
Abstract
Significance:
There is evidence to implicate reactive oxygen species (ROS) in tumorigenesis and its progression. This has been associated with the interplay between ROS and oncoproteins, resulting in enhanced cellular proliferation and survival.
Recent Advances:
To date, studies have investigated specific contributions of the crosstalk between ROS and signaling networks in cancer initiation and progression. These investigations have challenged the established dogma of ROS as agents of cell death by demonstrating a secondary function that fuels cell proliferation and survival. Studies have thus identified (onco)proteins (Bcl-2, STAT3/5, RAS, Rac1, and Myc) in manipulating ROS level as well as exploiting an altered redox environment to create a milieu conducive for cancer formation and progression.
Critical Issues:
Despite these advances, drug resistance and its association with an altered redox metabolism continue to pose a challenge at the mechanistic and clinical levels. Therefore, identifying specific signatures, altered protein expressions, and modifications as well as protein–protein interplay/function could not only enhance our understanding of the redox networks during cancer initiation and progression but will also provide novel targets for designing specific therapeutic strategies.
Future Directions:
Not only a heightened realization is required to unravel various gene/protein networks associated with cancer formation and progression, particularly from the redox standpoint, but there is also a need for developing more sensitive tools for assessing cancer redox metabolism in clinical settings. This review attempts to summarize our current knowledge of the crosstalk between oncoproteins and ROS in promoting cancer cell survival and proliferation and treatment strategies employed against these oncoproteins. Antioxid. Redox Signal.
Introduction
I
ROS and Their Intracellular Sources
ROS are a group of partially reduced molecules derived from molecular oxygen, O2. They are categorized as free radicals such as superoxide (O2 .−), hydroxyl radical (OH−), or nonradical such as hydrogen peroxide (H2O2) (53). These ROS are capable of inducing oxidative modification that includes the modification of cysteine/thiol groups to sulfenic, sulfinic, sulfonic, or S-glutathionylated form (91, 101). Intracellular sources of ROS include the various oxidases, such as nitric oxide synthase, xanthine oxidase, nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX), cytochrome P450 (CYP), and the electron transport chain (ETC) of the mitochondria, particularly complexes I and III (77, 136, 159, 179, 227 –229, 244, 369, 370, 386). The cellular levels of ROS are kept in check by ways of a number of antioxidant defense systems, such as the glutathione (GSH) system, superoxide dismutases (SOD1, 2, and 3), catalase, thioredoxins (Trxs), and peroxiredoxins (Prxs). As such, the term cellular redox status refers to a state that reflects the balance between ROS generation and antioxidant defenses and is a critical determinant of normal cellular homeostasis. Consequently, an imbalance in ROS production and detoxification could result in a multitude of events ranging from oxidative stress-induced biomolecular damage such as DNA modifications, lipid peroxidation, to changes in protein structure and function (50, 102, 115, 144, 181, 186, 206, 379). Although an overwhelming increase in intracellular oxidative stress results in the activation of various modes of cell death, there is also convincing evidence that a mild to moderate increase in intracellular ROS promotes cell proliferation, differentiation, and survival by acting as secondary messengers (44, 145, 155, 162, 181, 273, 292).
From the standpoint of tumorigenesis, a pro-oxidant state (mildly elevated ROS) has been shown to provide cancer cells with an intracellular milieu that favors cell growth and proliferation by impinging on networks that either increase cell cycle progression and survival and/or impede execution (46, 80, 145, 262). It is, therefore, not surprising that the growth/survival promoting activities of a number of cancer-associated genes and their protein products (oncoproteins) have been linked to a change in the cellular redox environment, and as a consequence there is heightened interest in the design and development of therapeutic strategies to favorably alter the cellular milieu for drug-induced execution. Although there is definitive evidence linking “oncogene addiction” to an altered redox status (69, 194, 198, 233, 285, 331), the challenge remains in the identification of druggable targets that are directly or indirectly involved and/or promote the process of cellular transformation and its progression. It is highly plausible that redox heterogeneity within a bulk tumor serves as an evasive strategy to promote cell survival, but by the same token lends itself as the Achilles heel, which could be exploited for specific drug design and development.
The three major intracellular sources of ROS production, that is, mitochondrial ETC, NOX, and endoplasmic reticulum (ER) have not only being implicated directly or indirectly in the prosurvival activity of several oncoproteins but more importantly in drug-induced modulation of cellular redox status (34, 35, 37, 46, 92, 113, 156, 260, 342, 376, 397). The mitochondria are the main powerhouse of the cell wherein adenosine triphosphate (ATP) is generated during the sequential shuttling of electrons through a string of mitochondrial protein complexes, termed complexes I, II, III, and IV (122). Of note, during the shuttling of electrons, as mitochondrial respiration increases (increased oxygen consumption), the probability of electrons leaking onto O2 could potentially increase, thereby resulting in the reduction of O2 to O2 .− (Fig. 1a) (34). Some notable sites of O2 .− production are mitochondrial complexes I and III (77, 179, 230, 335). Various intracellular conditions such as hyperoxia, hypoxia, serum withdrawal, glucose deprivation, and drug treatment as well as genetic manipulation have been documented to enhance the production of O2 .− at the mitochondria (23, 34, 35, 96, 111, 205). One example is the germline deletion of complex II subunit B (SdhB) in hereditary paraganglioma (214). Empirical studies to inhibit or knockdown SdhB were shown to augment cytosolic and mitochondrial O2 .− as well as stabilize hypoxia-inducible factor and increase cell growth (111), thereby suggesting a role of ROS in paraganglioma. Another notable example is the treatment of antimycin A to induce mitochondrial O2 .− production (34, 77). Importantly, some of these intracellular conditions have also been hijacked by oncoproteins as a means to regulate ROS production to promote tumorigenesis (34, 35, 46, 85, 184, 205).

Apart from the mitochondria, NOX is another prominent source of ROS. Comprising of six different subunits that interact to form an active enzyme complex, including the membrane-bound (gp91phox and p22phox) and cytosolic (p40phox, p47phox, p67phox and Rac) factors, NOX produces O2 .− through a series of redox cycling processes from the donation of electron by NADPH to the production of O2 .− as shown in Figure 1b (12, 174). The phagocytic burst is the most commonly described NOX-mediated generation of ROS that utilizes extracellular O2 .− production as an antimicrobial defense (151). Interestingly, NOX-induced ROS production is not exclusive to phagocytes as evidenced by the identification and demonstration of similar NOX-like activity in a variety of cancer cell types, which has been associated with ROS-mediated prosurvival/growth signaling (38, 136, 342). For example, ROS produced from NOX2 at the endosomes promote the binding of TRAF2 to TNFR1/TRADD complex, thereby activating the IκB kinase complex and downstream nuclear factor kappa B (NF-κB) pathway (195). In addition, knockdown of the small GTPase Rac1, a component of NOX, prevents M2 macrophage differentiation, suggesting a role for O2 .− in promoting the acquisition of the M2 phenotype and the occurrence of tumor-associated macrophages (393).
The ER is another unique organelle that deliberately provides an oxidative milieu to aid the folding of proteins and formation of disulfide bonds. It is reviewed and suggested that ROS are produced through the disulfide bond formation in the ER during oxidative protein folding in yeast studies (29, 334). At the ER, protein disulfide isomerase (PDI) facilitates the process of oxidative protein folding by the concurrent oxidation of dithiol groups (target protein) and reduction of its own protein disulfide bonds. PDI is then recycled back to its oxidized form by ER oxidoreductin-1, which utilizes a flavin adenine dinucleotide-dependent reaction to catalyze the oxidation reaction of PDI. Through this process, O2 is used as a terminal electron acceptor, thereby producing H2O2 (Fig. 1c) (9, 29, 60, 93 –95, 182, 266, 333, 334). Besides the essential role of oxidoreductases in the ER, the presence of ROS in ER is further corroborated by evidence indicating that NOX4 is localized to the ER and that overexpression of NOX4 and its subunit p22phox increase the level of ROS in ER (366).
Cellular antioxidant defense systems
With ROS generation on one side of the coin, ROS detoxification on the other is, therefore, crucial to keep the intracellular ROS level in check. To this end, several antioxidant enzymes have been shown to detoxify intracellular ROS (97, 100). These enzymatic systems include SOD, catalase, glutathione peroxidase (GPx), glutathione reductase (GR), Prx, and Trx. SOD comprises three isoforms: SOD1 or Cu/Zn-SOD, localized primarily to the cytosol but also found in nuclei, lysosomes, and peroxisomes; SOD2 or MnSOD, localized in the mitochondria matrix; and SOD3, located in the extracellular matrix. SOD catalyzes a stepwise oxidation and reduction reactions, termed as dismutation, for affecting the conversion of O2 .− to H2O2 [extensively reviewed in (99)]. Subsequently, H2O2 is converted to H2O and O2 by the antioxidant catalase, which is primarily localized to the peroxisome and the cytosol (126, 290). Another major antioxidant enzyme involved in the detoxification of H2O2 is GPx (277). GPx reduces H2O2 by the oxidation of GSH to produce oxidized GSH and H2O. Oxidized GSH is then recycled to form reduced GSH by GR (267). The Trx-dependent Prx is also a major player in the antioxidant defense system. Prx is found in the nucleus, mitochondria, ER, peroxisome and the cytosol, depending on its various isoforms. The antioxidant protein functions to reduce peroxides, H2O2, and peroxynitrite (ONOO−) by using its thiol groups. For example, its thiol groups are oxidized to form disulfides, which are subsequently reduced by Trx to regenerate a functional Prx (121). These antioxidants function to ensure that a homeostatic redox milieu is in place to prevent excessive accumulation of ROS that could carry detrimental effects to the cell. As such, alterations in the cellular antioxidant defense systems, in addition to the aberrant generation of ROS, contribute significantly to the generation of the pro-oxidant intracellular environment that is associated with various processes of tumorigenesis. For example, this has been elegantly demonstrated by studies linking the varied expression of SOD2 to the different stages of tumorigenesis and its progression (73, 177, 203). Similar effects of Prxs have been documented and elegantly reviewed in Refs (116, 239). And perhaps the most commonly observed differences between transformed cells and noncancerous cells relate to the GSH metabolism with profound effects on cellular redox metabolism and cell fate (39, 82). These are just a few examples to highlight the importance that the balance between cellular ROS generation and antioxidant defense systems plays in altering cellular homeostasis and promoting abnormal cell growth and/or proliferation, thereby endowing cells with two critical hallmarks of cancer, that is, increased proliferation and death resistance (119).
The dichotomy of redox signaling in cell fate decisions
Depending on the concentration, ROS can evoke a myriad of downstream events ranging from cell proliferation and survival to cell death (Fig. 2) (13, 14, 155, 188, 240, 246, 254, 330, 332). Although sublethal levels of ROS have been shown to increase proliferation and enhance cell survival through the modulation of various phosphatase and protein kinases (14, 90, 136, 150, 197), an overwhelming increase in ROS is associated with macromolecular damage, such as lipid peroxidation, protein oxidation and inactivation, and DNA damage (115, 188, 197, 293, 294). The latter overwhelming increase in ROS, in the presence of intact tumor suppressor p53, further triggers apoptotic cell death (210), thus facilitating the removal of cells with irreparable lesions. Apart from the induction of p53-mediated apoptosis, ROS could also induce cell death via the sustained activation of c-Jun N-terminal kinase (JNK). The latter involves ROS-dependent inactivation of the mitogen-activated protein kinase (MAPK) phosphatase (MKP) that regulates the phosphorylation/activation of JNK by catalytic oxidation of cysteine to sulfenic acid (155). In a separate study, ROS-induced activation of apoptosis signal-regulating kinase-1 (ASK1) has also been implicated in the downstream activation of JNK/p38 MAPK (246, 330). In addition, a recent study further provides support to ROS-induced cell death via the disruption of iron metabolism, in which the use of pharmacological ascorbate was capable of increasing redox active labile iron for ascorbate toxicity via the H2O2-mediated disruption of iron–sulfur clusters (287). These results serve to reinforce the notion that ROS-induced changes in cellular kinase(s) and/or protein phosphatase(s) are critical determinants of a favorable response to death stimuli.
