
Abstract
Significance:
The concentrations of reactive oxygen/nitrogen species (ROS/RNS) are critical to various biochemical processes. Small-molecule fluorescent probes have been widely used to detect and/or quantify ROS/RNS in many redox biology studies and serve as an important complementary to protein-based sensors with unique applications.
Recent Advances:
New sensing reactions have emerged in probe development, allowing more selective and quantitative detection of ROS/RNS, especially in live cells. Improvements have been made in sensing reactions, fluorophores, and bioavailability of probe molecules.
Critical Issues:
In this review, we will not only summarize redox-related small-molecule fluorescent probes but also lay out the challenges of designing probes to help redox biologists independently evaluate the quality of reported small-molecule fluorescent probes, especially in the chemistry literature. We specifically highlight the advantages of reversibility in sensing reactions and its applications in ratiometric probe design for quantitative measurements in living cells. In addition, we compare the advantages and disadvantages of small-molecule probes and protein-based probes.
Future Directions:
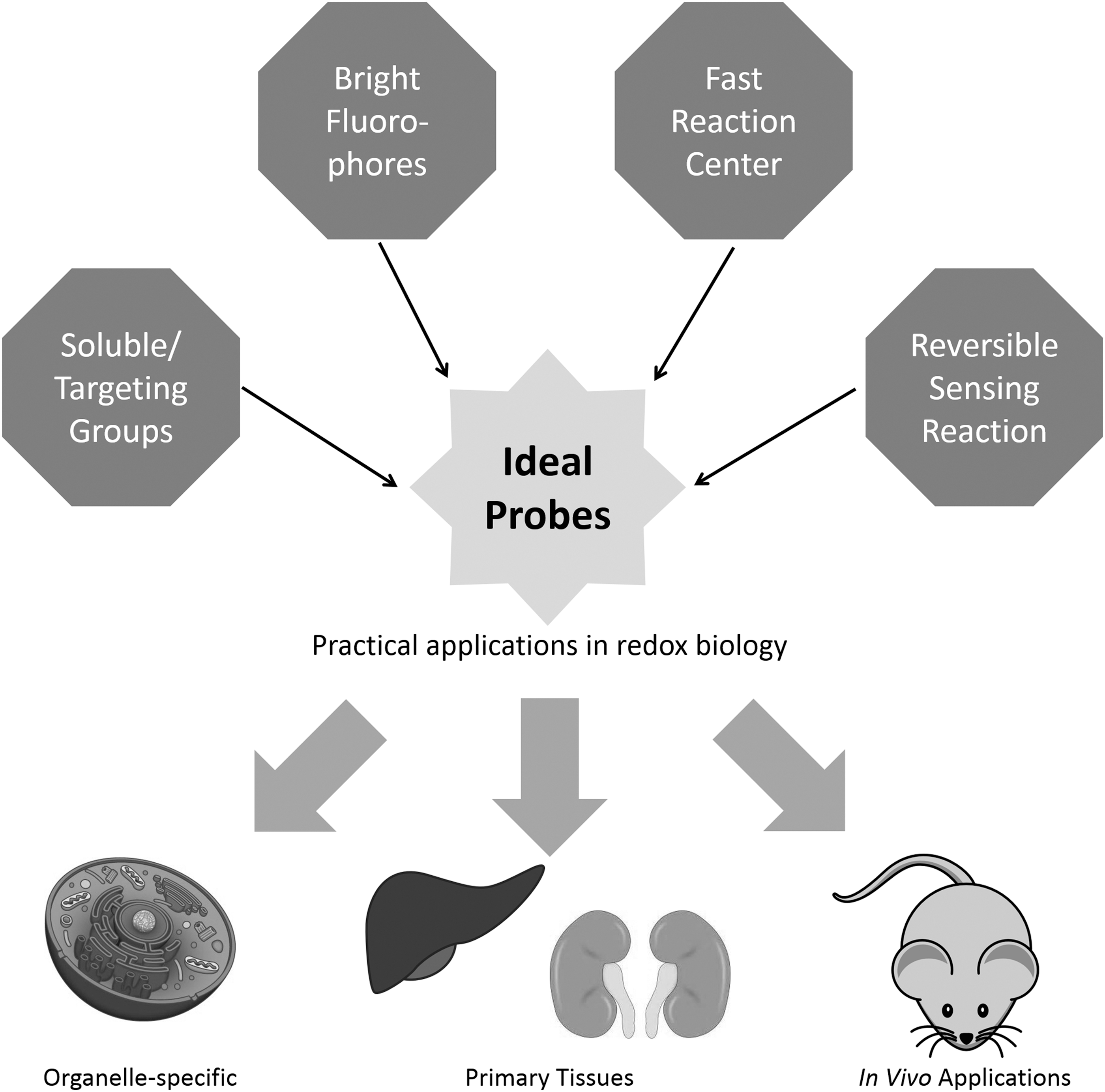
The low physiological relevant concentrations of most ROS/RNS call for new sensing reactions with better selectivity, kinetics, and reversibility; fluorophores with high quantum yield, wide wavelength coverage, and Stokes shifts; and structural design with good aqueous solubility, membrane permeability, low protein interference, and organelle specificity. Antioxid. Redox Signal. 29, 518–540.
Introduction
R
In general, there are two classes of methods used for detecting hydrogen peroxide (H2O2) or other redox signaling molecules in living cells: genetically encoded protein probes and small-molecule-based probes. Genetically encoded protein probes utilize naturally occurring redox-responsive domains for sensing. Several recent review articles, including the ones in this issue, have comprehensively summarized genetically encoded probes for H2O2, GSH, and thioredoxin (6, 9, 12, 44, 46, 47, 105, 108, 114, 125).
The best known examples of reaction-based small-molecule probes are the calcium probes developed by Roger Tsien, such as Fura-2 and Indo-1 (150). Since the development of these probes, numerous fluorescence-based small-molecule probes with reaction centers have been developed, especially in the field of redox signaling (14, 22, 53, 68, 145, 159, 166). Due to the high reactivity of most of the redox signaling molecules, the selectivity of the corresponding small-molecule probes still has sufficient room for improvement. Therefore, it remains a challenge to chemically distinguish between similar redox-active small molecules using small-molecule-based probes (164).
As biological research has advanced, an increasing number of redox signaling molecules have been uncovered (130, 136). Popular probes that were once considered selective for their targeted analytes are now known to cross-react with newly discovered redox signaling molecules. For example, dichlorodihydrofluorescein (DCFH) and boronate-based probes were initially considered to be H2O2 specific probes. However, recent studies revealed that they cross-react with other species in the redox signaling chain, such as peroxynitrite (70, 137). This will be further discussed in later sections. Therefore, it is very important for redox biologists to fully understand the sensing mechanism and limitations of the probe to draw appropriate conclusions.
