
Abstract
Significance:
Cancer cells that are resistant to radiation and chemotherapy are a major problem limiting the success of cancer therapy. Aggressive cancer cells depend on elevated intracellular levels of reactive oxygen species (ROS) to proliferate, self-renew, and metastasize. As a result, these aggressive cancers maintain high basal levels of ROS compared with normal cells. The prominence of the redox state in cancer cells led us to consider whether increasing the redox state to the condition of oxidative stress could be used as a successful adjuvant therapy for aggressive cancers.
Recent Advances:
Past attempts using antioxidant compounds to inhibit ROS levels in cancers as redox-based therapy have met with very limited success. However, recent clinical trials using pro-oxidant compounds reveal noteworthy results, which could have a significant impact on the development of strategies for redox-based therapies.
Critical Issues:
The major objective of this review is to discuss the role of the redox state in aggressive cancers and how to utilize the shift in redox state to improve cancer therapy. We also discuss the paradox of redox state parameters; that is, hydrogen peroxide (H2O2) as the driver molecule for cancer progression as well as a target for cancer treatment.
Future Directions:
Based on the biological significance of the redox state, we postulate that this system could potentially be used to create a new avenue for targeted therapy, including the potential to incorporate personalized redox therapy for cancer treatment.
Introduction
T
Generally, oxidative stress can arise from (i) inefficient reduction-oxidation (redox) regulation of normal cellular physiologic processes, including mitochondrial respiration and signal transduction; (ii) decrease in Aps; and (iii) an increase in reactive oxygen species (ROS)/reactive nitrogen species (RNS). For these reasons, cancer cells are usually under a higher oxidative stress than normal cells, and the cellular redox state is believed to be an important factor in cancer cell fate. It was well documented that cancer cells are usually under a higher oxidative stress than normal cells and that an additional increase in prooxidant level can trigger cell death (12, 62, 92, 379). Research supporting the latter line of thinking led us to propose that the persistence of redox adaptation in cancers contributes to the development of aggressive phenotypes, including resistance to cancer therapies.
Recent preclinical studies and clinical trials using pro-oxidant compounds reveal noteworthy results that these compounds activate antioxidant protection response in normal cells while inducing cell death in various cancer cells (191, 296, 355, 379). Thus, the development of sensitive and practical methods to detect cellular redox status is essential for the success of pro-oxidants therapy and to facilitate the application of redox therapy to precision medicine.
The major objective of this review is to discuss the role of the redox state in aggressive cancers and how to utilize the shift in redox state to improve cancer therapy. The biological significances of redox state will create a new avenue for cancer precision medicine. We also discuss the paradox of redox state parameters; that is, H2O2 as the driver molecule for cancer progression as well as a target for cancer treatment. Specifically, an effort is made to discuss the potential of redox cycling compounds as a radical strategy for targeting cancer cells and the potential to incorporate personalized redox therapy for cancer treatment.
Redox State Regulation
Redox balance is the sum of the reducing and oxidizing equivalents within a cell. The levels of ROS/RNS, antioxidant proteins (APs), and redox thiol couples will alter the balance of the redox state. ROS/RNS are generated during normal physiological metabolism and in response to stresses, including exposure to xenobiotics, cytokines, growth factors, hormones, and invasion of bacteria (287). The majority of ROS/RNS are hydrogen peroxide (H2O2), hydroxyl radicals (OH•), superoxide radicals (O2 •−), nitric oxide (NO•), and peroxynitrite (ONOO−). ROS or RNS can damage macromolecules or react with sulfhydryl (sulfenylation), glutathione (GSH, glutanylation), and cysteine (oxidation) groups to activate/inactivate specific proteins (47, 134, 160, 162, 169). The median level of oxidized cysteine residues in the proteome is between 5% and 12%, which can increase to >40% by the addition of oxidants (73, 160, 161). ROS/RNS regulate many life and death events depending on the levels of generation, the spatial distribution, and subcellular compartment sites. APs are important parameters for regulation of ROS/RNS levels and govern target-specific transduction of redox signals. APs are compartmentalized and tightly controlled at both the genetic and activity levels. The major enzymatic antioxidants include superoxide dismutase (SOD), catalase (CAT), glutathione peroxidase (GPx), glutathione S transferase (GST), and glutaredoxin (Grx). These enzymes work in concert with thiol-redox couples to control ROS/RNS levels. A cell contains six major redox couples: NADH/NAD, NADPH/NADP, cysteine (Cys)/cystine (CySS), GSH/glutathione disulfide (GSSG), peroxiredoxin (Prx)-sulfiredoxin (Srx), and thioredoxin (Trx)/thioredoxin disulfide (TrxSS). Each of these redox couples is present in specific concentrations in subcellular and extracellular compartments for each cell type of the human body (Fig. 1). The redox couples ensure that electrons are available at specific subcellular locations. For instance, thiol systems fine-tune the production of H2O2 by limiting its diffusibility and stability in each subcellular compartment. The pKa of specific residues on proteins determines how sensitive these residues are to the available H2O2 (152, 271, 328). In addition, protein thiols are subject to other modifications (e.g., nitrosylation, sulfhydration, metal ion binding) that, in turn, regulate or act as signaling molecules to control cell function (220).
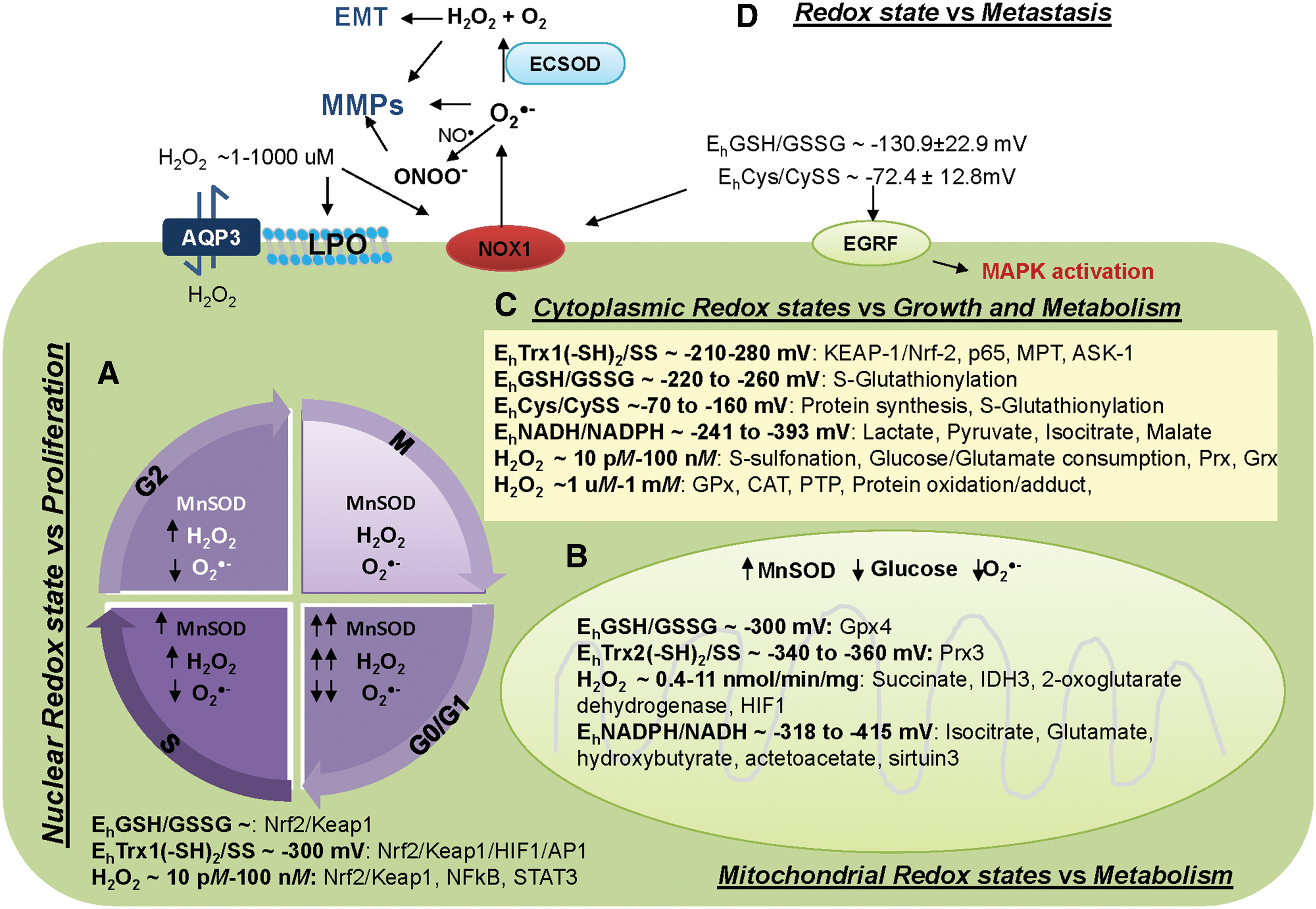
From a broader standpoint, in addition to these main parameters, DNA-repairing enzymes or proteins that respond to oxidative stress (i.e., hypoxia inducible factor-1α [HIF-1α], heat shock protein [HSP]) as well as metal storage and transporters (i.e., transferrin) work together to help maintain the redox state as a secondary mechanism for redox regulators (81, 135, 179). In summary, the ROS/RNS, APs, thiol couples, and other redox players provide an enriched diversity to the central redox organizational structure that establishes specificity in biological processes.
Healthy cells continuously produce or are exposed to ROS/RNS as byproducts of oxidative phosphorylation (OXPHOS), such as O2 •− or biological signaling molecules like NO•. Protective APs and thiol couples keep these ROS/RNS in a homeostatic steady state. The spatial distribution of ROS and thiol couples in cells is not uniform. The order of reducing redox status as ascertained by levels of redox potential value is mitochondria > nucleus > cytoplasm > endoplasmic reticulum > extracellular space (130). The range of redox potentials calculated from thiol couples, Cys/CySS, Trx/TrxSS, GSH/GSSG, and NADPH/NADP, and the range of H2O2 concentrations at each subcellular compartment are highlighted in Figure 1. For example, the redox potential of the GSH/GSSG couple has often been used as an indicator of the cellular redox environment. Buettner et al. reviewed that GSH/GSSG can turn molecular switches on and off, leading to different biological states of cells as follows (39): redox potential ∼−240, −200, and ≤ −170 mV, turn on cell proliferation, differentiation, and initiation of cell death, respectively. The GSH/GSSG is not the only parameter that has a role in the redox biology of cells; the redox potential of Trx in the nucleus (estimated to be ∼300 mV), thioredoxin reductase (TR), glutathione reductase (GR), and Prxs can regulate cell proliferation and apoptosis by a direct interaction/high affinity for binding partners/effector molecules, including AP-1, HIF-1α, NF-κB, Nrf2 (nuclear factor-erythroid 2-related factor 2), and Keap1 (Kelch-like ECH-associated protein 1) (232, 241, 322). Moreover, cytosolic H2O2 [∼10 pM–100 nM (131, 158, 310)] leads to the dissociation of transcription factor complexes, and it allows the transport of NF-κB and Nrf2 through nuclear pores to DNA binding sites (Fig. 1A). It has been suggested that an enhanced “reducing environment” provides the conditions that are necessary to optimize the electron transfer and enzymatic activity that are required for transcription factors to bind DNA in the nucleus (187, 376).
Generally, the mitochondrial redox state is mainly regulated by OXPHOS, glucose consumption rate (GCR), manganese superoxide dismutase (MnSOD), NADPH/NADP, Trx2/Trx2SS, and GSH/GSSG. The mitochondrial matrix NADH/NAD operates at a redox potential of −318 mV, which is necessary for the reductive force of mitochondrial ATP production (38, 161). Correlatively, the mitochondrial NADPH/NADP system operates at −415 mV and this system functions at a lower redox potential than the NAD system (161, 369). The NADH/NAD couple is essential to catabolism and energy supply (36). It regulates the conversion of lactate and pyruvate in the cytoplasm while regulating TCA cycle metabolites (isocitrate, hydroxybutyrate, acetoacetate) in the mitochondria (138, 369) (Fig. 1C). Based on cellular metabolism, mitochondrial ROS, including H2O2, are derived from mitochondrial respiration, which depends on NADH. Isolated mitochondria indicate that H2O2 in mitochondria is about 0.4–11 nmol/min/mg (8, 175, 238, 381). Evidence indicates that metabolites, APs, HIF-1α, and TCA- and OXPHOS-associated proteins, as well as signal transducer and activator of transcription 3 (STAT3), are regulated by the mitochondrial redox state (219, 230) (Fig. 1B). In aggressive cancer cells, such as chemo-resistant acute myeloid leukemia (AML) and radioresistant melanoma, OXPHOS and TCA activities are upregulated, suggesting that the OXPHOS-derived mitochondrial redox state of cancer cells is more oxidized than in normal cells (91, 265, 377). We have further addressed the difference of the mitochondrial redox state in the Rewired redox state section.
Remarkably, studies of subcellular compartments show that the redox potential of the cytosolic NADH/NAD is ∼−258 mV, which is more oxidized than in mitochondria by about −60 mV (358). Similarly, the cytosolic NADPH/NADP system operates at −393 mV, which is also more oxidized than mitochondria by about −22 mV (146, 161). Interestingly, the physiological concentration of H2O2 in the cytosol appears to be maintained in the submicromolar range (144). At this submicromolar concentration (0.01–100 nM), H2O2 regulates the redox switch of selected proteins, that is, Prx, Keap1 (9). Mechanistically, the oxidation of redox switches for highly reactive proteins, such as thiol-based antioxidants, is needed to transmit information along with a signaling cascade quickly. For instance, Prx2 acts as a sensor, reacts with H2O2, and finally relays the oxidation to form disulfide links between STAT3 monomers, which are addressed in Ref. (9). The response time observed for the Prx2 sensor is below 60 s (268, 313, 330). Intriguingly, the central positioning of H2O2 and thiol redox states in space and time is emphasized by the circadian variation of Prxs, and this evidence endorses the sensor role of Prxs (86, 171). Because Prxs are reduced quickly, H2O2 > 25 μM could lead to diminished Prx2 dimerization as a result of overoxidation (259, 330). Subsequently, this evidence supports the role of high H2O2 (at the micromolar range) as an APs inhibitor and inducer of oxidative damage products (195) (Fig. 1C). In addition, GSH serves as a main intracellular thiol redox buffer that maintains the cytoplasmic environment in a reduced state. Assessment of the GSH concentration revealed that it exists intracellularly at millimolar concentrations and is about 2–4 mM in plasma (234). Because GSH degradation occurs extracellularly, the export of GSH, GSSG, and GSH adducts is an important step in GSH turnover. Herein, we have reviewed in the H2O2: A Bona Fide Molecule for Cancer Treatment section, that levels of cellular H2O2 are higher in cancer cells, which suggested a pro-oxidant status of the cytoplasmic redox state.
