
Abstract
Significance:
NAD+ is an essential redox cofactor in cellular metabolism and has emerged as an important regulator of a wide spectrum of disease conditions, most notably, cancers. As such, various strategies targeting NAD+ synthesis in cancers are in clinical trials.
Recent Advances:
Being a substrate required for the activity of various enzyme families, especially sirtuins and poly(adenosine diphosphate [ADP]-ribose) polymerases, NAD+-mediated signaling plays an important role in gene expression, calcium release, cell cycle progression, DNA repair, and cell proliferation. Many strategies exploring the potential of interfering with NAD+ metabolism to sensitize cancer cells to achieve anticancer benefits are highly promising, and are being pursued.
Critical Issues:
With the multifaceted roles of NAD+ in cancer, it is important to understand how cellular processes are reliant on NAD+. This review summarizes how NAD+ metabolism regulates various pathophysiological processes in cancer, and how this knowledge can be exploited to devise effective anticancer therapies in clinical settings.
Future Directions:
In line with the redundant pathways that facilitate NAD+ metabolism, further studies should comprehensively understand the roles of the various NAD+-synthesizing as well as NAD+-utilizing biomolecules to understand its true potential in cancer treatment.
Introduction
In the early 20th century, Arthur Harden and coworkers described NAD+ as a cofactor in fermentation; later, a Nobel laureate, Otto Warburg, isolated NAD(P)+ and discovered its key role in hydrogen transfer in several key reactions (61, 184). NAD+ is an essential redox regulating moiety, which acts as a key element for a wide range of signaling pathways (10, 13, 71). For example, NAD+ functions as a substrate in deacetylation reactions carried out by the deacetylase enzymes such as sirtuins (SIRTs) and poly(adenosine diphosphate [ADP]-ribose) polymerases (PARPs). Therefore, NAD+ has the ability to affect the stability and activity of various proteins by modifying SIRT and PARP function and has a special role in various pathological conditions (10, 13, 18, 71). Due to the central role of NAD+ in important cell signaling pathways, NAD+-dependent processes have emerged as highly promising targets for therapeutic applications to treat diseases such as cancer.
Cancer cells undergo uncontrolled proliferation through subversion of characteristic physiological rules of growth regulation (58). This uncontrolled growth of cancer cells requires an increased energy demand (58). To meet the increased energy demands, cancer cells strive to acquire additional nutrient by using multiple mechanisms (74). The most well-characterized adaption is the Warburg effect, which involves a reliance on aerobic glycolysis for energy and biosynthesis of macromolecules. Importantly, aerobic glycolysis requires NAD+ for function of glyceraldehyde 3-phosphate dehydrogenase, and lactate dehydrogenase (LDH) consumes NADH to regenerate NAD+ for the continuation of glycolysis. Recently, inhibitors of NAD+ biosynthesis have been developed. Specifically, an inhibitor (FK-866) of an NAD+ synthesis enzyme, nicotinamide phosphoribosyltransferase (NAMPT), has potent anticancer activity in cell culture (70, 180) and, as such, is in early clinical trials as an anticancer agent (70).
In clinical trials, FK866 showed thrombocytopenia as the dose limiting toxicity, and steady-state concentrations increased with the dose of FK866. However, FK866 had low bioavailability and no significant tumor regression was observed (70). Later, clinical trials of another NAMPT inhibitor, CHS 828, found similar results regarding toxicity and efficacy (180). Novel NAMPT inhibitors such as MS50 are still being developed (187). KPT-9274 is an NAMPT inhibitor undergoing clinical trial and was shown to effectively inhibit tumor growth in vivo, with no toxicity (1).
While inhibiting the synthesis of NAD+ appears to be an attractive target for anticancer therapy, NAD+ may have a beneficial post-therapeutic role by enhancing the function of normal cells. Dietary supplementation of NAD+ precursors has been shown to increase NAD+ levels, enhance normal cell functions, and improve life span in mice (10, 45). Therefore, restoration of NAD+ during post-treatment recovery may promote tissue regeneration and healing of healthy cells following surgery, radiotherapy, or chemotherapy. Here, we give an overview of the current knowledge of the role of NAD+ in cancer.
NAD+ Biosynthesis
Biosynthesis of NAD+ is accomplished through multiple routes in mammals. These routes of NAD+ synthesis can be classified as either de novo or salvage pathways (Fig. 1). De novo synthesis of NAD+ starts with the essential amino acid L-tryptophan and proceeds through the kynurenine pathway (Fig. 1). In fact, 90% of the cell's free tryptophan is metabolized through the kynurenine pathway, which leads to production of NAD+ (19). In the first and rate-limiting step of the pathway, tryptophan is converted to N-formylkynurenine by the action of indoleamine 2,3-dioxygenase (IDO) or tryptophan dioxygenase (TDO). IDO is the main enzyme used in most tissues, with notably high activity in the lung, spleen, and small intestine, while hepatocytes in the liver rely mainly on TDO (94, 188). After a series of subsequent enzymatic conversions, α-amino-β-carboxymuconate-ɛ-semialdehyde (ACMS) is formed at a branchpoint in the pathway. At this point, ACMS may be diverted from NAD+ synthesis by ACMS decarboxylase (ACMSD) where it will be completely oxidized to carbon dioxide and water via the glutarate pathway or enter the tricarboxylic acid cycle (TCA) after being converted to acetyl-CoA following several enzymatic reactions (71, 151). Alternatively, ACMS can be directed toward the NAD+ synthesis pathway, where it spontaneously undergoes cyclization to form quinolinic acid (QA) (Fig. 1). Therefore, the formation of QA is inversely correlated with ACMSD activity, which exerts a major regulatory mechanism on the de novo pathway (76). Once QA has been committed to the NAD+ synthesis pathway, it is converted by QA phosphoribosyltransferase (QAPRT) to nicotinic acid mononucleotide (NAMN) (Fig. 1). QAPRT is the second rate-limiting enzyme of the de novo pathway and is most highly expressed in the liver and kidney (150). NAMN is then adenylated to nicotinic acid adenine dinucleotide (NAAD) by one of the three nicotinamide mononucleotide adenylyl transferase isoforms (NMNAT1-3) (Fig. 1). Finally, the last step in the de novo pathway involves amidation of NAAD to NAD+ by NAD+ synthetase (NADS) (Fig. 1), which is mainly expressed in the small intestine, liver, kidney, and testis (60).

