
Abstract
Significance:
Increasing evidence has indicated critical roles of nicotinamide adenine dinucleotide, oxidized form (NAD+) in various biological functions. NAD+ deficiency has been found in models of a number of diseases such as cerebral ischemia, myocardial ischemia, and diabetes, and in models of aging. Applications of NAD+ or other approaches that can restore NAD+ levels are highly protective in these models of diseases and aging. NAD+ produces its beneficial effects by targeting at multiple pathological pathways, including attenuating mitochondrial alterations, DNA damage, and oxidative stress, by modulating such enzymes as sirtuins, glyceraldehyde-3-phosphate dehydrogenase, and AP endonuclease. These findings have suggested great therapeutic and nutritional potential of NAD+ for diseases and senescence.
Recent Advances:
Approaches that can restore NAD+ levels are highly protective in the models of such diseases as glaucoma. The NAD+ deficiency in the diseases and aging results from not only poly(ADP-ribose) polymerase-1 (PARP-1) activation but also decreased nicotinamide phosphoribosyltransferase (Nampt) activity and increased CD38 activity. Significant biological effects of extracellular NAD+ have been found. Increasing evidence has suggested that NAD+ deficiency is a common central pathological factor in a number of diseases and aging.
Critical Issues and Future Directions:
Future studies are required for solidly establishing the concept that “NAD+ deficiency is a common central pathological factor in a number of disease and aging.” It is also necessary to further investigate the mechanisms underlying the NAD+ deficiency in the diseases and aging. Preclinical and clinical studies should be conducted to determine the therapeutic potential of NAD+ for the diseases and aging.
Introduction
Accumulating evidence has indicated critical roles of nicotinamide adenine dinucleotide, oxidized form (NAD+) in a variety of biological functions, including energy metabolism, mitochondrial activity, DNA repair, immunological functions, and calcium homeostasis (122, 123). In 2003, a study reported profound cytoprotective effects of NAD+, showing that NAD+ treatment can both restore the intracellular NAD+ level and decrease the astrocyte death induced by alkylating agent N-methyl-N′-nitro-N-nitrosoguanidine (MNNG) (124). Since 2004, a number of studies have suggested that NAD+ is highly protective in a number of models of diseases such as cerebral ischemia (125), myocardial ischemia (129), type II diabetes (T2D) (126), and glaucomas (117), as well as in models of aging (30, 70).
Increasing evidence has suggested that NAD+ is one of the fundamental factors in life that can significantly affect virtually all major biological processes (122). During the past 10 years, a large number of studies regarding the roles of NAD+ in diseases and aging have been reported. These studies have provided compelling evidence supporting the proposal that NAD+ deficiency is a common central pathological factor in a number of neurological diseases, cardiovascular diseases, metabolic diseases, degenerative diseases, and aging. The goal of this review is to provide generalization of the evidence supporting the proposal that NAD+ deficiency is a common central pathological factor in a number of diseases and aging, and to generalize the mechanisms underlying the beneficial effects of NAD+ administration. The generalization may provide important insights and guidance for the future studies in this highly promising research field.
General Information About NAD+
Nicotinamide adenine dinucleotide (NAD) exists in either its oxidized form (NAD+) or reduced form (NADH), which are critical cofactors of a number of oxireductases (46, 122, 123). In a number of redox (short for reduction-oxidation) reactions in energy metabolism, NAD+ is used by multiple dehydrogenases that catalyze substrate oxidation, in which NAD+ is converted to NADH by accepting two electrons and one H+ (46, 95, 122, 123). Moreover, NAD+ also acts as a substrate for several families of important enzymes, including sirtuins, poly(ADP-ribose) polymerases (PARPs), ADP-ribosyl cyclases (ARCs), and mono-ADP-ribosyltransferases (ARTs) (122, 123). Since NAD+ is impermeable to mitochondrial membranes, there are two NAD+ pools in cells, including cytosolic NAD+ pool and mitochondrial NAD+ pool (22, 23, 46, 53, 79). Under physiological conditions, the cytosolic NAD+/NADH ratio is ∼700 (96, 104, 127), whereas the mitochondrial NAD+/NADH ratio is approximately 7–8 (96, 104). NADP+, a molecule required for NADPH generation, can only be generated from NAD+ by NAD+ kinase (79, 122).
NAD+ synthesis
In animals and some bacteria,
Three isoforms of human NMNATs have been found in the nucleus, the Golgi complex, and the mitochondria, respectively (8, 62, 82). The nuclear enzyme NMNAT-1 is a key enzyme in both the de novo pathway and the salvage pathway of NAD+ synthesis (62). Mammals use nicotinamide as the precursor for NAD+ synthesis in the salvage pathway (85): Nicotinamide is converted to nicotinamide mononucleotide (NMN) by nicotinamide phosphoribosyltransferase (Nampt), which is, subsequently, converted to NAD+ by NMNATs (85).
NAD+ catabolism
NAD+ is used by multiple NAD+-dependent enzymes, including sirtuins, PARPs, ARCs, and ARTs (123). The catabolic products of sirtuins, PARPs, ARCs, and ARTs are nicotinamide plus O-acetyl-ADP-ribose, poly(ADP-ribose), cyclic ADP-ribose, and ADP-ribose, respectively (99, 123). Numerous biological functions of sirtuin family proteins, including SIRT1–SIRT7, have been found, which include regulation of apoptosis, inflammation, and energy metabolism (15, 42, 50, 81, 102). The most well-studied PARP is PARP-1, which plays important roles in DNA repair, genomic stability, cell cycle, and programmed necrosis (21, 123). A diagrammatic presentation of the major pathways of NAD+ metabolism is shown in Figure 1.

Future studies on NAD+-consuming ecto-enzymes, including the major ARC CD38 and ARTs, are of particular interest. CD38 plays important roles in the regulation of intracellular NAD+ levels under physiological conditions (20). Increased CD38 activity is also causative to age-related NAD decreases and mitochondrial dysfunction (12). CD38 uses extracellular NAD+ to generate cyclic ADP-ribose, a major endogenous agonist of ryanodine receptors (75). Mammalian ARTs constitute a family of ecto-enzymes that catalyze the transfer of the ADP-ribose group from NAD+ onto amino acid residues of target proteins (90). With increasing evidence indicating important roles of these ecto-enzymes (60, 90, 115), the answers of the following two key questions are needed: (i) What are the sources of the extracellular NAD+ that can act on the ecto-enzymes? (ii) Is it possible that some of the beneficial effects of NAD+ administration in disease models may result from the interactions between the extracellular NAD+ and the ecto-enzymes? Two studies have provided interesting information for answering the first question: It was reported that the NAD+ released during inflammation can lead to induction of ART2-mediated death of naive T cells (1); and stimulation of enteric nerves in gastrointestinal muscles can also lead to NAD+ release, which acts as a neurotransmitter that contributes to enteric inhibitory regulation of visceral smooth muscles (73). The studies suggesting significant roles of CD38 in such diseases as cerebral ischemia (58) and myocardial ischemia (35) have also highlighted the significance to search for the answer for the second question.
