to TNB produces a maximum absorption peak at 412 nm. The levels of total DTNB reduction by the sample and the amount of DTNB reduction by the sample in the presence of the reductase inhibitor solution were determined. The difference between these two results indicates the DTNB reduction, which is attributed to the level of TrX reductase activity.
Western blot analysis
Mouse brain tissues and cultured cells were collected and homogenized by sonication in ice-cold RIPA buffer (150 mM NaCl, 1% Nonidet P-40, 0.5% deoxycholic acid, 0.1% SDS, and 50 mM Tris, pH 8.0) with a protease inhibitor cocktail addition (Sigma-Aldrich). After centrifugation at 15,000 g at 4°C for 30 min, the supernatants were collected, and protein concentrations were measured using the UV 1700 PharmaSpec ultraviolet spectrophotometer. Equal amounts of protein (20 μg) were separated by 10% SDS polyacrylamide gels. The protein samples were transferred to PVDF membranes (Millipore, Billerica, MA) and probed with antibodies as follows: rabbit monoclonal antibody to caspase-1 (1:1000, ab179515; Abcam, Cambridge, MA) for recognizing procaspase 1 and the cleavage fragment, p10 (p12); rabbit anti-caspase-3 (1:1000, 9662; Cell Signaling Technology, Danvers, MA) for detecting full-length caspase-3 and the large cleaved fragment from caspase-3; rabbit anti-mature IL-1β (1:1000; Abcam), mouse anti-IL-1β (1:1000; Cell Signaling Technology), mouse anti-TXNIP (1:800; Santa Cruz Biotechnology), rabbit anti-ASC (1:1000; Cell Signaling Technology), rabbit anti-NLRP3 (1:1000; Cell Signaling Technology), rabbit anti-Nrf2 (1:1000; Abcam), rabbit anti-TrX (1:1000; Abcam), rabbit anti-ASK1 (1:1000; Cell Signaling Technology), rabbit anti-phospho-ASK1 (Ser83, 1:800; Santa Cruz Biotechnology), rabbit anti-phospho-APP (Thr668, 1:1000; Cell Signaling Technology), rabbit anti-BACE1 (1:1000; Sigma-Aldrich), mouse anti-GAPDH (1:10,000; Kangchen Biotech, Shanghai, China), and rabbit anti-lamin B1 antibody (1:1000; Abcam). After incubation with the appropriate HRP-conjugated secondary antibodies, the immunological complexes were visualized with enhanced chemiluminescence (Amersham Biosciences).
Immunohistochemistry and confocal laser scanning microscopy
Four human postmortem brain tissues (AD: serial numbers T4304, T4339, and 235-95; normal: P535-00) were obtained from the New York Brain Bank. Another two human brain tissues (normal) were obtained from Fengtian Hospital of China (one from a 59-year old and the other from a 63-year old). Samples were incubated with rabbit anti-TXNIP antibody (1: 100; Abcam) for 16 h at 4°C. After thorough rinses, the samples were then treated with biotinylated IgG (1: 200) for 1 h and incubated with streptavidin peroxidase for 30 min. Following treatment with TBS containing 0.025% 3,3-diaminobenzidine and 0.0033% H2O2 for 5 min, the sections were dehydrated, cleared, mounted, and examined with an Olympus microscope (Model DP71). Coronal frozen mouse brain sections (6 μm) were first blocked with 3% normal bovine serum albumin at room temperature for 30 min. The samples were then incubated with a mixture of primary antibodies of either rabbit anti-mature IL-1β (1:200; Abcam) or mouse anti-Aβ (1:500; Sigma) and goat anti-Iba1 (1:100; Abcam), rabbit anti-Aβ (1:100; Abcam) and mouse anti-GFAP (1:200; Cell Signaling Technology), rabbit anti-TXNIP (1:100; Abcam) and mouse anti-GFAP (1:200; Cell Signaling Technology), rabbit anti-TXNIP and goat anti-Iba-1 (1:100; Abcam), rabbit anti-TXNIP and mouse anti-NeuN (1:100; EMD Millipore), rabbit anti-Nrf2 (1:100; Abcam) and mouse anti-NeuN (1:100) overnight at 4°C. After rinsing, the samples were incubated with the appropriate secondary antibodies for 2 h at room temperature. The images were acquired using a confocal laser scanning microscope (Model SP2; Leica) and analyzed with Nikon EclipseNet and ImageJ software (National Institutes of Health, Bethesda, MD). Dystrophic astrocytes and microglia were determined by calculating the debris score according to a subjective scale of 0-3 μm with NIH Image software. Polarization index of microglia to Aβ plaques was assessed as previously described (72).