Even though most cells can synthesize NAD+ de novo from tryptophan, the main source of NAD+ is supplied by salvage pathways that synthesize NAD+ from dietary vitamin B3, or niacin, as precursors. The NAD+ salvage pathway has a wide range of starting metabolites, including nicotinamide (NAM), nicotinic acid (NA), and nicotinamide riboside (NR), all of which are converted to either nicotinamide mononucleotide (NMN) or NAMN as intermediates in the NAD+ pathway (16) (Fig. 1). Fittingly, NAM is also the by-product of NAD+-consuming reactions and is therefore more readily available to be used as an NAD+ precursor. The recycling of NAM to synthesize NAD+ makes the salvage pathway more direct and more economical than other pathways. In the classical salvage pathway, NAM is catalyzed by the rate-limiting enzyme NAMPT to NMN, which is then adenylated to NAD+ by NMNATs (Fig. 1). NAMPT is found in most tissues and is commonly elevated in cancer (14, 181). NMNAT1 expression is highest in the heart, kidney, liver, pancreas, and skeletal muscle (26). NMNAT2 is highly expressed in the brain and pancreas (33, 144). High levels of NMNAT3 are found in erythrocytes, lung, spleen, skeletal muscle, and heart (26, 196). Because NAM is the predominant precursor in NAD+ synthesis (150), the role of NAMPT in cell physiology has been extensively studied and has led to the development of specific and potent NAMPT inhibitors. The application of NAMPT inhibitors in cancer therapy showed promising tumor regression effects in preclinical trials; however, disappointing clinical trials showed that treatment with these inhibitors only stabilized the disease for patients and did not result in any objective tumor regression (73, 147, 180). Thus, to target NAD+ availability as a strategy for cancer therapy, researchers are beginning to expand their investigations on NAD+ synthesis along with the pathways that rely on NAD+.
In the Preiss–Handler salvage pathway, the precursor NA is first converted to NAMN by nicotinic acid phosphoribosyltransferase (NAPRT), which could then converge with the de novo pathway, where NAMN is converted to NAAD and then to NAD+ (142) (Fig. 1). In tissues with high NAPRT activity, addition of exogenous NA has been shown to effectively elevate NAD+ levels, whereas supplementation of NAM did not affect NAD+ levels (59, 148). Therefore, in tissues such as small intestines, liver, kidney, and heart (36, 79, 163, 185), NAPRT is likely to play an important role in cellular NAD+ levels and may be preferred over NAD+ synthesis via the classical salvage pathway. Indeed, cytotoxicity of NAMPT inhibitors correlated with expression levels of NAPRT (138).
Synthesis of NAD+ from NR begins with the phosphorylation of NR to NMN by the rate-limiting NR kinases (NRK1 and NRK2) (Fig. 1), then metabolized via the classical salvage pathway as described above (16, 146). Expression of NRK1 is highest in the pancreas, thyroid, and white blood cells (33), while expression of NRK2 seems to be mainly found in the heart, brain, and muscle (19).
Another source of precursors for NAD+ synthesis is breakdown of NAD+ extracellularly. For example, exogenous NAM can be cleaved by extracellular NAMPT to NMN (77), and phosphorylated intermediates such as mononucleotides (NMN and NAMN) and dinucleotides (NAD+ and NAAD) can be processed to nucleosides before uptake by the cell (131).
NAD+ can be phosphorylated to NADP+ through the action of NAD+ kinase (NADK), which was originally identified to be localized in the cytosol although recent reports have also identified a mitochondrially localized NADK (170, 193). NADP+ is reduced to NADPH through various redox reactions in metabolic pathways, mainly by the action of glucose-6-phosphate dehydrogenase and malate dehydrogenase enzymes (170). NADPH can also be synthesized from NADH by the action of nicotinamide nucleotide transhydrogenase in the mitochondria (69).
Compartmentalization of NAD+ Synthesis and Metabolism
NAD+ synthesis is, in part, regulated through compartmentalization within cells. The resident enzymes of the nucleus and cytosol differ in that NMNAT1 is mainly found in the nucleus, while NMNAT2 is cytoplasmic (12) (Fig. 2). However, the nucleus and cytosol can be considered one entity with regard to NAD+ compartmentalization since the rate-limiting entities in NAD+ biosynthesis are believed to be freely exchangeable between the nuclear membranes, and NAMPT and NAPRT can be found in both regions (Fig. 2). Despite the fact that NMNAT1 is the most efficient of the NMNAT isoforms, NAD+ levels in the nucleus are lower than other compartments; although whether this is due to increased flux has not yet been examined (194). Meanwhile, NMNAT3 is the sole NAD+ synthesis enzyme that is mainly localized in the mitochondria (131) (Fig. 2).
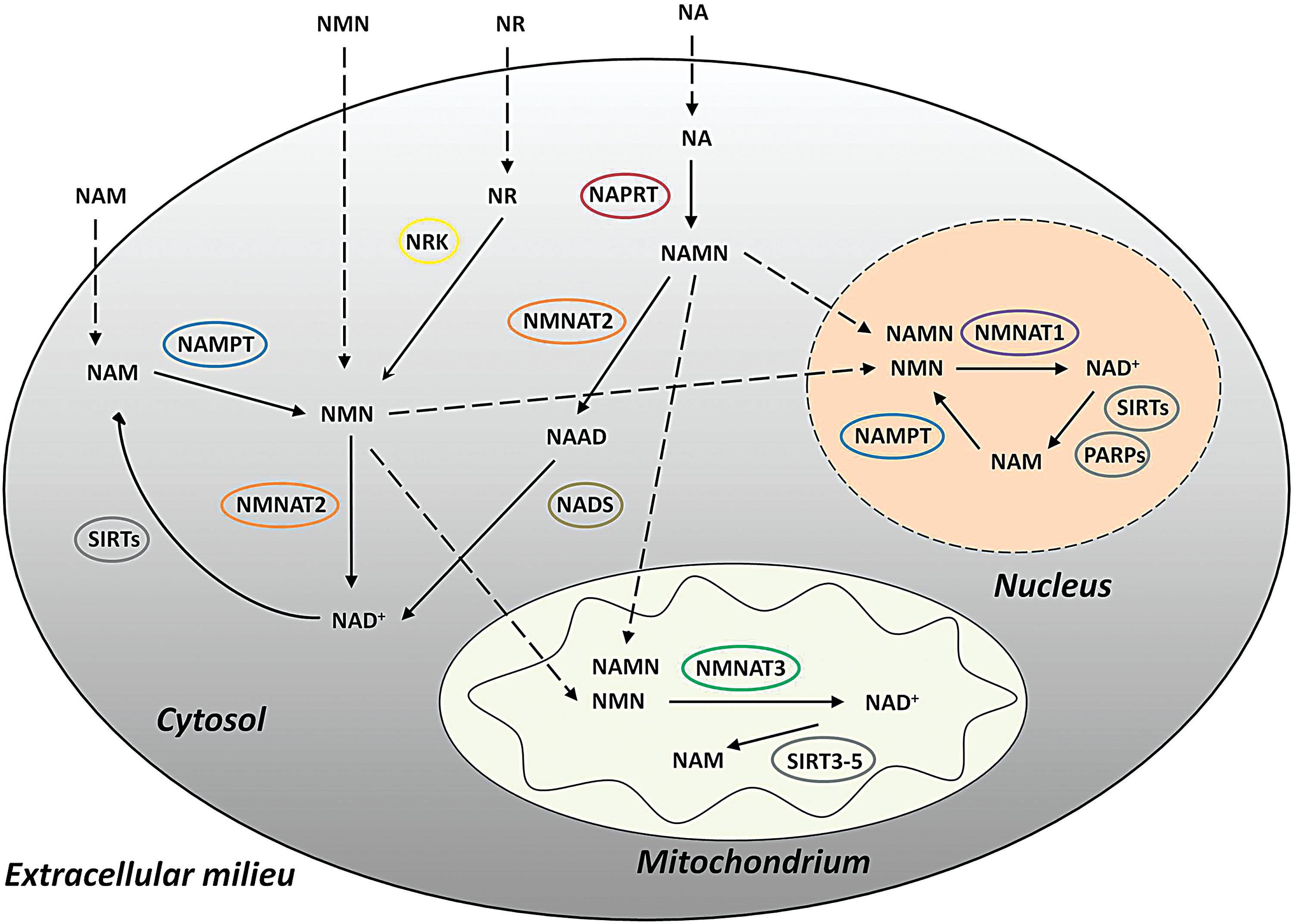
The mitochondrial pool of NAD+ contains up to 70% of the total cellular NAD+ since it is required not only for metabolic processes as a redox coenzyme but also as a substrate for the mitochondrial NAD+-dependent SIRTs that regulate metabolic enzymes (4) (Fig. 2). This pool of NAD+ in the mitochondria is crucial to sustain cell survival on the depletion of cytosolic NAD+ (189). Unlike the nuclear membrane, which is not a barrier for the diffusion of NAD+, the mitochondrial inner membrane is impermeable to polar molecules such as NAD+ (9). Although the malate/aspartate and glycerophosphate shuttles are well known to transport the equivalent of NADH across the inner mitochondrial membrane, there are no known NAD+ transporters in the mitochondria (171, 189). Since there is no means to directly import NAD+ into the mitochondrial matrix for use in metabolic processes, the mitochondrial NAD+ pool is isolated from the rest of the cellular NAD+ pool and mitochondria must synthesize their own NAD+ likely through NMNAT3 activity (189) (Fig. 2). Interestingly, the catalytic activity of NMNAT3 is the least efficient of the three isoforms (165).