NAD+ Deficiency Is a Critical Common Pathological Factor in a Number of Diseases
NAD+ deficiency is a key pathological factor in multiple neurological disorders
In 1997, we reported that intranasal NAD+ administration at 2 h after brain ischemia led to an ∼87% reduction of infarct size in a rat model of transient cerebral ischemia/reperfusion (I/R) (125). Using a mouse model of transient brain ischemia, we found that intraperitoneal NAD+ administration also profoundly decreased infarct formation and neurological deficits, which may be partially mediated by the NAD+-produced decrease in autophagy (132). Overexpression of Nampt (also called PBEF or visfatin) is also highly protective in models of cerebral ischemia (128). The beneficial effects of Nampt appear to be dependent on the enzymatic activity of NAD+ synthesis (112). These findings, together with the following three lines of observations, have suggested that NAD+ deficiency is a key pathological factor of ischemic brain injury: First, cerebral I/R leads to a significant decrease in the NAD+ levels in the brain (25, 58); second, excessive PARP-1 activation, a major NAD+-consuming enzyme (21), plays a key role in ischemic brain injury (123); and third, cell culture studies have indicated that NAD+ depletion mediates PARP-1 activation-induced neuronal death and astrocyte death (3, 4). Future studies are needed to determine whether NAD+ administration produces the beneficial effects by inhibiting the inflammation and/or enhancing the antioxidant capacity in the brains that were subjected to I/R.
Won et al. reported that head trauma led to a significant decrease in the NAD+ levels in the brain, which was restored by intranasal NAD+ administration (118). The NAD+ administration also produced a profound decrease in traumatic brain damage (118). The protective effects of the NAD+ administration appear to be at least partially mediated by its inhibitory effect on microglial activation (118). These findings have suggested that NAD+ deficiency is a critical pathological factor in traumatic brain injury. Future studies are warranted to answer the following key question: Does NAD+ produce its inhibitory effect on microglial activation by acting directly on microglia, or indirectly by decreasing necrosis?
NAD+ intervention significantly decreased the incidence of spontaneous recurrent seizure and abnormal electroencephalogram (EEG) activity, rescued contextual fear memory formation, and decreased neuronal loss in the CA1 region of the hippocampus (56). Exogenous supply of NAD+ also decreased neuronal apoptosis in the CA1 region of the hippocampus, and it reversed the increased Acp53/p53 ratio at the early stage of epileptogenesis (56). These observations have indicated that early-stage intervention with NAD+ prevents epileptogenesis in pilocarpine-induced Status Epilepticus mice by suppressing neuronal apoptosis. Another study has also suggested that NAD+ deficiency plays a crucial role in PARP-1-mediated acute epileptic neuronal death (110). These findings have collectively suggested that NAD+ deficiency plays a significant pathological role in epilepsy. Future studies are needed to determine whether NAD+ administration may produce the protective effects by inhibiting the inflammation and/or decreasing excitotoxicity in the brain.
Fang et al. reported that NAD+ can significantly enhance the lifespan and health span in models of Ataxia Telangiectasia (A-T), a rare autosomal recessive disease characterized by progressive neurodegeneration and cerebellar ataxia (28): Treatments that can restore intracellular NAD+ levels led to attenuated severity of A-T neuropathology, normalized neuromuscular function, delayed memory loss, and extension of the lifespan in animal models, which could be accounted for by the capacity of the restored intracellular NAD+ levels to stimulate neuronal DNA repair and enhance mitochondrial quality via mitophagy. Using primary rat neurons, the roundworm Caenorhabditis elegans, and a mouse model of A-T, Fang and Bohr further found that NAD+ insufficiency mediates the ATM/A-T mutated loss-induced mitochondrial dysfunction and compromised mitophagy (27). Collectively, these findings have suggested that NAD+ deficiency is a central pathological factor of A-T.
Two studies have suggested that NAD+ deficiency could play an important role in Parkinson's disease (PD) (52): PINK1 mutations-produced familial forms of PD are linked to mitochondrial impairment. Alterations of NAD+ salvage metabolism have been found in Drosophila pink1 mutants, whereas a diet supplemented with nicotinamide rescued mitochondrial defects and protected neurons from degeneration (52). A mutation of Parp, which can prevent excessive PARP-induced NAD+ depletion, also improved mitochondrial function in the pink1 mutants. It is warranted to further determine whether NAD+ administration can produce protective effects in other animal models of PD, such as the model of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced Parkinsonism (34).
There are studies suggesting that NAD+ deficiency may be an important pathological factor of Alzheimer's disease (AD) (111): NMN administration can significantly improve the cognitive functions of the rats in an intracerebroventricular Aβ oligomer infusion AD model. In Aβ oligomer-treated organotypic hippocampal slice cultures, NMN restored the levels of NAD+ and ATP, eliminated accumulation of reactive oxygen species (ROS), and decreased Aβ oligomer-induced neuronal cell death and inhibition of long-term potentiation. All of these protective effects were reversed by 3-acetylpyridine, which generates inactive NAD+.
It has been reported that NMNAT1 prevents axon degeneration by blocking the injury-induced, Sterile alpha, and Toll/interleukin receptor motif-containing protein 1 (SARM1)-dependent NAD+ consumption that is pivotal to axon degeneration (87). However, the study by Di Stefano et al. has suggested that accumulating NMN may account for the Wallerian degeneration: NMN deamidase, a bacterial enzyme, shares NMN-consuming activity with NMNAT2, but not NAD+-synthesizing activity, can delay axon degeneration in primary neuronal cultures, zebrafish larvae, and transgenic mice. NMN deamidase can also rescue axonal outgrowth and perinatal lethality in mice lacking NMNAT2 (24). Thus, future studies are needed to further determine whether NAD+ deficiency is a major pathological factor in axon degeneration.
In summary, a number of studies have suggested critical roles of NAD+ deficiency in multiple neurological disorders, including brain ischemia, traumatic brain injury, epilepsy, and A-T. NAD+ deficiency may also play significant roles in PD, AD, and axon injury.
NAD+ deficiency is a critical pathological factor in multiple metabolic diseases
There is evidence indicating a critical role of NAD+ deficiency in T2D (126): Nampt-mediated NAD+ biosynthesis is severely compromised in metabolic organs by high-fat diet (HFD). NMN can enhance hepatic insulin sensitivity and ameliorate glucose intolerance by restoring NAD+ levels in HFD-induced T2D mice. NMN can improve both glucose intolerance and lipid profiles in aging-induced T2D mice. NAD+ deficiency also contributes to the impaired endothelial progenitor cell mobilization in diabetes (108). Moreover, it has been suggested that SIRT1 activity-produced NAD+ loss may produce a significant pathological role in streptozotocin-induced animal models of type I diabetes (91). Collectively, these findings have suggested that NAD+ deficiency plays a significant pathological role in diabetes. Future studies are required to further investigate the roles of NAD+ metabolism in type I diabetes.