In contrast to the intracellular redox state, Cys/CySS is dominantly expressed in plasma. Blood and extracellular fluid possess major thiol-dependent redox nodes involving Cys and CySS, which have been associated with receptors and extracellular proteins, including matrix metalloproteinases (MMPs) (286, 327). The Cys/CySS couple may function as an oxidant–reductant in redox switching, thus providing a means to oxidize–reduce proteins without the direct involvement of more potent oxidants–reductants. The physiologic Cys/CySS redox potential (Eh) in healthy subjects is around −80 mV; whereas in subjects with idiopathic pulmonary fibrosis, the redox state becomes oxidized to between −62 and −20 mV (277). Several recent studies have demonstrated that modification of extracellular Cys/CySS could directly regulate or stimulate the differential expression of genes that control cellular activities, including cancer cell proliferation and invasion (48, 116, 158, 160, 162, 277) (Fig. 1D). We have performed a series of experiments modulating concentrations of Cys/CySS in tissue culture media and studied the effects on normal prostate epithelial cells (PrEC) and highly aggressive DU145 and PC3 prostate cancer cells. We found that media with oxidized Cys/CySS enhance prostate cancer cell growth, whereas media with reduced Cys/CySS increase prostate cancer cell invasion (47). In contrast, Nkabyo et al. found that media with more oxidized Cys/CySS result in less proliferation of colon cancer Caco-2 cells (247). However, neither oxidized nor reduced Cys/CySS affect normal PrEC growth or alter in vitro invasion assays (48).
In addition to Cys/CySS, full-length and truncated Trx have also been detected in plasma (263). Extracellular Trx levels usually range from 1 to 5 nM (297). They localize to the cell surface and serve as an electron donor for the potential peroxidase activity of albumin (44). Despite the high expression levels of Trx1, H2O2 is another redox state regulator that is abundantly present in the extracellular space, with its concentration in blood plasma being ∼1–1000 μM (98). The major sources of extracellular H2O2 are from NADPH oxidase (NOX)-mediated superoxide radical (242) and the diffusion of intracellular H2O2. Nevertheless, the absolute values remain unsettled due to the significant variability of published results and the level of H2O2 generated in cells that actually escapes to the plasma is not clear.
In summary, each cell type in the body is able to achieve its unique cellular function via a unique repertoire of proteins, each at a specific concentration, in a specific subcellular location. Redox state at each subcellular is distinctive regarding redox potential values, types and levels of APs, and thiol couple concentration. The redox potential value and redox state parameters of each subcellular compartment can turn on/off specific sets of proteins that contribute to distinctive physiological/pathological consequences (Table 1), giving the selective advantage for therapeutic design at the subcellular level. Thus, understanding the mechanism of the subcellular redox state will lead to the designing of a more effective therapy.
CAT, catalase, Cys, cystein; CySS, cystine; Eh, redox potential; EMT, epithelial-mesenchymal transition; GPx, glutathione peroxidase; Grx, glutaredoxin; GSH, glutathione; GSSG, glutathione disulfide; H2O2, hydrogen peroxide; HIF1α, hypoxia-inducibe factor-1α; Keap1, Kelch-like ECH-associated protein 1; LPO, lipid peroxidation; MnSOD, manganese superoxide dismutase; ND, non-determined; NO•, nitric oxide; NOX, NADPH oxidase; Nrf2, nuclear factor-erythroid 2-related factor 2; O2 •−, superoxide radical; Prx, peroxiredoxin; STAT3, signal transducer and activator of transcription factor 3; Trx, thioredoxin; VEGF, vascular endothelial growth factor.
Role of Redox State in Cancer Characteristics
ROS have both physiological functions and pathological effects within the cells (141, 301, 359). Cancer cells characteristically have a high antioxidant capacity that regulates ROS to levels that are compatible with cellular biological functions but still higher than those of normal cells (45, 349, 383). When the balance between pro-oxidants and antioxidants tips toward oxidants, oxidative stress occurs, causing adaptive responses that result in mutation and genomic instability. Researchers are particularly interested in redox states because of their clinical role in aggressive cancer regulation, specifically their post-transcriptional and translational modification of key regulators. In fact, redox state has been implicated in cancer treatments; for example, ionizing radiation (IR) and chemotherapy, both of which are cornerstones of cancer treatment. Radiation and cytotoxic chemotherapy are effective modalities that can kill most cancers if their use is not limited by concerns for injury to healthy tissues, a bystander effect. It is also important to recognize that the redox status of cancer cells can be heterogeneous depending on tumor type and degree of aggressiveness. Thus, the additional oxidative stress induced by these treatments may cause either further DNA damage or mutations that lead to the development of resistant cancer cells (264, 299, 317), that is, oxidative modification on 7, 8-dihydro-8-oxoG that leads to generation of Ras oncogene (294); subsequently, oxidized 8-oxoG becomes a cause for the development of several types of cancers. Redox imbalance has proved to be a contributing factor to numerous cancer phenotypes; that is, increased cell proliferation, invasion, and epigenetic changes that lead to pathologic and clinical progression of cancers. As emphasized by Forman et al., redox state alone will probably not be enough to predict cell behaviors (99, 100); one needs knowledge of the localized intracellular redox state and the biochemistry of specific subcellular niches within the cell to accurately predict cellular functions. Thus, it is important to understand how the redox state changes during cancer progression; how it activates or regulates different proteins during proliferation and metastasis; how it responds to cancer metabolism; or whether it is responsible for the resistant properties of cancers. The following section provides background information on redox state and its role in selective cancer characteristics.
Interexchange regulation between redox state and transcription factors
An increased oxidative status in cancer has been correlated with mutations of tumor suppressor genes and activation of redox-sensitive transcription factors (110, 239). It is well established that persistent elevation of ROS levels in cancers leads to constitutive activation of several transcription factors (151). ROS can also promote tumor formation by inducing DNA mutations and pro-oncogenic signaling pathways. For example, K-ras mutation, which is frequently identified in various cancers, is regulated by ROS. Concomitantly, once activated, K-ras mediated ROS production by promoting localization of p47phox, NOX1 component, to interact with protein kinase C (PKC) at the plasma membrane. Interestingly, a handful of investigations indicate that K-ras-dependent ROS generation appeared to be mitochondrial ROS. Regardless, these interactions mediate cell transformation and promote tumorigenesis (151, 157). Moreover, a mild pro-oxidant (O2 •−) inhibits the functional holoenzyme assembly of tumor suppressors, protein phosphatase 2A (PP2A) and prevents BCL-2 ser70 dephosphorylation in leukemia cells; thus, the pro-oxidant status stabilizes BCL-2-antiapoptotic activity and promotes chemoresistance of hematopoietic cancers (208).
Redox cycling of Cys residues is one of the important mechanisms of ROS-regulated activity of transcription factors and signaling molecules. Various redox-sensitive oncogene transcription factors contain residues in the region that are required for DNA binding, including NF-κB, Nrf2, AP-1, and p53 (117, 187, 376). Conformation is critical for proper protein–DNA and DNA–DNA interactions. One thing that Nrf2, NF-κB, and AP-1 have in common is that after activation and translocation into the nucleus, Trx1 and redox factor-1 (REF1) must reduce Cys residues in these transcription factors to bind with DNA and initiate gene activation. For instance, oxidation of Cys-38 in p65 or Cys-62 in p50 of NF-κB serves to control DNA binding (127, 130). Because of the functional importance of their downstream target genes in cancer development, NF-κB and Nrf2 will be addressed in this review.
NF-κB
NF-κB is particularly sensitive to cellular redox changes, in part due to the presence of multiple levels of activation. NF-κB binds to several promoter regions of genes that are involved in cancer formation and progression in several cancers (21, 272). NF-κBs are composed of members of the Rel family that has five members: p50, p65 (RelA), p52, c-Rel, and RelB. Activation of NF-κB in mammalian cells depends on two major NF-κB pathways: the p50:Rel A dimer-mediated classic pathway and the p52:RelB dimer-mediated alternative pathway (28, 74). NF-κB is usually sequestered in the cytoplasm as a result of its association with inhibitory proteins, such as the IκB family members. IκB sequentially sustains the p50:Rel A dimerization via the ubiquitination and proteasome-mediated degradation pathways (167). NF-κB is also subject to activation by mechanisms that involve the processing of p105 and p100 to release p50 and p52, respectively (28, 74). This relationship between tumor progression and NF-κB family activation extends to include several cancers. For example, NF-κB is constitutively activated in the androgen-independent prostate cancer cell lines PC3 and DU145 but not in the androgen-responsive LNCaP prostate cancer cell lines, suggesting that androgen exposure or androgen receptor, at least in part, modulates the expression of NF-κB (170, 256). A large-scale tissue array study of all members of the NF-κB family demonstrated that they are expressed in normal, HGPIN, and cancer tissues. With the exception of cRel, each member was also detected in the nucleus of cancer cells. Interestingly, nuclear localization of RelB correlated with a patient's high Gleason scores (Gleason score >8), suggesting that RelB may play an important role in the adaptive response to oxidative stress in prostate cancer (366). Although the traditional focus of studies that inhibit NF-κB for enhancement of cancer therapy has been on the canonical dimer p50/RelA (326), we recently identified the alternative dimer p52:RelB as a potential candidate for targeting radioresistant cancers.
Nrf2
It is well known that Nrf2 is the master transcription factor for APs through regulation of antioxidant responsive element (ARE). Similar to NF-κB, Nrf2 activity is regulated at multiple steps. Nrf2 usually binds to Keap1 in the cytoplasm, rendering it inactive. Additional factors involved in Nrf2 inactivation are the constitutive degradation of the Keap1-associated proteins, Cullin-3 and ring-box 1. These proteins form a core E3 ubiquitin ligase complex and are involved in the ubiquitination process and subsequent degradation by the 26S proteasome (149, 222). It was demonstrated that ROS modifies specific Cys residues (Cys151, Cys273, Cys288) on Keap1, which results in (i) release of Nrf2, (ii) inhibition of the constitutive degradation of Nrf2, and (iii) the translocation and facilitation of the binding of Nrf2 to the ARE (149). The Nrf2/Keap1 complex is regulated by variations in the redox potential of GSH/GSSG, Trx/TrxSS, and the concentration of H2O2 in the cytoplasm and nucleus (Fig. 1A). In turn, Nrf2 activation upregulated genes that are known to play roles in cancer survival, such as (i) antioxidant proteins; GSH synthesis enzymes (GCLC and GCLM), Trx1, cysteine/glutamate transporter (xCT), Prxs, TR1, NADPH:quinone oxidoreductase-1 (NQO1), GPx1, GST, heme oxygenase 1; (ii) metabolic enzymes; Glucose-6-phosphate dehydrogenase (G6PD), malate enzyme 1; and (iii) drug transporters; multi-drug resistance-associated protein 1 (MRP1) (52, 323). In addition, Nrf2 upregulates the level of NADPH and NADPH-generating enzymes that assist in quenching ROS and oxidized protein thiols (235). A study by Frohlich et al. showed that Nrf2 is downregulated in human prostate cancer and that loss of function reduces both expression and activity of GST, which, in turn, promote prostate tumorigenesis (102). Therefore, the increased oxidative stress in cancers most often results in a downregulation of Nrf2 (235). Conversely, numerous studies have demonstrated an oncogenic role of Nrf2. Nrf2 accumulation due to mutation of Keap1 was identified in nonsmall cell lung cancer, head and neck, and hepatocellular carcinoma (150, 155, 305, 311). The mechanism by which cancer cells are protected from an ROS-generating drug is often associated with Nrf2 and APs activation. For example, cancer cells with high Nrf2 were less sensitive to adriamycin, platinum-based drugs, fluorouracil (5-FU), and radiation. Inhibition of Nrf2 in these cancers leads to enhanced sensitivity to these treatments (289). Correlatively, Nrf2 deficiency in mice causes susceptibility to oxidative stress and carcinogen exposure (235). Together, this evidence suggests that Nrf2 could be considered a potential therapeutic target as well as a tumorigenesis inhibitor, probably based on the stimulus, environment, as well as the stage and type of cancers.
Redox state drives cancer growth via cell cycle regulation
It is widely accepted that ROS/RNS are involved in regulating cell growth and apoptosis. ROS/RNS act as an activator, co-factor, amplifier, or byproduct of these cell growth/survival signaling pathways. The redox state regulates cell cycle progression either by directly modifying cell cycle regulatory proteins or by induction of other signal transduction proteins. Further, ROS/RNS are involved in both cell membrane-to-nucleus and mitochondria-to-nucleus signaling pathways, which regulate critical biochemical effectors (89, 129). Halvey and Jones showed that ROS appears to be a critical component for growth factor signal transduction that activates cytoplasmic oxidation of Trx1 but not nuclear Trx1 (129). A series of studies since 1980 by Oberley et al. demonstrated that a decrease in MnSOD expression increased O2 •− production, which leads to an increase in glycolysis and cancer cell proliferation (207, 251). MnSOD protein levels and activity are significantly increased in quiescent (G0) compared with proliferating NIH 3T3 mouse fibroblasts and WI38 human lung fibroblasts (193). Overexpression of MnSOD extends the transit time of G1 and S phases without altering G2 transit time and, subsequently, suspends cell cycle progression in NIH 3T3 fibroblasts (193, 250, 252). Goswami et al. showed that synchronized HeLa cells exhibit a more oxidized environment during mitosis compared with interphase (119). They also demonstrated that the cellular redox state shifts toward a more oxidizing environment during S, G2, and M phases. A transient increase in pro-oxidant levels during the G1 phase is required for the mouse embryonic fibroblasts (MEFs) to initiate DNA synthesis (228). They further showed that inhibition of this oxidation state using N-acetyl-L-cysteine (NAC) before the S phase negatively impacts DNA synthesis, and the cellular redox state is reset to that found in the G1 phase (228). Therefore, significant inhibition of the pro-oxidant status prevents cells in the G1 phase from entering into the S phase. In line with this, we found higher levels of ROS/RNS in S and G2 M phases of the cell cycle compared with G0 phases in prostate cancer LNCaP cells; these data indicate a strong correlation between intracellular ROS/RNS and cell cycle progression. ROS/RNS levels in the lag phase of LNCaP cells are low, correlating with a decrease in PCNA staining, and strongly suggesting the possibility that ROS/RNS levels drive cell cycle progression in LNCaP cells (45). Together, Figure 1A exhibits an increase in intracellular ROS levels during progression from G1 to S to G2 and M phases. Overall, these data indicate that the redox state regulates the cell cycle machinery of each phase, from G0 through M phases, during the physiological condition and for cancer progression.
Since SODs convert O2 •− to H2O2, it is reasonable that SOD can regulate the redox cycle and processes that facilitate progression from G0/G1 to S to G2 and M phases. The periodic modification of MnSOD activity during the cell cycle phase is also evident under conditions of oxidative stress. For instance, MnSOD activity in MCF-10A human mammary epithelial cells decreases from 120 U/mg in a quiescent state to 30 U/mg in proliferating cells (292). Sophisticated experiments performed by Sarsour et al. demonstrated that MEFs with wild-type MnSOD show a typical growth curve consisting of a lag, an exponential, and plateau phases (293). However, exit from the exponential to the plateau phases are delayed in MEFs that are heterozygous for MnSOD whereas MEFs with MnSOD knockout fail to exit the proliferative cycle. Inhibiting cellular proliferation of MEFs with heterozygous MnSOD is associated with a delayed transit through G2 M phase. Overexpression of MnSOD facilitates the exit of heterozygous MEFs from the proliferative to the quiescent state. In addition, overexpression of MnSOD in prostate cancer PC3 cells results in an inhibition of PC3 cell proliferation by retarding the G1 to S transition of the cell cycle (45). These studies, combined with the recurring pattern of MnSOD activity during the cell cycle, suggest that MnSOD activity regulates a mitochondrial ‘‘ROS Switch’’ favoring O2 •− signaling (Fig. 1A). The relationship of redox state and cell cycle progression/proliferation has been extensively studied. Additional details on this topic can be found in a recent review by Menon and Goswami (291).
