
Abstract
Significance:
Genetically encoded hydrogen peroxide (H2O2) sensors, based on fusions between thiol peroxidases and redox-sensitive green fluorescent protein 2 (roGFP2), have dramatically broadened the available “toolbox” for monitoring cellular H2O2 changes.
Recent Advances:
Recently developed peroxiredoxin-based probes such as roGFP2-Tsa2ΔCR offer considerably improved H2O2 sensitivity compared with previously available genetically encoded sensors and now permit dynamic, real-time, monitoring of changes in endogenous H2O2 levels.
Critical Issues:
The correct understanding and interpretation of probe read-outs is crucial for their meaningful use. We discuss probe mechanisms, potential pitfalls, and best practices for application and interpretation of probe responses and highlight where gaps in our knowledge remain.
Future Directions:
The full potential of the newly available sensors remains far from being fully realized and exploited. We discuss how the ability to monitor basal H2O2 levels in real time now allows us to re-visit long-held ideas in redox biology such as the response to ischemia-reperfusion and hypoxia-induced reactive oxygen species production. Further, recently proposed circadian cycles of peroxiredoxin hyperoxidation might now be rigorously tested. Beyond their application as H2O2 probes, roGFP2-based H2O2 sensors hold exciting potential for studying thiol peroxidase mechanisms, inactivation properties, and the impact of post-translational modifications, in vivo. Antioxid. Redox Signal. 29, 552–568.
Introduction
H
In recent years, H2O2 has emerged as an important cellular signaling molecule (7, 47, 99, 108, 116, 117, 125, 132). Superoxide anions (O2 •−) and H2O2 are not only accidentally generated by selected flavoenzymes (64, 87) but also specifically produced by a variety of O2 •−-generating NADPH oxidases, H2O2-generating superoxide dismutases, and enzymes such as the sulfhydryl oxidase endoplasmic reticulum oxidoreductin 1 (Ero1) in the endoplasmic reticulum (78, 105, 107, 123). Proteins that were previously considered H2O2 detoxifiers are now realized to additionally serve as transducers and transmitters of H2O2 signals, resulting in the oxidation of specific target proteins (15, 18, 32, 45, 67, 121, 129, 132, 133, 135). Indirect evidence hints that what has been discovered so far may represent only the “tip of the iceberg” (49). H2O2 signaling and redox regulation may play a central role in cellular and organismal physiology, perhaps equivalent in importance to Ca2+ and phosphorylation-dependent signaling (79). However, it remains to be determined whether this is really the case or whether H2O2 signaling plays a more minor or specialized role in biological regulation, perhaps, for example, working in cooperation with other signaling pathways to “fine-tune” cellular responses.
The most likely mechanism for H2O2 signaling would be via the oxidation, either direct or indirect, of target protein cysteinyl thiol groups and perhaps also methionine residues in some cases. Protein cysteine oxidation can lead to a change in the activity, function, or subcellular localization of the target protein, thereby mediating a biological response (36). With the benefit of hindsight, it is obvious that H2O2 is the best suited of the various ROS for cellular signaling. H2O2 is relatively long-lived and stable compared with most other biologically relevant ROS, including O2 •−, hydroxyl radicals, peroxynitrite, and singlet oxygen (16, 19, 50). Further, H2O2 is poorly reactive with most biological molecules, including proteinogenic amino acids. Nonetheless, it is clear that a few proteins have evolved to harbor cysteine residues that are especially reactive with H2O2, thereby providing an important means of enabling signaling specificity (16, 50). Examples of such proteins include thiol peroxidases, which contain so-called peroxidatic cysteines that are located in dedicated H2O2 interaction sites and are surrounded by amino acids that serve to facilitate the reaction with H2O2 by stabilizing the reaction transition state and promoting leaving group departure. Similar catalytic mechanisms have recently been identified in other H2O2-reactive enzymes, for example, glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (62, 103). It is difficult to envisage how such specificity could evolve for other ROS, for example, hydroxyl radicals, which react indiscriminately with most biological molecules at diffusion limited rates (16, 19, 50).
The earlier considerations raise the questions of why it took so long to appreciate the biological importance of H2O2 and why, after realizing that H2O2 may have key biological roles, so much still remains unknown. At least a part of the answer to this question clearly lies with the difficulty of specifically monitoring H2O2 levels inside living organisms. Widely used small chemical “ROS” dyes have proved useful in many contexts but suffer from a range of issues, including poorly defined specificity for redox species, poorly characterized in vivo reaction mechanisms, susceptibility to photo-oxidation, and concentration-dependent kinetics owing to variable rates of cellular uptake and efflux (51, 141).
There was, and still remains, a clear need for novel tools, methodologies, and experimental approaches to facilitate the investigation of all aspects of biological H2O2 production, turnover, and signaling mechanisms.
In the past decade, considerable progress has been made with respect to the specific monitoring of cellular H2O2 levels. Advances have come mainly on two major fronts in the form of novel, specific, small chemical sensors such as boronate caged probes (1, 3, 20, 21, 27, 37 –39, 91, 122) and the development of genetically encoded H2O2 sensors, which utilize naturally H2O2 reactive proteins as their sensing moieties. The first such genetically encoded sensor was HyPer, which is based on the bacterial H2O2-sensing protein OxyR (12). These sensors are covered in another review in this issue and will not be discussed here. More recently, H2O2 sensors have been developed that utilize thiol peroxidases as their sensing moiety (58, 90, 93, 94, 112). These probes exploit the mechanistic principles of naturally occurring thiol peroxidase-based redox relays. The probes in this category, notably redox-sensitive green fluorescent protein 2 (roGFP2)-Orp1 and the recently developed roGFP2-Tsa2ΔCR sensor, are the main focus of this review.
The Conundrum of H2O2 Signaling: How Are Protein Thiols Oxidized?
Redox proteomic screens have revealed that many cellular proteins contain cysteine residues that are susceptible to H2O2-induced oxidation. However, with a few notable exceptions, most apparently “H2O2-sensitive protein cysteine residues,” as with most protein cysteine residues in general, are remarkably unreactive with H2O2 in vitro. Proteinaceous cysteine residues typically react with H2O2 at a rate similar to that of free cysteine, that is, in the range of 1–10 M −1·s−1. This stands in stark contrast to the reactivity of cysteine residues in proteins with highly evolved H2O2 active sites, such as the peroxidatic cysteine residues found in thiol peroxidases. Such peroxidatic cysteines exhibit second-order rate constants for the initial reaction with H2O2 in the range of 105–108 M −1·s−1 (16, 34, 48, 50). Moreover, some thiol peroxidases are among the most abundant proteins in the cell. The apparent paradox is, therefore, how “normal” protein cysteine residues can ever be oxidized by H2O2 given their extremely low relative reactivity, particularly in the context of the high abundance and reactivity of thiol peroxidases. There are two main models that seek to explain H2O2-induced cysteine oxidation, namely the floodgate model and the thiol peroxidase-based redox relay model. Thiol peroxidases play a central, although very different, role in both models.