Zhang et al. reported that troxerutin, a trihydroxyethylated derivative of natural bioflavonoid rutin, effectively restored NAD+ level and SIRT1 activity by decreasing oxidative stress. These effects rescued defects in lipin 1 signaling, resulting in improvement of the hepatic lipid homeostasis in diet-induced non-alcoholic fatty liver disease (NAFLD) (130). The study of Zhou et al. has also provided evidence that aging-associated NAD+ deficiency is a critical risk factor for NAFLD, suggesting that restoring NAD+ levels may be a promising therapeutic strategy for NAFLD (133): Both the hepatic NAD+ level and the Nampt-controlled NAD+ salvage were decreased in aged mice and humans. Given normal chow, middle-age DN-NAMPT mice displayed systemic NAD+ reductions and had moderate NAFLD phenotypes, which were deteriorated further under HFD challenge. Oral administration of nicotinamide riboside, a natural NAD+ precursor, completely corrected these NAFLD phenotypes induced by NAD+ deficiency alone or HFD, whereas SIRT1 overexpression partially rescued these phenotypes. These studies have suggested that the age-dependent NAD+ deficiency is a key pathological factor of NAFLD.
NAD+ deficiency has been found in the models of obesity (44), which may result from the HFD- or aging-induced decreases in Nampt activity and oxidative stress-induced activation of PARP-1 (43); for example, it was reported that SIRT1, SIRT3, SIRT7, and NAMPT expression was significantly lower, whereas total PARP activity was increased in obese human subjects compared with lean human subjects (84). Restorations of NAD+ levels have also been shown to significantly decrease obesity (13). Collectively, these studies have suggested that NAD+ deficiency is a critical pathological factor of obesity.
NAD+ deficiency is a key pathological factor in heart diseases
Our study showed that NAD+ administration can significantly decrease myocardial I/R injury in a rat model of myocardial ischemia (129). NAD+ was shown to decrease the myocardial I/R-induced apoptotic changes, including increased levels of active form of caspase-3 and decreased levels of anti-apoptotic Bcl-XL (129). NAD+ also decreased oxidative damage by attenuating the myocardial I/R-induced decrease in the superoxide dismutase 2 (SOD2) levels (129). We have found that myocardial I/R leads to a significant decrease in the NAD+ levels in the heart, which was restored by the NAD+ administration (unpublished results). It was also reported that NMN can protect the heart from I/R injury in both ischemic and reperfusion phases, which appears to be partially mediated through SIRT1 activation (120). Moreover, expression of Nampt in the heart was significantly decreased by ischemia, I/R, and pressure overload (41); whereas cardiac-specific overexpression of Nampt in transgenic mice increased NAD+ content in the heart and decreased the size of myocardial infarction and apoptosis after myocardial I/R (41). Collectively, these findings have suggested that NAD+ deficiency plays a critical role in myocardial I/R. Future studies are warranted to determine whether NAD+ administration produces its beneficial effects by inhibiting inflammation and/or decreasing oxidative stress, due to the critical roles of inflammation (103) and NAPDH oxidase-induced ROS (67) in ischemic myocardial injury.
Increased NADH/NAD+ ratios and protein hyperacetylation have been found in human failing hearts and in mouse hearts with pathologic hypertrophy (51). Increases in the NAD+ levels by stimulating the NAD+ salvage pathway can decrease mitochondrial protein hyperacetylation and cardiac hypertrophy (51). These observations have suggested a significant role of altered NAD+ metabolism in cardiac hypertrophy (51, 69).
A recent study has suggested that the decreased Nampt-induced NAD+ deficiency plays a significant role in human thoracic aortic aneurysm (114): The Nampt levels of the ascending aortas from patients with dilated aortopathy are inversely correlated with their aortic diameters. Smooth muscle cell (SMC)-Nampt knockout mice have mildly dilated aortas that had a 43% decrease in NAD+ content in the media. The SMCs also showed premature senescence, oxidized DNA lesions, and double-strand DNA (dsDNA) strand breaks.
NAD+ deficiency is a critical pathological factor in degenerative diseases
Using glaucoma-prone mice, it was found that NAD+ decreases with age, which renders aging neurons to become vulnerable to insults (117). Oral administration of the nicotinamide (vitamin B3) and/or gene overexpression of Nmnat1 was highly protective against glaucomas by modulating mitochondrial vulnerability (117) Nicotinamide at the highest dose can prevent glaucoma by ∼93%. This study has suggested that the age-dependent NAD+ deficiency plays a key role in the pathogenesis of glaucomas. Future studies are needed to determine whether prevention of NAD+ deficiency leads to prevention of glaucomas by such mechanisms as inhibiting inflammation and decreasing oxidative stress.
In the mdx mouse model of Duchene's muscular dystrophy, Ryu et al. found a significant decrease in the muscle NAD+ levels, a concurrent increase in PARP activity, and decreased expression of Nampt (86). Restoring NAD+ levels by dietary nicotinamide riboside supplementation improved muscle function and heart pathology in mdx and mdx/Utr−/− mice. These beneficial effects of NAD+ in the mice appear to be mediated, at least partially, by its effects on mitochondrial function, poly(ADP-ribosyl)ation, inflammation, and fibrosis (86). Collectively, these studies have indicated NAD+ deficiency as a pivotal pathological factor in Duchene's muscular dystrophy.
NAD+ deficiency may be a critical pathological factor in radiation-, drug-, and inflammation-induced tissue injury
Sheng et al. reported that NAD+ can greatly decrease the synchrotron radiation (SR) X-ray-induced increase in the levels of dsDNA damage and abnormal cell nuclei, as well as decreases in the levels of the cell layers of the seminiferous tubules in testes (92). PARP-1 activation resulting from SR X-ray-induced oxidative stress appears to play a key role in the SR X-ray-induced tissue damage (61). Because PARP-1 is the major NAD+-consuming enzyme that plays a key role in SR X-ray-induced tissue injury (93), these findings have collectively suggested that NAD+ deficiency is a key pathological factor in ionizing radiation-induced tissue injury. This proposal has been further supported by the following finding: In tissues exposed to γ-radiation, dietary nicotinamide supplementation can restore DNA excision repair activity via prolonged activation of PARP1 and PARG activities (6).
Wang et al. reported that NAD+ can decrease doxorubicin (DOX)-induced liver damage (106), including the DOX-induced increases in the levels of dsDNA damage, TUNEL signals, and active caspase-3. DOX was shown to significantly decrease the NAD+ levels of the livers, which was ameliorated by NAD+ administration (106). Since the study showed that the NAD+ administration can attenuate the decreases in both the glutathione (GSH) levels and the GSH reductase activity of the DOX-treated liver, NAD+ administration could produce protective effects, at least partially, by restoring the antioxidant capacity of the liver. Collectively, this study has suggested that NAD+ deficiency may be a critical pathological factor in DOX-induced liver injury.