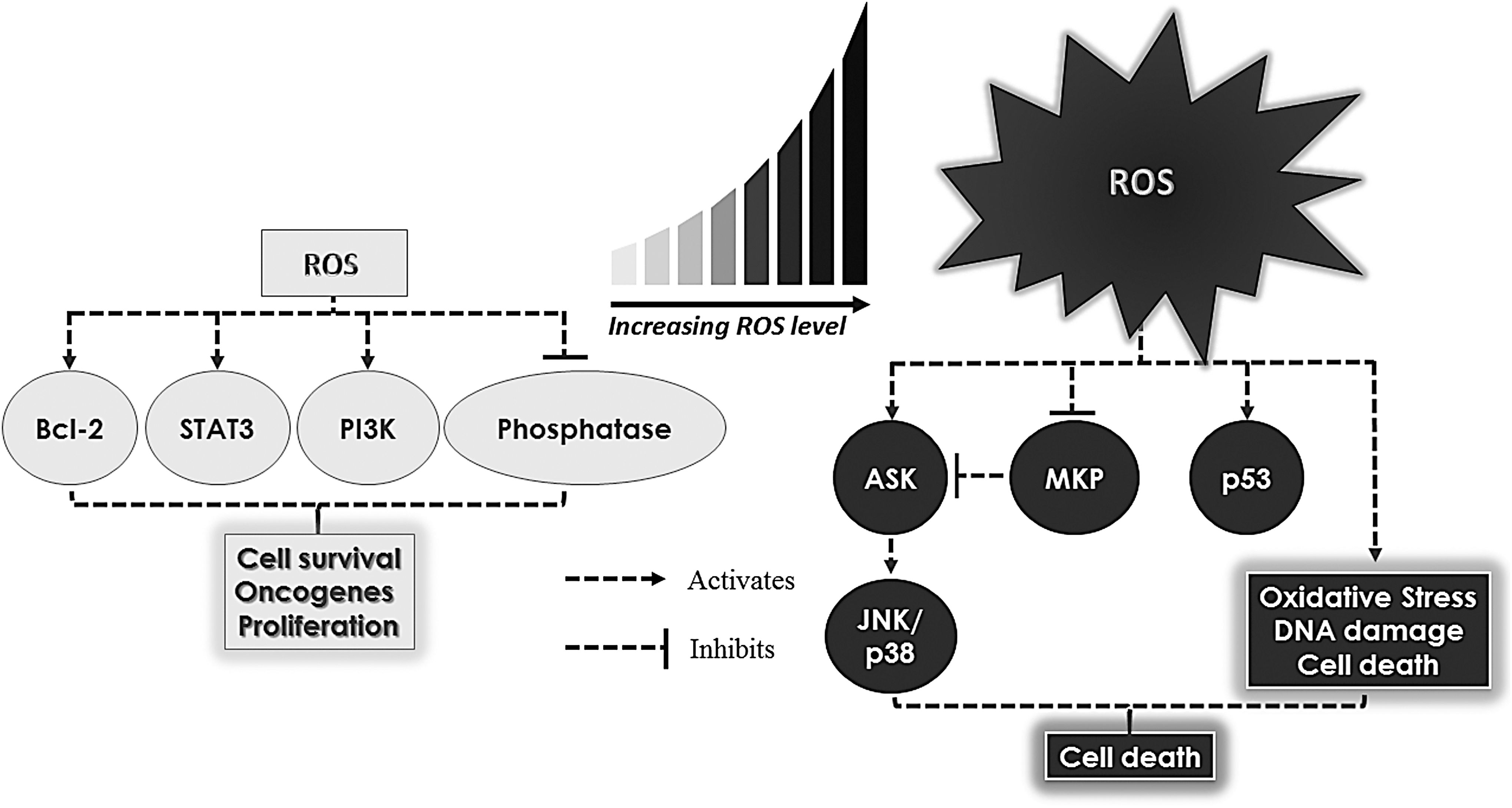
The dogmatic view of ROS from the standpoint of cell fate determination has undergone a radical change over the past couple of decades, thanks to a number of convincing reports providing clear evidence that ROS can also function as second messengers at concentrations that do not overwhelm the cells' antioxidant defense systems (13, 14, 188, 197, 216). Interestingly, the prosurvival effects of ROS, much in common with the prodeath functions, also involve activation of downstream signaling via oxidation of thiol groups of kinases or phosphatases. However, such modifications are often reversible, thereby showcasing their deliberate purpose in cell signaling (188, 216). For example, H2O2 could oxidize cysteine residues within the tumor suppressor phosphatase and tensin homolog (PTEN), thereby resulting in the inactivation of PTEN and consequential activation of the phosphoinositide 3-kinase (PI3K) signaling pathway. This oxidative modification could be reversed by Trx-mediated reduction of PTEN, thereby resulting in the restoration of PTEN phosphatase activity (188). Corroborating that, another study showed that elevated O2 .− could similarly induce a reversible oxidization of PTEN, which leads to an increased phosphatidylinositol (3,4,5)-trisphosphate and phosphorylation of protein kinase B (AKT), a mechanism similarly observed upon growth factor induction (197). Further evidence by another group also demonstrated that H2O2 could reversibly induce oxidation and inhibition of SH2-containing protein tyrosine phosphatases, which is crucial for mitogenic signaling by growth factor such as platelet-derived growth factor (PDGF) (216). These studies, therefore, support the role of ROS in signaling transduction independent of apoptotic pathways.
The strong evidence depicting the biphasic signaling of ROS, namely growth signaling or oxidative damage/cell death (155, 188, 210, 216, 254), therefore, begs the question: does there exist a fixed threshold of ROS across all cell types that could distinctly differentiate the prosurvival from the death-inducing activity of ROS? The presence of a uniformly fixed ROS levels across different cell types is highly unlikely and implausible, due to the differences in the ROS generating and detoxifying systems in different tissues of origin as well as the environmental factors that impact cellular redox state(s). It is more likely that each cell type possesses a distinct threshold, which could potentially be governed by multiple factors such as the spatial proximity of ROS and its target(s) and the production and turnover rates of ROS. Therefore, it is imperative to unravel the crosstalk between intracellular redox status, genetic and epigenetic factors, their products (oncoproteins), and environmental cues that are involved in the acquisition and progression of the cancer phenotype to be able to identify specific targets for intervention.
Bcl-2 Family: Their Canonical and Redox-Dependent Regulation of Cancer Cell Fate
The antiapoptotic protein Bcl-2
The Bcl-2 family consists of proteins that have a significant impact on cell death and apoptosis. They can be categorized into two groups, namely the antiapoptotic and proapoptotic groups. The antiapoptotic proteins comprise Bcl-2, B-cell lymphoma extra large (Bcl xL), and induced myeloid leukemia cell differentiation proten (Mcl-1), whereas the proapoptotic proteins comprise Bcl-2 associated X protein (BAX), Bcl-2 homologous antagonist killer (BAK), p53 upregulated modulator of apoptosis (PUMA), phorbol-12-myristate-13-acetate-induced protein 1 (NOXA), Bcl-2 associated death promoter (BAD), BH3 interacting domain death antagonist (BID), Bcl-2-like protein 11 (BIM). The canonical activity of the antiapoptotic members of the Bcl-2 family is associated with their ability to bind and sequester the proapoptotic family members, thereby inhibiting their recruitment to the mitochondria and preventing mitochondria outer membrane permeabilization (MOMP) and the release of apoptosis amplification factors (i.e., cytochrome c) from the mitochondria (19, 36, 63, 76, 104, 153, 225, 232, 328, 355, 359). For example, study has reported that antiapoptotic Bcl-2, Bcl-xL, and Mcl-1 could sequester proapoptotic truncated-BID, BIM, or PUMA (161). Similarly, others have shown that Bcl-2 and Bcl-xL/Mcl-1 could inhibit BAX and BAK, respectively, which prevents MOMP and cytochrome c release (76, 359). In contrast, the proapoptotic PUMA and NOXA could sequester Bcl-xL and Mcl-1, respectively, thereby releasing BAX and BAK to induce apoptosis (161, 218). By dint of their opposing functions, this suggests that a balance between the pro- and antiapoptotic proteins dictates cellular response to death stimuli, such as chemotherapy-induced cell death that is dependent on MOMP (291). Since the discovery of the prototype antiapoptotic protein Bcl-2, which is frequently overexpressed in hematopoietic cancers such as follicular lymphoma t(14;18)(+), acute lymphoblastic leukemia, chronic lymphocytic leukemia (CLL), non-Hodgkin's lymphoma, and diffuse large B-cell lymphoma (103, 131, 215, 226), a number of related proteins with similar functions have been identified, such as Bcl-xL and Mcl-1 (19, 63, 104, 341, 359). Overexpression of Bcl-2 or other functionally similar family members tilts the balance between the anti- and proapoptotic proteins in favor of the former, which in the clinical setting is associated with poor disease prognosis, tumor progression, and/or aggressive disease (103, 131, 367, 392).
Although the overexpression of Bcl-2 appears to play a critical role in the acquisition of the chemoresistant phenotype (46, 85), there is accumulating evidence to indicate that post-translational modification of Bcl-2 profoundly impacts its antiapoptotic activity. To that end, Bcl-2 phosphorylation was shown to increase its binding to Bax and Bak as well as reduce drug-induced cell death (49, 70). Other studies have also shown that increased Bcl-2 phosphorylation is capable of conferring resistance to chemotherapeutic drugs such as etoposide, doxorubicin, and paclitaxel (68, 206).
Intriguingly, a number of reports attributed the death-regulating activities of the pro- and antiapoptotic members (i.e., Bcl-2, Bcl-xL, and Bax) to the modulation of cellular redox metabolism (3, 46, 162, 165). The first observations linking Bcl-2 to cellular redox environment came from the report suggesting an antioxidant activity of Bcl-2 by virtue of its ability to regulate cellular antioxidant pathway(s) as well as induce resistance to ROS-induced cell death (135). Specifically, Bcl-2 was shown to protect cells from H2O2- and menadione-induced oxidative cell death as well as dexamethasone-induced lipid peroxidation (135). A separate study showed that Bcl-2 overexpression transcriptionally upregulated the mitochondrial antioxidant, SOD2 (163). These observations suggested two possible scenarios, first that Bcl-2 itself harbored antioxidant activity or second that the induction of antioxidant systems, such as SOD2, could be a feedback activation or adaptive response triggered by the pro-oxidant milieu supported by Bcl-2. Although there was scant evidence to support the former, work from Steinman provided support for the latter by demonstrating a significantly higher (up to 73% increase) SOD activity in a murine B-cell line overexpressing Bcl-2 in the absence of an exogenous stimulus, indicating an endogenously higher O2 .− scavenging rate. In a different bacterial cell system, Steinman also showed that Bcl-2 overexpression increased the mutation rate (up to threefold) as well as increased transcription of KatG catalase–peroxidase (enzyme with catalase and peroxidase activities) (311). These findings provided support to the hypothesis that the increase in cellular antioxidant capacity upon overexpression of Bcl-2 was not a direct effect of Bcl-2 as an antioxidant, but rather a feedback response to the pro-oxidant milieu afforded by Bcl-2 overexpression. The latter was corroborated by findings in human leukemia cells that provided evidence that overexpression of Bcl-2 inhibited death receptor- and drug-induced apoptosis by inhibiting upstream caspase 8 activation through the intermediary involvement of intracellular O2 .− (46). Furthermore, pharmacological inhibition (diphenyliodonium: DPI) of NOX, an O2 .− producer, or transient transfection with the dominant negative form of Rac1 (Rac1 N17) that blocks NOX activity restored apoptosis sensitivity of Bcl-2 overexpressing cells. Conversely, the apoptosis sensitizing effect of Rac1 N17 in Bcl-2 overexpressing leukemia cells was blocked upon an increase in O2 .− upon pharmacological inhibition of SOD1 (diethyldithiocarbamate). These robust findings not only indicate that Bcl-2 overexpression could induce a pro-oxidant milieu but also demonstrate a redox-dependent antiapoptotic activity elicited by Bcl-2 (46). To that end, Chen and Pervaiz showed that Bcl-2 overexpression induced a pro-oxidant milieu (increased O2 .− production) by engaging mitochondrial respiration; Bcl-2 overexpressing cells displayed higher mitochondrial cytochrome c oxidase (COX) enzymatic activity, oxygen consumption rate, and ATP synthesis (34, 35). This implies that Bcl-2 overexpression is capable of increasing the intrinsic electron transport across the ETC, and hence the probability of electrons leaking onto O2 to generate O2 .−. Furthermore, increased localization of the subunit Vα of COX (COX Vα), the rate-limiting enzyme of the ETC, to the mitochondria, as well as its interaction with Bcl-2, was demonstrated in cells overexpressing Bcl-2 (35). The link between Bcl-2 and a pro-oxidant tumor intracellular milieu was further reinforced by the discovery of a novel interaction between Bcl-2 and the small GTPase Rac1, which is involved in NOX assembly and O2 .− production (342).
In light of the evidence linking Bcl-2 overexpression to an increase in intracellular O2 .− and attributing a noncanonical antiapoptotic activity to the protein, how does one reconcile with the earlier findings that Bcl-2 inhibited oxidative stress-induced death signaling? One plausible explanation, presented in a preceding section, could be the feedback induction of antioxidant defense systems in cells overexpressing Bcl-2; however, a more convincing and direct evidence was provided by observations indicating a “redox rheostat” activity of Bcl-2 (Fig. 3d). Although Bcl-2 overexpression supported a pro-oxidant state under states of normoxia, this effect was remarkably reversed in the face of overt oxidative stress; Bcl-2 overexpression prevented further increase in mitochondrial O2 .− production by decreasing COX activity and clamping down oxygen consumption in cells exposed to exogenous stimuli that induced oxidative stress such as antimycin A, hypoxia, serum withdrawal, and glucose deprivation (34, 35). From the standpoint of the functional biology of Bcl-2, the opposing redox modifying activities of the protein could be critical in promoting the process of tumorigenesis and its progression. A mild increase in ROS levels during states of normoxia could provide cancer cells with a survival advantage via a stimulatory effect on cell proliferation, migration, and invasion (198, 331), whereas in the setting of overt oxidative stress (such as induced upon exposure to DNA damaging agents and γ-irradiation (11, 85, 270, 293, 294)), the ability to clamp down on a further build up of ROS could provide a protective mechanism for cancer cells to evade chemotherapy-induced execution.