A Brief Introduction to Thiol Peroxidases
Thiol peroxidases can be broadly separated into two categories, the peroxiredoxins and the cysteine-dependent glutathione (GSH) peroxidases. On the basis of their reaction mechanism and number of redox-active cysteines, peroxiredoxins can be further subdivided into three categories: the 1-Cys peroxiredoxins, the atypical 2-Cys peroxiredoxins, and the typical 2-Cys peroxiredoxins. All peroxiredoxins have in common a peroxidatic cysteine residue, which is a highly H2O2-reactive cysteine that performs the initial nucleophilic attack on H2O2, leading to the formation of a cysteine sulfenic acid and the release of water. In the case of 1-Cys peroxiredoxins, this sulfenic acid may subsequently directly react with other thiols, for example, GSH, leading to the formation of a disulfide bond. In the case of atypical 2-Cys peroredoxins, the second “resolving” cysteine residue is located in the same polypeptide chain as the peroxidatic cysteine. Usually, the resolving cysteine residue reacts with the peroxidatic cysteine sulfenic acid, leading to the formation of an intramolecular disulfide bond. Finally, in the typical 2-Cys peroxiredoxins, both a peroxidatic and a resolving cysteine are found in the same polypeptide chain. However, in contrast to atypical 2-Cys peroxiredoxins, these proteins form “head-to-tail” dimers, with the peroxidatic cysteine of one subunit forming an intermolecular disulfide bond with the resolving cysteine of the other. This can happen on both ends of the dimer. Further, typical 2-Cys peroxiredoxins can assemble into higher-order oligomers, for example, decamers and even stacks of decamers. On the basis of current knowledge, it is difficult to draw firm conclusions regarding the relative importance of the different thiol peroxidases for H2O2 signaling. However, it is clear that thiol peroxidases play a central role in both the floodgate model (144) and the thiol peroxidase-based redox relay model of protein thiol oxidation (16, 50, 59).
The Floodgate and Redox Relay Models for Explaining H2O2-Induced Thiol Oxidation
In the floodgate model of cysteine thiol oxidation, it is posited that peroxiredoxins first need to become inactivated by either hyperoxidation or phosphorylation before oxidation of target protein thiols can occur (144). The concept is that, on peroxiredoxin inactivation, H2O2 can accumulate to levels at which direct oxidation of “normal” cysteine residues is possible. Most likely, this would take place in specific cellular microdomains, perhaps close to significant sources of H2O2 such as NADPH oxidases (143) or at the centrosome (82, 109). In this model, peroxiredoxin inactivation is a prerequisite to protein thiol oxidation. One criticism of this model is that it does not solve the problem of the innate unreactivity of most protein thiols toward H2O2 and it is unclear whether H2O2 levels can really accumulate to the levels that are necessary to facilitate direct oxidation of a “typical” target protein cysteine residue.
In the thiol peroxidase-based redox relay model (16, 50), thiol peroxidases are not part of the problem of explaining H2O2-mediated oxidation of specific protein cysteine residues, they are the solution. The model proposes that thiol peroxidases initially react with the vast majority of H2O2 that is produced within the cell (or enters the cell), leading to the transient formation of a sulfenic acid group on their peroxidatic cysteine. Classically, it would be expected that a second cysteine, the identity of which varies between different thiol peroxidases, would then react with the sulfenic acid, leading to the formation of a disulfide bond. Thioredoxins would, subsequently, reduce the disulfide bond, thereby enabling another round of H2O2 scavenging. However, in the thiol-peroxidase redox relay, thioredoxin is replaced by a specific H2O2 target protein. In other words, oxidation is transferred from the thiol peroxidase to specific H2O2 target proteins instead of, or as well as, to thioredoxin. As a consequence, thiol peroxidases no longer compete with H2O2 target proteins for oxidation but instead act as essential mediators. Therefore, this model makes a diametrically opposed prediction to that of the floodgate model, namely that peroxiredoxin hyperoxidation would lead to a loss of target protein cysteine oxidation by physiologically relevant concentrations of H2O2.
Examples of Naturally Occurring Thiol Peroxidase-Based Redox Relays
The first example of a thiol peroxidase-based redox relay was identified in the yeast Saccharomyces cerevisiae by the group of Michel Toledano (32, 129). They observed that the GSH peroxidase homolog Orp1 passes oxidation from H2O2 to the transcription factor yeast antioxidant protein 1 (Yap1), which drives the expression of an array of antioxidant and detoxification enzymes. Yap1 usually moves back and forth across the nuclear envelope. Once in the nucleus, the nuclear export factor, chromosomal region maintenance 1 (Crm1) recognizes the nuclear export sequence of Yap1 and mediates its rapid nuclear export (145). It is believed that disulfide bond formation in Yap1 masks its nuclear export sequence, therefore preventing recognition by Crm1 (76). Oxidized Yap1 accumulates in the nucleus, where it can fulfill its function (31, 32). Endogenous Orp1 and Yap1, thus, constitute a redox relay that mediates a transcriptional oxidative stress response. In the cell, the interaction between Orp1 and Yap1 is facilitated by a third protein, Yap1-binding protein (Ybp1), which appears to act as a “scaffold” protein to bring Orp1 and Yap1 into close physical proximity and facilitates the reaction between Orp1 and Yap1 by stabilizing the sulfenic acid group formed on Orp1 (13, 56, 134). Scaffold proteins, in general, could serve to solve the “specificity” problem, that is, how does thiol peroxidase efficiently pass on oxidation to specific target proteins? Further, by ensuring a tight co-localization, scaffold proteins may prevent or limit a reduction by thioredoxin and/or GSH/glutaredoxin (Grx) that might otherwise compete with the target protein for receiving oxidation.
The first indication that typical 2-Cys peroxiredoxins might function in thiol peroxidase-based redox relays arguably came from S. cerevisiae, where it was demonstrated that the peroxiredoxin termed thiol-specific antioxidant 1 (Tsa1) is important for the induction of Yap1 gene expression (110). This concept was reinforced by later studies (100, 126), although the importance of Tsa1 for Yap1 oxidation is very limited in comparison to Orp1 and only becomes apparent in strains with mutations in Ybp1. Work in the fission yeast Schizosaccharomyces pombe also found evidence for a thiol peroxidase (Tpx)1 (the fission yeast typical 2-Cys peroxiredoxin)-dependent oxidation of the oxidant-responsive transcription factor Pombe antioxidant protein 1 (Pap1) and the mitogen-activated protein (MAP) kinase suppressor of tyrosine phosphatase 1 (Sty1) (15, 133, 135). More detailed insight into the Tpx1-mediated oxidation of Pap1 has subsequently been gained (18).
Recently, direct evidence has been found for the existence of typical 2-Cys peroxiredoxin-mediated redox relays in mammalian cells, in which typical 2-Cys peroxiredoxins are believed to oxidize target proteins, including the mitogen-activated protein kinase kinase kinase (MAP3) kinase apoptosis signal-regulating kinase 1 (ASK1), the transcription factor signal transducer and activator of transcription 3 (STAT3), and the cardiac protein protein deglycase 1 (DJ-1) (45, 67, 121). These natural relays provided the inspiration for the development of the GSH peroxidase-based H2O2 sensor roGFP2-Orp1 (58) and, recently, typical 2-Cys peroxiredoxin-based H2O2 sensors, most notably roGFP2-Tsa2ΔCR (94).
Mechanism of roGFP2-Based H2O2 Sensor Oxidation
roGFP2 is based on an enhanced GFP that has been modified to contain two cysteine residues on parallel beta strands, adjacent to the chromophore (60, 90, 93, 112). These two cysteine residues can form a disulfide bond, leading to minor structural rearrangements that alter the protonation state of the chromophore. This results in a change in fluorescence excitation wavelength between 405 nm (for the neutral form, which dominates in oxidized roGFP2) and 488 nm (for the anionic chromophore, which dominates in reduced roGFP2). Both the protonated and anionic forms of the chromophore emit light at ∼510 nm due to excited state proton transfer (2, 70, 106, 124). Importantly, changes in environmental pH do not affect the protonation of the chromophore as the β-barrel in roGFP2 is completely intact, which shields the chromophore (113). The two excitation wavelengths allow for ratiometric measurements, which renders the probe readout independent of changes in probe concentration.
