
Abstract
Epidemiological, preclinical and interventional clinical studies have demonstrated that environmental stressors are associated with health problems, namely cardiovascular diseases. According to estimations of the World Health Organization (WHO), environmental risk factors account for an appreciable part of global deaths and life years spent with disability. This Forum addresses the impact of the environmental risk factors such as traffic noise exposure, air pollution by particulate matter (PM), mental stress/loneliness, and the life style risk factor (water-pipe) smoking on health and disease with focus on the cardiovascular system. We will critically discuss the use of observatory/modifiable biomarkers of oxidative stress and inflammation in environmental research on the aforementioned risk factors highlighting the need of exposome studies. Another focus will be on the epigenetic regulation via microRNAs in environmental stress upon exposure to noise and toxins/heavy metals as well as mental stress conditions, providing mechanistic insights into the modulation of microRNA signaling by oxidative stress, and vice versa the contribution of microRNAs to oxidative stress conditions. We will also provide an in-depth overview on the mechanistic pathways that lead to health problems (e.g., cardiovascular diseases) in response to environmental psychosocial stress, air pollution exposure in the form of ambient PM and diesel exhaust, traffic noise exposure, and the life style drug (water-pipe) smoking. Almost all stressors share the activation of the hypothalamic-pituitary-adrenocortical axis and of the sympathetic nervous system with subsequent onset of inflammation and oxidative stress, defining the here proposed therapeutic (antioxidant and exercise) strategies. Antioxid. Redox Signal. 28, 735–740.
Introduction
O
According to the World Health Organization (WHO), air pollution clinically the most relevant environmental risk factor accounts for several million global deaths per year, most of them due to cardiovascular or cerebrovascular disease. The potency of air pollution as a trigger of health problems is underlined by the findings that short-term pollution-control activities (mainly reduction in traffic and industrial air pollution) applied during the 2008 Beijing Olympic Games led to a substantial reduction in the concentrations of air pollutants, which were perfectly mimicked by similar effects on biomarkers of inflammation, oxidative stress, and thrombosis in healthy adults.
According to the WHO, a total sum of more than 1 million healthy life years are lost every year from environmental noise exposure in the Western European countries and this risk was identified earlier by Robert Koch, who postulated in 1910 that “One day mankind will have to fight the burden of noise as relentless as the pest and cholera.”
Mental stress affects a large part of the population and it may be assumed that mental stress has also large impact on public health and global mortality. Environmental toxins such as heavy metals are known since long time to contribute to the development of different diseases, including cardiovascular events. Especially among young people, water-pipe smoking is spreading like a pandemic all over the world and is, according to the WHO, even more dangerous than normal cigarettes.
There is consensus that oxidative stress and inflammation play a key role in mediating the hazardous effects of aforementioned environmental stressors, but partially due to lack of well-established models for translational research in humans and animals, the detailed mechanisms responsible for their contribution to the global burden of disease and mortality are still elusive.
The focus of this Forum is to discuss recent advances and identify knowledge gaps in research on the impact of environmental risk factors on health and disease. Our aim was to review the current state of knowledge but also to provide new concepts to understand the vital crosstalk of the different systems (stress response pathways, redox signaling machinery, and immune system) involved in the pathophysiological processes triggered by environmental stressors and to reveal common disease pathways that may explain synergistic effects of simultaneous exposure to more than one of these environmental stressors. Besides the direct toxic effects of particles from air pollution or water-pipe smoking, heavy metals, and other environmental toxins, chronic stress reactions in response to noise exposure and psychosocial stress/annoyance lead to hormonal changes and alterations of the autonomic and endocrine system, sleep deprivation, and depression that are associated with pathophysiologic systemic alterations and disease development.
Topics Covered in This Forum
Ghezzi et al. provide a critical assessment of biomarkers of oxidative stress and inflammation in the context of environmental risk factors (1). The authors begin with a historical overview on these biomarkers as well as their definition and categorization into chemical reaction products (e.g., lipid peroxides) and biological (stress) responses (e.g., induction of nuclear factor erythroid 2 [NFE2]-related factor 2 [Nrf2]). They also discriminate between observational (e.g., oxidized proteins) and modifiable biomarkers, the latter representing potential targets that can be used for therapeutic approaches (e.g., cytokine modulation by monoclonal antibody therapy). Oxidative stress reactions and inflammatory pathways are discussed in detail for ultra-fine particles as contained in particulate matter (PM), for noise-triggered hearing loss, and mental stress/disorders. Suitable biomarkers of oxidative stress and inflammation for each environmental stressor are discussed. Special attention is paid to the induction of Nrf2 as a biomarker for oxidative stress but also pharmacological modulation of Nrf2 for antioxidant and anti-inflammatory therapy. The aforementioned environmental stressors, as well as heavy metals or cosmic irradiation, lead to an induction of Nrf2, which can be regarded as a compensatory stress–response reaction. Pharmacological activation of Nrf2 can protect from environmental stressors, whereas genetic Nrf2 deficiency can increase the susceptibility to them. Finally, the authors discuss how to assess the human exposome (also in the context of environmental stress) by transcriptomic, proteomic, and metabolomic methods.