Umapathy et al. reported that NAD+ can attenuate lipopolysaccharide (LPS)-induced acute lung injury in mice (101): NAD+ treatment significantly attenuated the inflammatory response by such pathways as blocking the parenchymal neutrophil infiltration and preventing capillary leak. NAD+ treatment was also shown to downregulate the messenger RNA (mRNA) levels of the proinflammatory cytokines.
Altered NAD+ metabolism plays a significant role in cancer
Multiple studies have indicated that NAD+ metabolism plays a significant role in cancer cell survival. NAD+ is mainly synthesized by the NAD+ salvage pathway in cancer cells (47). Pharmacological or genetic inhibition of Nampt has been shown to produce cancer cell cytotoxicity both in vitro and in vivo (47). Multiple studies have indicated that NAD+ depletion can lead to decreased ATP levels by lowering glycolytic rate, citric acid cycle activity, and oxidative phosphorylation (47). NAD+ depletion also causes sensitization of cancer cells to oxidative damage by manipulation of cell signaling pathways (47). For example, Chini et al. reported that both pharmacological and genetic inhibition of Nampt leads to decreased growth and survival of pancreatic cancer cells, which were accompanied by decreases in the intracellular NAD+ levels and glycolytic flux (18).
Several studies have also suggested that NAD+ plays a significant role in cancer cell survival and proliferation due to NAD+ kinase-mediated conversion of NAD+ to NADP+ (98). The demand for NADPH is particularly high in proliferating cancer cells due to the two major functions of the molecule: First, NADPH acts as a cofactor for the synthesis of nucleotides, proteins, and fatty acids (95); and second, NADPH is required for decreases in the high levels of ROS in cancer cells that result from increased metabolic activity. NADPH production requires NADP+, whereas NADP+ synthesis requires NAD+ (95); thus, inhibition of NAD+ synthesis and inhibition of NAD+ kinase could become promising therapeutic strategies for cancer.
There are also reports suggesting that NAD+ treatment can decrease cancer cell survival, despite the fact that NAD+ can enhance the survival of normal cells: NAD+ treatment significantly decreased the survival of various types of tumor cells such as C6 glioma cells, whereas NAD+ treatment did not impair the survival of normal primary astrocyte cultures (131). The study has also indicated that oxidative stress mediates the effects of NAD+ on the survival of tumor cells. P2X7 receptors, altered calcium homeostasis, and autophagy are also involved in the effects of NAD+ on the cancer cell survival (36, 46, 131).
NAD+ deficiency is a key detrimental factor in aging
Age-dependent decreases in NAD+ levels have been found in the skeletal muscles (30, 86, 126), liver (126, 133), pancreas, and white adipose tissue (126) in animal models. Intracellular NAD+ has been shown to decrease with aging in humans and mice tissues/cells (32, 71, 126). For example, it was found that the NAD+ levels in the skin tissues of human newborn, young adult, and elderly are 8.54 ± 1.55, 2.74 ± 0.41, and 1.06 ± 0.91 ng/mg protein, respectively (65). PARP activity significantly increased with age in males (p < 0.0001; r = 0.768), which was inversely correlated with tissue NAD+ levels (p = 0.0003; r = −0.639). This result has implicated that oxidative DNA damage during aging may account for the age-dependent decreases in NAD+ levels. Increased CD38 activity can also lead to age-related NAD decreases and mitochondrial dysfunction. The study by Yoshino et al. has also suggested that the age-dependent decreases in NAD+ levels could be associated with the age-dependent decreases in the levels of Nampt (126). Future studies are necessary to further elucidate the mechanisms underlying the age-dependent decreases in NAD+ levels.
Multiple studies have suggested that restorations of the NAD+ levels are highly beneficial in aging models: Orally administered NMN was quickly utilized to synthesize NAD+ in tissues, which effectively mitigates age-associated physiological decline in mice (70). NMN enhanced energy metabolism, promoted physical activity, prevented age-associated body weight gain, improved insulin sensitivity and plasma lipid profile, and ameliorated eye functions. NMN also enhanced mitochondrial oxidative metabolism in skeletal muscle, and it prevented age-associated gene expression changes in key metabolic organs. These findings have suggested the preventive and therapeutic potential of NAD+ intermediates for aging (70).
It was reported that Nampt knockout mice exhibited an 85% decrease in the intramuscular NAD+ content, accompanied by fiber degeneration and progressive loss of both muscle strength and treadmill endurance (30). Administration of the NAD+ precursor nicotinamide riboside rapidly restored muscle mass and ameliorated functional deficits. Lifelong overexpression of Nampt led to preservation of muscle NAD+ and exercise capacity in aged mice. The findings have indicated a critical role of NAD+ deficiency in the age-dependent degeneration of muscle mass and function (30).
Son et al. has found that maintenance of mitochondrial NAD+ levels is critical for reversing cellular aging (94): The enzymes that control mitochondrial NAD+ levels, such as NMNAT3 and nicotinamide nucleotide transhydrogenase (NNT), appear to be susceptible to aging. Mitochondrial NAD+ levels decrease in aged cells, accompanied by reduced SIRT3 activity, which severely impedes cell fate transition. Restoring mitochondrial NAD+ levels by overexpressing NMNAT3 and NNT increased reprogramming efficiency of aged somatic cells and extended the lifespan of human mesenchymal stem cells by delaying replicative senescence (94). Premature aging in cockayne syndrome (CS) mice, nematodes, and human cells results from aberrant PARP activation due to deficient DNA repair, which leads to decreased SIRT1 activity and mitochondrial dysfunction (88). It has been reported that NAD+ supplementation can activate SIRT1 and rescue CS-associated phenotypes (88).
Collectively, these studies have suggested that NAD+ deficiency plays a critical role in aging, and prevention of NAD+ deficiency may be a highly promising strategy for delaying aging.
Mechanisms Underlying the NAD+ Deficiency in Multiple Diseases and Aging
Cumulative evidence has suggested that the NAD+ deficiency found in a number of diseases and aging results from excessive consumption of NAD+ and/or decreased NAD+ synthesizing capacity. For example, both increased PARP-1 activation and decreased Nampt activity were found in the models of obesity (44) and the mdx mouse model of Duchene's muscular dystrophy (86). Notably, the NAMPT expression was significantly lower, whereas total PARP activity was higher in obese human subjects compared with lean human subjects (84).
The major NAD+ consumption pathways include: (i) Excessive PARP-1 activation has also been found in numerous disease models (26, 52, 54, 64, 66, 116); whereas cellular studies have demonstrated that excessive PARP-1 activation, induced by such factors as oxidative stress, is a major cause of NAD+ depletion (21). (ii) The NAD+-consuming enzyme CD38 could also play an important role in NAD+ consumption (2, 20). It has been suggested that CD38 plays a key role in the NAD+ deficiency in cerebral ischemia (58). Increased CD38 activity also causes age-related NAD decreases and mitochondrial dysfunction (12). (iii) The NAD+-consuming enzymes sirtuins may also play an important role in NAD+ consumption: SIRT1 may mediate the NAD+ loss in streptozotocin-, Zn2+-, and cytokine-produced cytotoxicity, and in the tissue injury in streptozotocin-induced animal models of type I diabetes (91). (iv) It was reported that injury-induced SARM1 mediates the NAD+ consumption in axon degeneration (87). (v) Increased mitochondrial permeability transition (MPT) also contributes to the decreased NAD+ levels in the hearts that underwent myocardial I/R (89).