The basic functional premise of roGFP2-thiol peroxidase fusion probes is to reconstitute an entire peroxidase redox relay, complete with a “scaffold protein,” in one polypeptide chain. For example, in roGFP2-Orp1, Orp1 fulfills the same function as it does in the natural Orp1-Yap1 relay, that is, it serves to sensitively react with H2O2 and to, subsequently, transduce and transmit the oxidation from H2O2 to the target protein in the form of a disulfide bond. roGFP2 replaces Yap1 in receiving oxidation from Orp1 with oxidation of roGFP2, leading to a ratiometric change in the fluorescence excitation spectra. Consequently, a transcriptional response (in the context of the natural relay) is replaced with a fluorescence response (in the context of the probe). Finally, the short polypeptide linker serves the function of a scaffold protein, for example, Ybp1, and acts to hold the thiol peroxidase and roGFP2 moieties in close physical proximity. Although the general mechanistic principle of all roGFP2-based H2O2 probes is similar, the mechanistic details are quite different as outlined in the following paragraphs.
Probes based on GSH peroxidases
RoGFP2-Orp1 was the first reported roGFP2-based H2O2 sensor. It is based on the yeast thiol peroxidase Orp1, which was initially characterized as a GSH peroxidase homolog and, subsequently, more specifically as a phospholipid hydroperoxide GSH peroxidase (5, 6, 65). Mechanistically, Orp1 functions similarly to an atypical 2-Cys peroxiredoxin in that a reaction with H2O2 results in the formation of a disulfide bond in Orp1 between a peroxidatic cysteine (Cys36) and a resolving cysteine residue (Cys82) that are located on the same polypeptide chain (32). This disulfide bond is predominantly reduced by thioredoxin (32, 85).
Presumably, the initial step in the oxidation of roGFP2-Orp1 is the nucleophilic attack of the Orp1 peroxidatic cysteine thiolate on an H2O2 molecule (Fig. 1). This will result in the transient formation of a sulfenic acid group on the peroxidatic cysteine. Subsequently, there are two possibilities: Either the resolving cysteine can condense with the peroxidatic cysteine sulfenic acid, leading to the formation of a disulfide bond in Orp1, which can subsequently be transferred to roGFP2, or an roGFP2 cysteine may directly attack the sulfenic acid group. In the presence of a resolving cysteine, it would be expected that the kinetics of intramolecular disulfide bond formation between the resolving and peroxidatic cysteine would be much quicker than the formation of a disulfide between roGFP2 and the Orp1 peroxidatic cysteine. Consequently, the most likely oxidation mechanism of roGFP2-Orp1 involves the initial formation of an intramolecular CP–CR disulfide in Orp1 followed by a thiol-disulfide exchange reaction to roGFP2.

Interestingly, it was observed that an roGFP2-Orp1 construct with a mutated resolving cysteine (roGFP2-Orp1ΔCR) also responds efficiently to H2O2 (58). In fact, in vitro, the kinetics of roGFP2 oxidation in this probe were found to be much quicker than for roGFP2-Orp1. Nonetheless, the value of this probe for measurements inside living cells is probably very limited as it was shown that GSH inhibits transfer of oxidation from Orp1ΔCR to roGFP2. This is presumably because GSH reacts efficiently with the peroxidatic cysteine sulfenic acid group of Orp1. Consistent with these observations, in the cytosol of mammalian cells, roGFP2-Orp1ΔCR was found to be non-functional (58). As an important note, the domain order of Orp1 probes is apparently crucial for functionality. Although roGFP2-Orp1 functions well as an H2O2 sensor, Orp1-roGFP2 is non-functional (Morgan and Dick, unpublished data).
A probe based on the mammalian GSH peroxidase, GPX4 (roGFP2-GPX4), was also found to be functional in mammalian cells. However, this selenocysteine-containing probe was not extensively characterized and to the best of our knowledge has not been employed in any further studies since its development (58).
Probes based on typical 2-Cys peroxiredoxins
Orp1 is involved in the response to “oxidative stress.” Although the exact second-order rate constant for the initial reaction between the peroxidatic cysteine of Orp1 and H2O2 is unknown, it is likely to be in the order of 105 M −1·s−1 (14, 120). This is about two to three orders of magnitude slower than for the fastest typical 2-Cys peroxiredoxins, which may be as high as 107 M −1·s−1 or perhaps even 108 M −1·s−1 (16, 46, 50). Given the markedly increased H2O2 sensitivity of a typical 2-Cys peroxiredoxin compared with Orp1, it is feasible that there can be changes in intracellular H2O2 levels that are sufficient to mediate physiological responses but that are below the detection limit of either roGFP2-Orp1 or HyPer. Consequently, we recently asked whether it was possible to develop an roGFP2-based sensor employing a typical 2-Cys peroxiredoxin as the sensing moiety. The resultant probes include roGFP2-Tsa1, roGFP2-Tsa2, and roGFP2-PRDX2 (94). These probes have additionally been modified with respect to their peroxidatic and resolving cysteines, for example, to generate roGFP2-Tsa2ΔCP, roGFP2-Tsa2ΔCR, and roGFP2-Tsa2ΔCPΔCR. These cysteine mutants have dramatically modified probe properties. Next, we describe the mechanism of action of these probes, explain the rationale behind the creation of their cysteine mutant variants, and explore the mechanistic underpinnings of cysteine-dependent functional probe changes.
As with the roGFP2-Orp1 sensor, presumably the initial step in the oxidation of all typical 2-Cys peroxiredoxin probes must be a nucleophilic attack on an H2O2 molecule performed by the peroxidatic cysteine thiolate, leading to the formation of a sulfenic acid group. Subsequently, this oxidation must be transferred to the roGFP2 moiety, although exactly how this happens will differ dependent on the probe variant in question. Typical 2-Cys peroxiredoxins usually assemble in head-to-tail dimers, which can further come together to form a decameric ring. In each dimer, the peroxidatic cysteine of each peroxiredoxin molecule sits adjacent to the resolving cysteine of the other, thereby allowing for a total of two intermolecular disulfide bonds to form per dimer (59, 144). It would be expected that the kinetics of the reaction between the peroxidatic cysteine and the resolving cysteine will be much quicker than the kinetics of the reaction between the peroxidatic cysteine and one of the roGFP2 cysteines. RoGFP2-Tsa2 probes were shown to be able to dimerize with either another probe molecule or endogenous Tsa1 or Tsa2 to form probe–endogenous Tsa1/2 heterodimers. Consequently, for an roGFP2-Tsa2 probe, the most likely oxidative mechanism is the initial formation of an intermolecular disulfide bond between the two partners in the peroxiredoxin dimer, followed by a thiol-disulfide exchange reaction, which transfers the disulfide bond from the peroxiredoxin dimer to roGFP2 (Fig. 2). This conclusion is strongly supported by experiments with roGFP2-Tsa2 probes in which the peroxidatic and resolving cysteine have been mutated, as described later.