With the absence of upstream NAD+ enzymes, the mitochondria are limited, in which precursors can be used to synthesize NAD+. Yet, exogenous precursors of all known NAD+ metabolism intermediates are capable of increasing mitochondrial NAD levels, indicating that mitochondria must import NAD+ intermediates for conversion to NAD+ by NMNAT3 (140). The precursor NMN is present inside mitochondria at higher levels than in the cytoplasm (48) and can cross the mitochondrial membrane to be used for NAD+ synthesis (Fig. 2). Thus, NAD+ precursors and intermediates are first converted to NMN in the cytoplasm and then transported into the mitochondria to be converted to NAD+ (9, 131). The mechanism of NMN transport into mitochondria remains unknown and is a major area in the field that should be focused on to gain a comprehensive understanding of mitochondrial NAD+ synthesis.
The points where different NAD+ synthesis pathways merge make interrelationships between pathways possible, and this is significant for mitochondrial NAD+ synthesis as various precursors need to be converted to the NMN intermediate to be shuttled into the mitochondria. While mitochondria lack the NRK needed to metabolize NR, NR is still the preferred NAD+ precursor for mitochondrial NAD+ synthesis, where it only needs to be converted in the cytoplasm to NMN before uptake into mitochondria (26, 131) (Fig. 2).
Mitochondria also lack NAPRT so they cannot synthesize NAD+ from NA via the Preiss–Handler pathway (Fig. 2). Yet, it is possible to increase mitochondrial NAD+ levels from exogenous NA. The cytosolic NAD+ must be first degraded to NMN before being imported into the mitochondria, where NMNAT3 converts it back to NAD+ (Fig. 2). Despite the roundabout way of this mitochondrial NAD+ synthesis route, the reaction still occurs at a surprisingly fast rate due to the efficiency of the rate-limiting enzyme NAPRT (131).
Cleavage of NAD+ to NMN to be used for mitochondria can occur via various processes. Most commonly, NAD+-consuming reactions in the cytoplasm will release NAM, which can be salvaged to NMN by NAMPT (Fig. 2). To some extent, NMNATs can catalyze the reverse reaction from NAD+ to NMN (12, 98).
Role of NAD+ in Cancer-Related Signaling Pathways
NAD+, CD38, and calcium signaling: role in cell motility, proliferation, and immune system regulation
Not only is NAD+ a necessary cofactor for various metabolic reactions but is also a major regulator of cellular signal transduction pathways. NADP+ is a precursor for the second messengers cyclic ADP-ribose (cADPR) and NAAD phosphate (NAADP), which are potent regulators of intracellular calcium (Ca2+) mobilization (52, 100, 101) (Fig. 3). Ca2+ signaling, one of the most important signal transduction mechanisms, is tightly controlled by coordinated mobilization mechanisms of Ca2+ release from Ca2+ stores (35). cADPR is derived directly from NAD+, whereas NAADP is a derivate of NADP+ (100) (Fig. 3). Both molecules interact with different Ca2+ channels (116), cADPR can trigger Ca2+-induced Ca2+ release by interacting with the ryanodine receptor, whereas NAADP triggers Ca2+ release from lysosomal stores via two-pore channels (106, 136) (Fig. 3). Moreover, CD38 can also produce ADPR from NAD+, which binds to the plasma membrane calcium channel transient receptor potential cation channel, subfamily M, member 2 (TRPM2) and regulates extracellular Ca2+ influx (137) (Fig. 3). Ca2+ signaling is fundamental for regulating various physiological processes, including gene expression, cell cycle control, cell motility, energy metabolism, inflammation, cell survival, and cell death (35) (Fig. 3). Hence, it is not surprising that deregulated Ca2+ signaling has been associated with cancer initiation, progression, persistence, and metastasis (37). Therefore, NADP+-mediated regulation of Ca2+ mobilization provides an important mechanism of NAD+ regulation of cancer.