The major reported mechanisms for the compromised NAD+ synthesis capacity include: (i) Decreased activity of Nampt, which has been found not only in the metabolic organs that are exposed to HFD (126) but also in aging (43, 126). Notably, Nampt-controlled NAD+ salvage was compromised in the liver of both elderly mice and elderly humans (133). NAD+ deficiency has also been found in the models of obesity (44), which may result from both the HFD- or aging-induced decreases in Nampt activity and oxidative stress-induced activation of PARP-1 (43). The Nampt protein level in the diabetic db/db mice was also significantly lower than that of control animals. Although decreased Nampt activity has been shown under multiple pathological conditions, the mechanisms underlying the decreased Nampt activity require intensive investigation. One study has suggested that such oxidants as tert-butyl hydroxyl peroxide can induce decreased Nampt activity (119). Because both myocardial ischemia and aging can produce oxidative stress (5, 49, 67), the myocardial ischemia- and aging-induced decrease in the Nampt activity may be accounted for by the effects of oxidative stress on the Nampt activity. (ii) Decreased activities of the enzymes control mitochondrial NAD+ levels, such as NMNAT3 and NNT, which appear to be susceptible to aging (94). A diagrammatic presentation of the mechanisms underlying the NAD+ deficiency in multiple diseases and aging is shown in Figure 2.

Mechanisms Underlying the Roles of NAD+ Deficiency in Multiple Diseases
NAD+ deficiency plays critical roles in the mitochondrial damage in multiple diseases
It is established that mitochondrial alterations, such as MPT and mitochondrial release of multiple pro-apoptotic factors including apoptosis-inducing factor (AIF) and procaspase-3, play crucial roles in cell death and tissue injury (9, 113). It has been demonstrated that decreased NAD+ is sufficient to lead to mitochondrial alterations (3): Alano et al. reported that a selective decrease in the intracellular NAD+ level of primary neuron/astrocyte co-cultures using Bioporter-carried NAD+ glycohydyolase, a NAD+-hydrolyzing enzyme, is sufficient to induce MPT, mitochondrial depolarization, and nuclear translocation of AIF. Mitochondrial Bnip3 may also mediate the NAD+ depletion-induced mitochondrial alterations and subsequent neuronal death (59): Hypoxia-produced PARP-1 activation can lead to NAD+ depletion, resulting in SIRT1 inhibition and subsequent hyperacetylation and nuclear localization of the forkhead box protein FoxO3a, followed by increased levels of mitochondrial Bnip3, resulting in mitochondrial AIF release and neuronal death.
Restoration of tissue NAD+ levels has been shown to significantly attenuate the mitochondrial alterations in multiple disease models. For example, in the mdx mouse model of Duchene's muscular dystrophy, restoration of NAD+ levels by dietary nicotinamide riboside supplementation improved muscle function and heart pathology in mdx and mdx/Utr−/− mice (28). These beneficial effects of NAD+ in the mice appear to be mediated, at least partially, by its capacity to improve the mitochondrial quality via mitophagy. NAD+ was also shown to decrease myocardial I/R-induced changes of the levels of mitochondrial apoptosis-associated proteins, including caspase-3 and Bax (129). Collectively, these studies have suggested that NAD+ deficiency could play an important role in the mitochondrial damage in multiple diseases, and prevention of mitochondrial damage is a critical mechanism underlying the protective effects of NAD+.