IP analyses
IP assays were performed as previously described (67). Protein from mouse brain tissue or cell lysates was incubated with primary antibodies against TXNIP, NLRP3, or TrX at 4°C for 12 h. Immune complexes were captured with TrueBlot IP beads (eBioscience, Hatfield, United Kingdom) at 4°C for 4 h. After rinsing three times with IP lysis buffer, the bound proteins were boiled for 5 min and then eluted before performing Western blot analysis. Proteins bands were resolved and blotted with antibodies anti-NLRP3, anti-ASC, or anti-ASK1 at 4°C for 12 h.
Chromatin IP assay
ChIP was performed as previously described (67). Briefly, mouse brain tissues were treated with 4% paraformaldehyde for chromatin crosslinking. After neutralization in 0.125 M glycine, the samples were homogenized by sonication and were lysed in SDS lysis buffer (pH 8.1) containing protease inhibitors (Roche Molecular Biochemicals). One-third of the protein samples were saved as the input genomic DNA control. The remaining lysates were mixed with an antibody against Nrf2 (1:40; Abcam), MondoA (1:20; Proteintech), or nonimmune IgG (negative control). The IPs containing protein-DNA complexes were washed and then captured with protein A-agarose beads. After thorough rinsing, the precipitates were incubated with elution buffer (pH 8.0). To reverse crosslinking, the input tissue and elutes were added to 20 μL of 5 M NaCl and heated at 65°C for 4 h (27). The protein-DNA complexes were treated with 20 μg/mL of proteinase K (1 h, 45°C). DNA was purified with a PCR purification kit (Qiagen, Valencia, CA). Real-time PCR was performed using samples of ChIP DNA with the primers for amplifying TXNIP ARE or TXNIP ChoRE-a.
Cell culture and treatment
Human glioblastoma A172 and human SH-SY5Y neuroblastoma cell lines were obtained from the American Type Culture Collection (ATCC, Manassas, VA). A172 cells were cultured in Dulbecco's modified Eagle's medium (DMEM)/Ham's F-12 medium (Life Technologies) at a ratio of 1:1 supplemented with 10% fetal bovine serum (FBS; Hyclone, UT) and 1% penicillin/streptomycin (Thermo Fisher Scientific). SH-SY5Y cells stably transfected with the human APPswe or empty vector (neo) (74) were grown in DMEM containing 10% FBS and 40 μg/mL gentamicin. Cells were incubated in a fully humidified, 5% CO2 incubator at 37°C. When cells were ∼80% confluent, the cells were synchronized by overnight exposure to fresh media without serum or antibiotics. The cells were then treated with LPS, Dl-NBP, and Nrf2 siRNA using Lipofectamine 2000, trigonelline, or adenoviral gene transfer (Ad-TXNIP) as indicated. Untreated cells were used as vehicle control. The cells and culture supernatants were harvested for analyses.
Measurement of Aβ1–40 and Aβ1–42 levels
Soluble Aβ1–40 and Aβ1–42 levels in the culture medium of human APPswe and neo cells were detected by sandwich enzyme-linked immunosorbent assay (ELISA). The samples were loaded onto 96-well plates, and the levels of Aβ1–40 and Aβ1–42 were measured according to the manufacturer's instructions of the ELISA kit (Invitrogen, Carlsbad, CA). The absorbance was measured at 450 nm.
β-secretase and γ-secretase activity analyses
The activity of β-secretase and γ-secretase in the lysates of APPswe or neo cells was determined using specific peptides that were conjugated to the reporter molecules EDANS and DABCYL (R&D Systems, Minneapolis, MN). Briefly, after the indicated intervention, cells were collected and treated with extraction buffer. After homogenization, the supernatants were harvested. The samples were mixed with the reaction buffer and substrate, and were incubated in the dark (2 h, 37°C). The absorbance was measured using a fluorescent microplate reader.