In this review, we will not only summarize redox-related small-molecule fluorescent probes but also lay out the challenges for probe design to help redox biologists independently evaluate the quality of reported small-molecule fluorescent probes.
Key Challenges for Designing Small-Molecule Fluorescent Probes
Discovering sensing reactions
Discovering a reaction specific to the intended analyte is fundamental to designing a reaction-based small-molecule probe. In the case of protein-based probes, such as redox-sensitive green fluorescent protein (roGFP) or hydrogen peroxide sensor protein (Hyper), scientists can take advantage of existing sensing moieties from naturally occurring protein structures (105). In contrast, the sensing reactions for small-molecule-based probes mostly originate from “intelligent design.”
Taking hydrogen peroxide as an example, only a few chemical reactions have been reported to detect hydrogen peroxide in living cells. Among the reported H2O2 probes, the majority of them are based on the oxidation of boronate esters (111, 145). Other sensing reactions for hydrogen peroxide include oxidation-induced C-C bond cleavage of benzils (2), C-S bond cleavage of perfluoro-benzyl sulfonates (106), and C-N bond cleavage of anilines (82); direct oxidation of phosphorous (138), tellurium (78), and selenium (90, 179); and direct metal chelation by hydrogen peroxide (42). Most of the reaction-based probes have been extensively reviewed (14, 55, 145). Here, we discuss several aspects of the design criteria for ideal probes, including selectivity, kinetics, and reversibility of the sensing reactions (Fig. 1).
Selectivity of sensing reactions
A selective sensing reaction is the key to designing small-molecule fluorescent probes. It should be noted that we prefer the term “selective” over “specific.” Although specific probes, which react with nothing but the analyte of interest, are always desired, hardly any probes are truly specific due to the complexity of biological systems. Therefore, we prefer to use the term “selective” to describe probes that preferentially react with the desired analyte at physiological conditions. It is important to fully evaluate the reactivity of the designed probe and any analytes that could potentially interfere under physiologically relevant conditions.
Taking the development of H2O2 probes as an example, DCFH was widely used as an H2O2 probe (23). However, the complete mechanism for the oxidation of DCFH to dichlorofluorescein (DCF) involves multiple steps and many oxidants are involved, including oxidized glutathione (GSSG), NAD, oxygen (O2), iron, and nitric oxide (NO·) (70, 71). Therefore, the fluorescent signal changes of DCFH to DCF cannot be solely attributed to changes in H2O2 levels and, indeed, reflect the overall redox state of the cells.
Advancing from DCFH, several oxidation-induced C-X (X = C, S, or N) bond cleavage reaction-based H2O2 probes were developed. The first reactive center used for H2O2 sensing was perfluoro-benzyl sulfonate (106, 172). Fluorophores are linked with quenchers through this cleavable ester bond, enabling fluorogenic responses in the presence of H2O2. Unfortunately, the reactivity of these acyl sulfonates toward other ROS was not extensively evaluated, partially due to the lack of knowledge of other redox signaling molecules at the time of probe development. Perfluoro-benzyl sulfonate is prone to reductive and nucleophilic reagents from an organic chemistry perspective. Therefore, strong reductive redox signaling molecules such as hydrogen sulfide (H2S) or strong nucleophilic molecules such as cysteine or glutathione are highly likely to react with this reactive moiety.
Recently, Chang and co-workers pioneered the use of boronate esters as selective reductants for H2O2, which led to a series of new H2O2 probes (4, 14, 28, 31, 34, 37, 111). The selectivity of this reaction has been examined against a series of oxidants in cells, including superoxide (O2·−), NO·, hydroxyl radical (OH·), hypochlorite (OCl−), ozone (O3), and singlet oxygen (1O2). It should be noted that peroxynitrite (ONOO−), which is generated intracellularly by nitric oxide and hydroxyl radical, was later shown to react with boronates at a much faster rate (over a million times) than H2O2 in a pH 7.4 buffer (137). Although the intracellular concentration of peroxynitrite is much lower than that of H2O2, care should be taken when interpreting results from boronate probes.
The development of GSH probes faces a similar challenge. There are many free thiol-containing species in the intracellular milieu, including small-molecule thiols and protein thiols. Glutathione, which is usually in the millimolar concentration range, accounts for the majority of small-molecule thiols in eukaryotic cells. However, the concentration of free thiols in proteins can also be as high as the millimolar range, potentially interfering with the readout of GSH probes. To provide a definitive assessment of the selectivity of GSH probes, particularly the intracellular selectivity, our group developed a gel permeation chromatography (GPC)-based assay, which we used in the study of our RealThiol probe (66).
In our protocol, live cells were treated with RealThiol first, then lysed in trichloroacetic acid to prevent the dissociation of RealThiol from protein thiols or small-molecule thiols under diluting conditions. GPC with fluorescence detection was then used to separate probes that had reacted with proteins from those that had reacted with small-molecule thiols. We found that the majority (>90%) of RealThiol had, indeed, reacted with GSH. However, this method cannot be easily applied to H2O2 probe development as the final products of the reacted probes are the same regardless of the nature of the oxidants. A possible experiment to test H2O2 probe selectivity is to mix a panel of physiologically relevant oxidants to determine the reaction selectivity. If possible, cell lysate should also be added to the mixture to evaluate any potential interference from intracellular proteins.
Kinetics of sensing reactions
Ideal sensing reactions should have relatively fast kinetics because the analytes are usually only present in the μM or even nM concentration range. The physiologically relevant concentration of H2O2, for example, is 1–100 nM (136). Considering a typical probe staining concentration of 10 μM, the reaction should have a second-order rate constant of at least 278 M −1·s−1 (assuming a constant 100 nM concentration of H2O2) to ensure sensing results will be available within 1 h, and given the detection limit of the instrument toward the corresponding fluorophore is 1 μM. Longer incubation times usually result in more artifacts, such as re-synthesis of the signaling molecule being detected, metabolic inactivation of probes, probe clearance from cells, and photobleaching of the fluorophores (164).
An ideal sensing reaction should occur in real time or at least be able to achieve a reasonable signal readout within 5 min. Unfortunately, it is often a tradeoff between reaction selectivity and kinetics. Faster reactions are usually correlated with lower energy barriers for oxidation, meaning it may be difficult to maintain reaction selectivity, especially in cases where the sensing reaction is irreversible and there are several molecules in the environment with similar reactivity. Such sensing reactions have not been well established in the development of small-molecule-based H2O2 probes. Most of the H2O2 probes reported based on boronates have second-order rate constants of 0.1–1.0 M −1·s−1, indicating a typical incubation time of >30 min for detecting a change in H2O2 concentration of 100 μM, far higher than its physiological concentration (110).