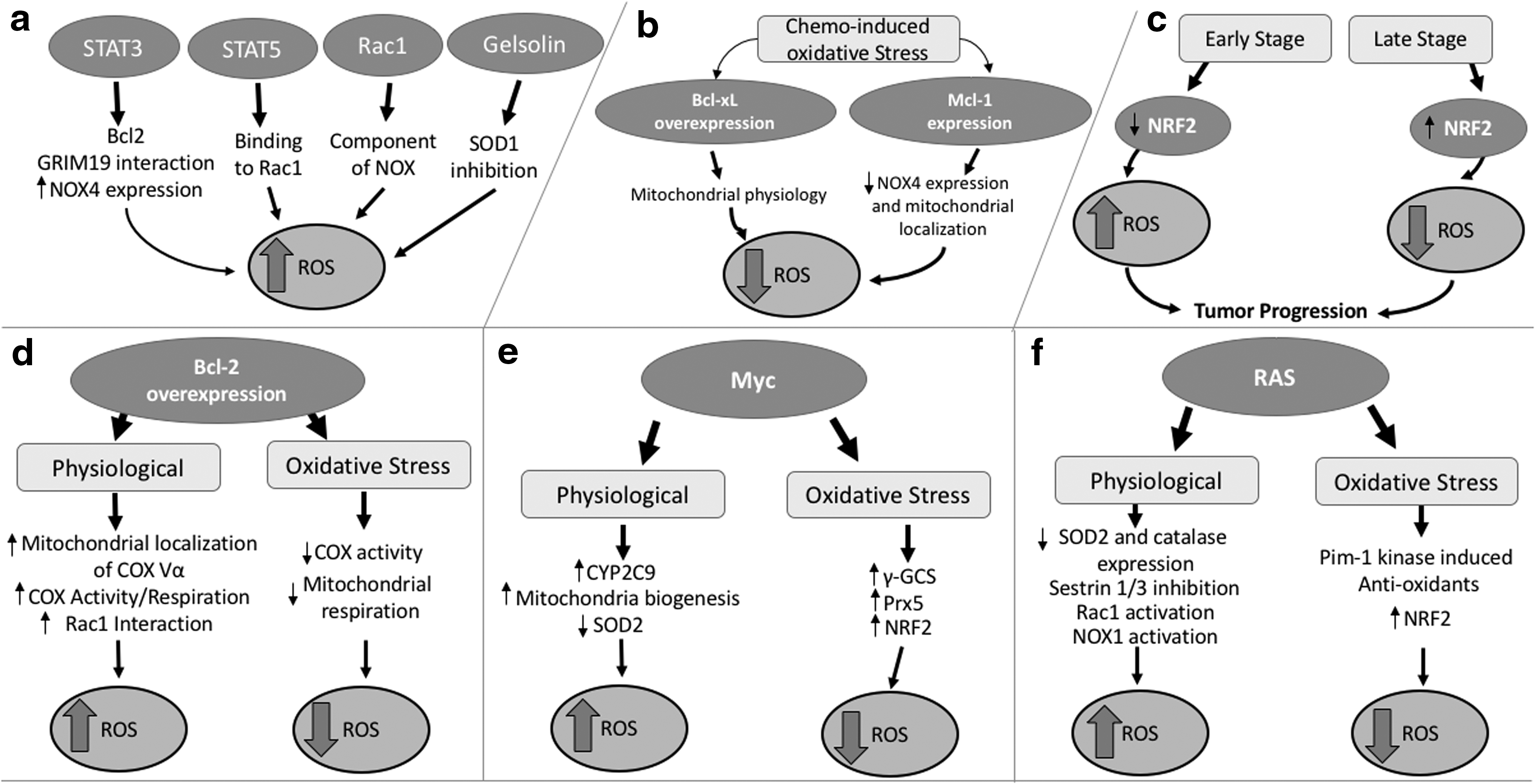
Interestingly, there appears to be a reciprocity vis a vis the relationship between the Bcl-2 family and cellular redox status. Not only does overexpression of Bcl-2 impact intracellular redox status by regulating O2 .− production under different metabolic states but also an increase in intracellular ROS has been linked to the post-translational modification of Bcl-2. In this regard, there is evidence to indicate direct oxidative modification of Bcl-2 (208), as well as our recent work underscoring the effect of an increase in intracellular O2 .− on the sustained phosphorylation of Bcl-2 at serine 70 via nitrative modification and inactivation of the phosphatase protein phosphatase 2A (PP2A) subunit B56δ, responsible for Bcl-2 dephosphorylation. Notably, the redox-mediated sustained phosphorylation of Bcl-2 at serine 70 not only induced resistance to apoptosis in vitro, but was also associated with poor prognosis and higher disease grade in a small group of lymphoma patients (206). The phosphorylation of Bcl-2, in turn, could regulate intracellular ROS by influencing the expression of antioxidant enzymes, as demonstrated by Deng et al. (69). These findings collectively suggest distinct functional outcomes of Bcl-2 overexpression, whereby (i) under physiological states, Bcl-2 overexpression supports a pro-oxidant intracellular milieu, which influences its phosphorylation status by redox-dependent inactivation of PP2A (particularly at serine 70) and stabilizes its antiapoptotic activity and (ii) sustained serine 70 phosphorylation of Bcl-2 prevents the deleterious build up of ROS particularly during oxidative stress, thus blunting oxidative stress-induced execution that underlies the mechanism of anticancer activity of a number of clinically employed chemotherapeutic drugs and γ-irradiation. These observations lend credence to the hypothesis that Bcl-2 serves a “redox rheostat” function contingent upon the cellular redox environment and its phosphorylation status.
Death inhibitory function of Mcl-1 or Bcl-xL is associated with redox modulatory activity
While discussing the antiapoptotic activity of the Bcl-2 family, two other members deserve special mention, namely Mcl-1 and Bcl-xL. Overexpression of Mcl-1 or Bcl-xL, similar to Bcl-2, is a frequent finding in aggressive and chemoresistant cancers, and associated with poor disease prognosis/outcome. Mcl-1 is overexpressed in hepatocellular carcinoma, multiple myeloma, high-grade B-cell lymphomas, and plasma cell myeloma, and its expression in multiple myeloma is linked to disease relapse and shorter life span (42, 299, 367). In contrast, Bcl-xL is overexpressed in colorectal and tongue carcinoma, in which its expression is also positively correlated with malignant tumor staging as well as metastasis in tongue carcinoma (284, 392). Interestingly, in addition to antagonizing the proapoptotic proteins of the Bcl-2 family (19, 41, 63, 104, 302, 341, 359, 391), Mcl-1 and Bcl-xL have also been implicated in the regulation of ROS (Fig. 3b). For example, Mcl-1 was shown to suppress doxorubicin-induced increase in intracellular ROS production (H2O2 and mitochondrial O2 .−) in p53-negative HCT116 colon carcinoma cells, which could be alleviated by gene knockdown of Mcl-1 (22, 64). The same research group further showed that knockdown of Mcl-1 increased NOX4 expression and NOX4 mitochondrial localization upon doxorubicin treatment, indicating that trafficking of NOX4 to the mitochondria may be crucial for drug-induced increase in ROS production in the absence of Mcl-1 (64). These findings provide testimony to the ROS regulating activity of Mcl-1 via mechanisms that suppress NOX4 expression, its mitochondrial localization, and ultimately mitochondrial ROS generation. Mcl-1 protein expression was also shown to be affected upon hypoxia-induced cell death. It was reported that hypoxia could reduce Mcl-1 level (33). As hypoxia induces ROS production such as mitochondrial O2 .− (34), it could be hypothesized that hypoxia-induced reduction of Mcl-1 is a mechanism that contributes to the rise in ROS level and subsequent cell death. With evidence showing that Mcl-1 overexpression could reduce hypoxia-induced cell death, it is likely that the antioxidant role of Mcl-1 could play a part in reducing ROS, thereby protecting cancers cell from ROS-dependent hypoxia-induced cell death. Similarly, overexpression of the antiapoptotic protein Bcl-xL significantly inhibited intracellular ROS in response to a variety of ROS-inducing triggers such as tumor necrosis factor alpha (TNF-α), serum, or glucose deprivation and exogenous H2O2 (106, 162, 189, 251). Although the precise mechanism underlying the redox regulatory activity of Bcl-xL remains undefined, Bcl-xL does not behave as an antioxidant, but instead maintains mitochondrial physiology, thereby controlling mitochondrial ROS production (106). Supporting the latter is the finding that TNF-α-induced ROS and subsequent cell death require the protein ROS modulator 1 (Romo-1). Romo-1 was shown to decrease mitochondrial membrane potential and subsequently induce ROS production (CM-H2DCFDA [2′7′-dichlorofluorescin diacetate]) through its inhibitory interaction with Bcl-xL. Importantly, overexpression of Bcl-xL could reverse TNF-α-induced ROS and cell death (162). These observations suggest a possible involvement of Bcl-xL in maintaining mitochondrial homeostasis by preventing excessive generation of stimulus-induced ROS, and in doing so inhibiting death execution.
Targeting the antioxidant side of Bcl-2 as a novel strategy to overcome chemotherapy resistance
There appears to be some degree of overlap in the noncanonical redox modulating activity of the major antiapoptotic proteins of the Bcl-2 family, which presents as a natural target for the development and design of therapeutic strategies against cancers rendered refractory by the aberrant expression of these prosurvival proteins. This acquires even more relevance given that a number of chemotherapy agents induce execution of cancer cells via mechanism that potentially involve a significant increase in intracellular ROS (270, 293, 294). It is plausible that the aberrant expression of proteins such as Bcl-2, Mcl-1, or Bcl-xL promotes drug resistance by impeding chemotherapy-induced oxidative stress. Therefore, it is highly desirable to identify novel therapeutic strategies and drugs that directly and/or indirectly inhibit the expression, phosphorylation (specifically serine 70), and activity of Bcl-2. To that end, the heightened interest and focus on the development of novel Bcl-2 inhibitors are a promising step in the right direction with potential therapeutic implications. Multiple Bcl-2 homology (BH) mimetics such as ABT737, ABT263, and ABT199 have been used empirically as well as in clinical trials to target cancers such as lymphoma, myeloma, and leukemia (1, 32, 56, 132, 146, 160, 167, 171, 252, 258, 276, 307, 317, 339, 344, 361). Although the success of some of these trials was compromised by the nonspecific targeting of Bcl-xL and the development of severe thrombocytopenia, the development of a more selective inhibitor, ABT199 (venetoclax), which does not target Bcl-xL and spares platelets, is extremely encouraging and has received FDA approval as a monotherapy for CLL (62, 307, 339, 344). Interestingly, a recent study linked the Bcl-2 inhibitory activity of ABT199 to an increase in ROS production (Carboxy-H2DCFDA), which further supports the redox regulatory role of Bcl-2 in cancer cells (376). Given these data, it is tempting to speculate that the increase in oxidative stress observed in cancer cells upon exposure to ABT199 could be a function of inhibiting the redox rheostat activity of Bcl-2 by preventing its phosphorylation at serine 70, thereby restoring apoptosis sensitivity of cancer cells rendered refractory to chemotherapy by the sustained expression for Bcl-2. Similarly, as alluded to the preceding section, given that Mcl-1 and Bcl-xL are upregulated in many human cancers and their ability to induce resistance to ROS-mediated execution (22, 42, 64, 106, 162, 189, 251, 284, 299, 367, 392), it is only logical to design and develop small molecules that specifically target the redox modulating activity of these proteins. Some examples of selective inhibitors for Mcl-1 include A-1210477 and S63845, whereas those for Bcl-xL include WEHI-539 and A-1155463 (175, 190, 191, 326). It remains to be seen whether the death sensitizing effects of these selective compounds also involve neutralizing the redox regulatory functions of these proteins, similar to what has been reported for the Bcl-2 inhibitor ABT199.
The STAT Family of Transcription Factors and Their Role in Tumorigenesis
STAT3 promotes survival and regulates ROS in a noncanonical manner
The signal transducer and activator of transcription (STAT) proteins belong to a family of transcription factors involved in the regulation of several cellular processes, including apoptosis and cell cycle progression (123, 306, 375). Specifically, the STAT proteins are involved in the JAK-STAT pathway and activated by factors or ligands such as growth factors (e.g., epidermal growth factor [EGF] or PDGF) and cytokines (e.g., interleukin-6 [IL-6]). Upon ligand binding, the receptor subunits dimerize, thereby recruiting the Janus kinase (JAK) family proteins to proximity for their transphosphorylation and activation at the cytoplasmic domain. Upon activation, JAKs phosphorylate STAT proteins, which upon phosphorylation form hetero- or homodimers, translocate to the nucleus wherein they bind consensus DNA-recognition motifs, found in the promoter region of the target genes, thus activating gene transcription (54, 55, 142). One notable activator or ligand for one such family member, STAT3, is IL-6 (Fig. 4) (172), a cytokine associated with poor prognosis in breast cancer patients and implicated in the progression of lung carcinoma (282, 384).