However, the relationships between NAD+ deficiency and the mitochondrial alterations have not been elucidated in a number of diseases. Considering the crucial roles of mitochondrial alterations in cell death and tissue injury (7, 113), it is warranted to further investigate the relationships between NAD+ deficiency and the mitochondrial alterations in the diseases.
Previous studies have suggested the following mechanisms underlying the NAD+ deficiency-induced mitochondrial impairments: (i) NAD+ prevents glycolytic inhibition at the GAPDH step (3): Because NAD+ and NADH are the critical co-enzymes of the activity of GAPDH (95), cytosolic NAD+ depletion can lead to glycolytic blockage, resulting in the loss of pyruvate supply to mitochondria, which is the driving force of mitochondrial tricarboxylic acid (TCA) cycle; and (ii) hypoxia-induced NAD+ depletion can produce increased levels of mitochondrial Bnip3 and subsequent mitochondrial alterations. Future studies are warranted to further investigate the mechanisms underlying decreased NAD+-induced mitochondrial alterations.
NAD+ deficiency plays critical roles in the compromised DNA repair in multiple diseases
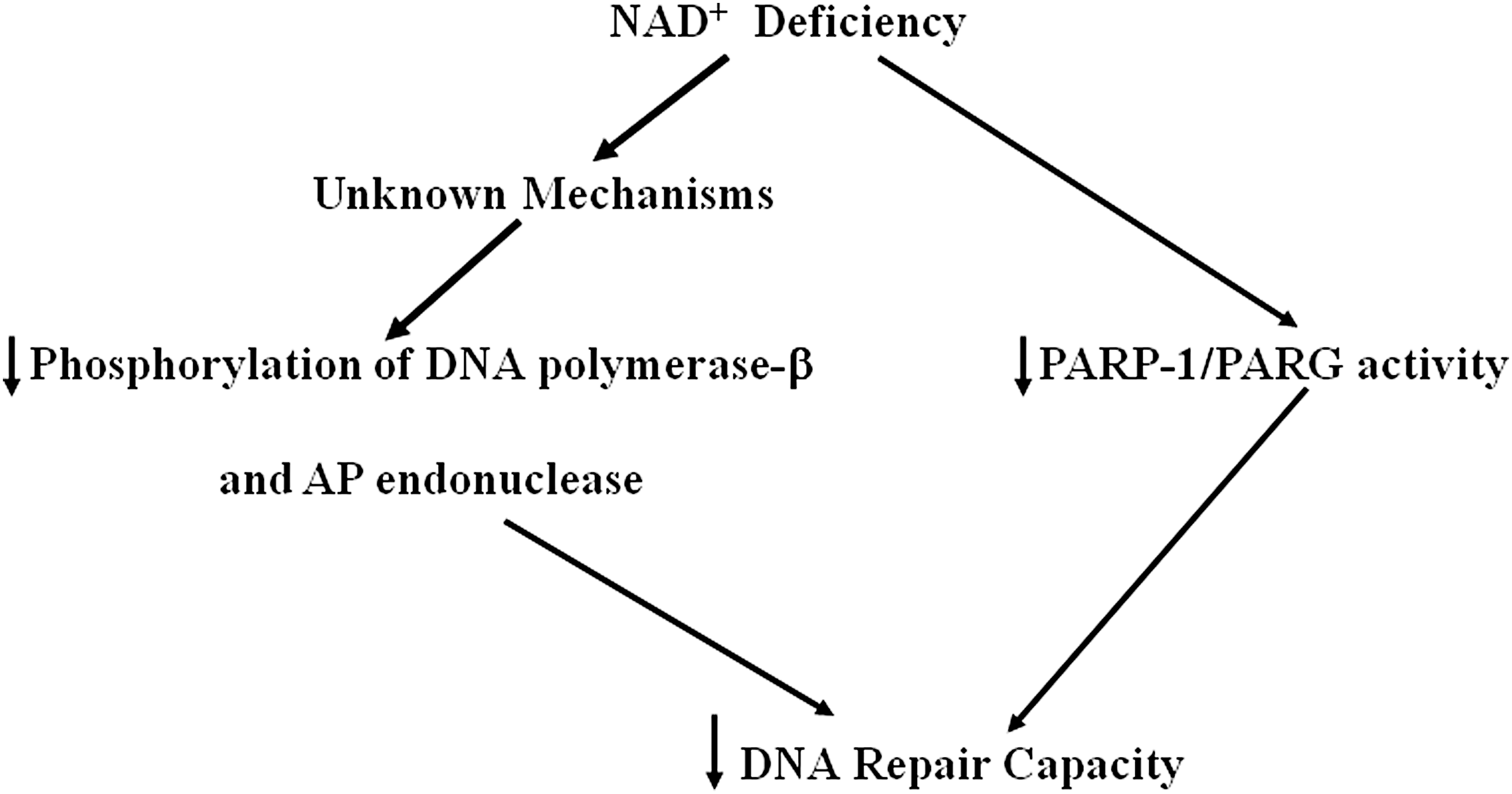
In a neuronal culture study, NAD+ replenishment restores DNA repair activity after oxygen-glucose deprivation (OGD) (109): Direct NAD+ repletion in neurons either before or after OGD markedly decreased OGD-induced DNA damage and cell death. Inhibition of serine-specific phosphorylation of the essential BER enzymes DNA polymerase-β and AP endonuclease appears to mediate the NAD+ restoration-produced enhancement of DNA repair activity, which has suggested that the NAD+ deficiency may lead to compromised DNA repair by altering serine-specific phosphorylation of the essential BER enzymes DNA polymerase-β and AP endonuclease. Dietary nicotinamide supplementation can also restore DNA excision repair activity via prolonged activation of PARP1 and PARG activities in tissues exposed to γ-radiation (6). A diagrammatic presentation of the NAD+ deficiency-produced decrease in DNA repair capacity is shown in Figure 3.
NAD+ deficiency plays critical roles in multiple diseases by altering the functions of sirtuins
The study of Pillai has suggested that NAD+ depletion mediates the link between PARP activation and decreased SIRT1 activity, contributing to myocyte cell death during heart failure (78): The PARP activation and the SIRT1 decrease in failing hearts also appear to regulate p53 acetylation. NAD+ supplementation can also activate SIRT1 and rescue the phenotypes associated with CS (88). SIRT1 activation can restore ATP levels, enhance mitochondrial biogenesis, and decrease renal injury after ischemia-reperfusion, which may be mediated by SIRT1-induced expression of peroxisome proliferator-activated receptor gamma coactivator 1-α (PGC1-α) (48).
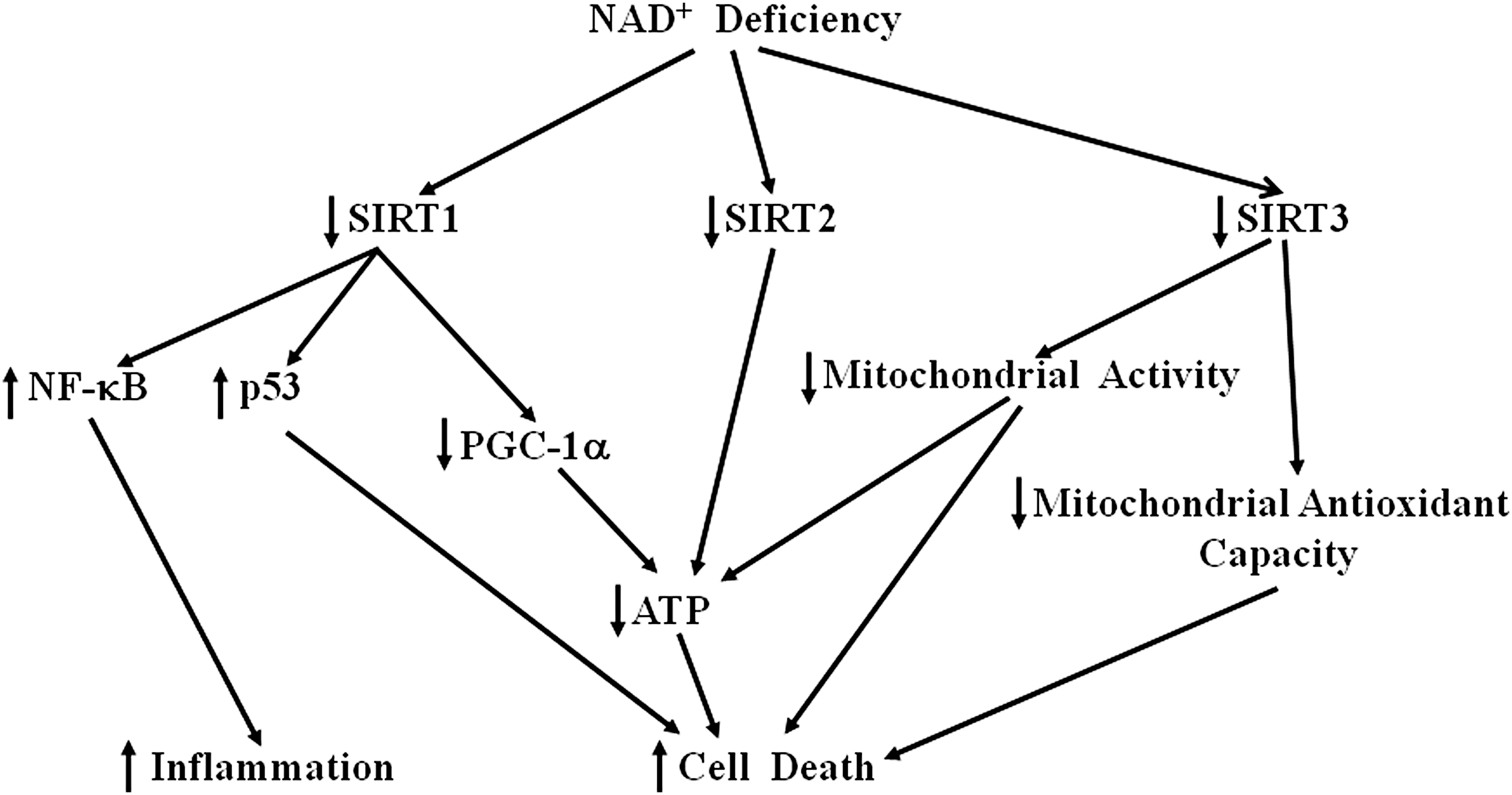
Magnifico reported that local application of pro-apoptotic stimuli on the somatodentritic compartment triggers a dying-back pattern involving caspase-dependent axon degeneration (63). NAD+ can prevent axonal apoptosis, which appears to be mediated by mitochondrial SIRT3 activation in axons (63). Collectively, these studies have suggested that NAD+ deficiency plays critical roles in multiple diseases by altering the functions of sirtuins, and the protective effects of NAD+ require its effects on sirtuins under certain circumstances. A diagrammatic presentation showing the pathological effects of the NAD+ deficiency-produced decreases in sirtuin activities is shown in Figure 4.