The ultimate goal for redox molecule sensing is to find or design a fast but specific chemical reaction against the analyte of interest (10). Another possibility is to design reversible sensing reactions with proper equilibrium constants to ensure sensors only respond to analytes at their physiologically relevant concentrations. A good example of the implementation of this strategy is the development of glutathione probes. Because glutathione is among the very few nonprotein thiol species present at >1 mM concentration in eukaryotic cells, it is feasible to develop a reversible sensing reaction that quickly and selectively responds to GSH with little interference from other free thiols. Several groups, including our own, have reported fluorescent probes for real-time imaging of GSH in living cells (17, 66, 101, 151).
Reversibility of sensing reactions
The reversibility of sensing reactions is a relatively complicated issue, especially for H2O2 and other ROS/RNS. By definition, a reversible chemical reaction, depending on conditions, can proceed in both the forward and reverse direction. A chemical equilibrium is reached when forward and reverse reactions proceed at equal rates (Fig. 2).
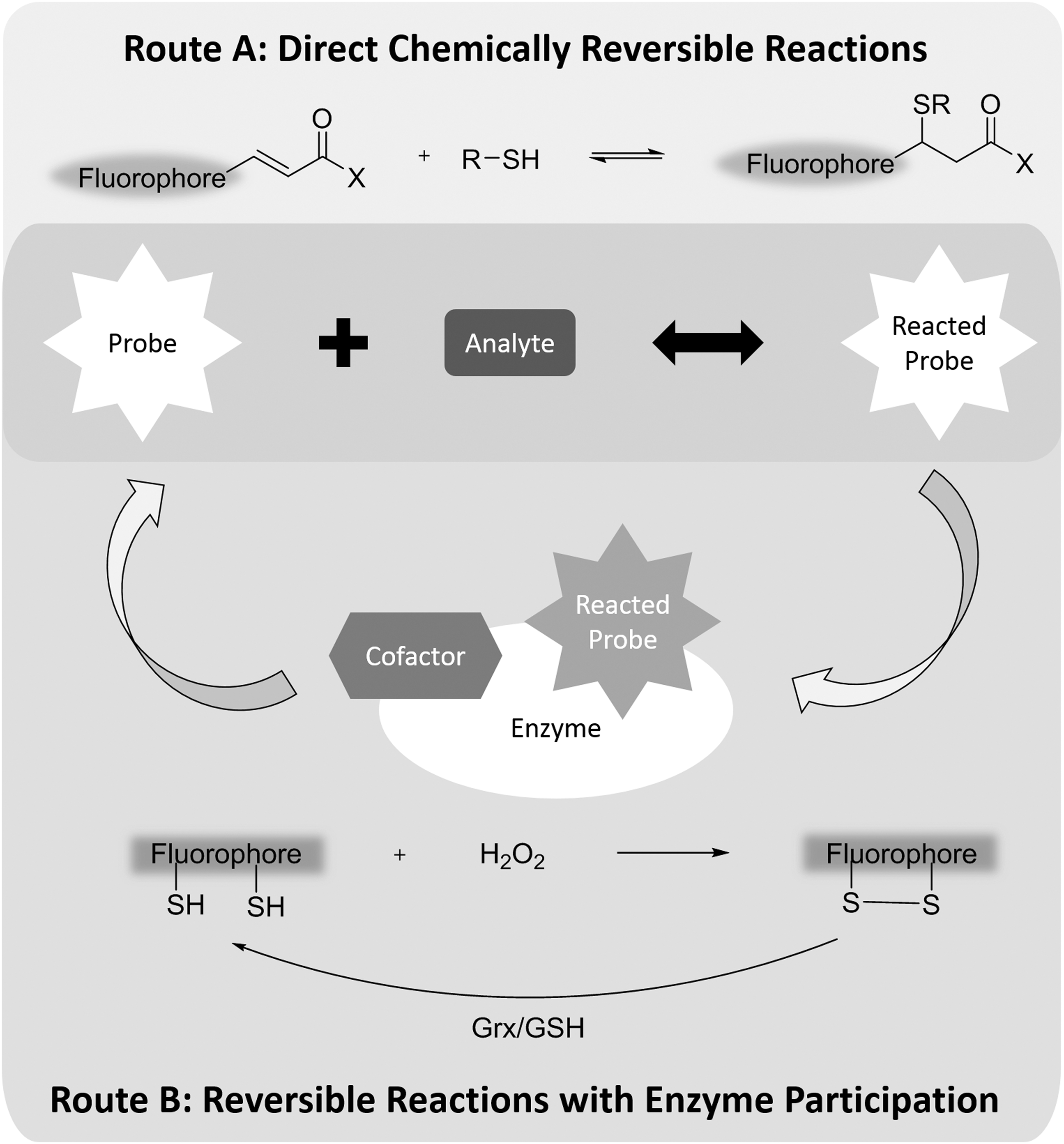
Although irreversible reactions are commonly used in the development of reaction-based fluorescent probes (14), there are a few major advantages to using reversible reactions for probe development. First, because equilibrium is driven by thermodynamics and can be established regardless of the concentration of reactants, it is possible to measure the concentration of an analyte using a probe with a much lower concentration, given there is an appropriate equilibrium constant between the analyte and the probe.
This concept has been well demonstrated in the development of reversible reaction-based GSH probes, in which probes with concentrations in the micromolar or nanomolar range establish equilibria with GSH in the millimolar concentration range (17, 66, 101, 151). If an irreversible reaction-based GSH probe were used, an excess of the probe (above the millimolar concentration of GSH) would have to be applied to react with all the GSH molecules to obtain a quantitative measurement. Complete scavenging of GSH in cells can trigger oxidative stress, which is undesirable, especially in redox biology studies.
Second, if both reacted and unreacted probes have different fluorescence responses, the concentration of the analyte can be calculated based on the fluorescence ratio of the reacted and unreacted probes at equilibrium conditions. This is usually referred to as “ratiometric” imaging. The key advantage of ratiometric measurement is that its quantification results are independent of the absolute probe concentration in cells. Last but not least, live monitoring or longitudinal tracking of the concentration of an analyte is only feasible by using reversible sensing reactions because irreversible reaction-based probes are saturated over time.
Reversible sensing reactions have been applied to develop probes for several biologically relevant molecules, including calcium and GSH. Because of the complexity of the cellular redox system, oxidation and reduction of these redox probes does not always proceed directly through the same reaction (Fig. 2). For example, H2O2 protein sensor Hyper transforms to its oxidized form after reacting with H2O2. But the oxidized form cannot return to the corresponding reduced form by oxidizing H2O to H2O2. Instead, Hyper utilizes the glutaredoxin (Grx)/GSH system for its reduction. As long as the reduction capability of the Grx/GSH system is stable, Hyper can be used to determine H2O2 concentration inside cells at the expense of consuming hydrogen peroxide (105).