Lamas and colleagues provide evidence for epigenetic regulation in the context of environmental risk factors (5). More precisely, the authors focus on the role of microRNAs in the modulation of human health and disease by environmental stressors such as chemicals (heavy metals, pollutants, and pesticides) but also exposure to noise or mental/psychosocial stress. They introduce the basis of epigenetic regulation of gene expression and the influence of the environmental exposome on this process. The authors discuss the impact of environmental chemicals, air pollution, noise-induced hearing loss, and mental stress on the microRNA profiles, their target genes, and thereby regulated cellular signaling pathways (e.g., inflammation), as well as associated pathologies and disease phenotypes. A major achievement of this review is the presentation of tables and figures with specific microRNAs being associated with specific environmental chemicals and risk factors such as air pollution, noise exposure, and mental stress/disorders. For hearing loss and mental disease, the role of oxidative stress for microRNA expression changes and vice versa the induction of oxidative stress by specific microRNAs is discussed in full detail. A strong translational aspect is the discussion on microRNAs as environmental stress–response biomarkers as well as potential therapeutic targets.
Meyer and Wirtz highlight the effects and underlying mechanisms of psychosocial stress (4). They give an introduction to the major pathways being operative in mental stress reactions such as the activation of the sympathetic nervous system (SNS) with increased release of catecholamines, which is followed by an activation of the hypothalamic-pituitary-adrenal (HPA) axis with time-delayed cytokine production (e.g., interleukins) and cortisol release. The involved basic processes such as steroid biosynthesis, physiological, and stress-induced functions of the HPA axis are explained in complete detail. The authors provide evidence for a vital crosstalk in this system as shown by interleukin-dependent stimulation of the HPA axis and cortisol-mediated anti-inflammatory effects. The impact of glucocorticoids in general and cortisol in particular on cell signaling, inflammation, and oxidative stress is discussed. Special attention is paid to interleukin-6 signaling that involves signal transducer and activator of transcription 3 (STAT3) activation, improved mitochondrial respiration, and overall energy metabolism. The authors thereby ascribe (short-term) mental stress challenges with subsequent cortisol/interleukin-6/STAT3 signaling pathways as protective mechanisms in stress adaptation.
Xia and Li give an overview on the effects of social isolation on cardiovascular health (9). Owing to the demographic shift (aging population) but also owing to our career and job-orientated societies, people have less social interactions, leading to social isolation and loneliness. Data from a meta-analysis (148 studies and 308,849 participants) revealed a correlation between the number and quality of social relationships and overall mortality that was independent of classical risk factors. Other studies identified loneliness as a cardiovascular risk factor, leading to increased incidence of hypertension in humans and aggravated atherosclerosis in different animal models. Loneliness and social isolation are associated with changes of behavior such as physical inactivity, smoking, and reduced sleep quality but also with psychological disorders such as dysphoria, anxiety, social withdrawal, and depression. On a mechanistic basis, loneliness and social isolation lead to an activation of the HPA axis (as described for mental stress in the preceding paragraph) with subsequent glucocorticoid (e.g., cortisol) release. Chronic loneliness and social isolation will lead to glucocorticoid resistance, leading to a loss of glucocorticoid-dependent suppression of inflammatory pathways, which results in excessive inflammation and increased oxidative stress with Nox2 as a key enzyme. At the same time, higher glucocorticoid levels are associated with reduced nitric oxide bioavailability, increased vasoconstrictor activity, and may also affect the SNS and the vagal system. Loneliness and social isolation have large impact on the immune system, creating a proinflammatory phenotype. Chronic low-grade inflammation may represent a key mechanism of loneliness-associated chronic diseases such as atherosclerosis, cancer, and neurodegeneration. The authors also discuss potential mitigation strategies to reduce loneliness (e.g., by stress management training, improvement of social skills, and meditation) or to prevent its adverse effects on health (e.g., by allopregnanolone, β-blockers, oxytocin, and NADPH oxidase inhibitors).