The rationale behind resolving cysteine mutation in roGFP2-Tsa2ΔCR
In a side-by-side comparison between roGFP2-Orp1 and roGFP2-Tsa2 expressed in the cytosol of yeast cells, it was unexpectedly observed that the two probes have very similar sensitivity toward H2O2 (94). The reason for this was found to be that thioredoxin very effectively competes with roGFP2 for reducing the Tsa2 disulfide bond. In an attempt to mitigate against thioredoxin-mediated reduction, the resolving cysteine of Tsa2 was mutated. The resultant sensor, roGFP2-Tsa2ΔCR, which can still non-covalently dimerize with endogenous Tsa1 or Tsa2, is ∼20-fold more sensitive than roGFP2-Tsa2 and permits measurement of “basal” endogenous H2O2 levels. Consistent with a thioredoxin-dependent effect, when roGFP2-Tsa2 and roGFP2-Tsa2ΔCR are expressed in a yeast strain in which the genes for both cytosolic thioredoxins are deleted, the H2O2 sensitivity of the two probes is very similar (94).
The main conclusion that can be drawn from the experiments with the roGFP2-Tsa2ΔCR sensor is that an roGFP2 cysteine residue can directly accept oxidation from a peroxidatic cysteine sulfenic acid group; no other factors or intermediate steps are required (Fig. 3). In the roGFP2-Tsa2ΔCR sensor, one of the two roGFP2 cysteine residues presumably performs a nucleophilic attack on the peroxidatic cysteine sulfenic acid group, leading to the formation of a transient intermolecular disulfide bond between the roGFP2 moiety and an endogenous Tsa1 or Tsa2 partner within the Tsa2 dimer or another probe molecule. Subsequently, the second cysteine of roGFP2 would attack the intermolecular disulfide bond, generating an intramolecular roGFP2 disulfide. In summary, the resolving cysteine of the typical 2-Cys peroxiredoxin is not required for the oxidation of roGFP2, and although proximal roGFP2 can apparently efficiently reduce the Tsa2 sulfenic acid group diffused yeast thioredoxin cannot.
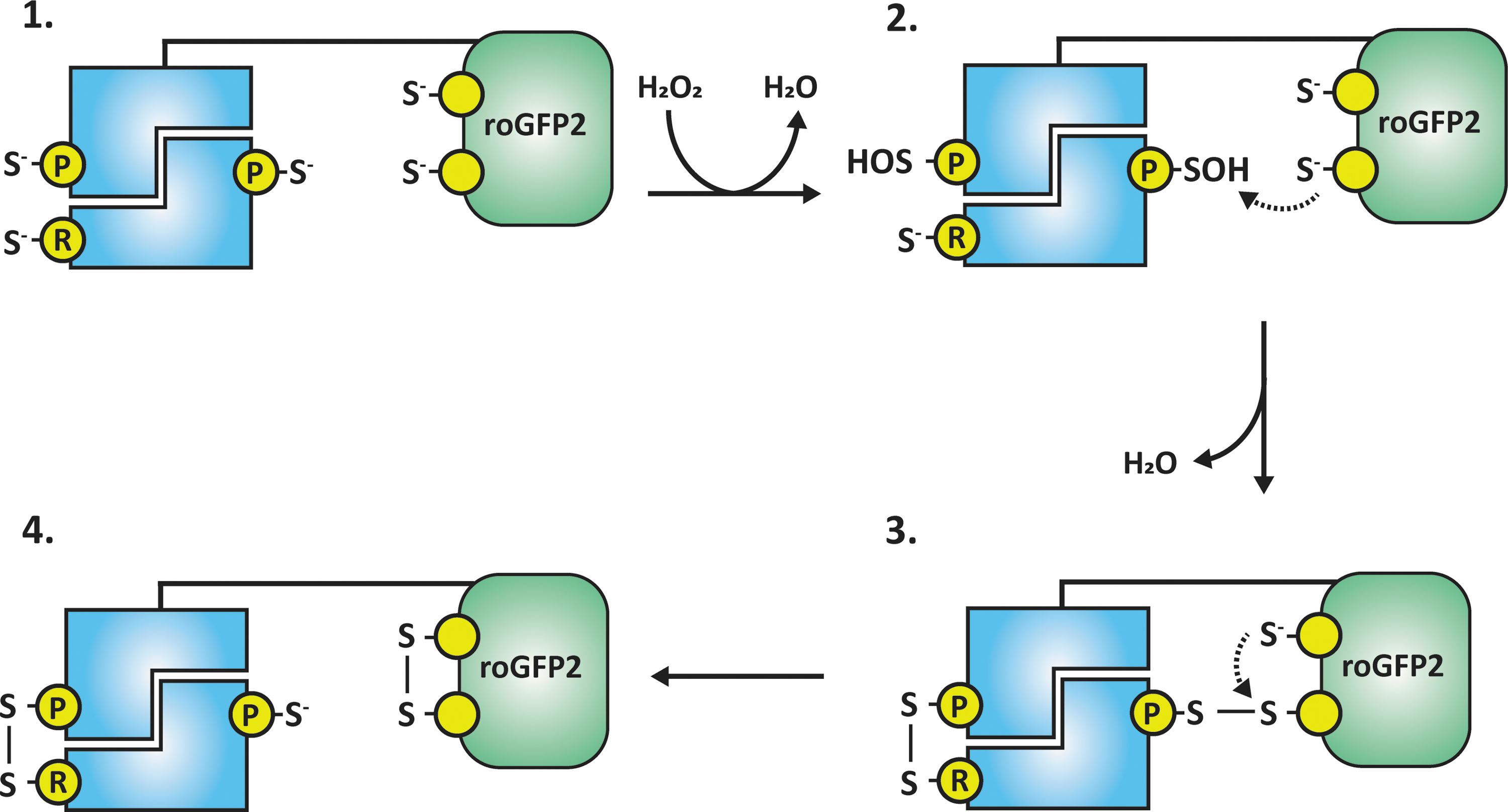
Decamerization of typical 2-Cys peroxiredoxin-based roGFP2 probes
Typical 2-Cys peroxiredoxins can assemble into decameric rings (59, 144). Likewise, roGFP2-Tsa2 and roGFP2-Tsa2ΔCR were also observed to exist predominantly in a decameric state in the cytosol of yeast cells (94) (and it is very likely that other typical 2-Cys peroxiredoxin-roGFP2 probes, for example roGFP2-PRDX2, also assemble into decamers). Although roGFP2-Tsa2 probes can assemble into homo-decameric rings, this is not the most common decameric form of the probe. Similar to the hetero-dimers in Figures 2 and 3, roGFP2-Tsa2 molecules were observed to form hetero-decamers in the presence of endogenous Tsa1 and Tsa2 (94) (Fig. 4). Interestingly, it was observed that the roGFP2-Tsa2 probes incorporated almost exclusively into endogenous Tsa1 decamers rather than endogenous Tsa2 decamers. This is perhaps not surprising as Tsa1 and Tsa2 are closely homologous (86% identity, 96% similarity) and Tsa2 and Tsa1 are probably present at a nanomolar versus micromolar concentration (34, 53). The available endogenous Tsa2 concentration may be even lower, for example, if it is bound to ribosomes (115). The number of roGFP2-Tsa2 molecules that can be incorporated into an endogenous Tsa1/2 decamer is apparently highly flexible. In the cytosol of wild-type yeast cells expressing roGFP2-Tsa2, Tsa1 decamers incorporating everything from one to nine probe molecules were observed. The different decameric species can easily be resolved by blue native-polyacrylamide gel electrophoresis as each extra probe molecule incorporated into the decameric ring will increase the molecular mass by ∼25 kDa due to the fused roGFP2 moiety (94). The fact that the roGFP2-Tsa2 probes can assemble into endogenous Tsa1/2 decamers raises further questions regarding the mechanism of roGFP2 oxidation in these sensors but simultaneously affords new possibilities to investigate such mechanisms. It was observed that roGFP2-Tsa2 probes in which either the peroxidatic cysteine or both the peroxidatic and resolving cysteine were deleted (roGFP2-Tsa2ΔCP and roGFP2-Tsa2ΔCPΔCR, respectively) were still efficient, functional H2O2 sensors. The roGFP2-Tsa2ΔCP sensor has comparable H2O2 sensitivity to an roGFP2-Tsa2 probe, whereas the roGFP2-Tsa2ΔCPΔCR probe is comparable to roGFP2-Tsa2ΔCR. This suggests that roGFP2 oxidation occurs in trans, that is, roGFP2 receives oxidation from a peroxiredoxin other than the one to which it is genetically fused (Fig. 4). Most likely, this would be the partner peroxiredoxin in the dimer, that is, oxidation is transferred across the B-type interface (between subunits of a dimer) analogous to the electron transfer between the resolving and the peroxidatic cysteine of a typical 2-Cys peroxiredoxin (48, 59). In the context of an roGFP2-Tsa2ΔCP sensor, this would still permit the formation of a disulfide bond between the Tsa2 sensor and the partner peroxiredoxin that can be reduced by thioredoxin, hence the decreased sensitivity; whereas in the roGFP2-Tsa2ΔCPΔCR probe, no disulfide bond formation is possible (Fig. 4B, C). It is currently unclear as to whether or not oxidation can also be transferred across the A-type interface (between dimers). Further experiments would be required to clarify this issue.