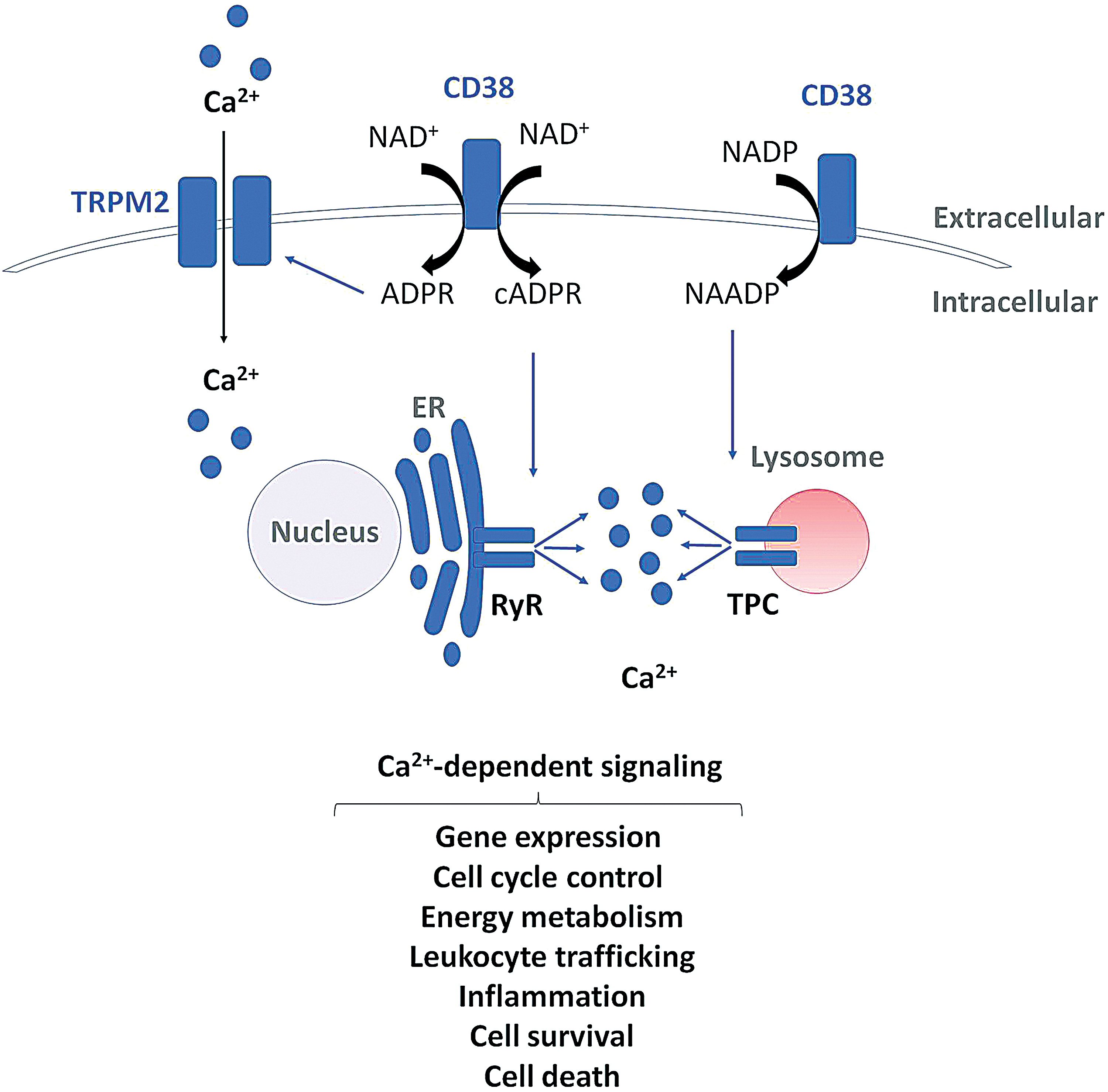
CD38 and its homologue CD157 are bifunctional ectoenzymes that are expressed on the surface of lymphoid and myeloid cells as well as other tissues (115). CD38-dependent Ca2+ mobilization is also important for leukocyte trafficking and chemotaxis (135, 154) (Fig. 3). CD38 is highly expressed in many hematological malignancies, including myelomas and leukemias, and is associated with poor prognosis (115, 149). Because of this, neutralizing anti-CD38 antibodies are currently undergoing clinical trials and show broad-spectrum antitumor activity in multiple myeloma (MM) patients (117). Anti-CD38 antibodies have been demonstrated to induce cell death of MM cells through several mechanisms, including complement-dependent cytotoxicity, antibody-dependent cell-mediated cytotoxicity, antibody-dependent cellular phagocytosis, and apoptosis (40, 134). Interestingly, CD38 has also been identified as a potential therapeutic target in other types of cancers such as cervical cancer, where enhanced CD38 expression has been shown to promote cancer cell proliferation and inhibit apoptosis (108). On the contrary, in prostate and pancreatic cancer, low CD38 expression has been associated with poor prognosis and decreased sensitivity to NAMPT inhibition (34, 110). It has also been demonstrated that specific subpopulations of stem-like progenitor cancer cells in hematopoietic malignancies, termed hematopoietic cancer stem cells (HCSCs), are negative for CD38 expression (27, 88). This revelation is of clinical importance since only small numbers of cancer stem cells (CSCs) are required to reinitiate tumor formation. If these HCSC populations do not express CD38, then additional therapies may be required to eliminate HCSC populations and prevent cancer relapse. Possible combination therapies could include treatment with all-trans retinoic acid to induce differentiation and upregulation of CD38 in CD38- HCSC populations before treatment with CD38-targeted therapies (130, 191). Interestingly, the natural flavonoid apigenin is an inhibitor of CD38 (44). Apigenin is commonly found in many fruits and vegetables and has been demonstrated to exhibit anticancer effects by activating cell cycle arrest and apoptosis via the p53 pathway (91, 173, 198). Recently, CD38 has also been demonstrated to play a major role in tumor immune evasion. High expression of CD38 has been detected on non-neoplastic myeloid-derived suppressor cells (86) and regulatory T and B cells (Tregs and Bregs) (46, 92), which are associated with suppressed immune response and increased cancer progression. Treatment with the anti-CD38 antibody daratumumab has been shown to strongly increase antitumor T cell immune responses by depleting CD38+ immunosuppressive cells (92).
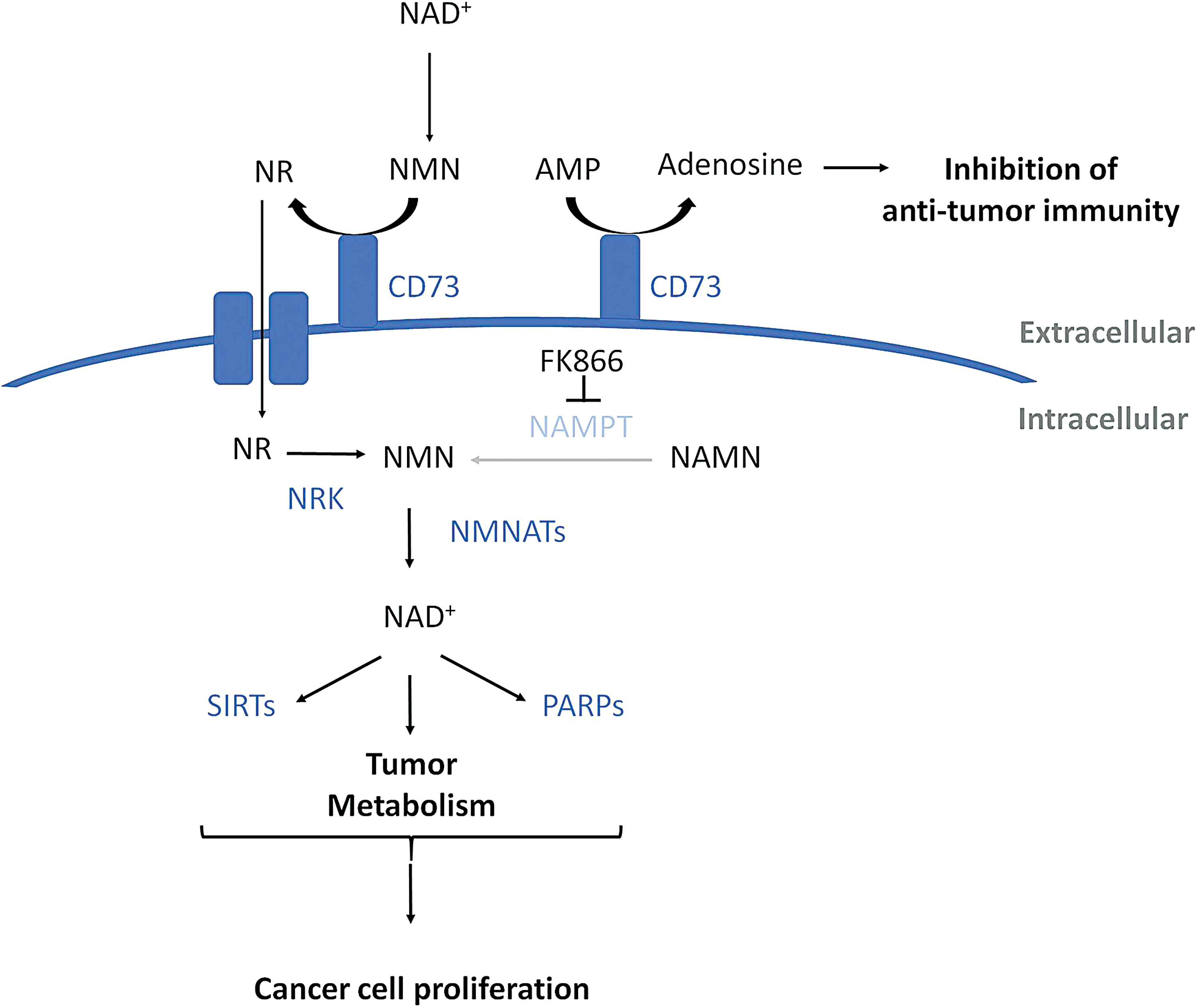
CD73 is another bifunctional ectozyme that can dephosphorylate extracellular NMN into NR (51) (Fig. 4). This NR can then enter the cell where it is converted back to NAD+ by the action of the enzymes in the NAD+ salvage pathway (Fig. 4). Because NR is converted to NMN by the action of NRK, bypassing NAMPT, CD73-mediated production of NR has been shown to sustain NAD+ biosynthesis in cancer cells treated with the NAMPT inhibitor FK866 (53) (Fig. 4). CD73 also catalyzes the conversion of extracellular adenosine monophosphate (AMP) to adenosine (Fig. 4). Extracellular adenosine produced by CD73 has been shown to inhibit T cell activation and suppress antitumor immune responses by binding to the A2A adenosine receptor on the surface of T cells (41) (Fig. 4).