Redox imbalance causes cancer metabolism adaptation
Due to rapid growth and limited availability of oxygen and nutrients, cancer cells developed alternative metabolic reactions that allow them to adapt and survive under stress conditions such as oxidative stress. Central reactions of energetics and metabolism are controlled by near-equilibrium NAD reactions (161). The interaction of metabolism and redox state is a two-way street in which they reciprocally regulate one other. It is now well accepted that the redox state regulates metabolism mainly via ROS-mediated glucose consumption and cellular catabolism (103), whereas metabolism regulates redox state via generation of APs, ROS-generating enzymes, and NADPH/NADH (2, 166, 258). Thus, oxidative stress can also be caused by a higher cellular metabolism. Both metabolism and redox state are crucial for cancer survival. The integration of metabolism and redox state allows cells to modulate activities such as cell survival and proliferation according to their stimulation factors.
Effect of redox state on metabolism
As outlined in the Redox State Regulation section, redox state governs the functioning of cell metabolism. The interplay between molecular redox switches and participation of redox-active metabolites has been intensively investigated. It is well established that mitochondrial ROS play a significant role in regulating cellular metabolism. In this regard, mitochondrial ROS control cellular catabolism via regulation of metabolism-related enzymes or their metabolites through NADH/NADPH production systems (387). Ahmad et al. showed that mitochondrial O2 •− and H2O2 significantly contribute to glucose deprivation-induced cytotoxicity and oxidative stress in prostate cancer PC3 cells (4). In contrast, mitochondrial CAT and MnSOD can partially inhibit glucose deprivation-induced cytotoxicity. Greater inhibition of glucose deprivation-induced cytotoxicity and GSSG accumulation is observed when both enzymes are overexpressed compared with either enzyme alone. It was demonstrated that mitochondrial redox imbalance in sirtuin (Sirt) 3 −/−MEFs results in phenotypes that promote tumorigenesis (172). The accumulation of mitochondrial ROS, due to loss of SIRT3 enzymatic activity, inhibits oxygen-dependent prolyl hydroxylases, which regulate HIF-1α stability. HIF-1α was recently shown to respond to stimuli such as 2-oxoglutarate (alpha-ketoglutarate), succinate, or fumarate (334). Consequently, loss of SIRT3 results in HIF-1α stabilization and promotes oncogenic pathways that correlate with increased accumulation of mitochondrial ROS and shift cancer metabolism. Based on this evidence, ROS may be regarded as a control of redox metabolites instead of aerobic metabolism by-products.
In addition, ROS-modified post-translational modifications, that is, S-glutathionylation or sulfenylation, are directly or indirectly responsible activities of several metabolic-related enzymes. For example, aconitase, the enzyme catalyzing the second reaction in the TCA cycle, reacts rapidly (3 × 106–3 × 107 M –1s–1) with O2 •− (210). Pyruvate dehydrogenase kinase 2 (PDHK2) activity, part of pyruvate dehydrogenase complex that converts pyruvate to acetyl-CoA and controls redox couple NADH/NAD, is inhibited by mitochondrial O2 •− mediated H2O2 (147). This occurs via reversible oxidation of cysteine residues 45 and 392 on PDHK2. Of the metabolism-related enzymes, the M2 isoform of pyruvate kinase (PKM2) has been characterized as a cancer-specific metabolism-related enzyme. PKM2 can be inactivated under conditions of high ROS and, subsequently, affects NADPH generation (295). As PKM2 is less efficient than PKM1 in converting phosphoenolpyruvate to ATP and pyruvate, upstream glycolytic intermediates can flow into the pentose phosphate pathway where NADPH-reducing equivalents can be synthesized (351). The inactivation of these metabolism-related enzymes is evidence that ROS regulate the TCA cycle reaction.
Other ROS-associated metabolic pathways, including fatty acid and cholesterol synthesis, glutamine metabolism, and the pentose phosphate pathway, are also altered and are proven key factors for cell survival by providing biosynthetic precursors for macromolecules, such as nucleic acids, lipids, and proteins. Several studies report that the redox state plays essential roles in these metabolisms, such as an insulin-like effect of H2O2 (221). Detailed studies about other metabolisms have been reviewed elsewhere and are not discussed here.
Effect of metabolism on redox state
Cancer cell metabolism (increased glycolysis, high lactate, and hypoxia) is related to oncogene activation and loss of tumor suppressor genes. Cellular catabolism is organized by high-flux thermodynamically controlled NADPH/NADH production systems that are regulated by metabolism-related enzymes and their metabolites (161, 363), as indicated in Figure 1. The metabolite concentration contributes to the NADH/NAD to NADPH/NADP ratio. For example, combining malate with glutamate induces rapid oxidation of respiratory substrates and shifts the redox state toward a more reduced state when compared with malate treatment alone, as indicated by relative increases in NADH/NAD and NADPH/NADP (63). The NADPH/NADP couple is a major co-factor in a variety of ROS-generating enzymes, such as NOX, as well as for antioxidant systems, that is, Prxs and GPx. The concentration of cellular NADPH/NADP is, in part, controlled by the lactate/pyruvate concentration. Deficient NADPH/NADP production due to irregular cancer metabolism could, subsequently, result in an aberrant redox status due to either a decrease in APs function (Prx, TR, GPx) or ROS-generating enzymes (NOX). Accordingly, if the NADPH concentration is low, the decrease in H2O2 production is much less pronounced than expected from in H2O2 consumption by APs, which results in a more oxidized state of the mitochondria matrix (33). Further, levels of metabolites (e.g., lactate, malate, and succinate) influence how much H2O2 is produced, varying from 49 to 490 nmol H2O2/min/g of liver wet weight (50, 61, 94, 255, 309). These studies support the role of metabolism by products, NADPH in the regulation of cellular redox state.
Moreover, the metabolism substrates (i.e., glucose and GCR) are associated with ROS and cell cycle in MnSOD wild-type MEFs (292). Sarsour et al. demonstrated that an increase in MnSOD activity and a subsequent decrease in GCR are accompanied by a reduction in cell proliferation. Notably, for MEFs with 10% S phase, GCR is approximately 40 pg cell−1h−1, which increases to 120 pg cell−1h−1 in cultures with 25% S phase cells. Likewise, malignant brain tumor tissues display high metabolic activity through an increased GCR compared with normal brain tissues (143, 374).
Further, TCA cycle-associated enzymes have been demonstrated to regulate cancer redox state. For instance, fumarate hydratase can serve as a tumor suppressor gene; mutations in the enzyme result in specific tumor formation via O2 •− mediated cell proliferation as well as S-glutathionylation on fumarate in renal cancer (298, 336). Subsequently, fumarate accumulation in renal cell carcinomas (RCC) enhanced ROS production by promoting conjugation between fumarate and GSH and disruption of GSH metabolism. This cascade event was inhibited by NAC treatment (318, 336).
In addition, SIRT- and NAD+-dependent histone deacetylases in mammalian cells have been shown to regulate aberrant cancer redox states (41). SIRT3, one of the seven human sirtuins, is involved in many aspects of cancer metabolism (331). It interacts the electron transportation complexes I and II, thus functioning as an energy-sensing protein that promotes efficient energy utilization. A recent study described the expression of SIRT3 as an event that occurs subsequent to varying the redox potential of NADPH/NADP (Fig. 1B). Metabolically, loss of SIRT3 enzymatic activity inhibits ATP synthesis through hyperacetylation of oligomycin sensitivity-conferring protein at lysine 139 and promotes aerobic glycolysis (344). Acetylation of isocitrate dehydrogenase 2 at lysine 413, due to loss of SIRT3 enzymatic activity, promotes ROS production, blocks the regeneration of GSH, and promotes B cell malignancies (371, 372, 388). SIRT3 is also involved in redox regulation through deacetylates and activates MnSOD (58). In addition to its roles in OXPHOS and ROS production, SIRT3 also promotes fatty acid oxidation; loss of SIRT3 enzymatic activity inhibits fatty acid oxidation through acetylation of long-chain acyl-coenzyme A dehydrogenase, which has been observed in many types of cancers (95, 354).
Overall, these studies indicated that increases in metabolism correlate with increases in the oxidized redox state, which potentially makes cancer cells more susceptible to additional increases in ROS levels. It is now accepted that aberrant metabolism and redox state are widely observed in human cancers. A detailed understanding of the mechanisms by which cancer metabolism induces cytotoxicity and oxidative stress in cancer cells, or vice versa, may be useful in developing biochemical rationales for novel therapeutic interventions for cancer treatment.
Rewired Redox State in Aggressive Cancers
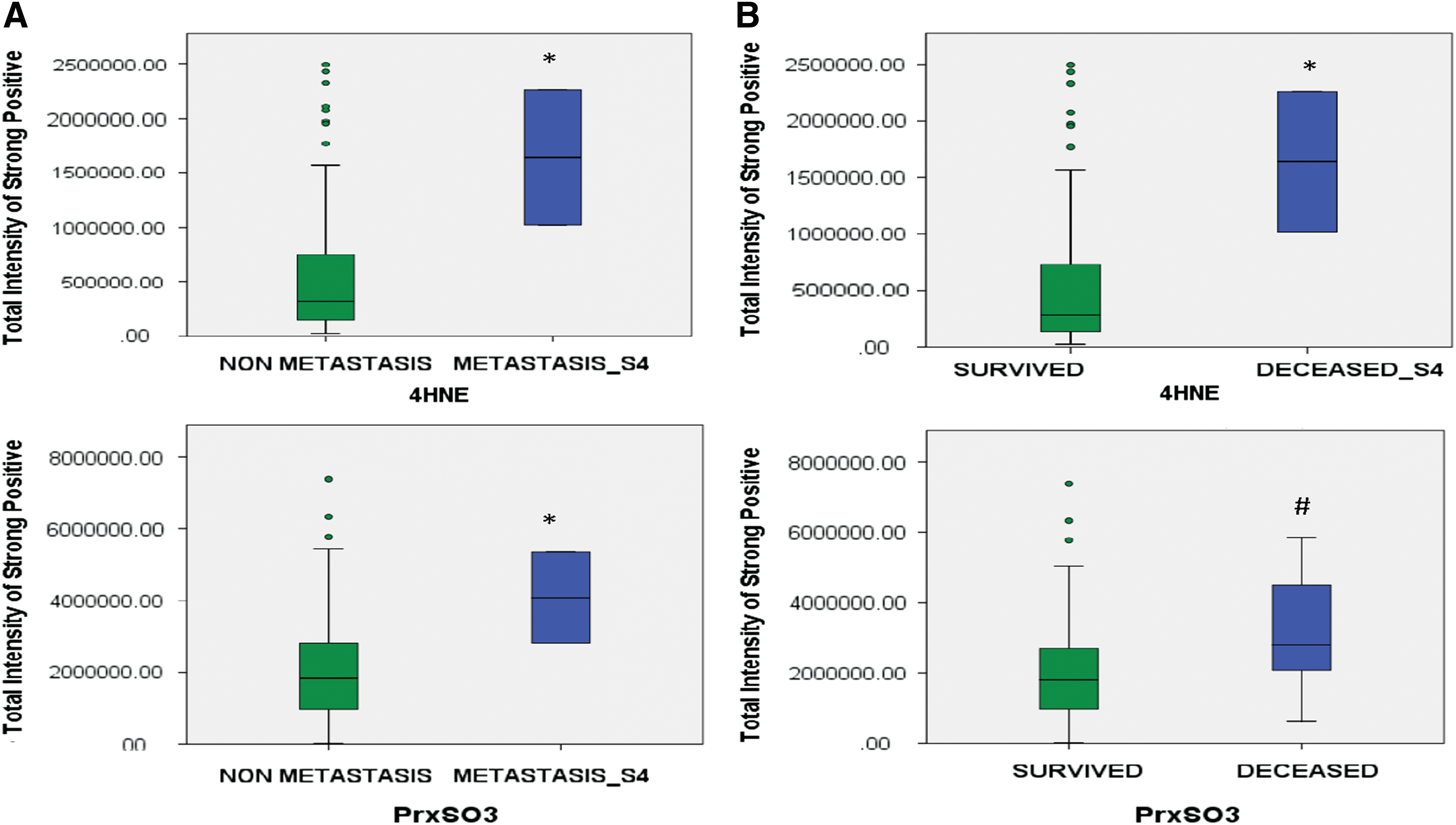
The main biological characteristics of cancers that influence the treatment outcomes are the differences acquired either intrinsically or extrinsically such as nutrition, stress environment, and the repopulation capacity of surviving cancer cells during the interval between treatments (104, 329). In fact, these factors could contribute to the variation of redox state of a cancer at each stage. Numerous human cancer cells harbor low levels of ROS, APs, and thiols at early stages. However, during cancer progression and treatment, especially as a result of redox-based therapy, cancer cells develop the ability to survive and sustain a high proliferative capacity by resetting their redox homeostasis and possessing high levels of ROS and APs (than their normal counterpart cells), a process called “rewired redox state” (Fig. 2) (12, 45, 92, 355, 379, 382). We propose that the rewired redox state is a protective response by cancer cells to defend against treatment-induced stresses, thereby leading to treatment resistance and metastasis. We have proposed that even aggressive cancers with high levels of APs are perilously close to an oxidative stress-mediated toxicity threshold (Fig. 2). Our laboratories are significantly committed to correlating the oxidative stress markers in aggressive cancers, including metastasis and radiation-resistant prostate cancer. Based on a prostate cancer tumor microarray that was constructed from 165 samples of prostate cancer patients and 34 samples of noncancer individuals, we established that the levels of 4-hydroxynonenal (4HNE) and the oxidation form of Prx (PrxSO3) are significantly increased in metastatic prostate cancer (Fig. 3A), an incurable disease that remains difficult to control. The increased levels of these oxidative stress markers inversely correlate with survival rates in prostate cancer patients (Fig. 3B). In addition, a review article by Mishra et al. reveals that the Prxs-Srx system functions as oncogenic signaling in various cancers, including breast, bladder, leukemia, lung, colorectal, ovarian, and prostate (232); therefore, the expression levels of Prxs are often upregulated in cancers and are likely to be modified by ROS. Overall, the data support the correlation of a rewired redox state with a more oxidized state in advanced stage cancer. From the clinical translation point of view, high levels and/or specific types of ROS may reveal a specific vulnerability of a malignancy or an insight into apoptosis pathways that could be used to selectively enhance cell death by further increasing the level of cellular ROS (Fig. 2). For example, Clement et al. demonstrated that overexpression of BCL-2 resulted in an elevated level of O2 •− and pH in leukemia cells and blocked caspase activation, along with H2O2-induced cytosolic acidification. Conversely, decreasing the O2 •− level increased the sensitivity of leukemia cells to a novel anticancer agent Merocil, which induces apoptosis (59, 60, 64). These findings suggest that manipulated ROS levels could switch cancer cells to a pathway that is responsible for cell death. It is noteworthy that ROS act as signaling molecules in diverse physiological processes; thus, increasing the steady-state levels of ROS beyond threshold limits in nontransformed cells could induce bystander effects, including chemoresistant phenotypes and radiation-induced injury in normal cells (140, 265). A significant characteristic of aggressive cancers is their ability to escape treatment and survive in a pre-existing stress environment. Further studies are needed to identify whether the rewired redox state is a pre-existing condition or a consequence of cancer treatment (Fig. 4).