These intriguing observations raise a number of questions regarding the situation in cellular compartments where more than one endogenous typical 2-Cys peroxiredoxin is present. For example, are all endogenous peroxiredoxin decamers homogenous, that is, consisting of only one peroxiredoxin type? At least in the case of roGFP2-Tsa2 probes, the answer is apparently not, as these probes readily incorporate into endogenous Tsa1 decamers in yeast cells. Alternatively, it may be that the roGFP2-peroxiredoxin fusions do not behave in a manner that is representative of endogenous peroxiredoxins. For example, if decamer homogeneity were ensured by differential subcellular localization of different peroxidoxin isoforms, it may be that the presence of an roGFP2 tag disrupts correct targeting.
If, in the more likely scenario, cells do not harbor any mechanism to ensure peroxiredoxin homogeneity and instead form peroxiredoxin decamers containing more than one type of peroxiredoxin, the question would be as to whether or not there is a functional relevance to this. Modular enzyme activities for various heterodimers have, for example, been previously demonstrated for GSH transferases (33, 86). It is thus possible that peroxiredoxin function or activity may be modulated by varying the subunit composition of the decameric rings. For example, although yeast Tsa1 is the dominantly expressed isoform, Tsa2 expression is markedly increased on oxidative challenge (142). Assuming that Tsa2 does, indeed, incorporate into endogenous Tsa1 decamers, it will be extremely interesting to address the functional relevance of the hetero-decamer formation. These studies provide a good example of how genetically encoded probes can be utilized to make observations that go far beyond their intended function as H2O2 sensors.
Probes based on atypical 2-Cys peroxiredoxins and 1-Cys peroxiredoxins
Probes based on atypical 2-Cys peroxiredoxins include roGFP2-alkyl hydroperoxide reductase 1 (Ahp1). This probe has been demonstrated to be functional, although its potential for measurements remains to be further explored (94). To date, probes based on 1-Cys peroxiredoxins have not been extensively characterized. It was reported that a probe based on the mammalian 1-Cys peroxiredoxin PRX6 (PRX6-roGFP2) was not functional as an H2O2 sensor (58). However, this may be due to the subunit orientation, as a probe based on the yeast 1-Cys peroxiredoxin (Prx)1 (roGFP2-Prx1) functions well as an H2O2 sensor (94). Alternatively, the difference in functionality between roGFP2 probes based on yeast and mammalian 1-Cys peroxiredoxins may be due to structural differences between the two enzymes that, for example, lead to steric hindrance in the case of the mammalian enzyme. It is possible that 1-Cys peroxiredoxin-based probes may have value for monitoring the response of endogenous mitochondrial matrix peroxiredoxins to changes in mitochondrial respiratory chain function, although this remains to be explored.
Mechanism of Probe Reduction
The oxidation reaction of the probe is irreversible per se, in the sense that a cysteine sulfenic acid group plus water will never give rise to a thiol and H2O2 under any physiological conditions. RoGFP2-based H2O2 probes, nonetheless, allow reversible monitoring as the roGFP2 disulfide can be reduced. The most likely mechanism by which this occurs is via GSH/Grx-mediated reduction of the roGFP2 moiety. Indeed, deletion of the yeast glutaredoxins Grx1 and Grx2 leads to full oxidation of an roGFP2-Tsa2ΔCR probe expressed in the cytosol (94). A second possibility that cannot be strictly ruled out is that the reduced peroxidatic cysteine may attack the disulfide bond on the roGFP2 moiety, leading to the formation of an intermolecular disulfide (the backward reaction of species 5 in Fig. 2). Such a disulfide bond might also be susceptible to reduction by either the GSH/Grx or the thioredoxin system. When a resolving cysteine is present, there is also a third possibility to form a disulfide bond between the peroxidatic and the resolving cysteine (backward reaction of species 4 in Fig. 2). This disulfide bond may, subsequently, be reduced by GSH/Grx or thioredoxin with additional options regarding whether mixed intermediate disulfide bonds are formed with the peroxidatic or the resolving cysteine residue.
RoGFP2-Tsa2ΔCR Does Not Significantly Contribute to Cellular H2O2 Scavenging
Clearly, any sensor that responds to H2O2 must consume some H2O2. In the case of roGFP2-Tsa2ΔCR, one H2O2 molecule leads to the oxidation of one roGFP2 moiety (94) and thus the contribution to H2O2 scavenging becomes a question of flux, that is, how quickly is the probe reduced after oxidation or in other words, to what extent does the probe contribute toward the cellular H2O2-scavenging capacity. Conceivably, such peroxiredoxin-based sensors may significantly influence cellular H2O2 handling. We have, therefore, previously employed several different experimental approaches to address this question. First, growth assays were used to assess the impact of roGFP2-Tsa2ΔCR probe expression on the ability of Δtsa1 Δtsa2 yeast cells to grow in the presence of different concentrations of H2O2. No effect of roGFP2-Tsa2ΔCR or roGFP2-Tsa2ΔCPΔCR expression was observed relative to cells containing an empty plasmid; whereas in contrast, plasmid-based expression of a wild-type unfused Tsa2 fully restored cell growth (94).
As a second means of testing the H2O2-scavenging capacity of roGFP2-Tsa2ΔCR, a mitochondrial matrix targeted roGFP2-Tsa2ΔCR sensor was expressed in Δtsa1 Δtsa2 yeast cells. These cells were further transformed with a cytosol-targeted non-fluorescent probe mutant, roGFP2-G67A-Tsa2ΔCR or were transformed with Tsa2 as a positive control or an empty vector as a negative control. The response of the mitochondrial probe to exogenous H2O2, applied at a range of different concentrations, was then measured. The rationale behind this experiment was that the amount of H2O2 that is able to pass through the cytosol and reach the matrix probe will depend on the H2O2-scavenging capacity of the cytosol. It was found that expression of Tsa2 in the cytosol strongly decreased the amplitude of the matrix probe response relative to the empty vector containing cells, at all H2O2 concentrations tested. In contrast, expression of the cytosol-targeted fluorescent probe mutant, roGFP2-G67A-Tsa2ΔCR had almost no effect on matrix probe response compared with empty vector containing cells, at all H2O2 concentrations tested. In conclusion, this assay further supports the notion that the roGFP2-Tsa2ΔCR sensor does not significantly contribute toward cellular H2O2 scavenging (94).