Wilson et al. summarize how diesel exhaust and the contained ultrafine particles contribute to the development and progression of cardiovascular diseases (8). According to an estimation of the WHO, air pollution caused up to 7.3 million premature deaths globally in the year 2012 (mostly from cardiovascular disease). Air pollution ranks among the 10 most important risk factors for global all-cause mortality. After an introduction on the hallmarks of diesel exhaust toxicity, the authors provide data for a correlation between diesel exhaust exposure and cardiovascular risk with special emphasis on the role of oxidative stress, based on epidemiological (field) studies but also on controlled interventional (exposure) studies. PM in general and diesel exhaust in particular cause vascular dysfunction characterized by increased blood pressure, impaired endothelial function, vasoconstriction, and arterial stiffness as well as cardiac dysfunction envisaged by arrhythmia, cardiac ischemia, and reduced heart rate variability but also systemic inflammation. Moreover, exposure studies in animals and humans revealed accelerated progression of atherosclerosis, platelet hyper-reactivity, and thrombus formation. As for the mechanism, the authors discuss the classical hypothesis that inhaled particles confer their adverse effects on the cardiovascular system primarily via immune cell activation in the lung with release of cytokines to the bloodstream, or alternatively via mechanic stimulation of alveolar sensory receptors with detrimental effects on the autonomic nervous system and cardiovascular function. Based on a more recent hypothesis, nanoparticles can directly cross the alveolar membranes and interact directly with vascular and blood cells, even by crossing the blood–brain barrier, leading to local inflammatory tissue damage. Animal and human studies demonstrated elevated levels of multiple markers of oxidative stress and inflammation upon exposure to PM. A key role of oxidative stress in the pathophysiology underlying PM exposure in mice and men was supported by amelioration of the phenotype by administration of antioxidant supplements (e.g., TEMPOL, fish oil with omega-3 fatty acids, and B vitamins).
Rao et al. provide an overview on the pathways by which ambient air pollution (PM) exposure confers systemic effects focusing on cardiovascular oxidative stress pathways (7). The authors critically assess the contribution of direct reactive oxygen species (ROS) formation by the particles (e.g., at reactive surfaces) versus endogenous ROS sources, activation of immune cells versus nonimmune cells, and the role of pulmonary oxidative stress and inflammation for the overall pathophysiology underlying PM exposure. In two tables, the authors summarize animal and human studies on PM exposure and the key findings with respect to markers of oxidative stress and inflammation. Differences in the experimental conditions of PM exposure studies are also critically discussed. Mechanistic studies in animals revealed that PM exposure led to vascular dysfunction, activation of leukocytes, progression of atherosclerotic plaques, increased vascular superoxide formation and expression of NADPH oxidase subunits, impaired glutathione metabolism, and loss of beneficial effects of high-density lipoprotein. Some hallmarks of PM pathology were normalized by the NADPH oxidase inhibitor apocynin and genetic p47phox deficiency. PM exposure may also lead to protein misfolding, unfolded protein response, and endoplasmic reticulum stress, which also interacts with apoptotic and inflammatory pathways. Although PM exposure leads to mitochondrial dysfunction and damage, so far no study proved a direct contribution of mitochondrial ROS formation to the observed phenotype. PM exposure also induced antioxidant defense systems such as Nrf2 and antioxidant enzymes regulated by these transcription factors. The authors highlight in detail the crosstalk between oxidative stress and inflammatory pathways in PM exposure models, a concept that was supported by prevention of vascular inflammation in PM-exposed p47phox knockout or TEMPOL-treated mice. The mechanism for the onset of inflammation is thought to comprise PM-dependent danger signals such as the formation of damage-associated molecular patterns that via specific receptors (e.g., CD44, TLR4, and CD36) trigger classical kinase signaling, NFκB activation, and cytokine synthesis. TLR4 signaling also directly activates gp91phox-dependent NADPH oxidase.