The involvement of the kynurenine pathway in cancer has been well documented, especially in brain cancers (2, 3, 64). In glioblastoma, IDO and TDO overexpression or stimulation generates toxic levels of downstream metabolites in the kynurenine pathway, QA, and 3-hydroxyanthranilic acid, which suppress T cell antitumor immunity function (166). In addition, kynurenine itself can modulate DNA polymerase activity, which contributes to genomic instability and cancer progression in gliomas (21). Thus, kynurenine metabolites have been the interest of anticancer drug developments. The novel IDO inhibitor epacadostat is undergoing clinical trials for cancer immunotherapy via dendritic cell activation and suppression of regulatory T cell proliferation (85). Combination therapies involving IDO inhibitors with IFNα, CD3/CD19, or paclitaxel have also been explored in kidney cancer, B cell lymphoma, and murine melanoma model, respectively (85, 121, 197). Bortezomib, a proteasomal inhibitor, has been shown to also downregulate IDO expression as a mechanism of its antitumor effects in nasopharyngeal carcinoma (83). These studies highlight that intermediates in the NAD+ synthesis pathways also contribute to cancer progression and must be considered during therapy.
NADP+ regulation of redox pathways
NAPDH, the reduced form of NADP+ , acts as an important cofactor for various enzymes in pathways, which promote cancer cell growth and the biosynthesis of macromolecules such as nucleotides, proteins, and lipids (97). As such, high levels of NADPH have been linked to cancer progression (170). Head and neck squamous cell carcinoma cell lines have been shown to exhibit upregulated NADPH-producing reactions, which contribute to their resistance against radiation (103). NADPH oxidases also depend on NADPH to produce reactive oxygen species (ROS), and have been shown to stimulate angiogenesis in human epithelial cells, increase melanoma metastasis, and dampen NK cell antitumor activity (7, 177). Furthermore, NADPH is also required for the regeneration of glutathione, the major cellular antioxidant (112). High levels of glutathione are cytoprotective and have been associated with cancer progression due to its capability to protect against oxidative stress-induced apoptosis (172).
Due to the procancer functions of NADPH, the enzymes that synthesize NADPH are being explored as therapeutic targets in cancer (139, 170). 6-amino nicotinamide is the only inhibitor of NADK, which has been tested in clinics thus far. While it demonstrated promising preclinical cancer cytotoxicity, it also displayed dose-limiting neurotoxicity in animal models (65). Due to the neurotoxic side effects, the clinical dose was limited and minimal anticancer activity was observed in patients at the maximum tolerated dose (68, 170).
Regulation of tumor suppressors and oncogenes by NAD+
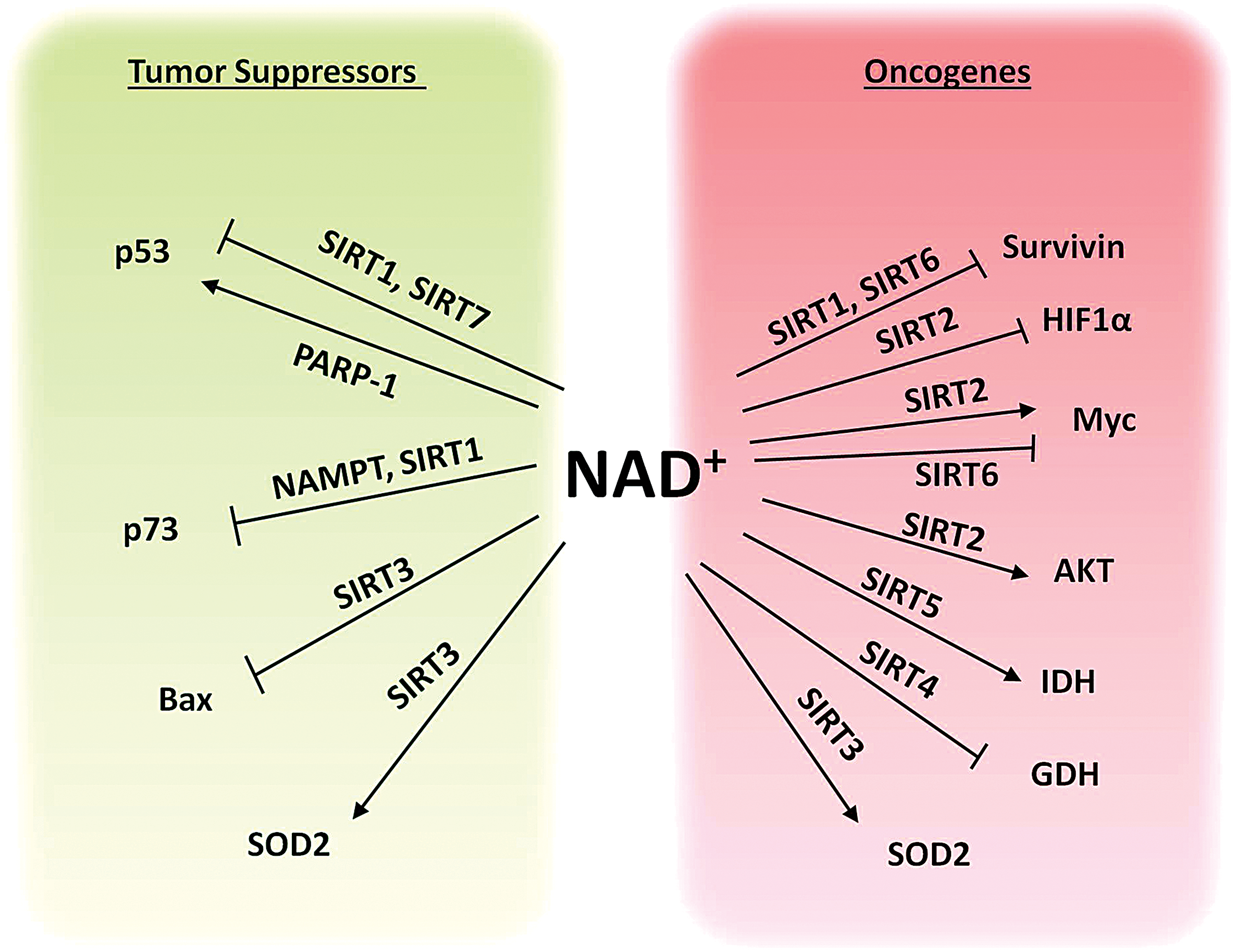
Physiologically, the largest consumers of NAD+ are the PARP family of enzymes. Two members of the PARP family, PARP-1 and PARP-2, have been extensively studied in the context of cancer. PARPs 1 and 2 are important for maintaining genomic integrity and act as DNA repair enzymes (5, 11). PARPs help repair nontoxic single-strand DNA breaks (SSBs) before they can develop into potentially toxic double-strand DNA breaks (DSBs) during S-phase. If these SSBs are converted to DSBs, they must be repaired via homologous recombination (HR) mechanisms. Therefore, it may appear as though PARPs behave as tumor suppressors by consuming NAD+ to prevent aberrant cell metabolism and inhibiting genomic instability to prevent potential oncogenic mutations. However, PARP inhibitors exhibit synthetic lethality in cancer cells with HR deficiencies such as those treated with genotoxic agents or mutations in the tumor suppressor gene, BRCA (23, 38, 80, 107). Inhibition of PARPs in rapidly proliferating cancer cells with insufficient DNA-repair mechanisms leads to the accumulation of DSBs that cannot be repaired through HR and subsequent cell death. More recently, it has been discovered PARP and BRCA work together to protect the degradation of stalled replication forks allowing the cell cycle to proceed (22, 167, 190). It is now proposed that blocking this function of PARP prevents cancer cell replication and is one of the main mechanisms of PARP inhibitors in BRCA-deficient tumors (63). PARP has also been identified as a potential therapeutic target in non-BRCA mutated cancers. For example, cancers that harbor mutations in isocitrate dehydrogenase (IDH) enzymes, an enzyme that when mutated consumes NADP+ and produces the oncometabolite 2-hydroxyglutarate (2HG), have enhanced susceptibility to PARP inhibitors due to decreased NAD+ availability and 2HG-dependent HR defects (113, 168). In line with a connection between DNA repair and NAD+, induction of DNA damage sensitized cancer cells to inhibition of NAMPT (15, 29, 49, 87, 119). PARPs can also regulate the function of transcription factors to modulate gene expression. For example, PARP1 has been shown to regulate p53 function by binding and promoting the poly ADP-ribosylation of p53 and may promote p53 tumor suppressing functions (102, 175) (Fig. 5).