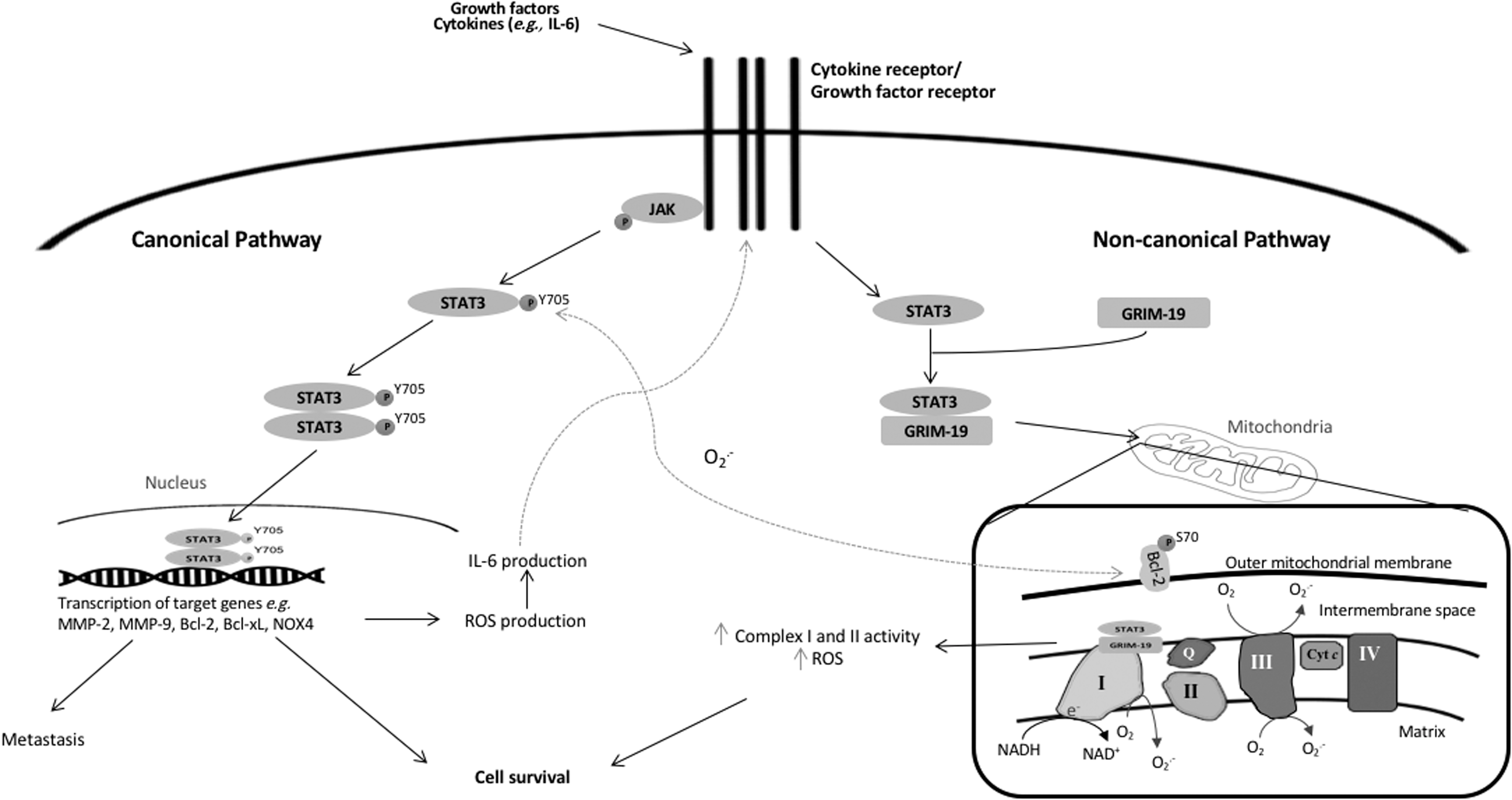
STAT3 has been most frequently associated with tumorigenesis and is widely documented as an oncoprotein. Aberrant STAT3 signaling, or the upregulation of STAT3 phosphorylation at tyrosine 705 (Y705), positively associated with JAK expression or human papillomavirus infection, is observed in a variety of cancers such as breast, lung, and cervix (139, 204, 298, 375). As a transcription factor, STAT3 induces the transcription of many target genes, most of which are crucial for growth, survival, cell transformation, angiogenesis, and metastasis in tumorigenesis (Fig. 4). One study showed that immortalized human breast epithelial cells could undergo cell transformation when a constitutively active STAT3 mutant (STAT3-C) is expressed. This was further shown to be dependent on the ability of STAT3-C to upregulate matrix metalloproteinase (MMP)-9 messenger ribonucleic acid (mRNA) and protein levels in these immortalized breast epithelial cells (61). Another group has shown that increased MMP-2 expression is also correlated with increased STAT3 activity in highly metastatic melanoma cells. They further demonstrated that transfection of STAT3-C and dominant negative STAT3 (STAT3-DN) reciprocally activated and inhibited MMP-2 promoter, respectively. In addition, the transfection of STAT3-DN in metastatic cells decreased the expression of MMP-2. Importantly, in vivo study through the injection of metastatic cells harboring STAT3-DN in mice also demonstrated a decrease in tumor cell growth and metastasis (372). In addition, STAT3 contributes to angiogenesis through the induction of vascular endothelial growth factor (VEGF), a key mediator of angiogenesis in cancer. The upregulation of VEGF was observed, concomitant with increased STAT3 activity in breast and head and neck cancers. This association was further corroborated when STAT3-C was stably expressed in murine NIH3T3 fibroblast and melanoma B16 cells. Importantly, the increased vascularization, as confirmed by higher hemoglobin content, was observed when STAT3-C-transfected B16 tumor cells were implanted in vivo (245). Corroborating that, another study also showed that targeting STAT3 with a small-molecule inhibitor could block VEGF expression in vitro as well as inhibit tumor growth and angiogenesis in vivo (245, 377). Apart from angiogenesis, constitutive STAT3 activation has been widely described to inhibit apoptosis through activation of antiapoptotic genes such as Bcl-2 and Bcl-xL (31, 98). Inhibition of STAT3 reduces expression of the antiapoptotic proteins and induces apoptosis (8, 84, 241). Intriguingly, Choi et al. reported the use of novel inhibitor of STAT3, tuberatolide B (TTB) to induce apoptotic cell death. TTB selectively inhibits STAT3 phosphorylation and transcriptional activity, reduces downstream targets of STAT3 as well as induces apoptotic cell death. Of note, the ROS scavenger, N-acetyl-L-cysteine (NAC), blocked the effects of TTB on ROS production (H2DCFDA), STAT3, and its target genes as well as cell death, indicating that the effect of TTB is dependent on ROS (45). Although a causal relationship between TTB-STAT3 apoptosis was not investigated here, these findings nonetheless suggest a role of ROS in regulating STAT3 activity. In this regard, several studies have demonstrated the effects of ROS on STAT3 as illustrated in Figure 3a. NOX has been reported to indirectly activate STAT3 by stimulating IL-6 production and subsequent activation of the JAK1-STAT3 pathway. Overexpression of NOX4 in nonsmall cell lung cancer (NSCLC) cells resulted in ROS-dependent IL-6 production as well as STAT3 phosphorylation at Y705, whereas NOX4 knockdown subsequently reduced ROS production (H2O2) and suppressed IL-6 production and STAT3 activity. Interestingly, the exogenous introduction of IL-6 and subsequent STAT3 activation could also upregulate NOX4. This collectively suggests a positive loop whereby increased IL-6 production and IL-6/STAT3 activation could increase NOX4 expression, which, in turn, upregulates ROS production to further enhance IL-6 production and IL-6/STAT3 activation (194). These data are further corroborated by a study that linked pharmacological inhibition of NOX (and downstream ROS production) (206) to a significant decrease in STAT3 activation in hepatocellular carcinoma cells (308). Corroborating that, our group demonstrated that exposure of cells to the NOX inhibitor, DPI, could reverse the induction of STAT3 phosphorylation in Bcl-2 overexpressing CEM lymphoblastic leukemia cells, which constitutively harbor elevated O2 .− levels (34, 156).
Although ROS have been linked to the activation of STAT3, STAT3 activation, in turn, could also regulate intracellular ROS production. In this case, a noncanonical role of STAT3, independent of its transcriptional activity, was implicated in the regulation of mitochondrial O2 .− production. Overexpression of wild-type STAT3 resulted in an increase in mitochondrial O2 .− production, which was reversed by a functional STAT3 mutant (STAT3-Y705F). Another group also demonstrated that gene associated with retinoid interferon-induced cell mortality 19 (GRIM-19) is capable of acting as a chaperone to recruit STAT3 to the mitochondria (318). This suggests that STAT3 could be inducing mitochondrial O2 .− via its localization to the mitochondria (Fig. 4). In support of this, STAT3-deficient cells (STAT3−/−) exhibit decreases in mitochondrial complexes I and II activities and reconstitution of STAT3 into STAT3−/− cells rescued the loss of ETC activity, thereby supporting the role of STAT3 in the regulation of mitochondrial redox metabolism (354). In addition, we previously reported a crosstalk between increased expression of the antiapoptotic protein, Bcl-2, cellular pro-oxidant milieu, and phosphorylation/activation of STAT3 (pY705); Bcl-2 overexpression induced an increase in STAT3 phosphorylation at Y705, which was reversed by DPI and Rac1 inhibitor. Of note, using site-directed mutagenesis (Y705F), STAT3 activation was associated with an increase in Bcl-2 phosphorylation at serine 70, which stabilized its antiapoptotic activity (34, 46, 156, 206, 342). Collectively, these data point to the existence of a positive feed forward loop between Bcl-2 and STAT3 phosphorylation, whereby an increase in mitochondrial O2 .− upon Bcl-2 overexpression induces STAT3 phosphorylation/activation, which, in turn, further augments ROS levels to promote serine 70 phosphorylation (and activation) of Bcl-2. These findings provide testimony that not only does cellular redox status positively impact STAT3 activation but also that the activated STAT3 amplifies this circuit by further fueling mitochondrial ROS generation (Fig. 4) (156).
Targeting the redox-modifying effect of STAT3 as an anticancer strategy
Based on the evidence linking STAT3 activation to a host of human cancers and its upregulation as a surrogate survival mechanism in cells rendered resistant to tyrosine kinase inhibitor (TKI), the transcription factor has been an attractive target for cancer therapy (139, 204, 298, 375). Intriguingly, STAT3 inhibitory effect was discovered as an off-target effect of some drugs developed for the treatment of unrelated indications, which led to the repurposing of these compounds for the treatment of cancers with elevated STAT3 expression (6, 235). One example is nifuroxazide, which has been in use since 1966 for the treatment of colitis and diarrhea. Notably, this antibiotic was shown to inhibit the phosphorylation of STAT3 at Y705 (236) and elicited antitumor and antimetastatic effects in colorectal cancer and melanoma cells (383, 398). Nifuroxazide reduced cell viability by triggering Bax-dependent apoptosis, as well as repressed two STAT3 target genes MMP-2 and MMP-9 that are involved in metastasis. It should be pointed out that the apoptosis-inducing activity of nifuroxazide was associated with an increase in intracellular ROS (mitochondrial O2 .− and H2O2); however, the precise mechanism underlying the involvement of ROS in this context is not well understood. A plausible scenario could be an increase in ROS triggered by an increased shift to a ratio of Bax to Bcl-2 upon inhibiting STAT3 activation. In this regard, mitochondrial translocation of Bax has previously been shown to trigger an increase in intracellular ROS (CM-H2DCFDA and dihydroethidium [DHE]) (3, 165).
Another STAT3 inhibitor, NSC-743380, has been shown to display antitumor effects against NSCLC cells by inhibiting STAT3 phosphorylation (Y705) (110) and inducing oxidative stress (201). Interestingly, the antioxidant, nordihydroguaiaretic acid, was able to reduce ROS level (H2DCFDA) and rescue cells from the death-inducing effects of NSC-743380, without affecting its STAT3 inhibitory activity, thus indicating a downstream signaling role for ROS in therapies aimed at targeting STAT3 activation. Indeed, knockdown of STAT3 increases ROS levels and suppresses tumor growth. More recently, Wong et al. reported promising antitumor effects of a first-in-man direct STAT3 inhibitor, OPB-51602, in a Phase I clinical study involving refractory solid malignancies (363). The molecular mechanisms underlying the antitumor activity of OPB-51602 also involve a strong mitochondrial redox component (Hirpara et al., unpublished results), thus underscoring the crosstalk between STAT3 and cellular redox metabolism.
Nevertheless, how does one reconcile with the similar effects on ROS production upon activation or pharmacological inhibition of STAT3? In light of the data supporting the translocation of activated STAT3 to the mitochondria and its downstream effect on mitochondrial metabolism, such as O2 .− production (156, 354), it is plausible that activated STAT3 amplifies mitochondrial ETC activity via its interaction with complex I component(s) (354), hence impacting oxygen consumption and as such increasing the probability of electrons leaking onto oxygen to generate O2 .−. This is similar to what has previously been shown by our group upon overexpression of Bcl-2 (34, 35). This assumption is further supported by the observation linking increased STAT3 phosphorylation to enhanced mitochondrial oxidative phosphorylation (OXPHOS) in drug-resistant cancer cells [(187) and Hirpara et al. unpublished results], which creates a synthetic lethal condition that relies on OXPHOS. This allows the targeting of mitochondrial ETC to shut down the forward transport of electrons (similar to rotenone) and in doing so triggers oxidative stress. Alternatively, the redox effect of shutting down STAT3 signaling could also be a function of inhibition of two transcriptional targets of STAT3, Bcl-2, and Bcl-xL. Downregulation of Bcl-2 or Bcl-xL could tilt the balance in favor of Bax, which could drive mitochondrial ROS production (3, 31, 98, 165), as discussed previously.