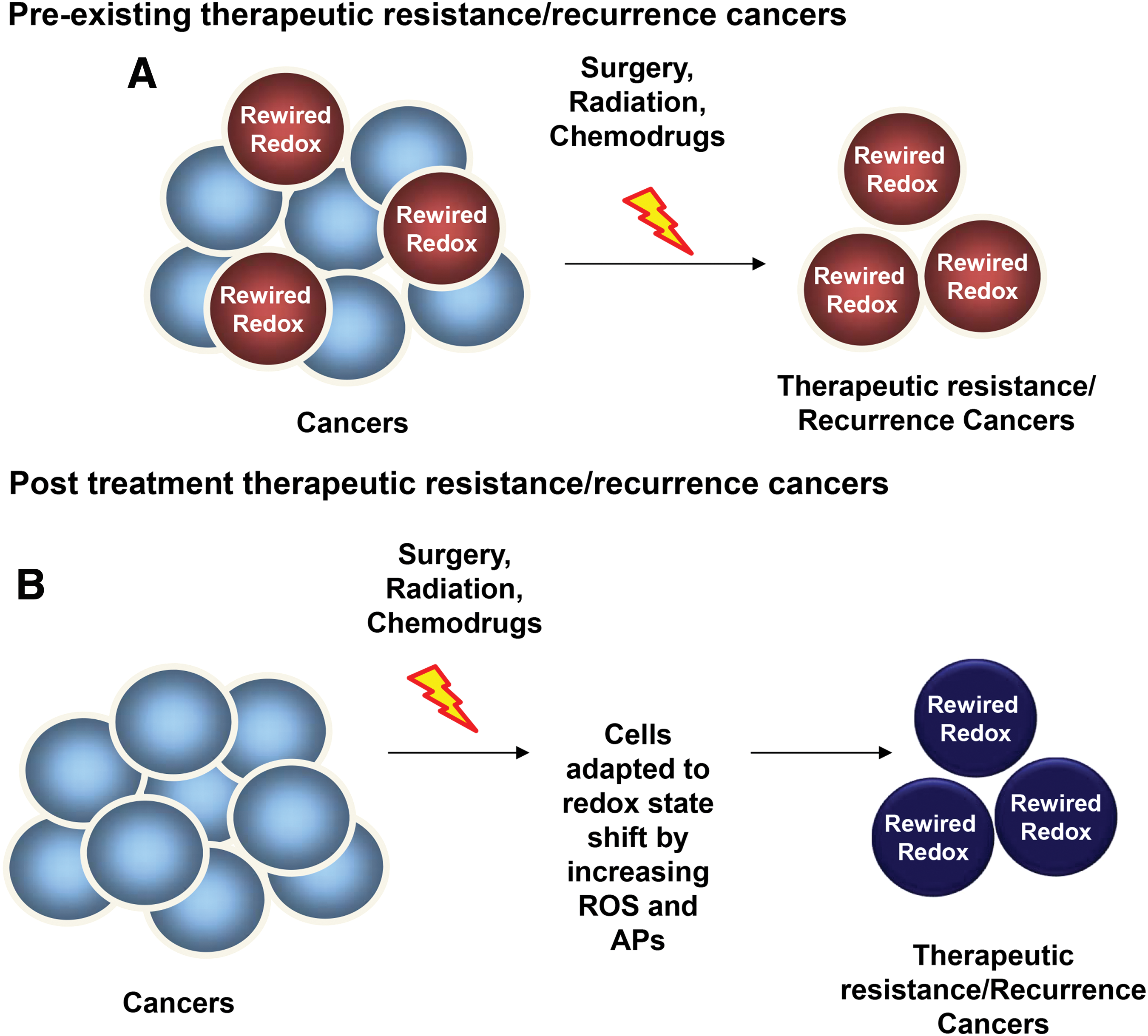
In response to an oncogenic or carcinogenic insult, mitochondria and cell function, in general, adapt by enhancing protective response mechanisms that promote cell survival and maintenance of cell function; thus, the rewired redox state could be correspondingly due to an upregulation of OXPHOS-mediated mitochondrial ROS. A recent study of naive AML patient-derived xenografts proposed that cells with pre-existing and persisting chemoresistant in cells display (i) high levels of ROS; (ii) increased mitochondrial mass; (iii) a high OXPHOS gene signature; and (iv) high OXPHOS activity (91). The upregulation of mitochondrial activities greatly influences cell survival rate. These features correlate with a chemoresistant clinical outcome (e.g., Cytarabine treatment) (91). Thus, pharmacologic manipulation (e.g., Tigecycline treatment) of mitochondrial energetic status toward low OXPHOS increases cell sensitivity to chemotherapy with Cytarabine. In addition, tumor biopsies from melanoma patients with disease progression and melanoma cell lines with acquired drug resistance demonstrate pre-existing high expression of mitochondrial biogenesis genes. Targeting mitochondrial biogenesis proteins using small molecules, such as the HSP90 inhibitor Gamitrinib, overcomes drug resistance in a subset of cell lines via the PK3/AKT-mediated mTORC1 signaling pathway (270, 377). Recent discovery indicates that Akt1 induced macrophage mitochondrial ROS and mitophagy (182); thus, targeting mitochondria could lead to mitochondrial ROS accumulation and induction of mitophagy-induced cell death, an alternative downstream pathway for ROS-induced cancer cell death.
Collectively, these pre-existing events in cancers are destined to recur before, during, and after cancer treatment. In this review, we gathered evidence that demonstrates a correlation between a rewired redox state and aggressive features of cancer, both pre-existing and acquired as consequences of treatment conditions. Special emphasis is given to radioresistant, chemoresistant, and metastasis cancers.
Radiation-resistant cancers
IR induces direct generation of ROS in large quantities (283). IR can increase ROS production both by inducing extracellular water radiolysis and by causing intracellular metabolic changes or damage to mitochondria (16, 24, 314, 352). Mitochondria and NOXs are important sources of ROS, which are also activated by IR, leading to persistent oxidative stress (352, 367). IR induces ROS production in water in the time-frame of seconds (1 × 10–9 s) (164, 337). In contrast, IR delays and persistently increases ROS production in the mitochondria for at least 24 h or longer (370). IR also induces a reversible mitochondrial permeability transition that stimulates ROS production (185). Deactivations of mitochondrial respiratory complexes I and III of the electron transport chain are associated with IR-induced mitochondrial ROS production (16, 370). These accumulations of ROS lead to an oxidative stress and a rewired redox state that is associated with higher AP levels to scavenge ROS (24, 93, 164). We reported that IR induces double-strand breaks that stimulate the NF-κB pathway, which generates positive feedback loops via cytokine production, and, in turn, activates DNA repair mechanisms (126). The cytokine-activated NF-κB pathway can also lead to induction of APs, which protect cancer cells against ROS-generating therapeutics.
To study the correlation of redox state and radioresistant cancers, a series of radioresistant cancer cell lines have been developed by exposing parental cancer cells to high doses of fractioned IR. These radioresistant sublines demonstrate higher clonogenic survival, proliferative activity, and motility than the parent line after radiation exposure. Interestingly, these radioresistant subclones demonstrate a significant change in redox state and redox-associated protein profiles. For example, radioresistant prostate cancer cell lines PC3RR exhibit higher ROS and MnSOD levels than parental cells (manuscript in preparation). Further, proteomics has identified an increase of Prx6 in PC3RR, DU145RR, and LNCaRR cells compared with the parental cells (51). MnSOD is located in mitochondria, whereas Prx6 is a 1-Cys Prx protein that is located exclusively in the cytosol. A high level of ROS from radiation could induce MnSOD and Prx6 expression as an adaptive response to protect radiation-mediated oxidative stress insult. In addition, increases of MnSOD and Prx6 have been associated with biochemical recurrence in prostate cancer patients after radical prostatectomy (275) as well as with fetal esophageal development (124, 257). Thus, these two APs could be used as markers for the radioresistant phenotype in cancers. Consistent with other studies, the radioresistant human esophageal adenocarcinoma cell lines (OE33R) demonstrate a slight increase in the G2 M phase with a significant increase in GSH level (211, 212) compared with parental cells (OE33). It has been established that 20 min post-irradiation, the level of GSH in the irradiated parent cell line is much lower than that of the radiation-resistant subline, suggesting that the newly produced subline retains a greater ability to preserve GSH level. Correlatively, inhibiting GSH synthesis with L-buthionine-sulfoximine (BSO) has been shown to restore the radiation sensitivity of the radiation-resistant human small cell lung cancer H69/R38 radiation-resistant subline (137). This evidence indicates that an increase of GSH levels could be a possible cause for radiation resistance.
As mentioned earlier, MnSOD is one of the APs that plays a role in the radiation sensitivity of cancer cells. Several studies suggest that the cell cycle/phase-specific radiation response is regulated via MnSOD activity (Fig. 1A). IR decreases MnSOD activity in human lymphocytes at G2/M phases whereas it increases MnSOD activity in the G0 phase (53). Treatment with SOD suppresses IR-induced chromosomal aberration at G2 M phases in human lymphocytes (26, 53). Based on cell cycle/phase-specific studies, periodic changes in MnSOD activity are higher during the G1 phase, which is associated with radioresistance. Further, radiosensitization of cells in the G2 phase correlates with lower MnSOD activity (26). Qu et al. (274) reported an increase in MnSOD activity and radioresistant properties in CNE1 human nasopharyngeal carcinoma when compared with the radioresistant CNE2 cell line. Inhibition of MnSOD expression in MCF7 human breast cancer cells decreased cyclin B1, cyclin D1, and p21 expression after IR (123, 194, 324). Interestingly, a study also reported that radioresistance is associated with a positive feed-forward cycle with H2O2 elevation after MnSOD expression (246). In a study by Josson et al., nuclear localization of RelB; which correlated with increased MnSOD expression and radioresistance (163), was significantly higher in aggressive PC3 prostate cancer cells compared with less aggressive LNCaP cells (163). Inhibition of MnSOD by RelB-specific siRNA, that is, overexpression of a dominant-negative p100 (163) or treatment with the peptide inhibitor SN52 (to prevent nuclear translocation of RelB) (365), resulted in a decrease in radiation-induced MnSOD expression and an increase in radiosensitivity in PC3 cells. Similarly, knockdown of MnSOD decreased radioresistance in CNE1, whereas scavenging of H2O2 by CAT expression or treatment with NAC abolished MnSOD-induced radioresistance (273, 274). Interestingly, Gao et al. recently reported that the overexpression of CuZnSOD in U118–9 human glioma cells increases radioresistance compared with vector control and parental cells (111), suggesting that CuZnSOD can also confer radioresistance in cancer cells. Due to their subcellular specificity, the effect of these SODs on radiation sensitivity could be due to cancer-type-specific and subcellular sites of ROS production.
Although the studies mentioned earlier suggest a radioresistance role for MnSOD, other studies show that targeting MnSOD could lead to radiosensitization of cancer cells. We had reported that when we used MnSOD-overexpressing Fsa-II cells implanted in mice, this resulted in a significant reduction in the radiation dose required to control one-half irradiated tumors compared with control mice (339). Overexpression of MnSOD by transfection with an MnSOD cDNA-expressing plasmid/liposome complex (MnSOD-PL) proved effective in increasing radiosensitivity of SCC-VII mouse SCC cells (D0 = 1.244 Gy compared with 3.246 Gy for control cells). The combination of MnSOD-PL with the EGFR inhibitor, Gefitinib, further increased radiosensitization (D0 = 0.785 Gy) (88). The administration of recombinant MnSOD (rMnSOD) was also effective in enhancing radiosensitivity of cancer cells (29). Together, these results suggest that cellular and mitochondrial ROS generations are essential components of IR and that the different redox states of cancer cells may contribute to the seemingly paradoxical role of APs, that is, MnSOD or GSH in radiosensitivity phenotype.
Chemoresistant cancers
Typically, the antineoplastic drugs that are currently used for cancer chemotherapy induce high levels of oxidative stress (17, 66). For example, anthracyclines (doxorubicin) generate ROS via the reduction of anthracycline to semiquinone-free radicals by the microsomal enzyme p-450 reductase, hypoxanthine-xanthine oxidase, cytochrome b5 reductase, nitric oxide synthase, or NADH dehydrogenase at mitochondrial complex I (71, 112, 343). In the presence of O2, redox cycling of semiquinone-free radicals results in the production of O2 •−. Taxanes (paclitaxel and docetaxel) interfere with the electron transport chain and result in the production of O2 •− (15). 5-FU generates mitochondrial ROS via a p53-dependent pathway (205). Platinum-related compounds (carboplatin and cisplatin) are converted into a highly reactive form on entering the cell, which can react rapidly with thiol-containing molecules and shift the cellular redox state to oxidative stress (66, 107). Platinum-related compounds may induce mitochondrial dysfunction and increase ROS production via the disrupted respiratory chain and cytochrome P450 (346). Accordingly, patients who receive chemotherapy often exhibit signs of ROS-induced lipid peroxidation (LPO) in their plasma (17). Elevated oxidants in the circulations of cancer patients have been reported after administration of the anthracycline epirubicin (217, 229). Markers of LPO are elevated in both plasma and intestines of rodents after irinotecan administration (347).
Chemoresistant mechanisms include enhanced expression of survival signaling transporters that increase drug efflux, alterations in drug metabolism, mutations of drug targets, and potential rewiring of the redox state. As mentioned earlier, some cancer cells can overcome drug-induced oxidative stress by enhancing their AP systems and establishing a higher ROS level (Fig. 2), especially after treatment with ROS-inducing anticancer drugs. For example, Kumar et al. demonstrated that doxorubicin resistance was associated with increased MnSOD expression in a model of basal breast cancer cell lines (180). Chen et al. indicate that MnSOD-mediated NF-κB activation confers cisplatin resistance in lung adenocarcinoma via the NF-κB/Bcl-2/Snail pathway (55). As a result, BCL-2 inhibitor (ABT-199) or NF-κB inhibitor (curcumin) may be potentially useful to improve tumor regression and chemotherapeutic response in patients with MnSOD/BCL-2-positive tumors. Collectively, MnSOD can serve as a biomarker for identifying drug-resistant cancer cells.
It has been shown that the more APs in the cell, the greater the resistance to the chemotherapeutic agents. Among APs, increased levels of GSH constitute a commonly recognized rewired redox state in a variety of chemoresistant cancers. In addition to antioxidant activity, GSH acts as a detoxificator agent by directly forming a GSH-drug conjugate, that is, paclitaxel, carboplatin, and bortezomib (6, 224). Accordingly, platinum drugs, which generate extremely high ROS levels, can be inactivated by GSH (224). Moreover, the increased GSH level can form glutathione S-conjugated molecules to facilitate drug efflux by MRP1 (14). Therefore, pretreatment with BSO significantly increases paclitaxel cytotoxicity through ROS accumulation (6).