NAD+ is also a required substrate for many important signaling enzymes such as the NAD+-dependent histone deacetylase (HDAC) family of SIRTs. SIRTs are a family of evolutionarily conserved proteins originally classified as NAD+-dependent class III HDACs (18). SIRTs control the physiological activity of their target proteins, which include both histone and nonhistone proteins, by promoting the deacetylation of various lysine residues. In this manner, SIRTs act as major regulators of a diverse range of biological processes (57). SIRTs cleave NAD+ to transfer the acetyl group from their substrate to the ADP-ribose moiety of NAD+ to produce O-acetyl-ADP ribose and a deacetylated substrate (20). Deacetylation increases the availability of lysine residues for ubiquitination and subsequent proteasomal degradation. Thus, SIRTs regulate target substrates through promotion of protein degradation.
SIRTs 1–3 possess robust deacetylase activity, however, the deacetylase activity of SIRTs 4–7 is low to none. SIRT4 is the only member of the SIRT family that has been determined to have no deacetylase activity (179). Recent evidence has shown that SIRTs with low deacetylase activity can remove other types of acyl groups from lysine residues on their target proteins (6, 43, 105). Most notably, SIRT5 has been shown to primarily promote demalonylation, desuccinylation, as well as deglutarylation (43, 199).
The target substrates of SIRTs include regulators, metabolic enzymes, histone proteins, transcription factors, proteins involved in proliferation, cell survival, and death, signal transduction proteins, DNA repair enzymes, regulators of circadian clock, inflammatory and immune response proteins, and many more (72). The dependence of SIRTs on NAD+ means that NAD+ levels can influence almost every cellular phenomenon. The roles and substrates of SIRTs have been extensively studied and thoroughly reviewed (28, 55, 72, 118, 155).
Interestingly, the NAM generated in deacetylase reactions by SIRTs acts as a negative feedback regulator of SIRT activity (17). This NAM is converted back to NAD+ by the action of the NAD+ salvage pathway enzymes NAMPT and NMNATs. Hence, NAD+ biosynthesis enzymes also regulate SIRT signaling (148, 194). We recently demonstrated that pharmacological inhibition or depletion of the NAD+ synthesizing enzyme NAMPT inhibits cancer cell viability by promoting the acetylation and stabilization of tumor suppressor protein TAp73 (Fig. 5). We attributed this increase in TAp73 acetylation and stabilization to decreased level of the NAD+-dependent deacetylase SIRT1 (159).
SIRT1 is the most well-known member of the SIRT family and by far the most extensively studied. It is well established that SIRT1 is an important regulator of carcinogenesis, however, there is much debate as to whether SIRT1 acts as a tumor suppressor or an oncogene. This is primarily due to conflicting evidence that has attributed both cancer-promoting and cancer-suppressing functions to SIRT1. It has been shown that SIRT1 targets and deacetylates the tumor suppressor proteins p53 and p73 (39, 178) (Fig. 5), therefore influencing oncogenesis. However, it has been argued that p53 deacetylation by SIRT1 does not affect many p53-mediated functions (164). SIRT1 overexpression has also been observed in several types of cancers and has been associated with poor prognosis (32, 75, 195). In contrast, there have been many in vivo studies demonstrating SIRT1's role as a tumor suppressor. For example, SIRT1 expression or induction in mice has been shown to reduce the incidence of spontaneous carcinomas and sarcomas and reduce tumor progression (67). Furthermore, SIRT1 levels are significantly decreased in some human cancers, including glioblastoma, breast carcinoma, and hepatic carcinoma (182). Moreover, SIRT1 has been shown to negatively regulate the oncogene survivin (Fig. 5), an inhibitor of apoptosis that maintains cell viability and promotes tumor growth in BCRA1-associated breast cancer (183). It is highly likely that due to SIRT1's diverse range of target substrates, the function of SIRT1 in carcinogenesis is both context and cell dependent.