Therefore, regarding redox probes, a practical definition of reversible probes should be probes that can monitor both increases and decreases of an analyte in biological systems on the minute-level time scale. Generally, the forward and reverse reactions should proceed at comparable rates under physiological conditions. Increasing the reaction rate between H2O2 and its protein sensor reduces the limit of detection that the sensor is able to achieve (114). However, it should be noted that blindly pursuing the lowest limit of detection for probe design is not advised. All probes have their own dynamic range of detection. Highly sensitive probes tend to have saturated readouts even at moderate concentrations of the analyte. Therefore, it is important to match the dynamic range of designed probes with the physiological concentration range of the analyte.
Of the small-molecule H2O2 probes, only a few probes claim applications in monitoring both increases and decreases in H2O2, including thiol-disulfide-based and Te/Se, P oxidation-based probes (78, 138, 179, 181). However, the selectivity of these sensing reactions has not been well characterized. As Kaur et al. pointed out, these probes would be better described as reflecting “global redox changes” instead of H2O2 changes (73). To the best of our knowledge, these H2O2 probes have not been used by redox biologists in their studies.
Several reversible redox probes targeting other ROS/RNS claim to monitor peroxynitrite (ONOO−)/GSH (103), O2·−/GSH (99, 103, 158, 179, 181), and hypochlorous acid (HClO)/H2S (158) “redox couples.” For example, the Han group reported several fluorescent probes for monitoring redox cycles between peroxynitrite and glutathione and defined peroxynitrite and glutathione as a redox couple (103, 179, 181). However, by definition, a redox couple is the two species of a half-reaction involving oxidation or reduction, for example Cu2+/Cu. It is questionable to define ONOO− and GSH as a redox couple.
In Han's study, the reversibility of the ONOO−/GSH probe was demonstrated by oxidizing the probe with peroxynitrite first, then reducing the probe with addition of GSH. However, peroxynitrite and GSH co-exist in cells. An appropriate test of the reversibility of Han's probe should be performed in a mixture of peroxynitrite and GSH. In fact, Han's probe could be potentially used as a reversible peroxynitrite probe. Nonetheless, although the biological meaning of these designed redox couples is elusive, they could be a good starting point for developing new reversible redox probes that oxidize and reduce through different mechanisms.
In summary, reversible sensing reactions have several distinct advantages, although the design of them can be challenging. GSH probes are a unique case; they have been successfully developed to reversibly react with their analyte to achieve quantitative real-time imaging. It may not be trivial to replicate the success of GSH probes to discover similar reversible reactions for highly energetic ROS/RNS. Chemists could potentially mimic protein sensors, such as Hyper, by designing small-molecule probes that can be oxidized by ROS/RNS and reduced by endogenous enzymatic systems. Regardless, new reversible reactions for the sensing of other ROS/RNS that enable real-time monitoring of redox signals are still in great demand.
Comparison between protein-based probes and small-molecule probes
Two types of protein-based probes are widely used in redox biology, roGFPs (39, 60) and Hyper (9)-based sensors. roGFPs are often used for measuring GSH redox potentials, and Hyper-based sensors are often used for measuring hydrogen peroxide. Their sensing reactions are based on thiol to disulfide transformations that induce changes in fluorescent protein conformations and, thus, spectral properties. Both reactions are intrinsically reversible and can be used for quantitative measurements.
There are several main advantages to protein-based genetically encoded probes, including high selectivity and sensitivity, amenability to subcellular targeting, and applicability to tissue-specific or whole-body imaging in animals. Despite these advantages, genetically encoded probes are relatively laborious to work with due to the cell transfection process, and it is challenging to apply them in primary tissues, such as patient samples. In addition, high expression of sensor proteins may alter cellular redox state due to rapid consumption of the corresponding analytes. Therefore, it is important to minimize the expression level of sensor proteins to avoid artifacts.
In contrast, small-molecule probes can be a great complement to genetically encoded probes due to their convenience, applicability in primary tissues, and minimal perturbance to the interrogated biological system (if a nanomolar to low micromolar probe concentration is used).
Since H2O2 probes are available in both protein- and small-molecule-based versions, it provides a fair battleground for comparing these two sensing strategies. Small-molecule H2O2 probes are mostly used to detect large H2O2 changes in cells, such as the change that occurs under strong exogenous stimulation with bolus treatment of highly concentrated H2O2 (15, 106). Although these probes provide information on cellular responses under extreme conditions, they do not have sufficient sensitivity to detect basal levels of H2O2.
Protein-based probe Hyper is much better at monitoring subtle concentration changes in H2O2 (9). In addition, Morgan et al. recently reported a highly sensitive peroxiredoxin-based probe that can detect basal levels of H2O2, in the low nanomolar to high picomolar range (114). Compared with small-molecule-based H2O2 probes, their protein-based counterparts have superior sensitivity, dynamic ranges, reaction kinetics, and selectivity. More importantly, protein-based H2O2 probes are reversible and can provide quantitative measurements of H2O2 levels in living cells, whereas all small-molecule probes are based on irreversible reactions and can only provide qualitative assessment of large changes of more than 100 μM increase in H2O2 levels. Therefore, it is not surprising that redox biologists prefer protein-based hydrogen peroxide probes in their studies.
Protein-based and small-molecule-based glutathione probes provide different but complementary measurements. RoGFP-based probes such as roGFP1/2 and Grx1-roGFP2 measure GSH redox potentials, which are proportional to the value of [GSH]2/[GSSG] (39, 60). In contrast, state-of-the-art small-molecule-based GSH probes provide direct measurements of [GSH] (66, 101, 151). To the best of our knowledge, there are no probes available that can quantify GSSG concentrations, either protein or small-molecule based. A possible workaround to measure [GSSG] in living cells is to apply both roGFPs and reversible small-molecule GSH probes simultaneously, then calculate [GSSG] based on the values of [GSH]2/[GSSG] and [GSH].
Protein-based redox probes have been further developed by incorporating unnatural amino acids. The Ai group has been a pioneer in this area and recently reported genetically encoded fluorescent probes for peroxynitrite (26) and hydrogen sulfide (19). It should be noted that the protein-based probes reported by the Ai group are based on irreversible reactions with the corresponding analytes, meaning that it would be difficult to obtain quantitative measurements from their readouts or to follow dynamic changes in analyte concentrations.
We have summarized some of the key factors that redox biologists need to consider while choosing between these two types of sensors in Table 1. In our opinion, for basic cell biology studies, protein-based reversible probes, if available, should be preferred due to their superiority in quantitative measurements and real-time monitoring. Small-molecule fluorescent probes become useful when studying primary tissues or when the corresponding protein-based probe is unavailable. Further, we believe that probe developers from both small-molecule and protein camps need to learn from each other and come up with new ideas for redox sensing that can address untapped biological questions.