A family of cell membrane transporter proteins has been implicated in chemoresistance, especially via promoting drug efflux (142). The elevation of oxidative stress in a chemoresistant cancer could regulate drug transporter expression at the transcriptional, translational, and post-translational stages. The ATP-binding cassette (ABC) transporter family, including multi-drug resistance protein 1 (MDR1), MRP1, and breast cancer resistance protein (BCRP), is the most extensively studied for chemoresistant cancer. All ABC transporters contain four domains: two nucleotide-binding domains and two transmembrane domains. It is noteworthy that at a lower dose, ROS increased MDR1 expression in Caco-2 colon cancer whereas at higher doses, ROS negatively regulated the MDR1 expression (204). Drug transport activity of human MDR1 is correlated with the redox states of its two cysteine residues (Cys431 and Cys1074) (156). Mutations at certain cysteine residues within MDR1 or dimerization of MRP1 drastically reduce drug-transport activities. Yang et al. investigated the roles of Cys7 and Cys32 in the MRP1 and found that the mutations at Cys7 caused conformational changes and prevented dimerization in MRP1 (368). Traditionally, Cys592 and Cys608 in BCRP are located on the extracellular face and are critical for protein stability by forming an intramolecular disulfide bridge in the ABC transporter (214). Liu et al. demonstrated that mutations at these two cysteine residues result in protein misfolding and degradation, thereby impairing the drug elimination systems (200). Recently, Cys284, Cys374, and Cys438 were reported to be involved in intramolecular disulfide bond formation and necessary for BCRP function (201). In addition to direct regulation by the redox state, MDR1 could be indirectly regulated through PKC and protein kinase A (PKA) (226). Brennan et al. demonstrated that the formation of PKA intramolecular disulfide bonds is activated by redox modifications that cause a subcellular translocation and result in phosphorylation of established protein substrates (34). Giorgi et al. further demonstrated that PKC catalytic properties could be altered by redox mechanisms, which, in turn, influence the activity of MDR1 (114). Activation of PKC has been reported to increase the phosphorylation of MDR1 in multidrug-resistant MCF-7 breast cancer cells (27), which results in a decrease in drug accumulation and sensitivity.
On the contrary, selected studies indicate a negative relationship between ROS and chemoresistant phenotypes of cancer. For example, overexpression of NOX1 in prostate cancer cells significantly decreases expression of the MDR transporter P-glycoprotein (353). Likewise, the addition of ROS-producing agent, emodin, inhibits MDR expression and increases retention of doxorubicin (145). Collectively, identifiable alterations of the redox state network in chemoresistant cancers offer excellent knowledge of the drug's mechanism as well as novel interventions that could counteract the off-target effects.
Metastatic cancer
In most cancers, the tumor environment is a key factor that controls metastasis and angiogenesis. Due to diffusibility and abundance, ROS potentially provide a permissive environment for cancer development. The metastatic abilities of many types of cancers types, including prostate, breast, and ovarian, positively correlate to cellular redox state (278). ROS generation commonly activates growth factors and integrin, a cell-surface-adhesion receptor that promotes attachment to extracellular matrix and assists in cell invasion. Redox imbalance can facilitate both growth factor and integrin signaling by redox-dependent activation of PKC, which leads to activation of the mitogen-activated protein kinases (MAPK) signaling cascade and tumor cell migration (168). Alternatively, PKC is involved in H2O2-dependent suppression of PTP, leading to MAPK pathway activation (105). In addition, accumulated evidence also indicates that mitochondrial ROS play a critical role in the regulation of integrin and MMPs. Nelson and Melendez have proposed a possible link between MMP1 and MnSOD in promoting cancer metastasis. MnSOD-derived mitochondrial H2O2 enhanced MAPK signals via Ras, which, subsequently, activated c-Jun, C-Fos, Fra-1, and Ets-1. These transcription factors increase the transcription of MMP1 (244, 357). Li et al. showed that overexpression of MnSOD in glioma cells resulted in elevated MMP1 and MMP9 levels (56, 192). Chen et al. reported that the upregulation of FoxM1-MMP2 axis by MnSOD promotes lung tumor invasion (56). These studies corroborate the role of mitochondrial ROS in the regulation of cancer invasion/migration.
An increasing body of evidence points to the importance of the extracellular redox state as a contributing factor to metastatic development. It is well known that O2 •− is produced at plasma membranes by NOX1 (97). O2 •− can also be produced when Cys is oxidized to become CySS (260). O2 •− can be converted to H2O2 or reacts with NO• to produce ONOO−; both molecules can act locally or diffuse across plasma membranes to cause oxidation of proteins and DNA damage. In line with this, OH•, which is the most highly active of ROS, has also been linked to invasiveness and metastasis in lung cancer cells by upregulating the expression of caveolin 1, which is a structural protein component of the plasma membrane that functions in vesicular trafficking (209, 280). Our study of prostate cancer cell lines demonstrated a slight increase in extracellular H2O2 levels in the highly aggressive WPE1-NB26 prostate cancer cells when compared with immortalized RWPE1 prostate epithelial cells (47, 49). Using xanthine/xanthine oxidase to produce O2 •−in the media, we demonstrated that O2 •− increases the invasive capability of WPE1-NB26 prostate cancer cells, at least partially through an induction of MMP2 and membrane type 1 (MT1)-MMP activities (47). MMPs can be activated by both intracellular and extracellular ROS. Induction of invasiveness by the cancer cells could be due to the production of O2 •− at the cell membrane, in the extracellular space, or both. In fact, we demonstrated that prostate cancer invasion and metastatic progression might, at least in part, correlate with overexpression of NOX1 and downregulation of ECSOD. Blocking NOX1, overexpressing ECSOD, and increasing the redox potential of Cys/CySS attenuated MMP activities of prostate cancer (48). Further, ROS-mediated induction of the cell adhesion molecule ADAMs via p38-MAPK was identified in prostate cancer (321). In line with this, insulin-stimulated ROS can activate the extracellular signal that regulates MEK/ERK and PI3K/AKT kinase signaling pathways and promote cancer metastasis via HIF-1α and vascular endothelial growth factor (VEGF) expression (106); these activities result in enhanced angiogenesis and vascular permeability. In addition, ECSOD-derived H2O2 can promote VEGF signaling in caveolin-enriched lipid rafts and stimulate endothelial cell migration and proliferation through oxidative inactivation of PTPs (254). Similarly, NOX1 induces an H2O2-mediated increased expression of VEGF, the VEGF receptor, and the activity of MMPs, thereby promoting the processes of neovascularization and metastasis in cancer (340). It has been reported that generation of H2O2 by NOX4 mediates endothelial cell proliferation, whereas NOX2 prevents apoptosis and promotes endothelial cell survival (70, 101). Interestingly, NOX4 contributes ∼30% of extracellular H2O2 in the vascular system (35). Xia et al. demonstrated that NOX4 knockdown decreased the expression of VEGF and HIF-1α and tumor angiogenesis in ovarian cancer cells (362). Regardless of the source of ROS production, several studies suggest that extracellular H2O2 plays a role in the regulation of vascular function, stimulation of smooth muscle cell proliferation, and angiogenesis in cancer formation (385). In cancer cells, NO• production may increase blood flow, resulting in increased oxygen delivery and increased angiogenesis. On the contrary, NO• production in cancer cells may also inhibit tumor cell growth and invasion through an inhibition of MMP activity (341). We demonstrated that the invasive ability of the highly aggressive WPE1-NB26 prostate cancer cells was decreased after treatment with SNAP, NO• -donor compounds (47, 49). However, administration of NO•-donor compounds to animals caused a quick decrease in blood pressure that was problematic for in vivo studies. At present, it is not clear whether increasing or decreasing NO• levels would be more beneficial to cancer patients; further and better-designed studies would help clarify this void.
ROS/RNS have been associated with invasion, angiogenesis, and acidosis in extracellular spaces. It is very likely that their contribution to metastasis could be rewired under the control of intracellular and extracellular redox states. For this reason, specific compartmental redox states in cancers need to be precisely defined to properly investigate their role in the regulation of metastatic cancer.
H2O2: A Bona Fide Molecule for Cancer Treatment
ROS have been implicated as primary and secondary messengers to regulate cancer cell growth. Among them, H2O2 is a potential candidate as a key molecule that decides the fate of cancer survival. H2O2 is the two-electron reduction product of O2. At least 30 enzymes have been identified as H2O2-generating enzymes (33, 190); among them are the enzymes xanthine oxidase, NOX, SOD, and flavoprotein dehydrogenase. Sites for H2O2 generation include mitochondria, peroxisomes, and cell membranes (33, 190). Because of its physiochemical properties, H2O2 is capable of serving as a messenger that carries a redox signal from the site of its generation to a target site. There are H2O2 gradients even within subcellular organelles. H2O2 on the external side of the mitochondrial inner membrane, as well as in the matrix, originates largely from mitochondrial complex III; whereas mitochondrial complexes I and II exclusively contribute to the mitochondrial matrix H2O2 (33). Overall intracellular H2O2 steady-state levels are provided in Figure 1 and Table. 1. Dr. Helmut Sies identified the physiological range of intracellular H2O2 concentration as approximately 0.01–10 nM (310). These concentrations are dependent not only on how much H2O2 is increased by stimulators but also on the steady-state levels and capacity of APs. The fluctuation in H2O2 concentration is reportedly regulated by the circadian rhythmicity of Prx (86, 171). However, overoxidation of Prxs by H2O2 could lead to inactivation of Prxs and form PrxSO3. H2O2 modulates the activity of the following transcription factors: AP-1, Nrf2, CREB, HIF-1α, p53, NF-κB, SP1, and signaling for epithelial-mesenchymal transition (EMT) (219). Increased H2O2 levels can have either a positive or negative effect on cell growth, depending on the levels and the subcellular location of increased H2O2. Laurent et al. (183) demonstrated that in nonmalignant NIH/3T3 cells, the basal levels of H2O2 are low and that increased levels of H2O2 correlate with cell growth (159, 219, 361). Increased levels of H2O2 at the cell surface are largely mediated by a growth factor mechanism(s), resulting in the activation of cell cycle progression (281, 282, 320). On the other hand, increased levels of H2O2 within mitochondria of SV-40 transformed fibroblast cells resulted in the inhibition of cell cycle progression (83). Increased levels of H2O2 in mitochondria inhibit cell cycle progression as a protective mechanism to prevent cell replication when the potential for DNA damage is high. Many cancer cell types display a similar dichotomy and are stimulated to undergo cell division at low concentrations of H2O2, whereas higher concentrations cause inhibition of cell growth and, eventually, cell death.
It has been shown that the ratio of O2 •− to H2O2 determines cancer fate. A predominant increase in O2 •− (oncogenic ROS) supports cell survival and promotes oncogenesis, whereas a tilt in favor of H2O2 (onco-suppressor) induces cell death signaling (266). Evidently, H2O2 can induce either cell proliferation or cell death, depending on its levels, with specific thresholds in specific cell types. H2O2 also acts as a regulator and sensor of Prxs and sulfiredoxins in cancer development. The review by Mishra et al. (232) addressed the fact that the hyperoxidation of Prxs, paradoxically by H2O2, acts as a chaperone as well as an antioxidant that regulates different signaling pathways, such as DEP-1 and VEGF, which may function as a double-edged sword in tumorigenesis. Reactivity of thiol proteins toward H2O2 spans several orders of magnitude from a low of 20 M −1s−1 for some protein tyrosine phosphatases such as PTP1B, to a high of 107 M −1s−1 for Prx2. For H2O2 signaling, concentrations lower than 1 μM are probably mediated by a highly reactive protein such as Prx2. Prx2 acts as a primary H2O2 receptor that specifically transmits oxidative equivalents to STAT3, thus forming a redox relay for H2O2 redox signaling (316). Certain cysteinyl residues in Prxs or selenocysteinyl residues in GPx are highly reactive to H2O2; the second-order rate constant for the reaction is ∼107 M −1s−1 instead of 1 M −1s−1 as in Trx or GAPDH (308, 360). For H2O2 concentrations higher than 1 μM, the mediation of signaling by proteins with different reactivity toward H2O2 is feasible, but mediation by a high or low reactive protein is not equivalent, as the response time changes. For instance, H2O2 in the micromolar range regulates PTP by prolonging activation of the growth factor, which leads to an aberrantly enhanced proliferation of tumor cells (186). The release of pico- to submicromolar H2O2 disrupts the tyrosine phosphorylation network but also participates in immune defense against infection (7).
However, the absolute concentration at which H2O2 activates these pathological effects has not been firmly established. Herein, we addressed the range of H2O2 concentration with a selectively assay method that could potentially be applied for determining the subgroup of cancers that is suitable for personalized redox-based cancer therapy. The dual roles of H2O2 as a source of oxidant stress at high concentrations and as a signaling molecule at low concentrations are analyzed in terms of threshold concentrations (Fig. 5). In view of the wide range of possible H2O2 concentrations in cancer cells, the measurement of H2O2 concentration has not been well established. Accumulated evidence suggests that H2O2 concentrations >100 nM are pathologic (9, 85, 144, 253, 310, 348). Several reasons are possible for the discrepant H2O2 levels in the cancer studies, including (i) a variety of different cell lines were used; (ii) variation in the method utilized; and (iii) intracellular compartmentalization of antioxidants could result in lower estimates of H2O2 concentration. Several studies, including ours, measure total intracellular H2O2 fluxes using 3-aminotriazole (3AT)-mediated inactivation of CAT, which is one of the few assays that quantitates the absolute concentration of H2O2. 3-AT forms a covalent bond with CAT (referred to as “compound I”), rendering it inactive. The extent of CAT inhibition in the presence of 3-AT is dependent on the initial concentration of H2O2. Since the reaction is typically described by a pseudo-first-order kinetics, the rate of CAT inhibition by 3-AT can be used to estimate H2O2 concentration in the cells (50). Because the only known molecule capable of converting CAT to Compound I is H2O2, this method provides a very specific estimate of steady-state H2O2 fluxes inside the cells. Rewardingly, H2O2 concentrations in normal counterparts and cancer cells measured by this method vary from 5 to 50 pM (4, 69, 85, 253, 348). Based on data from our laboratory, H2O2 steady-state levels of radioresistant prostate cancer PC3 cells are 1.8-fold higher compared with parental PC3. Similarly, H2O2 steady-state levels in carboplatin-resistant serous ovarian cancer OV-90CD cells are 1.7-fold higher compared with parental serous ovarian cancer OV-90 cells (manuscript in preparation). Metastatic bladder tumor cells display a nearly two-fold (18–31 picomolar [pM]) increase of H2O2 compared with their nonmetastatic parental counterpart (136). Remarkably, the H2O2 concentration in these cells is significantly increased after exposure to cytotoxic compounds. Based on the accumulated evidence, we propose that elevation of intracellular H2O2 ≥ 20 pM from H2O2 steady-state concentration or an increase in H2O2 concentration by at least 1.5-fold is required to induce toxicity to these advanced cancer cells (Fig. 5). Consistent with our speculation, Wagner et al. demonstrated that the steady-state H2O2 concentration in prostate cancer PC3 cells is 13 ± 4 pM; when the H2O2 concentration increases to 51 ± 13 pM after exposure to 1 μM Doxorubicin, the viability of PC3 cells is significantly decreased (348). Olney et al. established that the H2O2 concentration in pancreatic cancer MIA PaCa-2 cells was 43 ± 5 pM and it increased to 71 ± 15 pM after treatment with 20 mM Ascorbate acid treatment (253). Based on these studies, H2O2 can act as a tumor suppressor. However, these H2O2 concentrations are lower than H2O2 steady-state levels that were reported when other methods were used (10). We anticipate that it will be possible to predict the sensitivity of cancer cells to redox-based therapy based on the absolute change in H2O2 concentration or range of H2O2 concentrations using the same measurement methodology.