NAD+ deficiency plays a significant role in the oxidative stress in multiple diseases
Our study has shown that NAD+ can increase the GSH levels and GSH reductase activity in DOX-treated mouse liver, which provides the first evidence indicating the capacity of NAD+ to increase antioxidant capacity (106). We further used cell culture models to study the mechanisms underlying the effects of NAD+ on the antioxidation capacity, showing that NAD+ can increase both the GSH levels and the GSH/glutathione disulfide (GSSG) ratios of PC12 cells (unpublished results).
Collectively, these studies have suggested that NAD+ deficiency may play an important role in the oxidative stress in multiple diseases, and enhancement of antioxidant capacity may be an important mechanism accounting for the protective effects of NAD+. Future studies are needed to further determine the roles of NAD+ deficiency in the oxidative stress and antioxidant capacity in multiple diseases.
NAD+ deficiency plays critical roles in the ATP decreases in multiple diseases
Multiple studies have shown that NAD+ can prevent ATP depletion of cells exposed to oxidative stress or alkylating agents (3, 4, 124). However, since extracellular NAD+ may also affect cellular functions by such pathways as acting on CD38, it is required to conduct studies to exclude the possibility that NAD+ administration produces its protective effects by its extracellular actions. We have applied NAD+-carrying nano-particles to determine the roles of NAD+ decreases in H2O2-induced decreases in the intracellular ATP levels of primary astrocyte cultures and PC12 cells, showing that a small amount of NAD+-carrying nano-particles is sufficient to prevent the H2O2-induced decreases in the intracellular ATP (16). Because the concentration of the NAD+ in the cell culture media is so low that it is impossible for the free extracellular NAD+ to affect the intracellular NAD+ and ATP levels, our study has provided the first direct evidence demonstrating that NAD+ depletion is the pivotal factor mediating the oxidative stress-induced ATP depletion (16). Since oxidative stress is the common pathological factor in a number of diseases (105), our study has suggested that NAD+ deficiency may, at least partially, account for the ATP decreases observed in many diseases, and the protective effects of NAD+ on tissue injury may result partially from the capacity of NAD+ to attenuate the ATP decreases. It has also been shown that Nampt upregulation or downregulation can significantly increase or decrease the NAD+ and ATP concentrations in myocytes, respectively (41).
It is necessary to elucidate the mechanisms underlying the decreased NAD+-produced ATP reductions. Alano et al. has indicated that NAD+ attenuates glycolytic inhibition by preventing NAD+ depletion-produced blockage at the GAPDH step (3). Because de novo synthesis of NAD+ requires consumption of ATP, regeneration of NAD+ may also lead to decreased intracellular ATP levels (95). A diagrammatic presentation of the NAD+ deficiency-produced decrease in intracellular ATP levels is shown in Figure 5. Since sirtuins, a family of NAD+-dependent enzymes, play crucial roles in modulating energy metabolism (15, 74), future studies are warranted to determine the roles of NAD+ deficiency-induced changes of sirtuin activities in the NAD+ deficiency-produced ATP reductions.

NAD+ deficiency plays critical roles in multiple diseases by inducing necrosis, apoptosis, and autophagy
NAD+ treatment prevents rotenone-induced apoptosis and necrosis of differentiated PC12 cells (39, 40). There are also animal model studies showing that NAD+ can prevent apoptotic changes (106, 129). Accumulating evidence has also suggested a significant role of NAD+ in autophagy (36, 132). Collectively, these studies have suggested that NAD+ deficiency may play important roles in necrosis, apoptosis, and autophagy in multiple diseases.
Roles of NAD+ in inflammation
It has been reported that NAD+ administration can lead to inhibition of inflammation: NAD+ can decrease TBI-induced inflammation in the brains (118). Umapathy et al. also reported that NAD+ treatment significantly attenuated the inflammatory response by such pathways as blocking the parenchymal neutrophil infiltration, and preventing capillary leak (101). NAD+ treatment was shown to downregulate the mRNA levels of the proinflammatory cytokines (101). Because SIRT1 can inhibit NFκB signaling (17, 121), NAD+ may affect inflammation by modulating SIRT1 activity. Collectively, these studies have suggested that NAD+ deficiency plays an important role in the inflammation in multiple diseases, and attenuation of inflammation plays a significant role in the protective effects of NAD+.
NAD+ deficiency may play critical roles in multiple diseases by altering stem cell functions
It has been found that maintenance of mitochondrial NAD+ levels is critical for reversing the aging of human mesenchymal stem cells (94): Accompanied by reduced SIRT3 activity, mitochondrial NAD+ levels decrease in aged cells, which severely impedes cell fate transition. Restoring mitochondrial NAD+ levels by overexpressing NMNAT3 and NNT extended the lifespan of human mesenchymal stem cells by delaying replicative senescence (94). It was also reported that both NAD+ and NMN were capable of promoting proliferation and differentiation of neural stem cells in vitro. Collectively, these studies have suggested that NAD+ deficiency may play critical roles in multiple diseases and aging by altering stem cell functions.
Roles of extracellular NAD+ in the protective effects of NAD+ in multiple diseases
Hayakawa et al. reported that in a mouse model of transient focal cerebral ischemia, ischemia induced entry of astrocytic mitochondria into adjacent neurons, which amplified cell survival signals (37). Decreased CD38 by CD38 small interfering RNA (siRNA) led to decreased extracellular mitochondria transfer and worsened neurological outcomes (37). This study has implicated a critical role of extracellular NAD+ in cerebral ischemia, since CD38 is an ecto-enzyme using extracellular NAD+. Because it was reported that NAD+ administration can profoundly decrease ischemic brain damage (125, 132), the protective effects of NAD+ might partially result from the effects of extracellular NAD+. It is expected that future studies on this increasingly important topic would generate a number of significant findings.