FRET, Förster resonance energy transfer; Grx, glutaredoxin; GSH, glutathione; GSSG, oxidized glutathione; H2O2, hydrogen peroxide; Hyper, hydrogen peroxide sensor protein; OxyR-RD, regulator domain of E. Coli OxyR gene; PF1, peroxyfluor-1; roGFP, redox-sensitive green fluorescent protein.
Ideas for designing new reactions
As combinatorial chemistry and high-throughput screening has advanced in recent years, these cutting-edge techniques have been applied in probe design (152, 153). Ahn et al. pioneered the use of a target-based combinatorial rosamine library in searching for GSH probes (3). Schiedel et al. introduced a diversity orientated library for screening and discovered a set of bright coumarin-based fluorophores (131). Xie et al. designed a screen and discovered several new sensing reactions for biothiols, which brought insights into the future design of sensing reactions (168). In terms of H2O2 probe design, we are unaware of similar screening strategies being applied. In general, a well-controlled screen is much more efficient than testing sensing reactions one at a time, especially under physiologically relevant complex conditions.
Choosing Appropriate Fluorophores
Quantum yields of fluorophores
For any fluorophore, quantum yield is always among the most important characteristics. A high quantum yield can significantly reduce the amount of fluorophore that is necessary to administer to cells. In the case of detecting redox signaling molecules, low probe loading reduces consumption of the analyte of interest, minimizing interference to the biological system being interrogated. Currently, quantum yields of fluorophores have to be determined experimentally, which often requires laborious synthesis.
The Lavis group recently reported a general method to improve quantum yields in several of the most commonly used fluorophores, which greatly advanced the imaging field (52, 54). Their elegant strategy replaced the freely rotating di-alkyl groups, which occur in many fluorophores, with a structurally rigid azetidine group, reducing rotation-related nonradiative energy relaxation process to enhance quantum yields.
Several new strategies, including aggregation induced emission (AIE) and excited state intramolecular proton transfer (ESIPT), provide potentially useful mechanisms for altering quantum yields.
AIE probe molecules are nonfluorescent at low concentrations, but locally high concentrations of probe molecules induce and self-assemble through π-π interactions and trigger luminescence (63). Although sensitive detection of H2O2 has been demonstrated by using AIE probes in test tube experiments, these probes are difficult to apply in cells (85, 86, 90, 98, 160, 189). To achieve noticeable emission, AIE probes rely on high local concentrations of probe molecules, which can be toxic for cells. Some reported probes even require a loading concentration of up to 1 mM (189). AIE probes are also very hydrophobic due to the indispensable large conjugated building blocks of phenol rings. This can cause solubility and distribution problems in cells, which will be discussed in the following sections.
The ESIPT phenomenon was initially discovered in 3-hydroxyflavone, whose fluorescence relies heavily on intramolecular hydrogen bonding (148, 176). Based on the ESIPT mechanism, several H2O2 probes were developed and their selectivity was evaluated in test tube experiments (88, 94, 191). However, due to the complex environment inside cells with nearly ubiquitous hydrogen bond donors and acceptors, ESIPT probes may lose most of their excited state energies to the adjacent environment, instead of emitting photons. As Zhao et al. pointed out, ESIPT fluorophores prefer aprotic solvents, and “it is still a challenge to use ESIPT molecular probes in protic solvents due to the severe interference of solvents to perturb the ESIPT process” (190).
Current applications of ESIPT redox probes mostly require high percentages of co-solvents or surfactants, resulting in potential artifacts and limited biological significance (94). Further optimization is still required for this type of probe to become practically useful in biological systems.
Quantum dots are another set of emitters developed based on nanoparticle technology. Due to a different emission mechanism than small molecules, quantum dots have extremely high quantum yields and photostability, and their emission is relatively insensitive to environmental changes. Resch-Genger et al. extensively reviewed the applications of quantum dots versus small-molecule-based probes in cells in detail (126). Several H2O2 probes based on quantum dots have been reported (56, 89, 129, 133, 185). It should be noted that depending on their sizes and surface charges, quantum dots may enter cells through endocytosis, rendering them uniquely useful for studying H2O2 signaling in trafficking vesicles.
Stokes shift of fluorescent probes
Stokes shift, defined as the difference between excitation maximum and fluorescence emission maximum, is another key factor in determining probe quality. Fluorophores with small Stokes shifts are usually prone to self-quenching, whereas those with large Stokes shifts are usually desired for ratiometric or Förster resonance energy transfer (FRET)-based probes. Coumarin and cyanine fluorophores have intrinsic push-pull structures, leading to potentially large Stokes shifts. However, some of these structures have high molecular weights or high polarity, potentially limiting the cell permeability of the designed probes. A “prodrug” strategy can be applied to enhance cell permeability by converting polar probes into nonpolar pro-probes, which are then processed inside cells to regenerate the parent probes (66).
FRET provides another strategy to design probes with pseudo large Stokes shifts. Several FRET-based H2O2 probes were reported to have large Stokes shifts (5, 41, 163).
Design of ratiometric fluorescent probes
Calcium probe Fura-2, developed by Roger Tsien, was the first widely used ratiometric probe for biomolecule sensing (150). Ratiometric fluorescent probes show a shift in their excitation or emission spectra on reaction with their corresponding analytes. This feature is highly desirable in live cell imaging because it provides an internal standard that allows for the correction of artifacts due to photobleaching, variations in laser intensity, and inhomogeneous probe concentration inside cells. Figure 3 explains the chemical principle behind the ratiometric measurements and the strategy to quantify analyte under different scenarios. At equilibrium conditions, the concentration of an analyte is proportional to the ratio of reacted and unreacted probe independent of the absolute probe concentration, given that the probe concentration is much lower than that of the analyte (Eq 1, Fig. 3A).