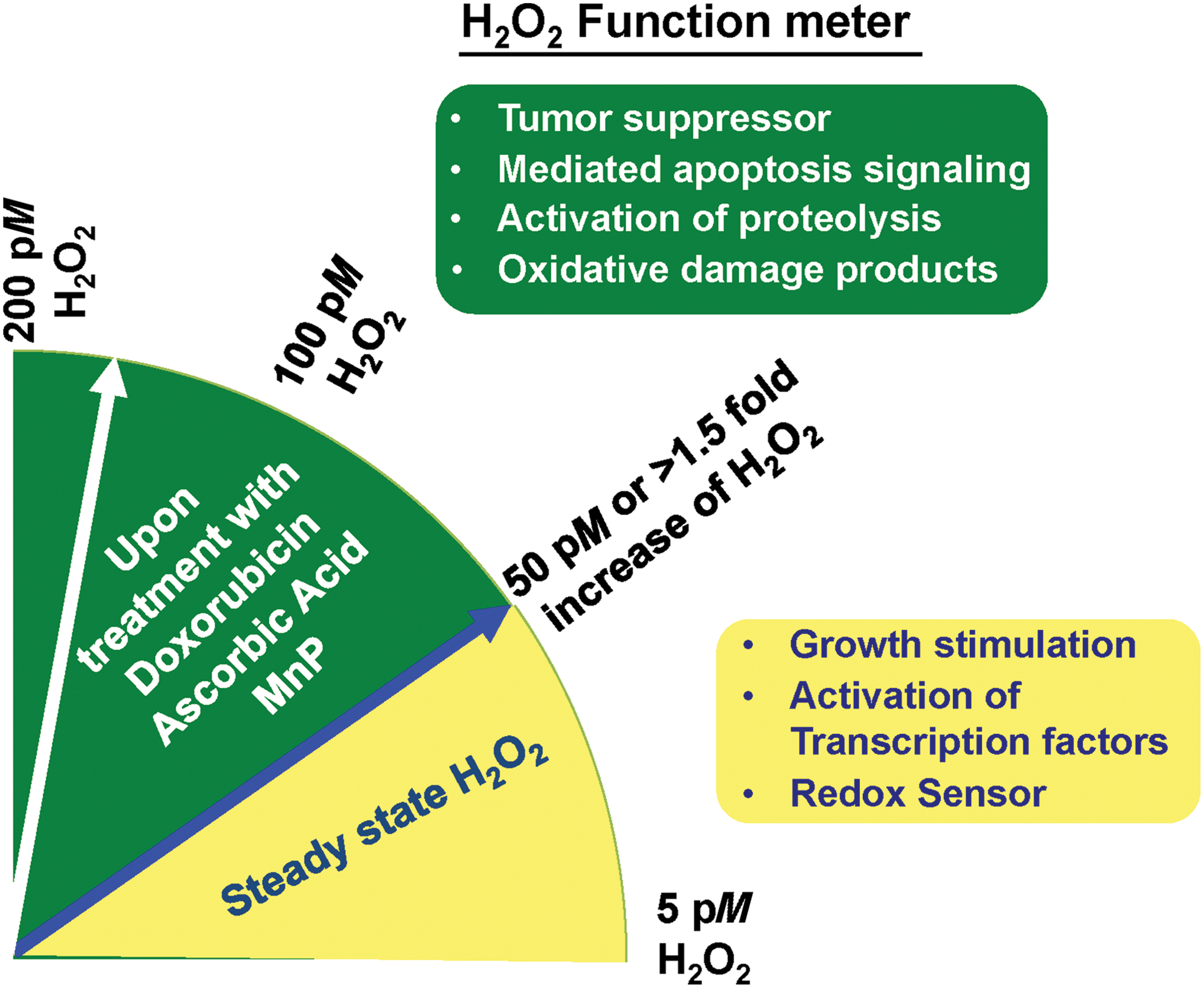
According to recent publications, H2O2 concentration in blood plasma is ∼1–1000 μM (98), which would be >100-fold higher than inside the cells. The assays and concentrations of extracellular H2O2 have been summarized elsewhere and will not be elaborated on here (98, 218). When H2O2 is added externally, the intracellular concentration of H2O2 is lower than that applied extracellularly, and a gradient across the plasma membrane is established (10). If an H2O2 gradient across the plasma membrane of 20 is established, the addition of extracellular H2O2 at 100 μM would correspond to an intracellular concentration of 5 μM. The effect of a gradient magnitude of this degree is unknown and is also dependent on the cell type and whether extracellular APs, that is, Prx, Cys, ECSOD, are active at the external H2O2 concentration applied in the experimental design. The presence of active APs increases gradients by approximately two orders of magnitude, from values lower than 10 to those in the 650–1000 range (3, 10, 72, 144). In this case, the APs in the extracellular space not only relay the oxidative signal downstream but also will trap H2O2. Further, it has been reported that adding H2O2 in the micro to millimolar concentrations leads to an increase of intracellular H2O2 only in the pM range (348). The same study also supports a proficient ability of thiol couples and APs inside cells to remove the H2O2 originating from the extracellular environment; alternatively, an inability of H2O2 to diffuse intracellularly or be transported across the plasma membrane may account for the minimal change in H2O2 concentration (3, 10).
Although it was initially believed that H2O2 could freely diffuse across the plasma membranes, recent genetic evidence suggests that some membranes are poorly permeable to H2O2 and that its transport may be regulated by specific aquaporins (AQP) channel proteins. Thus, extracellular H2O2 may remain outside the cancer cell because of limited diffusion, resulting in higher levels of extracellular H2O2 (25). AQP3 mediates H2O2 uptake to regulate downstream signaling at the cell membrane, whereas AQP8 mediates uptake at the mitochondrial membrane (40, 132). Knockdown of mitochondrial AQP8 in human hepatoma HepG2 cells causes loss of viability (40). Cellular stress conditions reversibly inhibit the permeability of AQP8, providing a novel mechanism for regulation of cell signaling (40). This discovery opened an exciting field for regulation of H2O2 transportation in cancer cells.
In summary, H2O2 is a possible candidate marker of redox shift therapy because of its relative stability. Traditional targeted cancer therapy routinely inhibits expression of a marker. Ironically, the use of H2O2 as a target molecule for cancer treatment by increasing its production beyond a cancer cell's antioxidant capacity is preferred for inhibiting its production. Many antitumor agents exhibit antitumor activity through H2O2-dependent activation of apoptotic cell death (90); thus, the use of an extracellular H2O2-generating system has been proposed as an alternative approach for the development of H2O2-dependent antitumor therapy. It is possible that chemotherapeutic strategies that both augment metabolic H2O2 production and limit ROS detoxification may allow for H2O2 to exceed these pico-nanomolar ranges and drive tumor cell death. However, if the APs for removing H2O2 are particularly efficient, the final concentrations of H2O2 that are needed to execute these advanced cancer cells will be significantly higher than pico-nanomolar ranges. Future research should include targeting AP systems that are responsible for removing H2O2 to determine whether the cytotoxicity induced by H2O2 can be increased further. Although encouraging progress has been made in understanding the biological significance of low levels of H2O2 in cells and tissues, much is yet to be discovered. Lastly, documentation of the effective final concentrations of H2O2 in cancer cells is pivotal for utilizing pro-oxidants in cancer therapy. Therefore, quantitative measurements of H2O2 with improved sensitivity, specificity, and selectivity promise better insights into H2O2 concentration.
New Prospect for an Old Concept: Shifting Redox State Using Redox Cycling Compounds as a Radical Strategy for Cancer Treatment
As discussed earlier, ROS can both assist and limit cancer cell proliferation. Although cancer cells rewire the redox state by increasing their production of ROS, some cell lines engage antioxidant networks to ensure that ROS levels do not surpass a toxicity threshold. Based on the distinctive differences in H2O2 and redox potential, past attempts have focused on using low-molecular-weight redox modulators to (i) increase ROS/RNS formation without changes in antioxidant defenses or (ii) sustain ROS/RNS formation but decrease antioxidant defenses. A number of agents and approaches have been developed to shift the intracellular redox state for cancer treatment. We previously demonstrated that 50 μM diphenyleneiodonium (DPI), 5 μM menadione, 50 μM H2O2, and 500 μM BSO significantly inhibited the viability of the prostate cancer cell lines, LNCaP and PC3 (45). Olney et al. (253) demonstrated that 25 μM BCNU, a GR inhibitor, significantly decreased colony formation in human pancreatic cancer (AsPC-1) cells from 45% to 18%. Iskandar et al. indicated that small compound, C1, preferentially targeted mutant K-RAS-driven cancers via induction of ROS and activation of AKT/PKB, which served as the signal for the simultaneous induction of autophagy and apoptosis (151). In this regard, a subset of mutant K-RAS cancer is more sensitive to increased intracellular ROS production. These studies are supportive of the central question as to whether low-molecular-weight redox modulators could be used for cancer treatment. Alternatively, redox-recycling compounds used as a single agent in cancer treatment may not be effective because of the decomposition of H2O2 by the APs system, including CAT, Trx, or GSH. Administration of an H2O2-generating system in combination with the AP inhibitor may improve antitumor efficacy. Zhou et al. demonstrated that the addition of arsenic trioxide, a compound capable of ROS production, and 2-Methyoxyestradiol (2-ME), an SOD inhibitor, significantly enhanced cytotoxic activity in primary and 2-ME-resistant chronic lymphocytic leukemia cells (384). Phase I/II trial of the ascorbic acid (AA) and arsenic trioxide combination (NCT00275067), which is associated with the depletion of GSH, is reported to be effective for relapsed or refractory multiple myeloma in a clinical study (13).
For redox-based therapy to be successful, it is important to use the appropriate ROS and ROS-generating system. Therefore, it is not surprising that the treatment of cancer with dietary antioxidants has been successful in some studies whereas it has been ineffective or detrimental in others (215). For example, treatments of cancer cells, including prostate cancers (DU145, PC3), bladder cancers (U20S, U987), leukemia (SUDHL.4, HBL-1), and ovarian cancers (OVCAR-3 and Caov-3) with the ROS-generating natural compounds plumbagin and resveratrol, were associated with cell death in these cancers (215). Interestingly, it was reported that resveratrol improved the therapeutic effects of arsenic trioxide in lung cancer cells via ROS-dependent ER stress and apoptosis (54). Noteworthy, the outcome of resveratrol treatment was also dependent on the concentration given and the genetic phenotype of cancers. For example, low concentrations of resveratrol (4–8 μM) activated NOX-mediated O2 •− production, which, in turn, blocked caspase activation, DNA fragmentation, and translocation of cytochrome c induced by anticancer drugs (5). A mini-review about resveratrol has been addressed by Pervaiz and Holme (267). Further, supplementation with carotenoids increases mortality in breast cancer patients (121), whereas supplementation with vitamins C and E is associated with a reduced recurrence rate (67). Supplementing with vitamin C also potentiates the anti-proliferative effect of doxorubicin in breast cancer (122), whereas supplementing with vitamin D is associated with increased survival in colorectal cancer patients (245). In contrast, the very large “Selenium and Vitamin E Cancer Prevention Trial” (SELECT), comprising 35,533 men from 427 study sites in the United States, Canada, and Puerto Rico, found no initial reduction in the risk of prostate cancer in healthy individuals taking either selenium or vitamin E supplements (338). Another study showed that NAC promotes melanoma progression by increasing melanoma metastasis in vivo through small guanosine triphosphatase activation (184).
In principle, the thermodynamic disequilibrium of redox couples, APs, and ROS/RNS levels within and between subcellular compartments is the determinant of cellular redox state. We believe that concurrently targeting the redox state of each subcellular compartment would provide ultimate redox-based targeted therapy for cancer treatment. Ideally, redox-recycling compounds would be used in combination with well-known chemotherapeutics to target the subcellular compartments with less effective or inaccessible chemotherapeutic drugs (Fig. 6). For example, cisplatin-resistant cancers exhibit a high level of GSH. Cisplatin-GSH complexes provide less effective cisplatin-induced DNA damage, accumulation of endogenous ROS, and rewired redox state within the cells, which are phenotypic features of drug-resistant cancer. To overcome this commotion, the combination treatment of mitochondrial SOD mimetic (MnP, regulated mitochondrial ROS) with platinum drugs would instantaneously induce both DNA damage and mitochondrial stress. The capacity of cancer cells to respond to oxidative stress is impaired when multiple subcellular compartments are involved and in response, several apoptosis-induced cell death pathways are activated concurrently. Further, MnP [Mn(III) meso-tetrakis(N-n-butoxyethylpyridinium-2yl) porphyrin] can conjugate with GSH (153, 154), thus potentially preventing the cisplatin-GSH conjugates by competitively binding GSH. It is conceivable that by increasing subcellular compartment ROS levels beyond the antioxidant capacity of cancer cells, MnP offers a therapeutic opportunity for treating cisplatin-resistant cancer.