The neglected STAT5 in tumorigenesis
In addition to STAT3, STAT5, referring to both STAT5a and STAT5b isoforms, has also displayed tumorigenic properties (368). Constitutive activation of both STAT3 and STAT5 has also been reported in breast cancer (346). With that in mind, STAT5 is postulated to function as an oncoprotein through regulation of several pathways. STAT5 activation has been described to inhibit apoptosis through its role as a transcription factor in regulating genes involved in antiapoptotic signaling, such as Bcl-2 (219). Knockdown of STAT5b and expression of dominant-negative STAT5b induced G1 arrest and subsequent cell death, highlighting the role of STAT5 in the inhibition of apoptosis (219). In addition, knockdown of STAT5 significantly reduced the expression of proteins involved in metastasis such as focal adhesion kinase, VEGF, and MMP-2 together with a reciprocal increase in E-cadherin. These findings underscore the involvement of STAT5 in the activation of cell invasion and metastasis (374).
One of the activators of STAT5 signaling pathway in acute myeloid leukemia (AML) is FMS-like tyrosine kinase 3-internal tandem duplication (FLT3-ITD). This strong association between STAT5 and FLT3-LM was observed in primary AML blast cells. In addition, introduction of the FLT3 mutants increased the transforming potential of interleukin-3-dependent murine pro-B cells (Ba/F3) in vitro (309). More relevantly, a separate study showed that FLT3-ITD expression in AML cells induced an increase in intracellular H2O2 in an FLT3- and NOX-dependent manner; PKC412, an inhibitor of FLT3, as well as NOX inhibitors DPI and VAS2870, significantly reduced ROS levels in FLT3-ITD-expressing AML cells. Importantly, knockdown of p22phox, a component of NOX, is capable of reducing STAT5 phosphorylation and signaling in FLT3-ITD-expressing AML cells, suggesting that FLT3-induced H2O2 via NOX/p22phox is crucial for STAT5 signaling (365). The latter is further supported by observations highlighting the critical involvement of the small GTPase Rac1 in the crosstalk between STAT5 and ROS; FLT3-ITD-induced ROS generation (H2DCFDA) was shown to require both activated STAT5 and Rac1, as silencing of STAT5, inhibition of Rac1, or expression of RacN17 alleviated ROS levels. In addition, the same study also reported that phosphorylated STAT5 could bind to Rac1, suggesting that this binding may be a mechanism of FLT3-ITD/STAT5-mediated ROS production (281). Furthermore, the binding of Rac1 to phosphorylated STAT5, in this case STAT5a, was believed to be a requirement for its nuclear translocation, as presence of active Rac1 (Rac1 V12) and depletion of Rac1 reciprocally affect the nuclear localization of phosphorylated STAT5a (157). Given the function of Rac1 in ROS production, the binding of two proteins strongly argues in favor of an involvement of ROS in the nuclear translocation of STAT5. Moreover, STAT5 activation has also been associated with increased ROS production (Carboxy-H2DCFDA) in FLT3-ITD-expressing AML cells via increased expression of its transcriptional target NOX4 (150). Of note, knockdown of NOX4 reduced ROS levels and attenuated FLT3-ITD-induced transformation. These findings point to yet again another positive loop between NOX and STAT5, whereby activated STAT5 is capable of increasing NOX expression, thereby increasing ROS level to activate STAT5, similar to STAT3 (Fig. 3a).
STAT5 is also one of the significant drivers of chronic myelogenous leukemia (CML) and was observed to positively correlate with BCR-ABL mutation rate. Intriguingly, STAT5 is proposed to enhance mutation rate of BCR-ABL via ROS production in CML cells, as shown by the increased ROS-dependent DNA damage. Overexpression of STAT5 was accompanied by an increase in ROS level (DHE) independent of JAK2 but dependent on BCR-ABL signaling. STAT5-induced ROS production subsequently resulted in DNA double strand breaks, indicated by increased γH2AX (phosphorylated histone H2AX) level, which could be rescued by NAC. As a result, the potential STAT5-induced ROS-mediated BCR-ABL mutation was proposed to reduce sensitivity to TKIs as a mechanism of TKI resistance in BCR-ABL-driven CML cells (352). These findings are corroborated by the significantly high STAT5 expression in patients with acquired resistance to BCR-ABL inhibitors, such as imatinib (353), thus suggesting the involvement of ROS in mediating STAT5-induced tumorigenesis.
Targeting STAT5-ROS nexus as an anticancer strategy
The crosstalk between STAT5 and intracellular ROS appears to play a critical role in STAT5-induced tumorigenesis and/or its progression, and as such offers a window of opportunity for the design and development of specific STAT5 inhibitors for targeting FLT3-ITD-expressing AML as well as restoring the sensitivity of BCR-ABL-driven CML cells to TKI. However, unlike the abundant availability of STAT3 inhibitors, few STAT5 inhibitors have been developed. Nevertheless, in a repurposing approach, the antipsychotic drug, pimozide, has demonstrated inhibitory effect on STAT5 phosphorylation, resulting in apoptosis of AML cells and reduced tumor burden in FLT3-driven AML mouse model. In addition, pimozide could enhance the reduction of STAT5 phosphorylation as well as cell viability when treated with PKC412 or sunitinib, both TKIs (237). Interestingly, pimozide inhibited binding of STAT5 to NOX4 promoter, leading to a significant decrease in NOX4 mRNA expression (150). Although there is lack of evidence to associate pimozide-mediated STAT5 inhibition to a change in cellular redox status, the observed decrease in NOX4 mRNA would be expected to decrease ROS levels after STAT5 inhibition. Despite the relatively modest advance in the design of STAT5 inhibitors, the redox modulating effects of STAT5 could be a potential novel strategy for the design and development of new pharmacological agents for the management of AML and CML.
RAS GTPase Family and ROS: Tumorigenic Partners in Crime
The RAS proteins belongs to the small GTPase superfamily and are involved in signaling and regulation of critical cellular functions such as cell survival, proliferation, motility, and cell proliferation (10, 16, 268, 312). RAS GTPases transduce a broad range of signals dependent on their activation upon binding to guanosine-5′-triphosphate (GTP), which is exchanged for guanosine diphosphate (GDP) in the inactive state (268). The rapid switch between GTP-bound active form and GDP-bound inactive form ensures an efficiently controlled and transient signal transduction. The transition between those two forms is further modulated by the functions of guanine nucleotide exchange factors (GEFs), stimulating GTP loading for RAS activation, and GTPase-activating proteins (GAPs) enhancing GTP hydrolysis for RAS inactivation (30, 202, 268).
The RAS subfamily comprises three genes HRAS, NRAS, and KRAS. These genes are mutated in about one-third of human cancers (30, 202, 213, 268, 312, 360), hence the heightened interest in understanding the functional biology of these proteins and development of specific targeting strategies. RAS proteins are involved during the early phase of transformation/tumorigenesis as well as play a critical role in the maintenance and progression of the disease (40, 183). RAS mutations mostly affect the regulation of RAS activation through impairing their ability to interact with GEFs and GAPs, thus resulting in prolonged RAS activation and stimulation of its downstream effectors such as RAF/MEK/extracellular signal-regulated kinase (ERK) and PI3K/AKT/mTOR pathways, which, in turn, promote cancer cells survival and proliferation (10, 16, 27, 86, 268).
There is a preponderance of evidence to indicate an intricate crosstalk between RAS-driven carcinogenesis and cellular redox status (Fig. 3f) (128, 129, 170, 313, 356). Not only has an increase in ROS linked to the regulation of RAS activity (57, 127, 129, 220), but RAS-induced carcinogenesis has also been associated with a change in cellular redox milieu (Fig. 5). For example, nitric oxide (NO)/O2 (via ·NO2) and O2 .− similarly promote the dissociation of GDP from RAS, therefore, facilitating GTP reloading and promoting its subsequent activation (127 –129, 220). Similarly, H2O2, in combination with transition metals such as copper (Cu2+) and iron (Fe2+), as well as ONOO−, a reaction product of nitric oxide NO and O2 .−, also promotes GDP dissociation (128). Furthermore, cysteine 118 (Cys118) within RAS is susceptible to S-nitrosylation through a reaction with ·NO2, thus resulting in guanine nucleotide dissociation (129, 358). The same Cys118 is susceptible to S-glutathionylation, which plays a part in inducing RAS activity through the formation of RAS-radical intermediates preceding the formation of S-glutathionylated RAS (129, 133, 358). Interestingly, oxidative modification at Cys118 has been implicated in RAS-driven carcinogenesis, as site-directed mutagenesis to C118S reduced carcinogen-induced lung tumorigenesis in mice (141).

Not only does ROS impact RAS structure and/or activity, but also RAS activation itself is a strong inducer of cellular ROS production, which has also been linked to the tumor-promoting activity of RAS (198, 221, 356). In this regard, it is worth mentioning that the major intracellular survival and proliferation networks involved in cellular redox metabolism such as NF-κB activity, hypoxia signaling, MAPK activation, and autophagy are also modulated by RAS (16, 27, 86, 90, 109, 198, 261, 268, 288, 312, 351, 356). Mutant KRAS was shown to induce the expression of miR-155 via MAPK and NF-κB pathways, which resulted in the downregulation of the transcription factor, Foxo3a, that transcribes for two major antioxidant genes, SOD2 and catalase (349) (Figs. 3f and 5). Similarly, the synergy between oncogenic RAS and ROS (CM-H2DCFDA) has been highlighted in active mutant KRASD12-driven lung carcinogenesis, in which a null mutation of Prx1 resulted in a strong enhancement of tumorigenesis through stimulation of ERK/cyclin D1 signal. The latter suggests that the ROS scavenging activity of Prx1 could act as a tumor suppressor mechanism in lung carcinogenesis (256). Moreover, the effect on Prxs is also manifested via the inhibitory effect of RAS on Sestrin 1 and 3, which function to suppress intracellular ROS (173). On the contrary, Prx1 was also shown to promote active mutant HRASV12-induced hepatic carcinogenesis, as removal of Prx1 decreased ERK activation, and increased ROS-induced DNA damage and apoptosis (117). A similar effect of RAS-induced ROS and tumorigenesis has been demonstrated on the master antioxidant transcription factor NF-E2-related factor (NRF2). Although NRF2 might act as a suppressor of malignant transformation, its role in later stages of KRAS-driven lung cancer progression was shown to be critical in cell survival and tumor growth (283). NRF2 has also been shown as an important mediator of drug resistance in NSCLCs harboring KRAS mutations (325). Furthermore, inhibition of NRF2 ubiquitination by the absence of Sag-E3 ubiquitin ligase was described as a critical mechanism in KRAS-driven skin carcinogenesis (371). Last but not least, Pim kinases, which control the expression of the antioxidant enzymes Prx3, GPx4, and SOD, are also downstream effectors of mutant KRAS-induced cell growth, as deletion of Pim kinases resulted in KRAS-induced ROS-dependent cell death (305) (Figs. 3f and 5). These observations lend credence to the hypothesis that oxidative stress triggered by RAS results in the induction/upregulation of cellular antioxidant defense systems, thus maintaining cellular redox levels sufficiently high to promote growth and proliferation but preventing the development of overwhelming oxidative stress.
Having presented evidence to link RAS-driven carcinogenesis to a feedback induction of antioxidant systems, the evidence linking ROS more directly to RAS-induced transformation is highly convincing. Activation of RAS is a stimulus for ROS generation through the intermediary involvement of Rac1, which was shown to be required for malignant transformation and cancer progression (198, 221, 356). Similarly, KRAS mutations strongly correlate with increased expression of the O2 .− generating enzyme, NOX1, in colorectal carcinoma (185). In another study, RAS-induced NOX1 expression and activation were linked to the downstream activation of the MEK/ERK pathway (2, 254). Corroborating these findings, in a model of pancreatic cancer, mutant active KRAS (GTP-bound) was shown to induce oxidative stress upon knockdown of tumor protein p53-inducible nuclear protein 1 (TP53INP1). Further evidence to link RAS signaling to carcinogenesis was provided by observations implicating active Rac1-mediated O2 .− production and the intermediary involvement of NOX in RAS-induced growth and proliferation (4, 78, 249). Finally, mutant KRAS-induced anchorage-independent proliferation via MAPK signaling pathway was shown to require mitochondrial O2 .− produced primarily from complex III (313, 356). Taken together, all these results demonstrate the strong links between oncogenic RAS and ROS production, which can act as a double-edged sword for cancer cells: ROS production is necessary to sustain mutant KRAS-driven cell proliferation but needs to be kept in check through oncogenic RAS-dependent regulation of antioxidant mechanisms. Therefore, oncogenic RAS can be considered as a dual redox regulator that stimulates ROS production to activate its downstream pathways while regulating antioxidant mechanisms to ensure that ROS level is maintained at prosurvival level.