Summary: NAD+ Deficiency Is a Common Central Pathological Factor in a Number of Diseases and Aging
Collectively, cumulating evidence has suggested that NAD+ deficiency can induce a number of pathological changes, thus acting as a common central pathological factor in multiple diseases and aging. NAD+ produces its beneficial effects by targeting at multiple pathological pathways, including attenuating mitochondrial alterations, DNA damage, and oxidative stress. These beneficial effects lead to prevention of cell death and inflammation, resulting in prevention of the tissue injury and degeneration in the diseases and aging. A diagrammatic presentation of the mechanisms underlying the NAD+ deficiency-induced pathological changes in multiple diseases and aging is shown in Figure 6. Further demonstration of this proposal would profoundly enhance our understanding regarding the common pathological mechanisms of a number of diseases and aging, which would establish a critical basis for novel therapeutic strategies for multiple diseases by targeting at NAD+ metabolism.

The major reasons accounting for why NAD+ deficiency plays critical roles in multiple diseases are the following: (i) It is established that NAD+ plays crucial roles in a number of major biological functions (122, 123). Therefore, it is conceivable that decreased NAD+ levels would produce significant and broad pathological changes of multiple major biological functions in cells and tissues. (ii) A major known cause of NAD+ deficiency is excessive activation of PARP-1 (21). Excessive activation of PARP-1 has been indicated to play critical roles in the animal models of a number of diseases, such as ischemic brain injury (26, 33), myocardial ischemic injury (76, 100), MPTP-induced Parkinsonism (64), diabetes (11, 31), hemorrhagic shock (55), asthma (10), hypoglycemic brain injury (97), and alcoholic and non-alcoholic steatohepatitis (72). Because NAD+ depletion has been indicated to mediate PARP-induced cell death (3), the studies indicating critical roles of PARP-1 activation in a number of diseases have also suggested critical roles of NAD+ deficiency in the diseases. (iii) Increasing evidence has indicated crucial roles of sirtuins, particularly SIRT1, in multiple biological functions, including energy metabolism, inflammation, and cell survival (15, 42, 50, 81, 102). Therefore, it is reasonable that NAD+ deficiency would lead to multiple pathological changes due to its effects on sirtuins.
Therapeutic Potential of Restoration of NAD+ Levels for Diseases and Aging
Restoration of NAD+ levels is a highly promising therapeutic strategy for a number of diseases and aging
Based on the information generalized earlier, NAD+ appears to hold great potential to become a therapeutic agent for a number of diseases and aging: (i) NAD+ administration has shown exceptional protective effects on multiple disease models, which can reach >80% protection in many cases; (ii) NAD+ can produce multiple protective effects by targeting at multiple pathological pathways of the diseases; (iii) NAD+ could have great therapeutic windows, for example, 9 months after the initiation of glaucoma, NAD+ still can prevent 93% incidence of the disease (117); (iv) NAD+ belongs to the few drugs that are protective in many disease models; and (v) compared with antioxidants, the distinct advantages of NAD+ include that it targets at more downstream components than antioxidants, and its multiple functions include its capacity to increase antioxidant capacity. Similarly, compared with PARP inhibitors, NAD+ targets at more downstream components than PARP inhibitors.
NAD+ transport: a key factor in the therapeutic potential of NAD+ for diseases and aging
A major known mechanism for regulating the levels of cytosolic and mitochondrial NAD+/NADH levels are NADH shuttles, including malate-aspartate shuttles (MAS) and glycerol 3-phosphate shuttles (29, 68). The NADH shuttles transfer the reducing equivalents of cytosolic NADH into the mitochondrial matrix (29, 68). Multiple biological functions of NADH shuttles have been reported, for example, MAS plays important roles in myelination in mouse brain (83), neuronal synthesis of glutamate and glutamine (77), and Ca2+-induced mitochondrial respiration activation in neurons (57). Our study has suggested a novel role of MAS in the survival of C6 glioma cells (107): The major function of MAS in C6 glioma cells is to prevent glycolytic blockage resulting from excessive accumulation of cytosolic NADH, which has suggested a new mechanism that may underlie Warburg effect (80).
There has been no information regarding the transport mechanisms of NAD+ across the blood-brain barriers (BBB) under normal and pathological conditions. The study showing that intraperitoneal injection of NAD+ can significantly decrease ischemic brain injury (132) has implicated that NAD+ may be capable of crossing the BBB, at least after cerebral I/R. Future studies are warranted to determine the capacity of NAD+ to transport across the BBB under both physiological and pathological conditions. There has been little information regarding the mechanisms and kinetics of NAD+ metabolism in the body after NAD+ is administered into the body by intravenous, intraperitoneal, or intranasal administration. Therefore, future studies on this increasingly important topic are also highly needed.
Conclusions and Perspectives
Accumulating evidence has supported the notion that NAD+ is one of the fundamental factors in life that can significantly affect nearly all major biological processes. In the “Hypothesis of Central Regulatory Network in life,” it was proposed that “NAD, ATP and Ca2+ may be the most fundamental components in life which mediate nearly all of the key biological processes. The close interactions among these components may constitute a Central Regulatory Network in life” (123). Several recent articles have suggested that NAD+ deficiency plays critical roles in age-related disease (19, 42) and heart and renal disease (38). In our current review, we have generalized the information supporting the proposal that NAD+ deficiency is a common central pathological factor in a number of diseases and aging. To solidly establish the key concept regarding the roles of NAD+ deficiency in diseases and aging, it is necessary to conduct future studies on the following major research directions: It still requires a number of studies to further solidly establish the concept that “NAD+ deficiency is a common central pathological factor in a number of disease and aging.” The establishment of this concept would become a milestone in mankind's search for the fundamental mechanisms and therapeutic strategies for diseases and aging. For the diseases in which the roles of NAD+ have not been investigated, studies should be initiated to determine whether NAD+ deficiency is involved in the pathogenesis of these diseases. It is expected that studies applying new experimental approaches, including novel imaging techniques and phenomics approach, could significantly improve our understanding on the roles of NAD+ in diseases and aging. Although multiple studies have suggested the mechanisms underlying the critical roles of NAD+ deficiency in a number of diseases and aging, the information on this important topic is far from sufficient. Future studies on this topic may suggest novel and critical pathological mechanisms of multiple diseases and aging. It is necessary to further investigate the mechanisms underlying the NAD+ deficiency in the diseases and aging. There is significant deficiency in the information regarding the mechanisms of NAD+ transport and NAD+ metabolism in various tissues and organs. The major unanswered questions include: What are the transport mechanisms of NAD+ across the BBB under normal and pathological conditions? What are the mechanisms underlying the NAD+ transport across plasma membranes and the membranes of various intracellular organelles? What are the mechanisms of secretion of both NAD+ and the other key molecules in NAD+ metabolism from cells? What are the metabolic mechanisms of NAD+ in the blood? Preclinical and clinical studies should be conducted to determine the therapeutic potential of NAD+ in the diseases. Based on the profound protective effects of NAD+ in multiple disease models, it is expected that NAD+ may be proved to be a highly powerful therapeutic agent for multiple diseases.
Footnotes
Acknowledgments
This study was supported by a major basic research grant from the Science and Technology Commission of Shanghai Municipality, Grant Nos. 16JC1400500 and 16JC1400502, and Chinese National Science Foundation Grants No. 81271305.