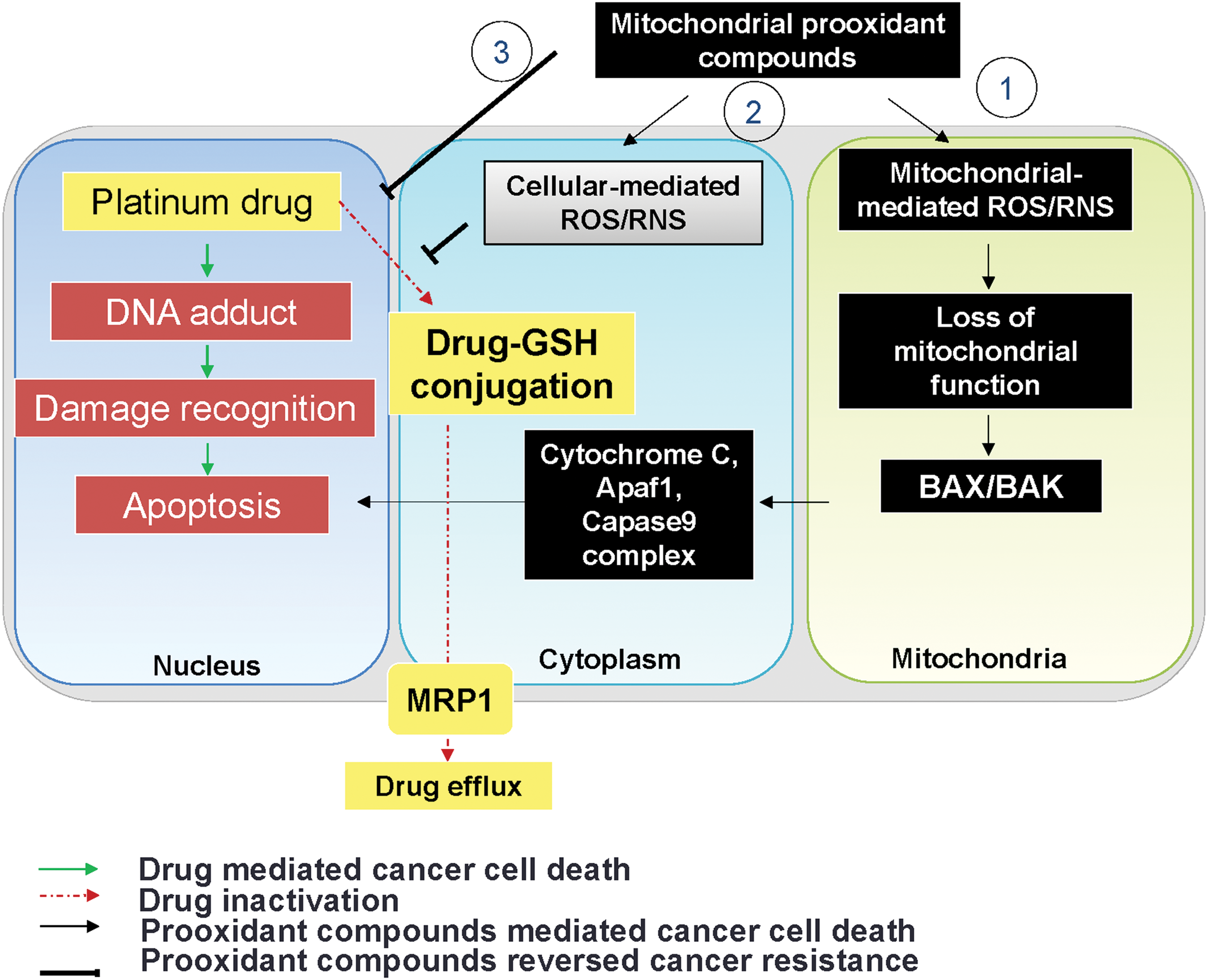
In this section, we review several redox-cycling compounds as prototypes that have the potential to become a pro-oxidant under oxidizing conditions to improve the efficacy of radio/chemo/target therapies by sensitizing cancer tissues while simultaneously protecting normal tissues against their side effects.
MnSOD
MnSOD is a mitochondrial enzyme that has at least two major functions. One is an antioxidant function, a defense mechanism that prevents the accumulation of damaged products that results from excess O2 •−. At the same time, it converts O2 •− to H2O2. The activity of MnSOD is a crucial element for controlling the amount of ROS in the mitochondria. The difference between aggressive cancer versus normal cells with respect to the increases of MnSOD and oxidative stress led to the recognition that antioxidants that undergo redox cycling can act as pro-oxidants and can be unique tools for stimulating different responses in normal and cancer cells. MnSOD has both tumor-suppressive and -promoting functions (174). The dichotomous role of MnSOD in cancer is primarily related to whether it functions as a mitochondrial ROS scavenger or an H2O2 generator at the certain stage of cancer. If MnSOD is an H2O2 generator, overexpression of MnSOD would inhibit cancer growth. However, if MnSOD is a mitochondrial superoxide scavenger, inhibition of MnSOD would enhance cancer cell death. For example, decreases in MnSOD's enzymatic activity and mRNA expression levels correlate with an early stage of cancer, whereas overexpression of MnSOD is correlated with aggressive cancers, that is, a drug-resistant subtype (30, 139). It has been shown that acetyl mimetics of MnSOD at lysine 122 (MnSODK122Q) increased mitochondrial ROS levels, resulted in genomic instability, and was associated with a Luminal B breast cancer malignancy risk (325, 386). Doxorubicin-resistant breast cancer cells exhibited a significant upregulation of MnSOD that regulated the switch between the EMT- and MET-associated phenotype by influencing the cellular redox environment via its effect on the intracellular ratio between O2 •− and H2O2 in breast carcinoma (206). Accordingly, knockdown of MnSOD through activation of the peroxisome proliferator-activated receptor gamma decreased colony-forming ability, switch-off the EMT characteristics, and sensitized the cells to doxorubicin-induced cell death (180). On the other hand, MnSOD overexpression significantly decreased growth and survival of breast cancer cells (22, 180), metastasis of fibrosarcoma (315), and androgen-independent prostate cancer cells (49, 249).
To directly test the role of MnSOD, we overexpressed MnSOD in normal MEF cells and found that increased expression of MnSOD reduces the frequency of neoplastic transformation caused by IR (75). Because overexpression of MnSOD suppressed this neoplastic transformation, we investigated whether MnSOD reduced cancer therapy efficacy. We implanted human MnSOD cDNA in syngeneic mice, which have intact immune function, and identified the radiation dose that controls one-half of the irradiated tumors (TCD50). As anticipated, the TCD50 obtained from the tumor control rate after irradiation was substantially lower for the MnSOD-overexpressing tumors compared with the control tumors (339). This study was the first to suggest that increasing MnSOD activity could be a tool to enhance radiation-induced cancer cell death. In line with this study, others demonstrated that overexpression of MnSOD suppressed cancer cell proliferation both in vitro and in mouse xenografts of human cancers (68, 345, 378). Mechanistically, the MnSOD-induced suppression of proliferation is probably associated with a delay in the progression from the G1 to the S phase (45, 345). To demonstrate the role of MnSOD as an antioxidant in normal tissues, transgenic mice expressing the human MnSOD gene were generated and treated with ROS-generating agents, including the anticancer drug doxorubicin. Study results support a major role for MnSOD in protecting normal tissue against ROS-mediated injury and suggest that mitochondria are the critical sites of cardiac and brain injury (46, 65). These findings on MnSOD function in normal and cancer tissues are the basis for a recent initiated research direction into pro-oxidant/antioxidant defense mechanisms, with the potential to address the challenge of serious normal tissue impairment following standard therapies.
SOD mimetics
Since it is impractical to genetically alter the expression of MnSOD in a clinical setting, we initially tested the effects of synthetic MnSOD. A variety of MnSOD mimics such as Mn-porphyrins have been developed and are being tested in preclinical studies for protecting normal tissues from treatment-induced injury (18, 20) and potentially killing cancer through their oxidant and reductant properties (MnIIIP/MnIIP redox couple). Under high oxidizing conditions in cancer cells, SOD mimetics kill glioblastoma, lymphoma, and prostate cancer (154, 223), both in vitro and in vivo. For combination treatments of SOD mimetics with radiotherapy, several reports indicate that SOD mimetics exhibit a radiosensitizing effect (231). Several members of this class of agents have been tested. In vitro data revealed that MnTnHex-2-PyP5+ (Mn(III) meso-tetrakis (N-n- hexylpyridinium-2-yl) porphyrin) attenuated DNA damage-repair after irradiation, suppressed phosphorylation of several MAP kinases, and enhanced radiation-induced apoptosis (304). Treatment with MnTnHex-2-PyP5+ resulted in a 10–20% reduction in the colony formation ability of 4T1 and B16 cells while IR treatment reduced colony formation up to 60–70% in both cell types (306). Combination treatment with MnTnHex-2-PyP5+ and IR further reduced colony formation, suggesting a synergistic effect. Interestingly, the administration of MnTnHex-2-PyP5+ (2 mg/kg/day) alone did not alter 4T1 and B16 tumor growth in vivo when compared with growth in the control group, but tumor growth was inhibited when combined with IR. MnTnHex-2-PyP5+ also decreases IR-mediated activation of AKT, ERK, JNK, and p38-MAPK, suggesting the possibility of a prosurvival role rather than a proapoptotic role (306). In a D245-MG glioblastoma (GBM) multiforme xenograft (patient-derived adult glioblastoma multiforme) mouse model, a subcutaneous injection of MnTnHex-2-PyP5+ twice daily at 1.6 mg/kg (24 h before IR and continued throughout the study) showed significant tumor radiosensitization. Likewise, a daily subcutaneous injection of MnTE-2-PyP5+ [Mn(III) meso-tetrakis (N-ethylpyridinium-2-yl) porphyrin] at 15 mg/kg as a single treatment caused an anticancer effect in a 4T1 mouse breast tumor xenograft model (276). MnTE-2-PyP5+ suppresses phosphorylation of ERK, which, subsequently, suppresses the activation of NF-κB (43). This suppression is related to the ability of Mn-porphyrin to oxidize cysteines of Keap1, thereby activating Nrf2, which, in turn, upregulates endogenous APs (332, 379). When MnTE-2-PyP5+ was combined with dexamethasone, MnTE-2-PyP5+ induced H2O2 production, which can interact with cysteines of different proteins, including a master transcription factor NF-κB, via S-glutathionylation (19, 37). Further, MnP, which is a member of the most potent group of SOD mimics and has an excellent safety profile, is reported to rescue radiation-induced white matter damage in cranially irradiated mice (356). Correspondingly, MnP enhances TRAIL-induced glioma cell death in the presence of a gap junction inhibitor. The mitigation ability of MnP after either temozolomide or cisplatin was also accessed; in combination treatment with temozolomide, MnP treatment mitigated the negative effects of both temozolomide and radiation on a rotorod performance scale (374). The therapeutic combination proved effective against many of the patient-derived glioma cells that are TRAIL resistant, with minimal cytotoxicity in immortalized normal human astrocytes. Efficacy is further enhanced by endogenous reductants, ascorbate, and thiols. Conversely, in normal tissues, these SOD mimics prevented IR-induced bone marrow suppression, erectile dysfunction, pulmonary fibrosis, brain damage, chronic proctitis, and doxorubicin-induced cardiac toxicity (191, 248, 356, 380). These data facilitated the initiation of a clinical trial at Duke University, where they are now tested as a radioprotector of normal brain (NCT02655601) in high-grade glioma patients. Another preclinical study on head and neck cancer patients, jointly conducted by Duke University and the University of Colorado (NCT02990468), showed that MnP mitigated radiation-induced normal tissue damage, including mucositis, xerostomia, and fibrosis, while augmenting the antitumor effect of radiation (11).
In conclusion, these studies validate that the use of synthetic mitochondria-targeted redox-cycling compounds to inhibit tumor cell growth and promote apoptosis could be incorporated into the current standard-of-care regime as an innovative treatment platform to improve treatment efficacy. Based on the clinical trials initiative and these evidence, we propose that MnP would be an ideal candidate for SOD mimetics for future use in cancer treatment intervention.
Ascorbic acid
AA or vitamin C is an essential dietary vitamin that humans and most mammals cannot synthesize due to an absence of the enzyme L-gulono-lactone oxidase (82). AA is a powerful reducing and oxidizing agent. It reacts with ROS to form the metabolite dehydroascorbic acid (DHA) via a semidehydroascorbate intermediate (42). In contrast, it acts as a pro-oxidant by producing AA radicals and promotes LPO. AA has a somewhat controversial history as a therapeutic for cancer treatment (84, 96). AA exhibits antioxidant properties when present at physiological concentrations (40–80 μM) (32). However, when used at pharmacological doses (≥20 mM), which can be achieved only through intravenous delivery, its oxidation can deliver a high flux of H2O2 (57, 85, 279). AA induces apoptosis in osteosarcoma, neuroblastoma, T cell leukemia, breast, pancreas, prostate, and colon cancers (62, 133, 269, 350, 355). Combination treatment of AA with several chemotherapy regiments has yielded positive outcomes, including improved efficacy of cisplatin and 5-FU against esophageal cancer (1), increased activity of doxorubicin, cisplatin, and paclitaxel in human breast carcinoma cells (181), synergistically enhancing the cytotoxicity of cisplatin against cervical cancer cells (188), and enhanced arsenic trioxide-induced cytotoxicity in multiple myeloma cells (120). In addition to its use with chemotherapy, a high dose of AA enhances the radiosensitivity of prostate cancer to radiation treatment (355). Recently, clinical trials of AA treatment in lung and brain cancers have demonstrated a significant decrease in cancer growth and metastasis (296). Considering the satisfactory results of high doses of intravenous AA but not oral AA, the complexity of the mechanisms involved in AA treatment deserves further investigation.
The mechanism by which AA induces cancer apoptosis has been extensively studied with the pancreatic cancer model in vitro, in vivo, and in clinical trials by Dr. Joseph Cullen's laboratory (62, 253). A recent study demonstrated that DHA, an oxidative product of AA, contributes to the selective antitumor effect of AA in K-RAS- and BRAF-mutant colorectal cancer cells via direct oxidation with GSH and GAPDH, subsequently depleting the cellular GSH pool (85, 375). Recent work from our laboratory showed that AA acts as a pro-oxidant at pharmacological doses and differentially modulates cellular responses to ROS in normal versus prostate cancer cells. The AA transporter SVCT2 was expressed at a low level, but the DHA transporter GLUT1 was expressed at a high level in cancer cells, with reverse results in normal cells. We propose that the high ROS environment in cancer cells rapidly oxidizes AA to DHA. The subsequent influx of DHA into cancer cells accompanied by GSH depletion suppresses RelB-induced SIRT3 expression (355). Interestingly, selectively sensitizing nonsmall-cell lung cancer and glioblastoma cells to AA via pro-oxidant chemistry involves a different schema, the redox-active labile iron pool (LIP) (237). H2O2 produced from AA oxidation selectively (i) increases the availability of LIP in the cancer cells, partially by disrupting Fe-S clusters, and (ii) mediates ascorbate toxicity in cancer versus normal cells (296). As recently mentioned by Schoenfeld et al., upregulation of LIP mediates Fenton chemistry and causes oxidative damage to cellular macromolecules (i.e., DNA, protein, lipids) (296). Based on the diffusion-limited kinetics of OH• species, this group proposed that redox-active iron chelated by these macromolecules most likely represents the most prevalent site of damage. In their model, endogenous ascorbate recycling mechanisms are driven by reducing equivalents from NADPH, GSH, and/or Grx, allowing for the continuous production of H2O2. The cytotoxicity induced by AA seems to be primarily mediated by H2O2 generated in extracellular fluids (296).
Due to successful AA treatment in animal models and preclinical studies, clinical trials in nonsmall cell lung cancer and glioblastoma are initiated, exhibit the feasibility, tolerability, selective toxicity, and potential efficacy of pharmacological AA, and show exciting potential.