These results are also in line with the observation that the mutation of the resolving cysteine increases probe sensitivity as it apparently prevents the thioredoxin-mediated reduction. Although there must be some flux through the sensors, due to the Grx/GSH-mediated reduction of the roGFP2 moiety, it seems that this is not fast enough to contribute significantly to cellular H2O2 scavenging.
What Are the Sensors Actually Measuring?
The oxidation of roGFP2-based H2O2 probes reflects a balance between H2O2-driven oxidation on the one hand and GSH/Grx-driven reduction on the other hand. However, as has been previously discussed, this is true for all of the genetically encoded H2O2 sensors currently available, including the HyPer family, and is representative of how the oxidation of endogenous cellular H2O2-responsive thiols must be regulated (thioredoxin is likely also important for reduction of many endogenous thiols). Nonetheless, it raises the possibility that a significant change in the cellular reductive capacity, for example, an increase or decrease in NADPH availability, will affect the probe oxidation. The more difficult question to answer is whether in this situation probe oxidation actually changes independently of H2O2 levels. One might argue probably not, as a decrease in NADPH availability would be expected to not only affect the reduction of the probe but also affect the reduction of endogenous thiol peroxidases. Thus, there really would be an increase in H2O2 levels, as reflected by the increased probe oxidation. At minimum, it could be argued that the probe oxidation reflects how the oxidation state of an H2O2-sensitive protein “perceives” cellular H2O2 levels to be. However, it is also pertinent to ask whether there may be physiologically relevant situations in which probe oxidation changes are independent of H2O2 changes.
Can probe oxidation change without a change in H2O2 levels?
Due to the complexity of the probe oxidation mechanism together with the reversibility afforded by the probe's interaction with other cellular redox systems, it is valid to ask whether there are situations in which changes in probe oxidation are uncoupled from changes in H2O2 levels. It is possible to imagine at least two plausible scenarios.
(i) Hyperoxidation of the sensing peroxidatic cysteine: When the thiol of the peroxidatic cysteine of the sensing thiol peroxidase becomes hyperoxidized to the sulfinic or even sulfonic acid, oxidation can no longer be transferred to roGFP2. Effectively, the sensor ceases to become an H2O2 probe. With the thiol peroxidase inactivated, the most likely scenario is that the roGFP2 would equilibrate with the prevailing glutathione redox potential (E GSH) provided that endogenous Grxs are available to catalyze the equilibration between the probe and GSH redox couples (73, 89, 90, 92, 93, 112). Hyperoxidation has been demonstrated for roGFP2-Tsa2ΔCR on exogenous application of H2O2 at mM concentration. However, to date, no evidence has been found for significant hyperoxidation under “normal” physiological conditions. Interestingly, it was observed that human PRDX2 is much more sensitive than Tsa2 to hyperoxidation in the context of an roGFP2 fusion (94). Therefore, caution should be applied if probes are developed that utilize alternative typical 2-Cys peroxiredoxins.
(ii) Expression in subcellular compartments or cell types where probe interactions are poorly characterized: Particularly, care should be employed when probes are to be utilized in a subcellular compartment or new cell type for the first time. As an extreme example, the secretory pathway of many organisms lacks thioredoxins and Grxs, in contrast to other highly abundant redox enzymes, whose interactions with roGFP2 and the fused thiol peroxidase domain are entirely uncharacterized. It is very unlikely in such a compartment that the probe oxidation state accurately reflects the H2O2 concentration. Even in compartments such as the mitochondrial intermembrane space (IMS), particular care should be taken. Although the GSH pool in the IMS is in equilibrium with the cytosol (73), Grxs and thioredoxins are highly limiting (74). Probes in the IMS will probably be much more oxidized than expected. Comparable to the effect of removing reductive pathways on cytosolic probe oxidation, IMS probes would be expected to lose a lot of their “reversibility” and accumulate in an oxidized state. In summary, the oxidation state of genetically encoded H2O2 sensors that are targeted to different subcellular compartments should not be used to draw conclusions regarding the inter-compartment similarity or difference of H2O2 levels.
How Do roGFP2-Based Sensors Compare with Small Chemical H2O2 Dyes?
To the best of our knowledge, no comprehensive side-by-side comparison of genetically encoded and small chemical H2O2 sensors has thus far been performed. This is a pity as such a study would undoubtedly be invaluable for the H2O2 research community. Nonetheless, here we briefly summarize the major advantages and disadvantages of the two classes of sensors.
There are now numerous examples of small chemical probes that are specific to H2O2 (1, 3, 20, 21, 27, 37 –39, 91, 122), although, in common with genetically encoded probes, reactivity with peroxynitrite may still be possible (95, 119). The majority of the sensors reported thus far are based on boronate cage protected fluorophores, with the boronate cage disrupting π-conjugation in the fluorophore, thereby leading to fluorescence quenching (51). Fluorescence is restored on a reaction of H2O2 with the boronate cage group. Probes in this group are available in a wide range of different colors (38), as ratiometic probes that are compatible with two-photon microscopy (27) and even as targeted probes for measurements in specific organelles (37, 39, 122). Further, the boronate cage principle has recently been extended to develop the positron emission tomography (PET)-compatible H2O2 sensor Peroxy-Caged-[18F]Fluorodeoxy thymidine-1 (PC-FLT-1) (20). In this sensor, the reaction of H2O2 leads to the generation of [18F]FLT, which can be transported into and retained within living cells in a peroxide-dependent manner. Thus, PC-FLT-1 is a PET tracer whose cellular accumulation is dependent on H2O2. The potential applications of such probes remain relatively unexplored as of yet. Other H2O2-specific chemical sensors, which are based on different mechanisms, for example dicarbonyl cleavage, are also available (1).
The major advantage of all small chemical probes is the ease of use and applicability relative to genetically encoded sensors. For example, there is no need for a priori genetic manipulations or generation of probe-expressing organisms and small chemical probes can readily be applied to cells, tissue sections, and organs. Disadvantages include a lack of reversibility and a relative lack of targetability. Further, newly developed small chemical probes may still be susceptible to differential cellular uptake and efflux, which is difficult to control for, particularly as most of the sensors lack the possibility for ratiometric imaging.
In contrast, genetically encoded sensors are readily targetable to specific subcellular locations and offer full reversibility, which permits dynamic measurements. Further, genetically encoded sensors also generally permit ratiometric, sensor concentration-independent measurements, which negates concerns surrounding differential probe expression between cells and cell types.
The relative sensitivity of small chemical probes compared with genetically encoded sensors remains unclear. However, given that small chemical probes are generally irreversible and thus tend to accumulate oxidation, one might predict that they are more sensitive toward H2O2 than genetically encoded sensors. Indeed, recently developed small chemical dyes have been shown to be able to detect signaling levels of H2O2. Another major question is the extent to which both small chemical probes and genetically encoded sensors are able to discriminate small, perhaps low nanomolar, changes in H2O2 levels. In our opinion, this remains unclear for any H2O2 sensor.