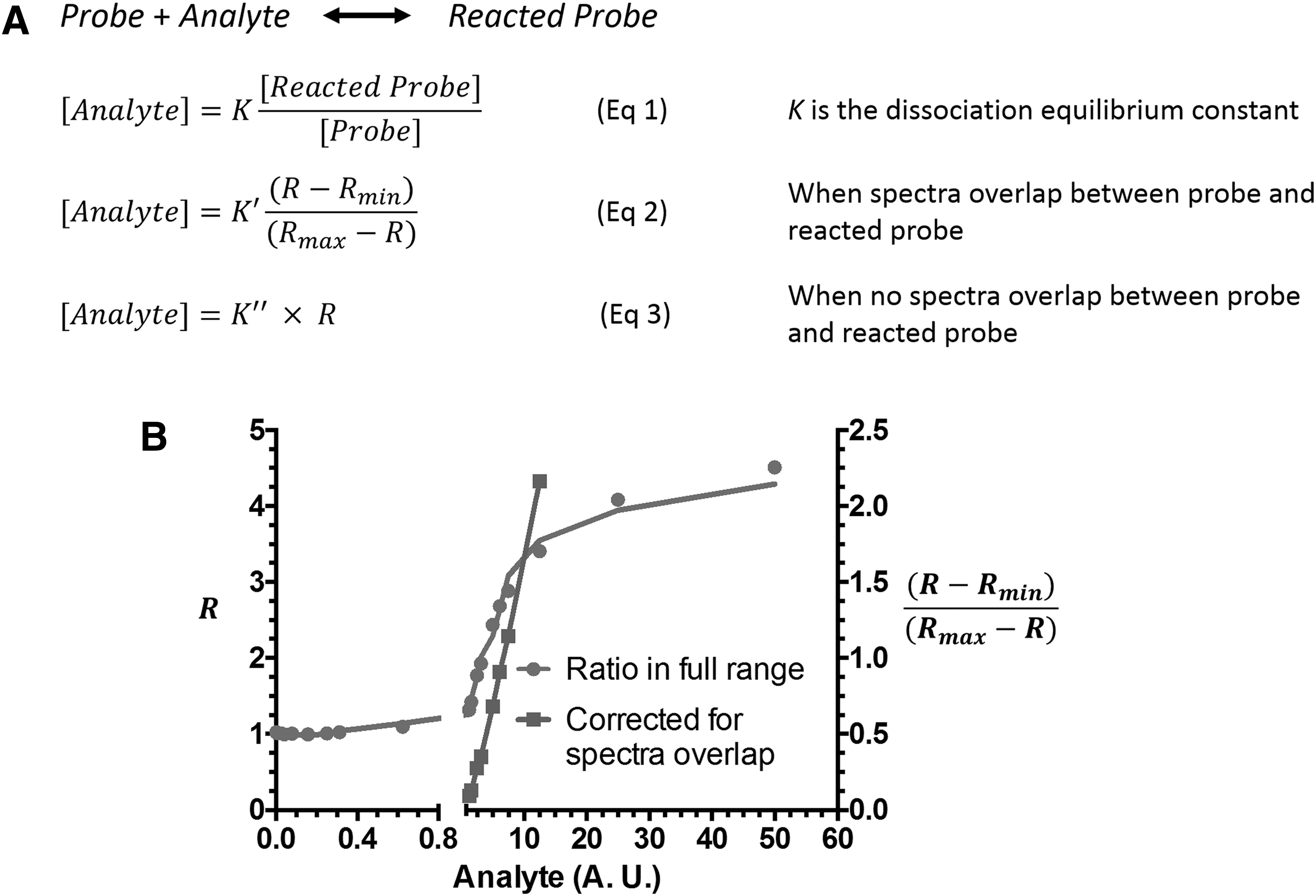
However, the quantitation equation can be complicated if there is spectral overlap or if the concentrations of analyte and probe are similar (Eq 2, Fig. 3A) (67). When there is no spectral overlap between reacted and unreacted probes, the analyte concentration is proportional to the ratio R between reacted and unreacted probes (Eq 3, Fig. 3A). It should be noted that all these calculations are based on the prerequisite that a chemical equilibrium is established between the analyte and the probe. Therefore, irreversible reaction-based probes are unsuitable for ratiometric-based quantitative measurements.
Wavelengths of fluorescent probes
When designing fluorescent probes for biological applications, it is important to consider appropriate excitation and emission wavelengths. Although some commercially available plate readers have light sources with tunable wavelengths, almost all laser-based instruments can only accommodate very limited excitation wavelengths. Commercially available laser sources usually cover 405, 488, 514, 543, 594, and 633 nm, with some high-end products also including 355, 441, 458, 532, 561, 611, 647, and 694 nm excitation wavelengths. Due to budget concerns, most instruments are equipped with no more than five laser sources at the same time. The efficiency of excitation decreases significantly when the excitation laser does not match the absorption maximum of a fluorophore.
Moreover, when a fluorophore has an excitation spectrum in between two laser wavelengths, it may create severe leaking artifacts, interfering with both channels simultaneously. The same concerns apply to emission wavelengths, although some instruments allow tunable emission bands. When designing probes, it is important to use fluorophores with appropriate excitation and emission spectra to minimize potential cross-talk between different fluorescence channels.
Developing a set of probes with different excitation wavelengths is advantageous due to the potential for multiplexing with other fluorescent probes or stains. For example, taking advantage of H2O2-responsive boronate chemistry, a series of H2O2 probes were developed based on fluorescein, rhodamine, and several other fluorophores (14, 163). These probes were multiplexed with a pH-sensitive probe to detect H2O2 under various pH conditions, as well as with an HClO probe to simultaneously detect HClO and H2O2 in live cells (36, 140).
Near-infrared (NIR) fluorophores gained special attention due to their potential in vivo applications and low phototoxicity (78, 169, 170, 181). As Wäldchen et al. pointed out, phototoxicity decreases exponentially when wavelength increases (157). NIR lasers also have excellent tissue penetration ability. Several cyanine-based H2O2 probes have been shown to be useful for in vivo applications (72, 120, 123). Silicon-rhodamine-based fluorophores, developed by the Nagano group, recently emerged as a new class of high quantum yield NIR fluorophores (80). H2O2 probes based on this class of fluorophore have also been reported (187).
Overcoming Cellular Barriers
Solubility and membrane permeability of probes
The intracellular environment is far more complicated than a homogenous mixture of proteins and small molecules, but it is spatially organized. The overall concentration of proteins can be as high as 5 mM, but these proteins are neither randomly nor evenly distributed inside cells. Instead, there are many organelles or compartments separated by lipid bilayers, creating hydrophilic pockets with hydrophobic boundaries. Therefore, the solubility of probe molecules can bias their localization and sometimes fluorescence properties as well.
For example, a hydrophobic fluorescent probe tends to show higher fluorescence readouts in lipid membranes than in the cytosol due to its preferential accumulation in hydrophobic environments. However, this does not necessarily mean that the analyte concentration is higher in membranes than in the cytosol. Therefore, to detect hydrophilic analytes, probes should have sufficient aqueous solubility to avoid potential artifacts. LogP values, which measure the partition of a molecule in an octanol-water biphasic system, are commonly used in medicinal chemistry to evaluate the solubility of drug molecules. We recommend integrating LogP measurements in the development of probes to ensure sufficient aqueous solubility.
Typical methods for increasing water solubility include adding carboxylic acid groups, hydroxyl groups, or other hydrophilic groups to the probe core structure. However, these functional groups are usually very polar, making it difficult for the probe to cross the hydrophobic plasma membrane. Several strategies have been developed to tackle this problem. The most well-known method is replacing the carboxylic acid groups with acetylmethoxyl (AM) esters, which are hydrophobic enough to cross the plasma membrane but cleavable by intracellular esterases so that the acid form of the parent probe is regenerated once inside the cell (69, 147, 149). In addition, the acid form of probes is usually cell impermeable, which prevents the probe from leaking out of the cells once it has been internalized.