Nrf2 DNA binding activity assessment
First, following the indicated treatment, nuclear Nrf2 was extracted from A172 cells with a nuclear extract kit (Active Motif, Inc., Carlsbad, CA) in accordance with the manufacturer's instructions. The samples were then treated with buffer A containing 40 μL of 10% Nonidet P (NP-40). Centrifugation was performed, and the pellets were resuspended in a prechilled, hypertonic, nuclear extraction buffer B containing 50 mM HEPES, 50 mM KCl, 300 mM NaCl, 0.1 mM EDTA, 0.5 mM dithiothreitol, 0.1 mM PMSF, and 20% glycerol (pH 7.9). The supernatant containing the nuclear proteins was obtained, and the Nrf2 DNA binding activity of nuclear extracts was determined with the TransAM ELISA kit (Active Motif).
Adenoviral gene transfer for TXNIP overexpression
An adenovirus vector encoding TXNIP was generated for adenoviral gene transfer (Ad-TXNIP) as previously described (62). Viruses were propagated in HEK293 cells, purified, and the stock titer was 109 infectious units/mL. A recombinant adenovirus encoding green fluorescent protein (Ad-GFP) was used as an internal control. After preliminary dose/response experiments for TXNIP expression, 1000 μL of stock in 1.75 mL of media was chosen for the infection in A172 cells, which were plated at a cell density of 1 × 105/cm2. The A172 cells were infected for 3 h and then recovered for 24 h in media without serum or antibiotics.
Determination of IL-1β secretion in the culture medium
The levels of proinflammatory cytokines IL-1β in the supernatants of the pharmacologically or chemically treated cells and the control cells were determined using the IL-1β ELISA kit (eBioscience, CA) in accordance with the manufacturer's instructions. The total amount of protein in the medium was used as the loading control.
Quantitative real-time PCR
Total RNA from mouse brain tissue and cultured cells was isolated using TRIzol (Invitrogen). A total of 2 μg of RNA from each sample was reverse transcribed using the Prime Script RT Reagent Kit (Takara, Otsu, Japan). Each gene amplification was performed using SYBR Green PCR Master mix (Applied Biosystems, Inc. [ABI], Foster City, CA) with an ABI 7300 Sequence Detection System. The following primers were used: mouse TXNIP: 5′-ATGGCCAGACCAAAGTGTTC-3′ (sense) and 5′-GGCTGTCTTGAGAGTCGTCC-3′ (antisense); mouse TrX: 5′-CAACAGCCAAAATGGTGAAGCTG-3′ (sense) and 5′-AGGTTTTAAACAGCTG-3′ (antisense); mouse NLRP3: 5′-AGCCTTCCAGGATCCTCTTC-3′ (sense) and 5′-CTTGGGCAGCAGTTTCTTTC-3′ (antisense); mouse ASC: 5′-GAAGCTGCTGACAGTGCAAC-3′ (sense) and 5′-GCCACAGCTCCAGACTCTTC-3′ (antisense); mouse β-tubulin: 5′-CTGGGCTAAAGGCCAC-3′ (sense) and 5′-AGACACTTTGGGCGAG-3′ (antisense); human TXNIP: 5′-TGGTGGATGTCAATACCCCT-3′ (sense) and 5′-ATTGGCAAGGTAAGTGTGGC-3′ (antisense); human TrX: 5′-AGCAGCCAAGATGGTGAAGCAGA-3′ (sense) and 5′-GCTCCAGAAAATTCACCCACC-3′ (antisense); human NLRP3: 5′-AACAGCCACCTCACTTCCAG-3′ (sense) and 5′-CCAACCACAATCTCCGAATG-3′ (antisense); human ASC: 5′-ACTCATTGCCAGGGTCACAGAAGTG-3′ (sense) and 5′-GCTTCCTCATCTTGTCTTGGCTGGT-3′ (antisense); and human β-tubulin: 5′-TCTGTTCGCTCAGGTCCTTT-3′ (sense) and 5′-TTCATGATGCGATCAGGGTA-3′ (antisense).
Statistical analysis
All data are presented as mean ± standard error of the mean. Statistical significance of the differences between groups was determined using one-way or two-way ANOVA. The genotype × treatment interaction, together with main effects of genotype and treatment, was analyzed. p < 0.05 was considered statistically significant.
Footnotes
Acknowledgments
This study was supported by the Natural Science Foundation of China (Nos. 81671041, U1608282, 81100808, 81372793), the China Postdoctoral Science Foundation (Nos. 2012M510849, 2013T60304), the Natural Science Foundation of Jilin Provincial Science & Technology Department in China (No. 20160101202JC), and the Science & Technology Research Fund of the Education Department in Jilin Province of China (2016, No. 235).
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.