Novel Insights Made with roGFP2-Based Redox Sensors
Two major advantages of genetically encoded H2O2 sensors compared with small molecule chemical “ROS” dyes are that the sensors are not only far more specific regarding the analyzed redox species but also allow dynamic measurements to be made in defined subcellular localizations. Since its development, roGFP2-Orp1 has been shown to be functional in a range of cell types and different subcellular compartments (9, 29, 30, 52, 54, 58, 68, 71, 72, 80, 81, 102, 114, 120, 127, 128, 136). The majority of work to date with roGFP2-based H2O2 sensors has seen them employed in much the same way as small chemical ROS dyes, that is, to resolve changes in H2O2 in a range of different experimental settings (although with the possibility to resolve changes with greater spatiotemporal resolution and specificity). Although roGFP2-Orp1 permits real-time measurements of changes in intracellular H2O2 levels, with a sensitivity that is apparently very similar to the HyPer probes (94), the full potential of genetically encoded H2O2 probes remains, in our opinion, far from being fully exploited (see Outlook section). Nonetheless, some interesting observations and technological advances have been made.
Whole organism imaging
One of the most exciting applications of roGFP2-based H2O2 probes to date has been the generation of transgenic mice and flies (Drosophila melanogaster) ubiquitously expressing roGFP2-Orp1. In the case of Drosophila, this enabled imaging of intact, living larvae (4). Further, in combination with specially developed dissecting protocols that “trap” the probe redox state and fix the tissue, imaging of dissected mature Drosophila imago organs was possible. This enabled the impact of diet, aging, pharmacological agents, and gene silencing on H2O2 levels throughout the organism to be monitored (4). Interestingly, subcellular compartment and tissue-specific changes in H2O2 levels were observed in response to several different treatments. For example, an age-related increase in H2O2 levels was observed most prominently in midgut enterocytes but to a lesser extent or not at all in other cell/tissue types. Different probe responses were also observed between male and female flies. Further, the effects of silencing the peroxiredoxin Jafrac-1 led to an increase in H2O2 levels in some tissues but not in others. Such observations are supportive of previous studies that, for example, report an increase in H2O2 levels with aging but suggest that the changes are much more nuanced and fine-grained than previously imagined (28, 40, 77, 88, 111). The causal relationships between H2O2 levels and aging, or indeed most (patho)-physiological responses, remain unclear and need to be further studied.
Recently, transgenic mice expressing the roGFP2-Orp1 sensors in different cellular compartments have been generated. As with Drosophila, probe measurements in mice are impeded by optical inaccessibility. Consequently, dynamic, real-time measurements are restricted to the surface of the skin or to tissues that can be surgically exposed (51, 52). To partially overcome these limitations, a chemical processing method for thin tissue sections was developed, similar to the procedure employed with Drosophila. This redox histology procedure “traps” the redox state of the H2O2 biosensors without affecting sensor fluorescence. The methodology permits the steady-state measurement of redox probes in different cell types within organs and tissues, providing an important approach for studying pathological and physiological H2O2 changes.
Applying the redox histology technique, it has been shown that during mouse embryonic development, cells with increased H2O2 levels are found in the embryonic liver, an event that coincides with stem cell differentiation and/or hepatocyte expansion. Both events have been previously implicated to be accompanied by redox changes (8, 101, 136). When adult hematopoietic stem cells were isolated from mice expressing roGFP2-Orp1 in the mitochondrial matrix, quiescent cells were found to have relatively low matrix H2O2, which increased as cells progressed to a fully differentiated form (17). The causal relationship between H2O2 and stem cell differentiation remains unclear.
Despite its limitations, redox histology using genetically encoded H2O2 sensors might now be applied to study a wide range of mouse model pathologies, for example, diabetes, cardiovascular and neurodevelopmental diseases, as well as cancer models for which H2O2 has been implicated in disease onset and progression.
Monitoring “basal” H2O2 levels
The recent development of peroxiredoxin-based H2O2 sensors now permits the measurement of “basal” cellular H2O2 levels (94). In other words, these probes are sensitive enough to monitor changes in H2O2 levels in unstressed, unstimulated cells. Quantitative insight into the relative contribution of the various possible sources of cellular H2O2 to total cellular H2O2 production is still lacking. However, multiple flavoenzymes as well as respiratory chain complexes likely all play a role. It may now be possible to address these questions in the future with peroxiredoxin-based probes. Further, it was observed that both cytosolic and mitochondrial matrix H2O2 levels respond strongly to changes in oxygen availability. Decreased oxygen availability leads to lower H2O2 and vice versa. It will, thus, be particularly interesting to apply these sensors to rigorously scrutinize and test long-standing hypotheses in redox biology, including those of hypoxia-induced ROS production and ROS increase after ischemia-reperfusion (10, 11, 23, 25, 26, 83, 96).
Outlook
Possible future applications of roGFP2-based H2O2 probes
There are several applications for which we consider that roGFP2-based sensors hold considerable potential to make novel insights.
Monitoring rhythmic redox changes
Recent reports point toward the existence of redox cycles associated with circadian and ultradian clocks and the cell division cycle (41, 97, 98, 131). In the case of circadian time-keeping, it has been proposed that an independent metabolic/redox clock may be fundamental to all life, evolutionarily predating clocks based on transcription translation feedback loops. These clocks were identified on the basis of periodic changes in thiol peroxidase hyperoxidation, although it remains unclear as to whether thiol peroxidases are players in the clock mechanism or merely a convenient readout. The capacity of genetically encoded H2O2 sensors to make long-term, non-disruptive, real-time, fully dynamic measurements in living cells renders them ideal to probe for cyclical H2O2 changes in a variety of settings, including circadian and ultradian cycles.
Scrutinizing competing models of H2O2-induced protein thiol oxidation
When characterizing the roGFP2-Tsa2ΔCR and roGFP2-PRDX2ΔCR probes, we observed not only that the H2O2-dependent oxidation of roGFP2 was, as expected, dependent on the presence of the fused peroixredoxin but also that peroxiredoxin hyperoxidation resulted in less efficient roGFP2 oxidation despite the higher H2O2 concentrations being used relative to conditions where peroxiredoxin hyperoxidation was not observed. Further, the effect was more pronounced in the roGFP2-PRDX2ΔCR probe, which is consistent with the fact that PRDX2 is more sensitive to hyperoxidation as compared with Tsa2. Only at very high, non-physiological concentrations of H2O2 (5–25 mM) was oxidation of an unfused roGFP2 observed. At these concentrations, it is known that glutathione disulfide (GSSG) production drives roGFP2 oxidation, and some direct oxidation of roGFP2 thiols may also be relevant. Although it would be imprudent to generalize observations made with a non-physiological protein to all cellular H2O2-reactive thiols, these observations, nonetheless, support the concept that peroxiredoxins are required for H2O2-dependent thiol oxidation rather than prevent it. Future experiments with roGFP2-based sensors, perhaps fused to other thiol peroxidases, may help us to shed more light on the conditions required for endogenous protein thiol oxidation.
Understanding in vivo enzymatic functions
A completely unexplored avenue of research is whether roGFP2-thiol peroxidase fusions can be used to investigate the applicability of in vitro measurements of thiol peroxidase enzyme kinetic parameters to the complex cellular environment. The vast majority of studies on kinetic parameters and mechanisms of thiol peroxidases have been performed in vitro, using recombinantly expressed proteins. Consequently, most mechanistic insights into thiol peroxidase function are obtained by using proteins in an environment that is far from the one in which they normally function. Further, it is now appreciated that thiol peroxidases may be subject to multiple forms of post-translational modifications, for example, limited proteolysis (75) or phosphorylation (24, 66, 82, 143), that can serve to modulate or regulate their activity and function. These considerations raise the obvious questions as to what extent mechanistic insights and in vitro-determined enzyme kinetic parameters are relevant and applicable to thiol peroxidase activity and function in vivo.