The AM ester strategy has been applied in many redox probes. It should be noted that the hydrolysis of AM esters releases formaldehyde, which may cause toxicity and/or alter the cellular redox state. Lowering the probe loading concentration can help to reduce the impact of the probe on cells.
Protein interference with sensing reactions
ROS/RNS are highly reactive small molecules, and their concentrations are tightly regulated in cells by a number of redox-active enzymes. To detect ROS/RNS, probe molecules must react quickly enough with ROS/RNS to compete with highly efficient ROS/RNS scavenging enzymes. For example, it has been reported that the most popular boronate ester sensing group for H2O2 can be outcompeted kinetically by proteins, such as glutathione peroxidases (GPx) and catalase (70). Therefore, the signal from these fluorescent probes only represents a small portion of the intracellular H2O2 pool.
In fact, most reported boronate ester-based H2O2 probes can only be used to measure increases in H2O2 concentrations in the micromolar range either from bolus administration of external ROS or under very special biological events, such as immune responses (36). To the best of our knowledge, we are unaware of anyone successfully using small-molecule fluorescent probes to measure physiologically relevant concentrations of H2O2 in cells.
In the development of glutathione probes, the highly abundant protein thiols could also react with the probes. To test this possibility, our group developed a GPC-based method with fluorescence detection to assess protein interactions with small-molecule GSH probes (67). This method could be modified and adapted to the development of other redox probes.
Probe turnover and clearance in live cells
Fluorescent probes are essentially xenobiotics to cells. Cells actively clear these xenobiotics through various mechanisms, including inactivation by P450 enzymes and excretion through ATP-binding cassette (ABC) transporters (65). Therefore, for live cell experiments, researchers need to take the intracellular turnover of probe molecules into consideration, preferably measuring the half-life of probe clearance. In the event of rapid probe clearance, time-lapsed experiments with relatively long incubation times may not be feasible. To address this problem, one could try to use ABC transporter inhibitors, such as probenecid, to reduce probe clearance (155, 156). If the clearance mechanism is unclear or the clearance rate cannot be reduced to the desired extent, the best approach would be to prepare a series of samples at different time points and stain each sample individually by using exactly the same conditions.
Designing experiments to perform quantitative detection of redox signaling molecules in live cells can be complicated and requires a number of controls; however, live cell experiments provide much more physiologically relevant information than bulk measurements using lysates.
Organelle specificity of small-molecule probes
The subcellular dynamics of redox signaling molecules have gained increasing attention in recent years. Redox probes targeting mitochondria (34, 41, 97, 107, 115, 162, 171), lysosome (76, 100, 124, 187), nucleus (37, 161), endoplasmic reticulum (7, 167), and plasma membrane (77) have all been reported.
For example, the triphenylphosphonium (TPP) group is commonly used in probe design for mitochondrial targeting, taking advantage of the highly negative mitochondrial membrane potential (Fig. 4A) (17). Probes with this group have been used to demonstrate how treatments with drugs, such as rotenone or antimycin A, can reduce the mitochondrial membrane potential. However, the reduction in fluorescence observed during these experiments could have been due to the probe dissociating from the mitochondria rather than from an actual decrease in the analyte concentration. Therefore, one must be very careful when interpreting the results of studies done with organelle-specific probes.
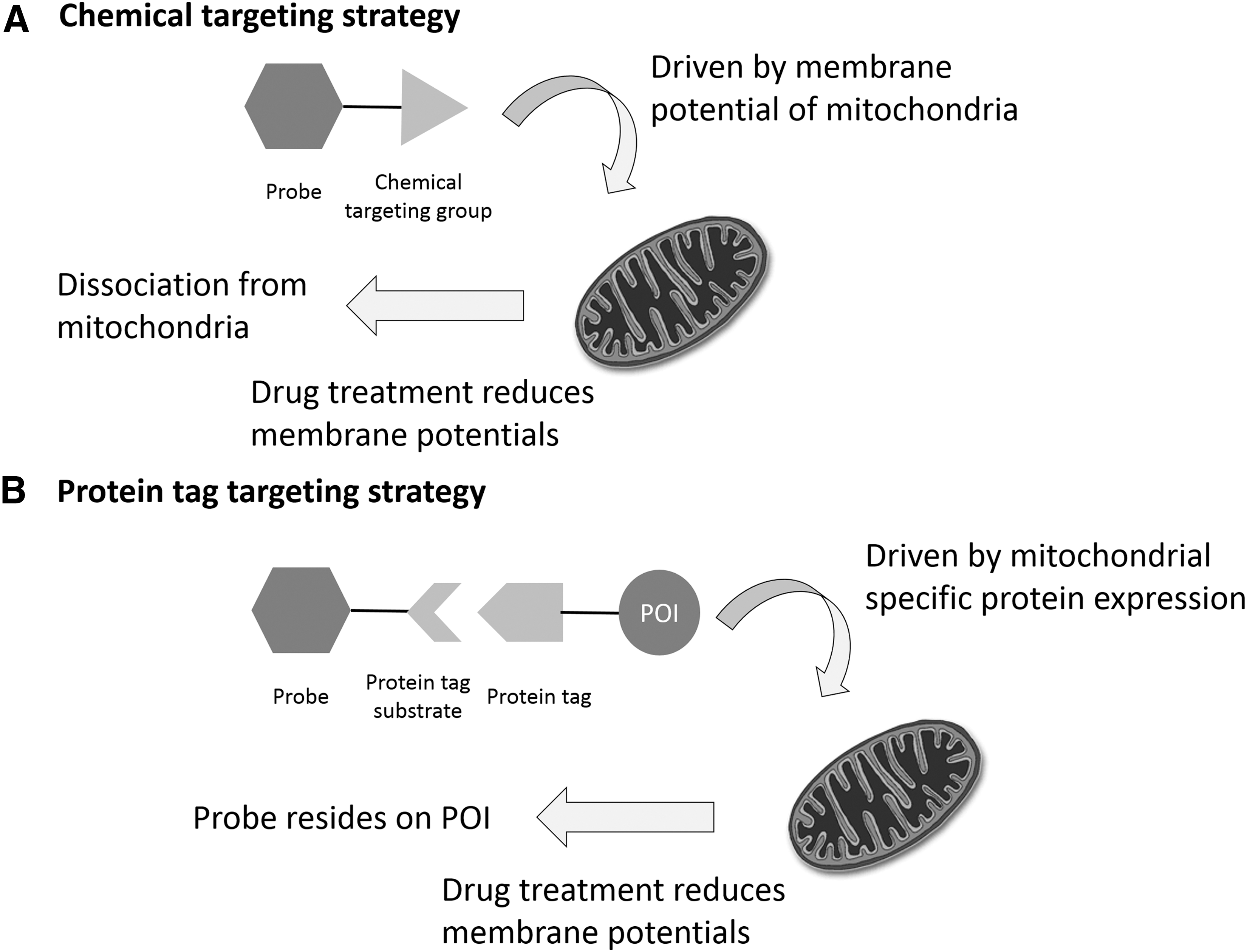
Protein tagging technologies, such as Halo-Tag (18, 102), SNAP-Tag (1, 50, 75, 113, 127), CLIP-Tag (50), and ACP-Tag (51, 177), can provide an alternative strategy for organelle targeting. Many small-molecule organelle targeting groups are synthetically difficult to produce, whereas protein tags are synthetically convenient and expandable to essentially all subcellular targets (Fig. 4B). For example, the Halo-tag protein can be fused with a nuclear-specific protein or nuclear localization sequence to specifically express the Halo-tag protein in the nucleus. Small-molecule probes can then be conjugated with the Halo-tag substrate to specifically react with the Halo-tag protein expressed in a particular organelle.