The therapeutic stakes of efficiently targeting mutant RAS-driven cancers
Inhibition of oncogenic RAS has become an important therapeutic strategy against RAS-driven cancers. However, past attempts at drugging RAS have not had the desired results. Nevertheless, continuous effort in understanding the protein as well as its structure for therapeutic intervention has been on-going (207, 213, 360). The efficacy of these strategies remains to be fully evaluated and the rather disappointing results with farnesyl transferase inhibitors have left an open area for developing novel strategic therapeutics (47, 86, 207, 360, 378, 381, 399). As mentioned previously, the ROS dependency of RAS-driven tumors may well present with a window of opportunity, which could be exploited for therapeutic management (79). The initial attempts in this respect involved alleviating intracellular ROS to levels that can no longer promote cellular proliferation. Interestingly, administration of antioxidant such as vitamin E for KRAS-pancreatic cancers elicited antiproliferative effects (347). The NOX inhibitor, DPI, has also been shown to deplete ROS level to promote apoptotic cell death in KRAS-mutated pancreatic cancer cell line PANC-1 through NOX4 inhibition (223). Similarly, metformin-induced growth inhibition of PANC-1 and Mia-PaCa cells carrying mutant KRAS was a function of decreased ROS production through upregulation of SOD2 and downregulation of NOX2 and NOX4 proteins (38).
The second type of strategy tested was to disrupt ROS homeostasis to reach lethal levels of ROS in oncogenic RAS-driven cancer cells (79, 279). Such a strategy was validated by a screening of >50,000 compounds, which resulted in the discovery of a class of molecules against mutant KRAS-expressing cells (289). Initially developed as a muscle relaxant, the lead molecule of this massive screen, lanperisone, induced cell death by increasing intracellular ROS (DCFDA), which was dependent on iron metabolism and activation of MAPK pathway (289). This molecule has been described to induce ferroptosis, a novel form of cell death that relies on iron-mediated ROS production, MAPK pathway, and mitochondrial porins voltage-dependent anion channel 2/3 (373). Another chemical library screening aimed at identifying compounds targeting lung adenocarcinoma cells harboring epidermal growth factor receptor or KRAS mutations identified a potential candidate, lung cancer screen 1 (LCS-1, which reduced proliferation and induced apoptosis through a mechanism that also involves inhibition of MAPK and PI3K pathways (304). Interestingly, further investigations into the biological activity of LCS-1 identified SOD1 as a likely target (303). Indeed silencing of SOD1 reduced cell growth of LCS-1-sensitive cells while its overexpression increased resistance to LCS-1. In addition, treatment with LCS-1 significantly reduced SOD1 activity (303). Interestingly, the use of vitamin C was shown to induce massive ROS accumulation (DCFDA) to kill mutant KRAS colorectal cancers. The effect was due to an increased uptake of oxidized vitamin C, termed dehydroascorbate (DHA), leading to oxidative stress via DHA depletion of GSH. Accumulated ROS subsequently inactivates glyceraldehyde 3-phosphate dehydrogenase, which in a highly glycolytic environment of KRAS mutant cells, leading to energetic crisis and cell death (388). Lastly, our group recently devised a novel strategy using a small compound with the ability to hyperactivate mutant KRAS harboring cells (as well as cells displaying HRAS and NRAS mutations), which resulted in AKT-dependent increase in mitochondrial O2 .− and intracellular H2O2 production and cell death (147). In addition, cell death induced by this small molecule displayed features of both autophagy and apoptosis mediated by activation of ERK and JNK (364). Taken together, these results show that ROS production might be the Achilles heel of RAS-driven cancers and could be exploited for the specific targeting of cancers carrying RAS mutations.
The Pro-Oxidant Effect of RacGTPase Promotes Tumorigenesis
Rac is another GTPase protein that belongs to the Rho family. It has been implicated in the regulation of cytoskeleton remodeling, chemotaxis, signal transduction pathways, and tumorigenesis (20, 25, 83, 158, 224, 233, 269, 301, 329, 382). The human Rac comprises three isoforms, namely Rac1, 2, and 3. Rac1 and 3 are ubiquitously expressed whereas Rac2 is specifically expressed in hematopoietic cells (74, 112, 212, 297). Similar to RAS GTPase, Rac activity is governed by the cycling of GTP and GDP (118, 178, 253).
An important role of Rac is its involvement in the assembly and activation of NOX1, a multisubunit complex that produces O2 .− (Fig. 3a) (37, 336); active Rac1 specifically binds to the NOX1 subunit, NOXA1. Mutation of NOXA1 that disrupts its interaction with active Rac1 results in reduced NOX1-dependent ROS production (37). Along similar lines, constitutively active Rac1V12 elevates the basal level of O2 .−, whereas dominant negative Rac1N17 or genetic knockdown of Rac1 decreases the basal level of O2 .− as well as prevents further ROS production upon growth factor or cytokine stimulation (43, 260, 342). Apart from NOX1, Rac1 also localizes to the mitochondria, the powerhouse of cells as well as a major source of intracellular ROS. Rac1 was shown to be coimmunoprecipitated with cytochrome c of the ETC, which suggests a potential interplay between cytochrome c and Rac1 in the production of mitochondrial ROS (H2O2) (250). Similarly, as discussed earlier, our group demonstrated that Rac1 is involved in the increase of mitochondrial O2 .− production through its interaction with Bcl-2 (342). Although observed from different studies, these protein–protein interactions between Rac1 and Bcl-2 or cytochrome c suggest that all three proteins may possess interdependent roles in mitochondrial redox metabolism.
From the standpoint of tumorigenesis, Rac1 overexpression has been documented in a host of human cancers, such as testicular, lung, and breast carcinomas (125, 154, 286, 396). These observations suggest that Rac1 may play an important role in promoting cellular transformation and tumor progression. Indeed, countless empirical studies have demonstrated that Rac1 is required for enhanced survival, proliferation, motility, and invasion in vitro (20, 25, 43, 125, 158, 260, 329, 337, 342, 382). For example, our group showed that genetic knockdown or inhibition of Rac1 resensitizes Bcl-2 overexpressing CEM cells to drug-induced cell death in vitro (342). In addition, a different study also demonstrated that Rac1 knockdown inhibited VEGF-induced vascular formation, cell migration, invasion, and proliferation in vitro. Corroborating these in vitro data, Rac1 knockdown in tumor-grafted or VEGF-supplemented mouse also showed reduced angiogenesis/tumor growth or angiogenesis, respectively (337). Apart from that, point mutation P29S and the splice variant Rac1b have been identified in melanoma and breast/colorectal/lung carcinoma, respectively (152, 176, 286, 301, 395). These aberrations have also been empirically characterized as gain-of-function in cell lines, thereby further supporting the oncogenic role of Rac1. For example, Singh et al. showed that Rac1b possesses enhanced intrinsic guanine nucleotide exchange activity and impaired intrinsic GTPase activity. Importantly, they further demonstrated that Rac1b could activate AKT as well as promote growth transformation of NIH3T3 murine fibroblasts (301). In contrast, Davis et al. demonstrated that, although mutant Rac1P29S maintains the intrinsic GTP hydrolysis, the active mutant possesses a substantially increased inherent GDP/GTP nucleotide exchange activity (58). Essentially, the functional outcome of Rac1P29S was further demonstrated by Krauthammer et al. to promote proliferation and migration of melanocytes (176). Aside from P29S mutation, other mutations that constitutively activate Rac1 such as Rac1V12 and Rac161L are also linked to increased proliferation, survival, and invasion (158, 260, 301, 329). It was revealed that NIH3T3 fibroblasts stably transfected with constitutively active form of HRASV12 was capable of enhancing unscheduled DNA synthesis, DNA repair capacity, and clonogenic cell survival after ultraviolet irradiation or cisplatin treatment. They further demonstrated meticulously that this was a downstream function of Rac1 as transfection with Rac1V12 alone could enhance DNA repair capacity and transient expression of loss-of-function variant Rac1N17 in HRASV12 cells could reciprocally diminish DNA repair capacity. The enhanced capability of DNA repair by activated HRAS and downstream Rac1 was further shown to be dependent on Rac1/NOX-induced ROS production as both HRASV12- and Rac1V12-induced DNA repairs were abrogated by NAC and DPI treatments (43). Although Rac1V12 and Rac161L were not identified in a sequencing study of breast tumors (286), these gain-of-function mutations nonetheless provide valuable information to further support Rac1 as an oncoprotein that drives tumorigenesis, migration, and resistance to cell death. Expanding on the role of Rac1-induced ROS and tumorigenesis, another group reported that expression of dominant negative Rac1N17 leads to decreased ROS levels (DCFDA), reduced NF-κB activation, and an increased susceptibility of human umbilical vein endothelial cells to TNF-α-mediated apoptosis. This suggests an oncogenic circuit, whereby Rac1-induced ROS production could activate NF-κB and thereby desensitize cancer cells to TNF-α-induced apoptosis (72). Along these lines, we previously demonstrated a direct link between Rac1-induced ROS and apoptosis resistance, in which the sensitization of cancer cells toward apoptosis upon Rac1N17-induced decrease in O2 .− production was reverted in the presence of an inhibitor of O2 .− scavenger (SOD1) (260). Corroborating that, genetic knockdown and inhibition of Rac1 were capable of resensitizing tumor cells to etoposide-induced cell death via alleviating intracellular O2 .− production (342).
It should be pointed out that Rac1-induced ROS have also been implicated in cellular cytoskeleton reorganization, cell motility, invasion, migration, and metastasis (20, 224, 269, 329, 382). For example, Rac1 plays a prominent role in the induction of epithelial–mesenchymal transition (EMT) by virtue of its involvement in regulating cell motility and actin dynamics. Constitutively active Rac1V12 was shown to induce actin reorganization, which is dependent on the production of O2 .− (224). Besides the direct regulation of the actin cytoskeleton by Rac1, another group under Martinez also reported that MCF-7 and T47D human mammary cells expressing either the active or dominant negative forms of Rac1 could affect ROS level (H2DCFDA) differently (329). The differing levels of ROS consequently regulate the invasive potential of these cells potentially via an NF-κB-dependent upregulation of urokinase, a serine protease involved in cell migration (329). Two other studies also reported that Rac1 and Rac1b are responsible for invasion with upregulated MMP-2- and MMP-3-induced EMT, respectively. The former study demonstrated that EGF treatment induced PI3K- and Src-dependent Rac1 activation and subsequent Rac1/NOX-induced ROS production (CM-H2DCFDA) in PANC-1 cancer cells, which resulted in increased secretion and activation of MMP-2 and, in turn, enhanced invasive capacity (20). The latter study similarly corroborates the redox, oncogenic, and metastatic roles of Rac1, wherein they specifically showed that treatment of mouse mammary epithelial cells with MMP-3 increased expression of Rac1b as well as consequentially augmented mitochondrial O2 .− production, which, in turn, is crucial for EMT (vimentin and snail upregulations) and migration (269). These observations collectively indicate the involvement of Rac1-induced ROS production as a crucial mediator of EMT, migration, invasion, and metastasis.
Intriguingly, as much as Rac1 could regulate signaling pathway via ROS production, it could also be regulated by ROS. Hobbs et al. reported a novel redox regulation of Rac1 by a selective thiol oxidation of a redox sensitive cysteine 18 (Cys18) under physiological state. The group also generated an Rac1C18D point mutant, an oxidized mimetic Rac1 at Cys18 that showed enhanced GDP dissociation rates in a cell-free system, contrary to the redox insensitive Rac1C18S point mutant. Although there was no change in the GTP hydrolysis rate in the presence of GAP in wild-type Rac1 (Rac1WT), Rac1C18S, or Rac1C18D in a cell-free system, transfection of these mutants and its wild-type in HEK293T cells demonstrated that Rac1C18D was hyperactivated as shown by the drastic loading of GTP compared with Rac1WT or Rac1C18S. Importantly, Rac1C18D could functionally increase lamellipodia formation in Swiss 3T3 cells (134). Knowing that redox-modified Rac1 has enhanced GDP dissociation in cell-free condition and GTP loading in vitro (134), it could be possible that a positive feed forward loop is in place for tumorigenesis wherein Rac1 could be susceptible to oxidative modification, further activating Rac1 and its functional role in ROS production. This may, in turn, drive mutation rate, genomic instability, survival, and metastasis in an ROS-dependent manner.
ROS-dependent approaches to targeting the tumorigenic activity of Rac1
Given the fact that overexpression or gain of function mutations enhance the redox effects of Rac1, there have been attempts to employ ROS scavengers as an indirect means to block the oncogenic effects of Rac1 (209, 255). One notable example is the use of NAC to inhibit Kaposi's sarcoma (KS)-like tumor development in constitutively active Rac1 (RacCa) transgenic mice. This study showed that developed tumors of RacCa mice possess significantly higher ROS (O2 .−) levels than those of wild-type mice. Although no direct measurement of ROS was employed after NAC treatment, RacCa mice supplemented with NAC did not develop any detectable tumor compared with their controls. In addition, supplementation of NAC after tumor onset was capable of deterring further tumor development. These observations, therefore, strongly suggest that ROS produced by constitutively active Rac1 fuel KS-like tumorigenesis and that antioxidants such as NAC could have potential chemopreventive value against ROS-induced tumorigenesis (209). Another notable mention is the Rac1 inhibitor, NSC-23766, which was shown to be effective in abrogating Rac1-induced resistance against cancer therapy. In a study involving breast cancer cells, the surviving fraction of cells after hyperfractionated radiation (HFR) was shown to harbor upregulated Rac1 expression and significant increases in the level/activation of prosurvival proteins, ERK1/2, NF-κB, Mcl-1, and Bcl-xL. A priori treatment with NSC-23766 resulted in a significant downregulation of prosurvival factors as well as inhibition in tumor colony forming ability after HFR (125). Although no ROS measurement was employed in this study, others have undoubtedly demonstrated that Rac1 inhibition with NSC-237766 could abrogate Rac1-induced ROS production (20, 83). These findings provide testimony that inhibition of Rac1 or its redox function could potentially shut down Rac1-dependent oncogenic effects.