Golbidi et al. summarize the unifying oxidative stress concept underlying noise exposure, mental stress- and (water-pipe) smoking-associated pathologies, and highlight possibilities to target the redox imbalance triggered by these environmental risk factors (2). The authors point out that water-pipe smoking exposes individuals to even higher amounts of toxic compounds and leads to comparable oxidative stress levels compared with conventional tobacco smokers. Likewise, noise exposure also increases oxidative stress and leads to subsequent hearing loss, whereas exposure to mental stress suggests a clear association with oxidative stress as documented by elevated levels of several oxidative stress markers and the amelioration of adverse phenotypes by antioxidant therapy (e.g., N-acetylcysteine and apocynin). In a large part of their review, the authors focus on strategies to overcome the redox imbalance triggered by these environmental risk factors, especially the beneficial effects of exercise, as a nonpharmacological strategy to activate endogenous antioxidant defense systems. Nrf2 plays a key role in the protective effects conferred by exercise and is important for the (often futile) compensation of environmental stressor-mediated oxidative stress and inflammation. Besides causing a direct upregulation of antioxidant genes such as superoxide dismutase and heme oxygenase-1 via Nrf2, exercise also downregulates inflammatory interleukins and chemokines and induces a family of universal stress response heat shock proteins having antioxidant and immunesuppressive properties (e.g., via inhibition of high-mobility group box 1 protein).
Münzel et al. provide a detailed overview on the historical development of noise research, the emerging socioeconomic impact of environmental (traffic) noise-related health problems, epidemiological studies on this important topic, clinical and preclinical noise research, and finally the synergistic effects of noise and PM exposure on the cardiovascular system (6). The authors were more interested in the indirect (nonauditory) systemic effects of low-intensity noise exposure [<85 dB(A) peak levels and <75 dB(A) mean sound pressure levels]. The direct (auditory) effects of high-intensity noise exposure [>100 dB(A) peak levels] are only briefly discussed. The main message of this comprehensive invited review article is that traffic noise exposure causes a primary (mental) stress reaction leading to the activation of the SNS (e.g., catecholamines) and the HPA axis (e.g., cortisol). As shown in noise-exposed animals, this initial phase was followed by inflammation and oxidative stress, which was envisaged by increased cytokines, infiltrated immune cells, oxidatively modified proteins, lipid peroxidation products, NADPH oxidase activation, nitric oxide synthase uncoupling, endothelial dysfunction, and increased blood pressure. This was also associated with substantial changes in gene expression profiles as measured by next-generation sequencing. In humans, this concept was supported by adverse effects of noise on vascular function, circulating stress hormones, and in some studies increased markers of systemic inflammation and oxidative stress. According to large-scale epidemiological studies, (traffic) noise exposure is associated with higher risk and incidence for cardiometabolic diseases such as hypertension, diabetes, obesity, stroke, and myocardial infarction. There is some evidence that noise and PM cause synergistic adverse effects on the cardiovascular system but this needs further preclinical and clinical investigation.
Conclusions and Outlook
As shown in Figure 1, pathophysiological processes in response to noise exposure and mental stress start at the neuronal and nervous level and necessarily involve the activation of the HPA axis and SNS followed by inflammation and oxidative stress. In contrast, heavy metals and other environmental toxins as well as fine and ultrafine particles from air pollution and (water-pipe) smoking can directly trigger oxidative stress and inflammation without primary involvement of the neuronal and nervous systems. Therefore, a major concept originating from the contributions of the present Forum is that environmental stressors share common pathways in their disease-triggering mechanisms that converge at the level of oxidative stress and inflammation, also defining potential therapeutic strategies. Of note, oxidative stress and inflammation are also hallmarks of all cardiometabolic as well as neurodegenerative and cancer diseases, providing multiple cut surfaces for their aggravation by the environmental stressors in a bonfire manner.