SIRT2 was originally thought to be primarily localized in the cytoplasm (72), however, it is also capable of shuttling to the nucleus, particularly during the initiation of mitosis (176). SIRT2 has been thought to act as a tumor suppressor by interacting with members of the anaphase-promoting complex and thereby regulating mitosis and cell cycle to maintain genome integrity and to suppress tumorigenesis (90). However, SIRT2 has also been shown to target cytoplasmic alpha-tubulin and it cooperates with HDAC6 to promote alpha-tubulin deacetylation, which can promote cell division and migration in cancer cells (133). SIRT2 has also been shown to bind and interact with the oncoprotein hypoxia-inducible factor 1 alpha (HIF1α), which is a transcription factor that promotes glycolysis and tumor growth under hypoxic conditions (158). In this context, SIRT2-mediated deacetylation of HIF1α promotes its hydroxylation and ubiquitin-mediated degradation, further supporting the notion that SIRT2 acts as a tumor suppressor (158) (Fig. 5). However, SIRT2 has also been shown to promote the stability of the major oncoprotein Myc by suppressing the expression of the ubiquitin ligase NEDD4 via H4 lysine 16 acetylation, thereby preventing Myc ubiquitination and proteasomal degradation (109) (Fig. 5). Moreover, SIRT2 also interacts with AKT/PKB and is required for AKT/PKB activation during normal insulin signaling (145). It has also been shown that SIRT2-mediated regulation of AKT/PKB promotes downstream GSK3beta/b-catenin-mediated epithelial–mesenchymal transition (31, 145) (Fig. 5). In line with the notion that SIRT2 acts as a tumor promoter, specific SIRT2 inhibitors have been shown to suppress the growth of various types of cancer (84). Taken together, it appears that the role of SIRT2 in cancer is also context and cell dependent.
SIRT3 is a major regulator of cell death and survival due to its prime location in the mitochondria, where most cell death signaling occurs. As with SIRTs 1 and 2, SIRT3 has also been designated as a tumor suppressor as well as a tumor promoter (89, 192, 200). SIRT3 has been shown to inhibit cell death during genotoxic and oxidative stress by deacetylating Ku70, which enhances Ku70-Bax interactions and prevents Bax translocation to the mitochondria, promoting the survival and persistence of cancer cells (169) (Fig. 5). Another major target of SIRT3 is the antioxidant enzyme superoxide dismutase 2 (SOD2). SOD2 is deacetylated and activated by SIRT3 (143), and this activation may have an oncogenic function that protects cancer cells from accumulating damage caused by ROS and ROS-mediated cell death; however, SIRT3-mediated activation of SOD2 may also have a tumor suppressive function that prevents the accumulation of ROS-induced malignant DNA mutations (Fig. 5). It is likely that a fine balance of SIRT3 activity is required to prevent mitochondrial dysfunction and tumorigenesis.
Arguably, SIRT4 remains the most elusive member of the SIRT family. SIRT4 has been deemed a mitochondrial tumor suppressor that is strongly induced during DNA damage and suppresses tumorigenic mitochondrial glutamine metabolism (81, 82, 200). SIRT4 promotes the ADP-ribosylation and downregulation of glutamate dehydrogenase, and is involved in glutamate and glutamine metabolism (56) (Fig. 5). This regulation of glutamine metabolism controls cell cycle progression and maintains genomic integrity to suppress tumor growth. Furthermore, low expression of SIRT4 is correlated with poor patient prognosis in several types of cancer (114, 124, 129, 162). While these results demonstrate a tumor suppressive role for SIRT4, the functional targets of SIRT4 remain largely unknown.
SIRT5 is a mitochondrial SIRT that has recently emerged as a major regulator of energy metabolism and oxidative stress (24, 128, 186, 199). It has been demonstrated that SIRT5 primarily acts by removing succinyl, malonyl, and glutaryl groups from its substrates to modulate their function (43, 132, 199). The targets of SIRT5 have mainly been identified as metabolic enzymes that are known regulators of cancer metabolism (132, 186, 199). One such substrate of SIRT5-mediated desuccinylation is IDH2, which catalyzes the oxidative decarboxylation of isocitrate to 2-oxoglutarate, and produces NADH in the TCA (199). IDH2 overexpression and gain-of-function mutations are common in cancers (50). SIRT5-mediated desuccinylation and deglutarylation activate IDH2, and downmodulation of SIRT5 inhibits IDH2 (Fig. 5), promotes the accumulation of ROS, and increases susceptibility to neuronal degeneration and ischemia/reperfusion injury of the heart (199). Another study demonstrated that IDH gain-of-function mutations promoted 2-hydroxyglutaric acid and hypersuccinylation in the mitochondria, enhancing mitochondrial dysfunction, apoptosis resistance, and tumorigenesis (104). These effects were reversed by overexpression of SIRT5, which relieved hypersuccinylation in IDH mutant cells and suppressed tumorigenic transformation (104). The role of SIRT5 in cancer is still unclear, and the role of SIRT5-mediated desuccinylation and activation of enzymes is still a very new area of research. Thus far, it appears that SIRT5 plays an important role in maintaining a balance of mitochondrial protein succinylation (104), however, the impact of SIRT5-mediated desuccinylation on the activity of potential target enzymes remains largely unexplored.
SIRT6 is most widely regarded as a tumor suppressor protein (78, 95, 96, 157). SIRT6 was originally described as a regulator of chromatin acetylation and genomic stability (122, 126), but recent studies have highlighted a novel role for SIRT6 in regulating cancer cell metabolism and signaling (78, 157). SIRT6 has been shown to prevent tumorigenesis by inhibiting anaerobic glycolysis. Moreover, it has been demonstrated that SIRT6 inhibits the expression of oncogenes c-Myc and survivin (123, 157) (Fig. 5). However, high expression of SIRT6 has also been correlated with poor patient prognosis in lung cancer (8). More research is needed to determine the precise role and regulation of SIRT6 activity in cancer.
SIRT7 was originally described as a promoter of rDNA transcription by regulating the activation of RNA polymerase I (47). Several targets of SIRT7 deacetylation have been proposed, including GABP-β1 and p53 (127, 153, 174) (Fig. 5); however, SIRT7 has also recently been shown to act through desuccinylation (105). It has recently been demonstrated that SIRT7 desuccinylates histone proteins to promote chromatin compaction and genome stability during PARP1-dependent DNA damage responses (105). The role of SIRTs in protein succinylation and malonylation opens up a novel and exciting area of research regarding the regulation of cancer-related proteins via NAD+-dependent SIRT-mediated post-translational modification.
NAD+ mediated metabolic reprogramming of cancer epigenetics
Epigenetic processes alter gene expression without any changes to the DNA sequence and comprise various mechanisms such as methylation of DNA or histone acetylation leading to repression or promotion of gene expression, respectively (161). Epigenetic processes themselves are greatly regulated by the metabolic status of the cells. One case is the removal of acetyl groups from the histones, leading to a tighter conformation of nucleosomes that inhibit access of transcription factors and promoters to the DNA sequence and hence limit the transcription and expression of target genes (Fig. 6). Hence, NAD+ can regulate the epigenetic status of cells via the NAD+-dependent sirtuin family of HDACs (Fig. 6). It has been shown that a metabolic shift from fatty acid oxidation to glycolysis leads to decreased levels of NAD+ and hence decreased SIRT1-mediated deacetylation of acetylated lysine 16 of histone H4 (H4K16ac) (152) (Fig. 6). Imbalance in the regulation of epigenetics can lead to activation or inhibition of certain cellular pathways leading to the development of cancer. An example of this is increased SIRT1 activity that has been linked with increased tumorigenesis, as it causes deacetylation of p53 tumor suppressor protein (178). The role of SIRT1 as an epigenetic modulator in regulating tumor suppressor and DNA damage repair genes has set this enzyme as a potential therapeutic target for cancer (156) (Fig. 6). SIRT1-dependent NAD+ activity can also induce epigenetics through DNA methylation pathways. Increased levels of SIRT1 have shown to enhance the stability of DNA methyltransferase, enhancer of zeste homologue 2 (EZH2) (111). DNA methylation has been recognized to play an important role in epigenetic repression of tumor suppressor genes in many types of cancer (66). One study suggests that EZH2 promotes breast tumor initiating cell expansion by downregulation of DNA damage repair system, which causes accumulation of RAF1 and hence activation of ERK-β-catenin signaling (30). NAD+ has proven to be a crucial link between epigenetics and cancer progression, and hence, it is an important area of study for therapeutic implications and better understanding of underlying cancer mechanisms.