There are several parameters of the roGFP2 oxidation that might be correlated with the enzyme kinetic parameters of thiol peroxidases and that might, therefore, be utilized to gain functional and mechanistic insights in vivo. Such roGFP2 parameters include (i) the maximum change in roGFP2 oxidation after the addition of a bolus of external peroxide, (ii) the sensitivity of roGFP2 oxidation to external peroxide, that is, the lowest detectable peroxide concentration, and (iii) the robustness of the roGFP2 response at high peroxide concentrations, which probably reflects the hyperoxidation susceptibility of the peroxidase. These parameters might be assessed for different thiol peroxidase-roGFP2 fusion constructs and peroxides. For example, as discussed earlier in the section Decamerization of typical 2-Cys peroxiredoxin-based roGFP2 probes, roGFP2 fusions can be used to determine the formation of typical 2-Cys peroxiredoxin hetero-decamers. Truncated fusion constructs might mimic the effects of limited proteolysis on enzyme catalysis and redox signaling. Moreover, glutamate or alanine mutants of roGFP2-thiol peroxidase fusions could mimic constitutively phosphorylated or dephosphorylated peroxidase species to characterize their relevance and properties. Hence, by monitoring roGFP2 oxidation in response to exogenous peroxides, it may be possible to gain qualitative and semi-quantitative insights into the impact of different post-translation modifications in vivo. Likewise, mutations that lead to different k cat or K m values in vitro could easily be tested in enzyme structure-function analyses in vivo.
Interrogating redox species cross-talk
A particularly interesting area for future study will be to apply genetically encoded H2O2 probes in combination with sensors for other redox couples, for example, the E GSH or NAD+/NADH (57, 63, 146). There are many questions that might be thus addressed, including the extent of cross-talk and interaction between the different redox couples and the identification of the most important enzymatic mediators. Importantly, these questions can be studied with high spatiotemporal resolution, meaning that it is possible to discriminate, for example, between changes in different redox species that occur concomitantly and situations where the change in one redox species is delayed relative to the other, even if this delay is only a few seconds. In addition, it is possible to interrogate the cross-talk between different subcellular compartments.
As an example of one potential application, it was recently shown in vitro that GSH/Grx can reduce the typical 2-Cys peroxiredoxin, PRDX2 (104). By monitoring the response of H2O2 and E GSH sensors to H2O2 in cells deleted for or overexpressing typical 2-Cys peroxiredoxins, one can determine the impact of changes in H2O2 levels on E GSH, which, due to the redox poise of the cytosolic and mitochondrial matrix GSH pools, is exquisitely sensitive to changes in GSSG levels. Consequently, it should be possible to monitor the extent of GSH/Grx-mediated thiol peroxidase reduction in vivo and to identify the most important thiol peroxidase mediators of this reaction.
Of course, it is not possible to simultaneously express different sensors with a significantly spectral overlap in the same cells as there is no way to disentangle the responses of each probe. Nonetheless, there are two possible ways to make such multi-parametric measurements. The first is simply to compare the responses of cell populations expressing different sensors with the same treatment. In this way, information can be gathered about the relative response of different redox species and, when combined with genetic manipulations, about the importance of different redox enzymes for mediating the interplay between these redox species.
The second possibility relies on the development of probes that fluoresce in different parts of the spectra, for example, red or infra-red probes. Red fluorescent H2O2 probes, for example HyPer Red (42), have recently become available and thus now permit simultaneous monitoring of H2O2 with other redox species such as the E GSH. Another possibility would be to create fusion probes that are based on a redox-sensitive red fluorescent protein. A redox-sensitive red fluorescent protein has recently been developed (43) and has already been fused to thioredoxin to create a reporter for the thioredoxin redox state (44). It would be intriguing to see whether these proteins can also be used to develop red E GSH sensors and peroxiredoxin-based ultra-sensitive H2O2 sensors.
Investigating redox microdomains and redox gradients
Another little explored area of cellular redox biology is the question of the existence of redox microdomains or redox gradients within cells, which may be highly relevant in explaining redox signaling specificity. As an example, it was observed that changes in mitochondrial respiratory chain function strongly affect levels of H2O2 in the mitochondrial matrix, whereas no significant change is seen in the cytosol (94). Nonetheless, this observation does not necessarily imply that no H2O2 escapes from mitochondria to the cytosol. On the contrary, there are several reasons to assume that it does (94). However, it is necessary to consider that genetically encoded H2O2 sensors are presumably freely soluble in the cytosol. In this respect, they report a spatially averaged cytosolic H2O2 level. Likely under many situations, there is a considerable flux of H2O2 emitted from mitochondria and subsequently passing into the cytosol. However, the very high abundance and H2O2 reactivity of the endogenous cytosolic thiol peroxidases would likely strongly limit the diffusion of this H2O2 through the cytosol, thereby establishing a steep concentration gradient of H2O2 surrounding mitochondria. This hypothesis is supported by recent in silico modeling studies (130). The roGFP2-Tsa2ΔCR sensor is probably sensitive enough to respond to locally increased H2O2 levels, whereas the reversible and dynamic nature of the probe combined with the rapid mixing of the entire cytosolic probe pool likely ensures that the spatially averaged probe response does not increase in response to localized H2O2 increases. It will thus be extremely interesting in the future to explore ways of more specifically localizing roGFP2-based probes to defined cellular subcompartments or microdomains. It may even be possible, for example, to anchor genetically encoded H2O2 sensors to the cytoskeleton, thereby more rigidly fixing their spatial localization within the cell (at least relative to a freely soluble sensor). In this way, it might be possible to monitor redox-signaling cascades with high spatio-temporality, such as the growth factor-dependent regulation of membrane-associated mammalian peroxiredoxin I (143) or the mitosis-dependent centrosome-associated peroxiredoxin I (82). Consequently, using fluorescent microscopy, it should be possible to observe whether there are differences in probe oxidation and thus in H2O2 levels in different locations within the cell. One might even try to monitor in real time the extent to which an externally applied H2O2 bolus penetrates into the cell and to what degree this is influenced by the presence or absence of different H2O2-scavenging enzymes, including thiol peroxidases and catalases.
Conclusions
RoGFP2-based H2O2 probes, as well as the HyPer series of H2O2 sensors afford extensive new possibilities for probing cellular and organismal H2O2 dynamics. Further, we anticipate that they will also allow new insights into the cross-talk and interplay of the different cellular redox couples and the identification of the most important enzymatic mediators. Finally, we predict that roGFP2-based sensors hold considerable potential for probing thiol peroxidase mechanisms and post-translational regulation in the cellular context. Although some work has started in some of these areas, the full potential of roGFP2-based H2O2 sensors remains far from being fully exploited.
Footnotes
Acknowledgments
Research in the authors' laboratories is funded by the German Research Council (DFG, RI2150/2-1 and 2-2, SFB1218 TP B02) to J.R. and the DFG SPP1710 grants DE1431/8-1 and DE1431/8-2 to M.D. B.M. was funded by the DFG SPP1710 grant MO 2774/2-1, as well as the Forschungsinitiative Rheinland-Pfalz BioComp and the University of Kaiserslautern Nachwuchsring.