On the flip side, studies have also taken the advantage of Rac1-induced ROS to induce apoptosis as a means to prevent tumor progression (164, 243). To that end, Rac1/NOX-dependent ROS production (DCFHDA) was shown to be the underlying mechanism of glutamate-induced execution of neuroblastoma cells. The authors demonstrated that glutamate-induced apoptosis was rescued upon treatment with either NAC, DPI, neopterine, or apocynin or upon transfection with the dominant negative Rac1N17, thus underscoring the involvement of ROS in glutamate-induced apoptosis of neuroblastoma cells (243). Similarly, knockdown of NOX1 or transfection of Rac1N17 not only reduced TNF-α-induced O2 .− but also blocked Rac1/NOX1-dependent necrotic cell death (164). Furthermore, our group also provided evidence, although indirect, that simvastatin, a 3-hydroxy-3-methyl-glutaryl-CoA (HMG-CoA) reductase inhibitor, -induced apoptosis involved non-canonical Rac1-induced O2 .− signaling that triggered downstream JNK activation and JNK-induced pro-apoptotic BIM extra long expression in HCT166 cells (397). Taken together, these data highlight the importance of Rac1-induced ROS production in the process of tumorigenesis and its progression as well as a potential target for therapeutic exploitation. Although there is conflicting experimental evidence vis a vis the involvement of Rac1-driven ROS production in promoting or inhibiting tumorigenesis, the contradiction could be explained due in part to the different cellular context, tumor microenvironment, the expression pattern of ROS producers, and detoxifier (antioxidant) and constitutive redox metabolism, which governs the ROS threshold of different cancer cell types in determining cell survival or execution.
Myc Oncoprotein, Its Role in Tumorigenesis and ROS Regulation
The oncogene MYC is frequently altered in different human cancer cells (18, 52, 199, 327). The MYC family of genes consists of c-MYC, N-MYC, L-MYC, and S-MYC (52, 234, 315). As a transcription factor, Myc forms heterodimers with Max and binds to a stretch of DNA belonging to a subset of general E-box sequence (7, 21, 52). Myc-Max heterodimerization has been shown to be essential for cell transformation (7). As a transcription factor, Myc controls the expression of a host of genes (389), of which some could potentially regulate cell fate, differentiation, proliferation, and tumor progression (259). Interestingly, Myc overexpression in cancer cells is frequently due to its gene amplification (52, 199, 238); however, despite the high frequency of Myc overexpression in human cancers, deregulated Myc alone is usually insufficient to induce tumorigenesis in most cases. Cooperation with other deregulated proteins such as Bcl-2 and Bcl-xL or loss of tumor suppressors alternative reading frame (ARF) and/or p53 is essential for malignant transformation of cells (81, 87, 259, 314). For example, Myc is capable of inducing apoptosis through activation of p53-dependent pathway; however, cells that survive the induction of apoptosis by Myc overexpression display loss of ARF, a tumor suppressor, or loss of function mutation(s) of p53 (400). As such, Myc-driven tumorigenesis requires the loss of tumor suppressors or its function such as p53 and ARF or overexpression of antiapoptotic proteins such as Bcl-2 or Bcl-xL (81, 87, 259, 314, 400). Nonetheless, with the high frequency of p53 mutations in human cancers, the role of Myc in tumorigenesis is potentially significant (137).
Loss of chromosomal organizational fidelity is a hallmark of cancer and oxidative stress is one of the proposed mechanisms of chromosomal instability (120, 180, 240, 269, 338). Of note, overexpression of c-Myc has been shown to induce DNA damage through the generation of oxidative stress in a model utilizing normal human fibroblasts with an inducible c-Myc transgene. Although the precise mechanism of ROS increase in this model was not clearly identified, the generation of excessive ROS (DCFDA) was indeed linked to DNA damage (338). Interestingly, Myc-induced oxidative stress and ROS-dependent DNA breakage, specifically DNA single-strand break, were only observed under conditions of serum starvation. The group led by Felsher provided further insight that significant elevation of ROS (DCFDA) upon low serum condition was due to CYP2C9 upregulation in Myc-overexpressing normal human foreskin fibroblasts. Inhibition of CYP2C9 by genetic knockdown attenuated Myc-induced ROS production (274).
Despite genomic instability being a hallmark of cancer, Vafa et al. (338) as well as Tanaka et al. (319) provided evidence demonstrating that Myc overexpression-induced ROS triggered growth inhibition and cell death, respectively (319, 338). According to Tanaka et al., c-Myc- and E2F-transcription factor 1 (E2F1)-induced O2 .− and H2O2 accumulation could enhance apoptosis of ARF-null NIH3T3 and p53-defective osteosarcoma Saos2 cells under conditions of serum deprivation. The underlying mechanism of Myc-induced ROS was linked to downregulation of SOD2 expression due to a decrease in NF-κB transcriptional/DNA binding activity (319). Although one would argue that the absence of ARF or p53 in NIH3T3 or Saos2 cells, respectively, would potentiate tumor progression upon Myc overexpression, an alternative reason for the observed cell death could potentially be due to an indirect effect of reduced transcription of p53-transcribed antioxidant genes in cells lacking functional p53 (278, 319).
Intriguingly, Myc has also been implicated in the biogenesis of mitochondria, the major source of ROS production. Through a series of experiments, Li et al. reported that Myc induction in the P493–6 B-lymphocytes enhanced oxygen consumption rate, mitochondrial mass, and function, thereby indicating that Myc could mediate mitochondrial biogenesis (192). Supporting that, rat fibroblasts (Rat1) lacking Myc were shown to possess lower mitochondria mass and number of normal mitochondria. Through chromatin immunoprecipitation experiments, the group ultimately discovered that Myc binds to mitochondrial transcription factor A, a nuclear encoded transcription factor that translocates to the mitochondria to activate mitochondria transcription and mitochondrial DNA replication, thus explaining the enhanced mitochondria DNA and biogenesis in Myc-expressing cells. Corroborating the findings of the previous study, Graves et al. demonstrated that depletion of Myc in rat fibroblasts resulted in decreased mitochondrial mass and defective structural integrity (108). In light of its role in mitochondrial biogenesis, Myc is indeed indicative of a causative relationship between the induction of oxidative stress in cells and the generation of mitochondrial ROS as a result of increased mitochondria mass.
Apart from Myc-dependent generation of ROS, Myc-dependent redox modulation has also been demonstrated. Graves et al. showed that Prx5 expression was dependent on c-Myc and that c-Myc could occupy the E-box elements in the PRX5 proximal promoter (107). This indicates that expression of Prx5 is dependent on the transcriptional activity of Myc. The role of Myc in redox modulation was further supported by findings that c-Myc positively regulated GSH synthesis via the transcription for γ-glutamyl-cysteine synthethase (γ-GCS), a rate-limiting enzyme that catalyzes GSH synthesis. In this study, exogenous H2O2 enhanced c-Myc recruitment to the γ-GCS promoter region to induce γ-GCS transcription. This recruitment was further shown to be dependent on ERK-mediated phosphorylation of c-Myc at Ser62, suggesting that Ser62 phosphorylation is a sensor as well as a switch to cellular response against oxidative stress (17). This highlights the importance of Myc in the regulation of redox balance in cells, which could ultimately impact upon cellular malignant transformation when a balance between oxidants and antioxidants is altered. Besides the transcription of Prx, the antioxidant role of Myc has also been demonstrated through its transcription of NRF2, which plays an important role in regulating intracellular redox milieu (71). Although Myc-induced tumorigenesis may require a second oncogenic hit such as the coexpression of Bcl-2 or Bcl-xL, the dual redox role of Myc, similarly to Bcl-2, suggests an important oncogenic role in established tumors, whereby it could maintain a moderate intracellular ROS environment suitable for malignant transformation while preventing excessive accumulation of ROS that could be detrimental to cancer cells (Fig. 3e). Nevertheless, the key question on what triggers its change from a pro-oxidant to an antioxidant role upon alterations in cellular redox status remains to be elucidated. The latter could potentially provide valuable information for targeting Myc-overexpressing cancer cells.
Drugging Myc: ROS dependent?
Due to the participation of Myc in tumorigenesis, it is again crucial to identify inhibitors or drugs that could shut down the oncogenic functions of Myc. A thorough review of Myc inhibitors was recently published by Whitfield et al., of which some are effective in vitro or in vivo as well as tested clinically (357). The compiled list comprises compounds that target Myc directly, via the interference of its transcriptional function, binding partner capability, and its expression, and indirectly, via the inference of its regulatory proteins and effect on Myc-driven tumors (357). Of these compounds and inhibitors, the compound 10058-F4 could relevantly and simultaneously affect redox status during its effect of decreasing ATP production and cell proliferation, arresting cell cycle and inducing apoptosis in ovarian cancer cells (348). A compound identified to inhibit the interaction of Myc and Max (385), 10058-F4, could significantly reduce intracellular and mitochondrial ROS levels (348). Although just an association between ROS reduction and apoptosis upon 10058-F4 treatment, this suggests that ROS is required for Myc-driven tumorigenesis as well as the manipulation of ROS level may be a major key to Myc-driven tumor execution.
NRF2: The Master Switch to Counter Oxidative Stress
NRF2 is a transcription factor that predominantly regulates the cellular antioxidant program. NRF2 binds to the antioxidant response element (ARE) and activates the transcription of antioxidant systems such as glutaredoxin, GPx, GR, Trx, and Trx reductase (124, 148, 324, 343). Under physiological states, NRF2 is kept at a constitutively low levels by its repressor protein, Kelch-like ECH associate protein 1 (Keap1) (149), a component of the Cullin3-based E3 ligase complex that ubiquitinates NRF2 for proteosome-dependent degradation (169). As such, an important step in the stability and activation of NRF2 as a transcription factor is its evasion from Keap1-dependent degradation. To that end, the cysteine residues of Keap1 play a crucial role in sensing oxidative stress, which induces the reduction of Keap1-dependent degradation of NRF2 as well as subsequent activation of NRF2 (345, 390). Once activated, NRF2 is responsible for reinforcing the cells' antioxidant machinery by binding to the ARE within the promoter of genes involved in buttressing the antioxidant defenses (124). The protective effect of NRF2-mediated inhibition of oxidative stress was elegantly illustrated in a study involving SOD1 mutant astrocytes (340). Along the line of tumorigenesis, NRF2-deficient mice were reported to be more sensitive to benzo[a]pyrene-induced neoplasia than the wild-type counterparts (271), and the incidence of bladder tumors in NRF2-deficient mice was also higher upon exposure to N-nitrosobutyl(4-hydroxybutyl)amine, a carcinogen specific for urinary bladder (143). Interestingly, NRF2 has also been reported to play a role in the anticancer effects of caloric restriction in mice (257). These findings point to a potential tumor suppressor effect of NRF2.
On the flip side, there are also reports suggesting a pro-oncogenic role for NRF2. In this regard, the antioxidant ability of NRF2 could be a survival strategy in established tumors to ward off chemotherapy-induced oxidative stress and execution (71, 325). Corroborating the redox-dependent prosurvival activity of NRF2 is the observation linking oncogene-driven tumorigenesis (such as KRAS and c-Myc) to upregulation and increased activity of NRF2, thereby contributing to a reduced cellular environment (71). Furthermore, Wang et al. reported that NAD(P)H quinone oxidoreductase 1 (NQO1), a transcriptional target of NRF2, was upregulated in advanced stages (Stages II and III) of lung cancer, and that knockdown of NRF2 sensitized A549 cells to chemotherapeutics such as cisplatin, doxorubicin, and etoposide (350).
Based on these data, it is plausible that the different expression patterns of NRF2 could drive different stages of tumorigenesis, as correlatively displayed by the different transcription activity of NRF2 in various stages of lung tumors (350). For example, during the phase of tumor initiation, NRF2 expression might be clamped down to provide a conducive pro-oxidant environment for cell proliferation and survival; however, in more advanced stages of the disease, NRF2 expression is significantly induced to prevent the development of overwhelming oxidative stress as a cell survival mechanism (Fig. 3c). A very similar pattern of expression during the various stages of cancer has been demonstrated for the antioxidant protein, SOD2 (73, 177, 203).
The conundrum: Inhibit or activate NRF2 as a therapeutic strategy?
The seemingly disparate roles of NRF2 at different stages of the tumorigenesis process present biologists and clinicians with the obvious quandary of identifying specific instances wherein inhibiting NRF2 could be a favorable anticancer strategy as opposed to other scenarios wherein induced activity could lend therapeutic relevance. The answer would plausibly be dependent on cell type, ROS level, and the stages of cancer. On the one hand, inducers of NRF2 may prove to be useful at early stages of tumorigenesis as ROS could potentially be a driving force for cell transformation and initiation of cancer. Indeed, multiple reports have suggested that activating the Keap1-NRF2 pathway is a viable option for cancer preventive therapy. For example, sulforaphane, an activator of NRF2 that was isolated from broccoli, was reported to have anticarcinogenic effect (296, 394). Another potent NRF2 inducer, oltipraz, was also shown to exert chemopreventive effect against urinary bladder carcinogenesis through activation of NRF2 (143).