Emerging Role of NAD+ in CSC Biology
CSCs are small population of pluripotent cells within tumors that display self renewal and multilineage differentiation capabilities (141). These characteristics allow CSCs to resist chemotherapeutic assaults or various treatments that are targeted toward extermination of cancerous cells. As a result, CSCs account for many cases of cancer relapse, aggressiveness, and metastasis (42). NAD+ has shown to be a link between metabolism and epigenetic modulation in cells, but its activity in the context of CSCs needs to be further investigated. Our recent study highlighted the important role of NAD+ in regulating pluripotency of CSCs through autophagic pathways (160). Autophagy is another major regulator of cellular processes and homeostasis, and it involves engulfment of proteins and organelles in autophagosomes and their delivery for lysosomal degradation (62, 125). Autophagy is believed to have a complex role in cancer cells. It can promote their survival by recycling cellular components to provide energy requirements of cancer cells, but defects of autophagy have also been linked with increased tumor progression (120). Previously, we showed that inhibition of NAMPT decreases the pluripotency of human NT2/D1 and murine P19 embryonic CSC-like cells, and induces their differentiation (160). This inhibition is accompanied by increase in markers for autophagy machinery, which suggests that NAD+ regulates CSC pluripotency through autophagy (160). Another study also highlights the importance of NAD+ in the context of CSCs by revealing that high expression of NAMPT in glioblastoma stem-like cells (GSCs) correlates with decreased patient survival, and the inhibition of NAMPT decreases the self-renewal capacity of these cells (54). The effect of NAMPT inhibition on GSCs is mediated by downstream transcription factor E2F2 whose inhibition causes downregulation of helix-loop-helix gene inhibitor of differentiation (54). Furthermore, inhibition of NAMPT effectively targeted human pluripotent stem cells (hPSCs) without affecting hPSC-derived progeny, revealing an advantage in dealing with the tumorigenic potential of hPSCs in hPSC-derived cell-based therapies (93). Together, these suggest that NAD+ is a potential target for anti-CSC therapy.
Future Directions
The major role of NAD+ in both energy metabolism and anabolic reactions makes it a crucial molecule required by cancer cells for their growth and proliferation (25). For this reason, NAD+ synthesis has been the interest of investigation for cancer therapeutic applications. So far, studies targeting NAD+ synthesis have shown promising antitumor effects. Importantly, interfering with NAD+ synthesis seems to preferentially target tumor cells over normal cells since cancer cells are more reliant on NAD+ metabolism for their survival; as is evident from the fact that NAMPT inhibition slows the growth of tumor xenografts without adversely affecting normal cells (14). While such tumor regression by NAMPT inhibition was not observed in clinical trials, the drug was nevertheless well tolerated by patients and had low toxicity (70). These results provide hope that treatments targeting NAD+ synthesis can be optimized with combination therapies to increase their antitumor efficacy.
One clear area needing advancement in the field is the development of NAD+ enzyme inhibitors. Our knowledge on the potential of NAMPT exploitation is plentiful due to the availability of potent and specific NAMPT inhibitors, but studies on other NAD+ enzymes such as NMNATs are lacking. If we wish to target NAD+ synthesis as a means of cancer therapy, inhibition of NMNATs should be further explored since they are the downstream enzymes of NAD+ synthesis for all the possible pathways. Currently, NMNAT inhibitors include gallotannin and the low-potency NAD analogs (12, 139), although these drugs affect the three NMNAT isoforms nonselectively. Thus, better and more selective inhibitors are needed to exploit NMNATs for cancer therapy and reduce side effects because a growing number of studies are showing that each NMNAT isoform has differential roles. Therefore, depending on the type of cancer alterations and NAD+-dependent processes, it will be of interest to target the compartmentalized NAD+ metabolism through inhibition of individual NMNATs.
The study of NAD+ synthesis and metabolism has been expanding to include the other less understood salvage pathways as well, and these pathways also provide new opportunities for clinical applications. As the reliance on certain NAD+ enzymes differs between cell types, it is possible that some tumors may be very susceptible to inhibition of NAD+ synthesis via the Preiss–Handler pathways, for instance. In that capacity, the efficacy and mechanistic aspects of targeting the Preiss–Handler pathway or NR pathway have not yet been elucidated in detail. Again, specific inhibitors of the rate-limiting enzymes NAPRT and NRK1/2 need to be developed. Alternatively, therapeutic strategies involving NAD+ metabolism may also explore combined targeting of multiple NAD+ synthesis pathways, such as inhibition of enzymes implicated in the classical salvage pathway and Preiss–Handler pathway. To this end, the effects of such combination approaches can be studied using NAMPT inhibitors on cells containing experimentally lowered expression of NAPRT or NMNAT.
Considering the current information on NAMPT inhibition in cancer therapy, further studies should explore the potential of interfering with NAD+ metabolism to sensitize cells to modulations of downstream pathways, such as autophagy, or processes that are reliant on NAD+ degradation. For example, NAMPT inhibition combined with inhibition of LDH A effectively reduced lymphoma growth in mice (99). As inhibition of NAMPT has been shown to play a role in autophagy (160), treatment could also be combined with activators or inhibitors of autophagy. This strategy involving autophagy modulation may be of particular relevance to the treatment of CSCs, which have been shown to be reliant on a careful balance of autophagy activity. In summary, with its multifaceted functions in carcinogenesis, NAD+ and its associated biomolecules may provide therapeutically targetable modalities that can be exploited for cancer treatment.
Footnotes
Acknowledgments
This work was supported by grants from the Canadian Institute of Health Research (CIHR), from the Canadian Breast Cancer Foundation–Atlantic (CBCF), and Breast Cancer Society of Canada (BCSC)/QEII Foundation Awards for breast cancer research through Beatrice Hunter Cancer Research Institute (BHCRI) to S.A.G. and P.W.K.L. T.S. is supported by the CIHR. B.K. is supported through a postdoctoral fellowship from Cancer Research Training Program (CRTP) of BHCRI. C.D. is currently funded through CIHR Masters Award and Nova Scotia Health Research Foundation (NSHRF) Scotia Scholar Award. E.M. is supported by Nova Scotia Graduate Scholarship (NSGS) as well as NSHRF Scotia Scholar Award. E.M. and C.D. were recipients of the BHCRI studentship award in the past.