Parthenolide
Parthenolide (PN) is a sesquiterpene lactone that is found in flowers and fruits of the traditional herbal medicine feverfew (Tanacetum parthenium). PN acts as a pro-oxidant/antioxidant that can elicit a protective response in normal cells but adds injury to tumor cells after radiation therapy (197, 202, 364). For instance, PN induced apoptosis in the AML-French-American-British subtype cancer while sparing normal hematopoietic cells (CD34+/CD38−) and progenitor cells (262). Likewise, PN can protect against radiation-induced cell death in normal PrECs but enhances radiation-induced killing in aggressive prostate cancer (236, 319). Mechanistically, PN activates NOX in prostate cancer cells but not in PrEC cells (236, 319). PN decreases the reduced form of Trx1 but increases the oxidized form of Trx, which is highly expressed in prostate cancer cells (319, 366). In contrast, PN increases GSH levels in PrEC cells, at least through the induction of the ROS-mediated Nrf2/ARE pathway. Combining PEG-SOD with PN treatment restores the basal level of O2 •−, confirming the SOD-induced inhibition effect on ROS signaling (319, 364). Importantly, PN has an opposite effect on the expression of RelB, a member of the alternative pathway of NF-κB, which is expressed at high levels in advanced prostate cancer (319, 364). PN suppressed RelB in prostate cancer cells but induced RelB in normal prostate epithelial cells. PN selectively targeted the progenitor and stem cell population of AML in the SCID mice xenograft model via the inhibition of NF-κB, activation of p53, and generating ROS production (173). A recent study by Pei et al. showed that a drug regimen containing PN, 2-deoxyglucose, and temsirolimus is a potent combination that can induce cytotoxicity to AML stem cells but not normal stem cells (261). Even though PN is very effective in inducing cancer cell death, its poor pharmacologic properties limit its clinical application. To improve the biologic availability of PN, a novel aminoparthenolide, DMAPT, has been developed and has the following unique characteristics: (i) favorable water solubility through the formation of an amine salt and (ii) prodrug characteristics in vivo, mediated by base-catalyzed cleavage to the parent compound (243). DMAPT, as a single agent, effectively inhibited prostatic tumor growth in a xenograft model when delivered at 100 mg/kg daily by oral gavage for 7 days; correlatively, it is effective as a radiosensitizing agent at the same dose when co-treated with radiation at 6 or 10 Gy (227, 236). Guzman et al. showed that an oral bioavailability of DMAPT is about ∼70% when compared with intravenous administration in mouse xenograft and canine leukemia models as determined by functional assays and multiple biomarkers (125). As reviewed by Siveen et al., DMAPT is currently being evaluated in a Phase II clinical trial in AML patients; thus, a further improvement in the bioavailability and selective toxicity of PN or DMAPT will promote promising therapeutic utilization of ROS-generating agents (312).
Forthcoming, we anticipate that other redox-based anticancer therapeutics with protective properties against cytotoxic therapy will be discovered and that they will have a significant impact on the care of cancer patients.
Developing Personalized Redox Therapy for Cancer Therapy
As the prospect of personalized medicine becomes more feasible, future therapies will integrate information about the individual patient's DNA sequence, RNA expression, proteomic expression profiling, and tumor metabolomes. The major barrier to the use of redox-active pro-oxidants/antioxidants to incorporate personalized redox therapy for precision cancer medicine is the lack of specific target gene/protein sets that can accurately inform the redox state of individual patients. Since a standard of ROS measurement in a clinical setting is not entirely practical, a redox state index based on a combination of APs, thiol couples, ROS/RNS-generating enzymes, and transcription factors is needed. Because H2O2 plays both physiological and pathological roles in cancer, redox-related proteins that produce or scavenge H2O2 and are upregulated in aggressive cancers would be likely candidates for a redox state index template. Herein, we propose the antioxidants or redox-related proteins that could be incorporated into a redox state index to determine whether antioxidant or pro-oxidant therapies would be effective in the treatment of an individual patient based on the availability of their expression levels (DNA, RNA, or protein) from a cancer database. Other redox-associated molecules would also need to be investigated to develop the redox state index approach.
Catalase
CAT is the major enzyme involved in the detoxification of high concentrations of H2O2, whereas GPx and the Prxs are responsible for removing low fluxes of H2O2 (128). The CAT activity of cancer cell lines from a variety of tissues reveal a wide differential in the ability of cells to remove H2O2. Doskey et al. showed that most cancer cell lines have low levels of both CAT and GPx (80). Their study suggests that the vast majority of cancer cells may lack the biochemical machinery needed to detoxify high H2O2 fluxes. As mentioned earlier, AA mediates H2O2 production and induces cancer cell death. Decreasing CAT activity increases sensitivity to AA (177). The differential sensitivity to AA of pancreatic cancer cell lines has been demonstrated to correlate with the capacity to remove H2O2. Thus, a treatment that combines AA and an inhibitor of H2O2 removal has the potential to be an effective therapy for pancreatic adenocarcinoma (62). Correspondingly, CAT overexpression in 4T1 cells significantly lowers ROS levels, restores IR-induced DNA damage-repair capacity, and diminishes IR-induced apoptosis (306). On the contrary, certain cancers demonstrate increased CAT activity, for example, the multidrug-resistant HL-60 leukemia cells (189). Targeting cancers that overexpress CAT results in inhibition in metastatic cancer growth and suppresses invasive breast cancer in mice (118). This extensive evidence indicates that anti-CAT agents, such as arsenic trioxide (currently employed against promyelocytic leukemia) (115, 203), might serve as a therapeutic target, particularly as a combination therapy to increase the efficacy of any H2O2-generating drugs.
Glutathione
Similar to CAT, GSH metabolism appears to be actively involved in protecting cancer cells from apoptosis and, additionally, is involved in mechanisms of multidrug and radiation resistance. Compared with normal cells, cancer cells contain a high GSH content (108). Increased levels of GSH within cancer cells are associated with resistance to platinum-containing anticancer compounds. Formation of GSH-platinum conjugation, mediated by glutathione S-transferase P1 (GSP1), is largely responsible for drug inactivation. In addition to GSP1, GSH production enzymes (GCLM and GCLC) and regeneration enzymes (G6PD and GR) are found at higher concentrations in several platinum-resistant cell lines (107, 224). These GSH-related metabolism genes were regulated by Nrf2. Potentially, Nrf2 genes could be used as a redox state index for platinum-resistant cancers. Thus, the expression of these genes may be considered a predictive tumor marker for platinum-resistant cancers and used for selecting the patients who would benefit from GSH inhibition as an additive treatment. BSO and phenylethyl isothiocyanate, which inhibit GSH synthesis and direct binding with GSH, respectively, have shown promise in selectively killing aggressive prostate cancer cells and in a preclinical mouse model of ovarian cancer (45, 49, 284, 285, 333). Supporting this notion, NOV-002, which is a GSSG mimetic liposomes that alters the intracellular GSH/GSSG ratio, has been tested in patients with HER2-negative breast cancers, which exhibit high expression of GSH. Administration of NOV-002 in combination with adjuvant chemotherapy (doxorubicin-cyclophosphamide followed by docetaxel) resulted in a favorable response rate and mitigation of side effects compared with adjuvant chemotherapy alone (176, 233, 290). Other GSH inhibitors have been tested in clinical trials, that is, Telcyta, Disulfiram, and sulforafane (176). For these reasons, combinations of GSH inhibitors with radiotherapy or chemotherapeutic drugs that cause cell death induced by oxidative stress may prove useful in targeting cancer cells. In particular, dual-function compounds such as MnP, which can generate ROS and conjugate GSH, would be a preferred choice for GSH-mediated resistance cancers (Fig. 6).
Iron metabolism-related proteins
Intracellular iron is an important element that may play a role in personalized redox-based therapy. Rewired redox state results in increased levels of O2 •− and H2O2, which are capable of disrupting intracellular iron metabolism (342). Intracellular iron is a double sword for cancer treatment; it can augment cancer treatment by amplifying the ROS-mediated cell death signal, alternatively, it can act as a mediator for nontarget injury for some chemodrugs, that is, doxorubicin. Therefore, disruption of iron hemostasis with iron overload could be a cause or consequence of cancer formation. The role of ferroptosis as a type of cell death that depends on intracellular iron levels was first proposed by Dixon et al. (77). Iron-dependent ROS formation and LPO are a major mechanism of ferroptotic-induced cell death. Recently, induction of the ferroptosis process by increasing iron overload has become an attractive method for redox-based therapy. For example, Sorafenib, a kinase inhibitor approved for the treatment of RCC and hepatocellular carcinoma, can induce ferroptosis in a number of different cancer cells, through inhibiting xCT-mediated cystine import and increasing mitochondrial ROS generation (78). Erastin (a Ras-selective lethal compound), which is a ferroptosis inducer, mediated LCL161 leukemia cell death by collapsing the permeability of the outer mitochondrial membrane and inhibiting Cys-dependent GSH regeneration (373). These events lead to the production of LPO and ROS (148). Silencing transferrin receptor encoding genes, which are involved in iron uptake, significantly abolished erastin-induced ferroptosis (87, 213). Lanperisone is another important FDA-approved drug shown to suppress the growth of K-ras-driven tumor cells through iron- and ROS-dependent mechanisms (302). Studies confirm that iron is needed to facilitate cancer cell death and the potential of iron homeostasis as a cancer treatment target. It is noteworthy that at higher doses iron could facilitate cardiotoxicity in doxorubicin-treated cancer patients via O2 •− generation (300). Thus, further experiments are required to understand the particular type of ROS important for ferroptosis induction.
NADPH oxidase
NOXs are composed of proteins that transfer electrons across biologic membranes. In the reaction catalyzed by these enzymes, the electron acceptor is oxygen, and the product of the electron transfer reaction is O2 •−. Owing to spontaneous and enzymatic dismutation, H2O2 is also rapidly generated (113). Several cancers overexpress the NOX family (NOX1-5, DUOX1-2), with each cancer overexpressing a different isoform of NOXs (23). NOX1 and NOX4 are widely studied (225). NOX1 is overexpressed in breast, colorectal, fibrosarcoma, head and neck, melanoma, renal, oral, and prostate cancers (165, 199). Expression of NOX1 is associated with growth, proliferation, and invasion. NOX4 is overexpressed in breast, colorectal, lung, ovarian, pancreatic, prostate, and thyroid cancers (199). Expression of NOX4 is associated with migration, angiogenesis, glycolytic switch, and poor prognosis (198). In prostate cancer, increased NOX1 mRNA and protein correlate with elevated H2O2 (196). Emerging evidence indicates that altered expression or activation of NOX enzymes contributes to alterations in mitochondrial metabolism; conversely, altered mitochondrial metabolism reforms NOXs expression in cancer cells. These data reveal cross-talk between NOXs enzymes and mitochondria (76). Several studies indicate that suppressing NOXs inhibits cancer growth and metastasis (165). Inhibition of NOX1 with siRNA in K-ras-transformed normal rat kidney cells resulted in a marked decrease in neovascularization (178). Inhibition of NOX1 by using the natural compound Cambogin in breast cancer decreased cancer metastasis (303). Inhibition of NOX1 with DPI decreased ROS production and resulted in an inhibition of the malignant phenotypes, growth, and invasion (307). Understanding the relevance of global changes in NOXs expression and/or activity in cancers would also require insight into their subcellular localization, which likely determines their distinct and/or unique roles in cancer characteristics. Overall, increasing expression or activation of NOX enzymes may be used as a marker to indicate which cancers would be susceptible to a given ROS-generating or enzyme-based anti-cancer treatment.
Future trend: a redox state index for cancer treatment
The diversity of agents and their modes of action in the modification of redox balance suggests that redox-based therapy will have different effects on individual cancers. Genetic profiling of clinical risk factors and an integrated approach using molecular imaging and redox state index may allow the recognition of patients who will benefit from redox-based therapy. The challenge for the physician and treatment team is to decide which strategy is appropriate for each patient. The redox state index is offered as a template that can be integrated with personalized redox therapy in the design of an optimum treatment for an individual patient. For example, if the patient's profile exhibits upregulated levels of CAT, Nrf2, and GSH-metabolism genes, the sole use of a redox-cycling compound such as AA would not be effective. Co-treatment with BSO or an anti-CAT compound, such as arsenic trioxide (115, 203) should be integrated for optimal impact for cancer. In contrast, if the patient's profile exhibits upregulated levels of NF-κB, transferrin receptor, and NOXs, co-treatment with a ROS generator compound, such as a SOD mimetic, would promote cancer cell eradication via oxidative stress overload. RNA expression of redox state index from The Cancer Genome Atlas (109) is presented in Figure 7. As expected, CAT, Nrf2, transferrin receptor, and NOX4 are overexpressed or mutated in a variety of cancers, which make them potential candidates for constructing a redox state index and incorporated into a treatment plan. Based on the redox state index, head and neck cancers exhibit high expression levels of NOX4 and transferrin receptor; whereas AML exhibits low expression of NOX4 but high expression of CAT. As a consequence, head and neck cancers are preferred candidates for personalized redox therapy with MnP. The prospective personalized redox therapy described earlier has not yet been incorporated into diagnostic, treatment, and surveillance practices for cancer patients. The potential strategy to apply these redox state indexes is summarized in Figure 8. As genetic and molecular biomarkers become better understood, the ultimate goal of precision medicine is to be more effective and less toxic, thereby improving the quality of life of surviving patients.


Conclusion
The advent of new strategies for early detection of cancer along with novel therapies makes this a time of extraordinary promise for patient survival, even though cancer remains a leading cause of mortality in the United States. ROS have been used for nearly a century to kill cancer, but novel approaches for using ROS in cancer therapy have recently gained attention. The involvement of redox mechanisms in cancer biology and anti-cancer treatments is a very active field of research. Our review is focused on a dual-purpose use of ROS, which is not only to kill cancer but also to protect normal tissues against the toxic effect of radiation/chemotherapy. This potential exists because malignant cells depend on elevated intracellular levels of ROS to proliferate and self-renew and because normal healthy cells do not have elevated basal levels of ROS. More importantly, the majority of chemotherapeutic drugs cause oxidative stress in noncancerous tissues that can activate an adaptive response in these normal cells. In addition to oxidative stress, there are other means of shifting redox state to reducing stress (79), some of which appear to have clinical potential but are not discussed in the current review.
It is important to establish the redox characteristics of subcellular compartments in various cancers at varying steps of progression to determine the appropriate redox therapies. Each cell type in the body utilizes different proteins for redox signaling. Thus, differences in redox biochemistry at the subcellular level between nonmalignant and cancer cells must be determined before rational therapies can be designed. Although most redox-modulated compounds work in more than one subcellular location, the one that modulates the redox state predominantly in mitochondria seems to provide the most positive response in the treatment of various advanced cancers. The ability to develop therapeutic strategies based on the intrinsic redox state of a particular cancer relies on precise monitoring of subcellular ROS levels. Therefore, continuing the effort to improve and expand the methods to quantify ROS in an effective, cost-permissive, and accurate manner, a daunting challenge for integrative redox state research into the current clinical setting, is needed. Finally, a major challenge emerging for clinical research on personalized redox therapy is that patient cohorts are smaller when stratified by multiple biomarkers. This warrants adaptation and further development of trial designs to gather solid evidence for new treatment options.
Footnotes
Acknowledgments
This work was supported in part by NIH grants CA188792, CA205400-02, and NIH/NIGMS COBRE program (P20 GM121327). The authors would like to thank Dr. Heidi Weiss, Dana Napier, and Rani Jayswal for technical assistance and statistical analysis of data in ![]() . This research utilized the service facilities of the Markey Biospecimen Procurement and Translational Pathology Shared Resource Facility as well as the Biostatistics and Bioinformatics Shared Resource Facility, and it was funded by a Markey Cancer Center support grant (P30 CA177558).
. This research utilized the service facilities of the Markey Biospecimen Procurement and Translational Pathology Shared Resource Facility as well as the Biostatistics and Bioinformatics Shared Resource Facility, and it was funded by a Markey Cancer Center support grant (P30 CA177558).