On the other hand, NRF2 has also been shown to be upregulated in head and neck cancer and play a role in chemoresistance, as well as its transcriptional activity has been positively correlated with the different stages of lung cancer (310, 325, 350). However, in comparison with the inducers of NRF2, the identification of small molecule inhibitors of NRF2 has not been overly successful. Nevertheless, as a repurposing strategy, dimethyl fumarate, approved for the treatment of multiple sclerosis and psoriasis, was reported to elicit strong antitumorigenic effect in vitro and in vivo at high dose by triggering oxidative stress via the reduction of protein deglycase (DJ-1)-mediated stabilization of NRF2 (280). Two other compounds, brusatol and luteolin, have also been reported to sensitize cancer cells to chemotherapeutic drugs, such as cisplatin, carboplatin, 5-fluorouracil, paclitaxel, etoposide, oxaliplatin, bleomycin, and doxorubicin, by reducing NRF2 protein level and its responses (275, 323). In addition, premalignant breast epithelial cells and breast cancer cells harboring loss/mutant BRCA1 were more susceptible to ROS accumulation and cisplatin-induced cytotoxicity due to the impaired NRF2-regulated antioxidant response (105). As several commonly used chemotherapeutic drugs trigger intracellular ROS generation/accumulation (270, 293, 294), the inhibition of NRF2 could possibly release the breaks on ROS-dependent execution (Fig. 6), thereby sensitizing cancer cells to chemotherapy as well as overcoming resistance to ROS-mediated therapies.
Gelsolin: A Novel Redox-Dependent Driver of Cancer Progression
Gelsolin is a widely distributed actin regulatory protein involved in cellular cytoskeleton organization. In vitro experiments have shown that gelsolin functions to sever actin filaments and cap the fast-growing end of actin filaments as well. These functions are regulated in response to changes in intracellular calcium concentrations as well as polyphosphoinositides (316). The role of gelsolin in tumorigenesis has been widely debated due to differences in expression levels in different tumor types (65, 196, 247, 321, 362, 380). Although a significantly reduced expression was reported in human ovarian tumors, gelsolin was extremely low to undetectable in primary tissue of bladder cancer patients (247, 321). Enforced expression of gelsolin in human bladder cancer cells inhibited colony forming ability as well as reduced tumor incidence in nude mice (321). Interestingly, a separate study by Deng et al. reported increased expression of gelsolin mRNA and protein in hepatocellular carcinoma tissue and cells, which was associated with enhanced growth and invasive capacity (65). In agreement with the latter, high gelsolin expression was associated with poor survival in NSCLC patients (380).
Despite the contradictory reports on the association between gelsolin and tumorigenesis, it is becoming clearer that gelsolin overexpression provides cells with increased migratory and invasive potentials. Specifically, overexpression of gelsolin in NIH3T3 fibroblasts demonstrated augmented motility, thus highlighting the involvement of gelsolin in the regulation of cell motility (48). In a separate study by Deng et al. (mentioned earlier), gelsolin overexpression also increased EMT markers such as MMP-2 and MMP-9 apart from the increased cell invasion (65). In light of these findings, De Corte et al. showed that gelsolin-induced epithelial cell invasion was dependent on the RAS–Rac signaling pathway. Transfection of dominant negative RASN17 or Rac1 N17 abolished gelsolin-induced collagen invasion, thereby suggesting that RAS and Rac1 are required for gelsolin-induced invasion (59). By implicating Rac1 in gelsolin-induced cell invasion, the role of ROS in gelsolin-induced invasion is further corroborated (see The Pro-Oxidant Effect of RacGTPase Promotes Tumorigenesis section). In support of the latter, a recent study uncovered a novel interaction of gelsolin with SOD1 (Fig. 3a), which resulted in reduced SOD1 activity and a resultant increase in intracellular O2 .− levels in HCT116 colon cancer cells. The resultant increase in O2 .− was also linked to an increased urokinase plasminogen activator (uPA) secretion, a protease involved in cancer invasion by degrading the extracellular matrix. Importantly, knockdown of gelsolin reversed the invasive capacity of HCT116 cells with a concurrent reduction in O2 .− level, which was reciprocally rescued with simultaneous knockdown of SOD1. These findings point to the involvement of O2 .− in gelsolin-induced tumorigenesis (331). Further supporting the role of gelsolin in promoting invasion and tumorigenesis, overexpression of gelsolin was shown to endow greater invasive capacity in oral squamous cell carcinoma cells (66). It would be tempting to speculate and worth exploring that ROS could be involved in the observed invasion-promoting activity of gelsolin. Despite mounting evidence supporting the proinvasive/tumorigenic activity of gelsolin, it has also been reported that gene knockdown of gelsolin induces EMT through a cadherin switch in human mammary epithelial MCF10A cells; increased expression of N-cadherin, Snail, Rac, and enhanced cell motility and invasion (320). These findings indicate the dichotomy of gelsolin during the process of tumorigenesis and its progression, potentially again from the standpoint of cellular redox status. It is possible that the apparently opposing effects reported in different cancer settings are a function of cell type and/or cell phase and require further investigations to justify targeting gelsolin as a potential therapeutic strategy.
Concluding Remarks and Take Home Message
Taken together, the crosstalk between cancer-promoting proteins and cellular redox status is emerging as a potentially important node in promoting the oncogenic phenotype, which could have implications for the design and development of novel therapeutic strategies. Based on the literature discussed in this work, a hypothetical network of proteins that could crosstalk directly or indirectly to regulate ROS and tumorigenesis is summarized in Figure 7. For example, overexpression of antiapoptotic Bcl-2 is capable of increasing intracellular ROS, which requires the involvement of COX Vα and Rac1 (35, 342). There is also indication for an interaction between cytochrome c and Rac1 to trigger intracellular ROS (250). Based on these data, one could conjecture the possibility that these four proteins exist in a functional complex to modulate cellular ROS. Similarly, the crosstalk between Bcl-2, ROS, and STAT3 is noteworthy (156) as it suggests a feed forward loop resulting in Bcl-2-driven STAT3 activation, which, in turn, transcribes for Bcl-2 (98, 156). In addition, the sustained activation of STAT3 could also enhance the expression of other antiapoptotic proteins such as Bcl-xL and Mcl-1 (31, 84, 200). Similarly, as discussed earlier, STAT5 interacts with Rac1 for its nuclear translocation, which in addition suggests an enhanced STAT5-induced expression of proteins involved in death inhibition such as Bcl-2, Bcl-xL, and Mcl-1 (193, 211, 219, 300, 387), also pointing to the potential involvement of ROS in this survival circuitry (157, 281). Rac1 is also a critical downstream mediator of the oncogenic signal of RAS, which includes the intermediary involvement of ROS. More interestingly, RAS–Rac signaling has been shown to drive gelsolin-induced collagen invasion (59, 78).
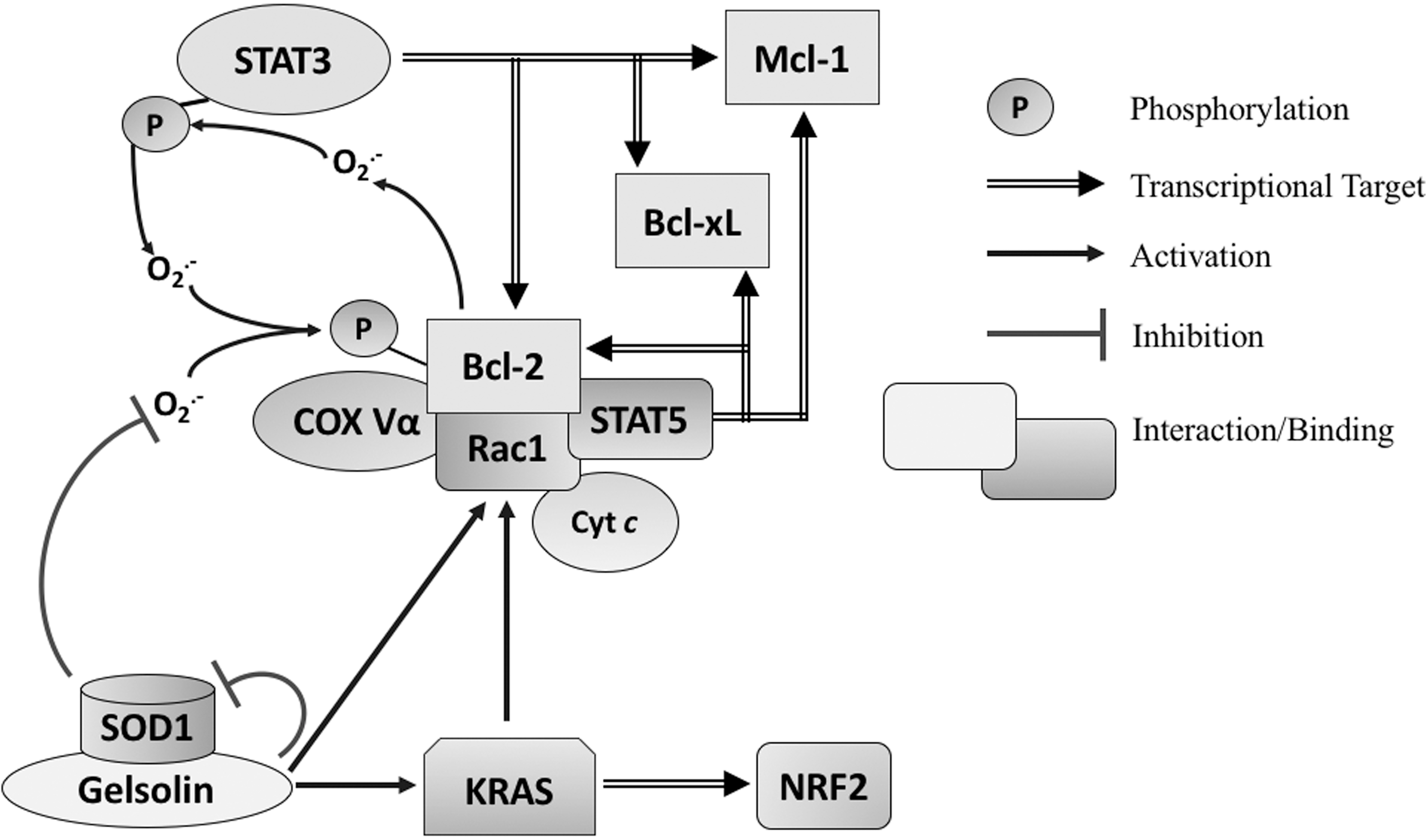
Although these findings implicate ROS in the initial process of tumorigenesis, we have also provided a discussion on the association of a robust antioxidant machinery with disease progression, invasion, and metastasis. The latter could be a feedback upregulation of the antioxidant systems, triggered by the pro-oxidant environment to ensure that the level of ROS is kept at a relatively low level to prevent oxidative damage. This has been exemplified in this review in the case of Bcl-2 and SOD2, whereby the two proteins behave differently under different states of redox milieu or stages of tumorigenesis (34, 35, 73). Similarly, cancer cells carrying mutant KRAS could upregulate NRF2 to potentially clamp down oxidative damage as a prosurvival mechanism (325). Nevertheless, it should be noted that the contribution of ROS in all cited works throughout this review may be governed by cell-type specificity as previously mentioned in The dichotomy of redox signaling in cell fate decisions section. Therefore, it is important to keep in mind that one or more pathways displayed in the hypothetical model may also be cell-type specific, which again warrants the use of comparable cell lines when tested empirically. Nevertheless, the hypothetical network model provides a stimulating platform to empirically investigate if some of the proteins included here (and perhaps others) function in an interdependent manner to regulate ROS and tumorigenesis (and even similar protein- or ROS-dependent pathologies). Given the possibility of this network, protein expression profiling could be employed on patients to determine unknown partner proteins aside from their common oncoprotein drivers such as KRAS or Bcl-2. This could have therapeutic implications for drugs currently being tested for targeting specific oncoproteins and their functions (Table 1) as well as for novel combinational therapy. In addition, the contribution of ROS in most of the published work as well as in the proposed model further warrants a personalized detection of ROS levels as well as redox enzymes expression levels of cancer versus normal cells in any given patient as indicators of potential hormetic ROS thresholds that could determine whether further ROS manipulation via drug or combinational therapy would lead to sensitization of cancer cells toward death execution. These various profilings, which include the determination of oncoproteins, antioxidant proteins, and ROS levels, would potentially pave a new personalized approach to cancer therapy/strategy.
BAK, Bcl-2 homologous antagonist killer; Bcl-2, B-cell lymphoma 2; Bcl-xL, B-cell lymphoma extra large; BIM, Bcl-2-like protein 11; HFR, hyperfractionated radiation; Mcl-1, induced myeloid leukemia cell differentiation protein; MMP, matrix metalloproteinase; mRNA, messenger ribonucleic acid; NOX, NADPH oxidase; N.R., not reported; NRF2, NF-E2-related factor; NSCLC, nonsmall cell lung cancer; ROS, reactive oxygen species; STAT, signal transducer and activator of transcription.
Footnotes
Acknowledgments
We would like to acknowledge the contributions of all authors whose work could have been unintentionally overlooked. S.P. is supported by grants from the National Medical Research Council (NMRC), Singapore.