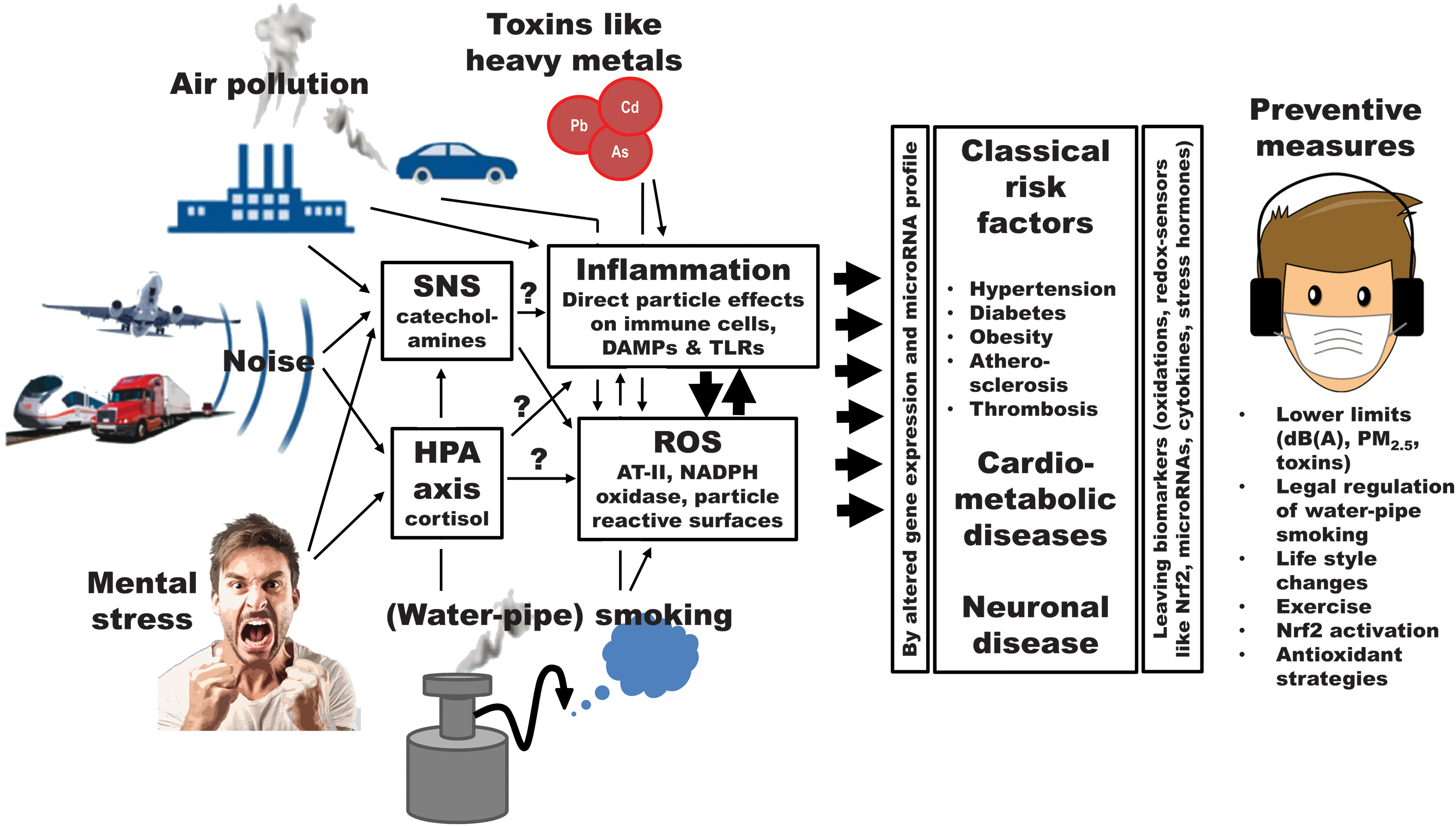
Of great importance for the future research on environmental stress-mediated (cardiometabolic) diseases is the choice of suitable biomarkers, which can be defined as signs that are just indicators of biological processes or proxies that one can interact with to modify the disease process. Based on these considerations, the redox sensor Nrf2, levels of specific cytokines, or the microRNA expression profile can be used as biomarkers to assess the degree of environmental stress, but can also be employed as targets for pharmacological modulation of the pathophysiological phenotype to prevent disease progression. Also nonpharmacological therapies such as physical exercise, caloric restriction, or life style changes could be meaningful in the prevention of environmental stress conditions and associated diseases.
Given the colocalization of noise, air pollution, and other environmental toxins but probably also psychosocial stress in big cities and large urbanized areas, the potential cobenefits of reducing several of these stressors should be considered. Until now, the health risk associated with each of these environmental stressors was assessed separately in most of the studies. However, according to the major concept of this Forum, these stressors may cause synergistic adverse effects and even act in a bonfire manner in people with pre-established cardiometabolic diseases. Therefore, the health problems and disease burden associated with the sum of these environmental stressors may even outperform all previous estimations, urging future research on potential additive effects of two and more environmental stressors.
Footnotes
Acknowledgments
Our studies on noise effects in mice and our Forum were supported by vascular biology research grants from the Foundation Heart of Mainz and the Boehringer Ingelheim Foundation for the collaborative research group, “Novel and neglected cardiovascular risk factors: molecular mechanisms and therapeutic implications” (A.D. und T.M.).