Although this method does require transfection of cells, similar to genetically encoded probes, this hybrid strategy provides a nice general method for positioning small-molecule probes in cells with subcellular resolution.
Small-Molecule Fluorescent Probes for Redox Signaling
To better assist readers in assessing the performance of redox-active fluorescent probes using the concepts of probe design, here we provide a brief summary of some of the representative probes and categorize them by their sensing reactions. We also excerpt available data including reaction selectivity, typical staining concentration, reaction kinetics, and wavelengths from these reports. Comprehensive summaries of more recently developed redox fluorescent probes can be found in these review articles (8, 14, 21, 22, 24, 55, 87, 92, 103, 111, 144, 145, 166, 183).
H2O2-responsive probes
H2O2-responsive probes are categorized into five groups: boronate-based probes (the most popular option), benzil-based probes, chalcogenides-based probes, metal-mediated probes, and other probes (Table 1).
As mentioned earlier, boronate-based hydrogen peroxide probes have good selectivity toward H2O2 at micromolar concentration levels (15, 25, 86, 98, 100, 116, 171, 189). The cross-reactivity is typically tested in a test tube experiment against a panel of individual ROS/RNS. The typical staining concentration used for cell experiments is in the low micromolar range, with the exception of the AIE-based probe that requires a staining concentration as high as 1 mM (189). To detect an increase of H2O2 of ∼100 μM, the typical incubation time for these probes is 30 min. Peroxyfluor-1 (PF1) only requires a 5-min incubation due to its fast reaction with H2O2 (15). For benzil-based probes, the reaction kinetics are generally faster than for boronate-based probes. An incubation time of only 10 min is needed before reading the signal (1, 2).
Testing of cross-reactivity between benzil-based probes and other ROS/RNS is performed in the same manner as that of boronate-based probes, and the results are similar. Chalcogenide-based probes take advantage of the affinity between oxygen and chalcogenide atoms, as well as the reversibility of such oxidation reactions. However, the selectivity profile for this kind of probe is not well established. To date, these probes seem to be more suitable as overall redox state probes instead of H2O2-selective probes. More detailed data from biological studies are required before we can fully assess their performance (74, 78).
Metal-mediated probes mainly rely on transition metals and their affinity toward oxygen under specific conditions. Few probes of this type have been reported, and there are few, if any, common properties within the group (62, 139). Other probes include the first H2O2-cleavable sulfonate probe (106) and the phenol oxidation-based probe (180). The common properties of hydrogen peroxide-responsive probes are summarized in Table 2.
AIE, aggregation induced emission; N/A, not available; NO, nitric oxide; O2·−, superoxide; OCl−, hypochlorite; OH·, hydroxyl radical; ONOO−, peroxynitrite.
Peroxynitrite-responsive probes
Peroxynitrite-responsive probes are categorized into three main groups: addition-based probes, oxidation-based probes, and other probes (Table 3). One type of addition-based probe relies on the addition of the NO moiety onto a secondary acyl amine (91, 109). Another type relies on the electrophilic addition of the whole molecule onto a positively charged acceptor (27, 192). Both types of addition-based probes showed good selectivity among a panel of ROS/RNS in in vitro experiments; however, the experiments were not performed by using physiologically relevant concentrations of all ROS/RNS. Specifically, the ONOO− was tested at a concentration of 10 μM or higher, which is hundreds of folds higher than its physiological concentration. These probes are administered to cells at low micromolar concentrations and require 20–30 min of incubation before acquiring signals.
Cys, cysteine; ESIPT, excited state intramolecular proton transfer; 1O2, singlet oxygen.
The last type of addition-based probes has a special design that incorporates an addition reaction with a cyclization step (142, 173). The authors claim that such a reaction design significantly improved selectivity, especially against hydrogen peroxide. However, because the physiologically relevant concentration of ONOO− is significantly lower than that of H2O2, selective detection of ONOO− is still challenging in cells. Even using these probes, the authors observed that signals generated from probes reacting with ONOO− were at most ∼200-fold higher than those generated by other ROS/RNS if all of them are at a similar concentration level. Considering the physiologically relevant concentration differences, at least 50% of their signal may come from nonspecific reactions between probes and other ROS/RNS inside a cell.
Oxidation-based probes can be further divided into three groups, phenol-based (88, 122), boronate-based (141, 182), and Te-based (181) probes. As mentioned earlier, most phenol-based and Te-based probes are best used as indicators of the overall redox state. However, some in vitro experiments reveal excellent selectivity between ONOO− and other ROS/RNS, with a difference in response of up to several thousand folds. This is potentially due to a kinetic difference (122). Boronate-based probes react with ONOO− quickly, but due to the low concentration of ONOO− inside cells, they still require a 30-min incubation time before optimal signals can be detected (84, 128, 141, 182). There are also a few other interesting mechanisms that have been applied to ONOO− sensing (13). The available common properties of peroxynitrite-responsive probes are summarized in Table 3.
HClO-responsive probes
HClO-responsive probes are all oxidation-based probes, and they are categorized into three sub-types (Table 4): chalcogenides-based (104, 154, 184), phenol/aniline-based (64, 81, 193), and thiol ether-based probes (11, 79). These oxidation-based probes, regardless of reaction reversibility, are mostly overall redox state indicators. The selectivity tests for these probes are usually performed by comparing probe responses with micromolar concentrations of all ROS/RNS including HClO, which do not represent their corresponding physiologically relevant concentrations. The staining concentration is usually in the low micromolar range, and the typical incubation time is 15–30 min. The available common properties of HClO-responsive probes are summarized in Table 4.
Thiol-responsive probes
Thiol-responsive probes are categorized into two main types: reversible and irreversible reaction-based probes (Table 5). The reversible probes are based on either Michael addition reactions or nucleophilic addition to rhodamines; whereas irreversible probes use several different reactions, including addition and substitution reactions.
A reversible Michael addition with a proper equilibrium constant can be applied to quantitative monitoring of glutathione concentrations in live cells. Since our group first introduced the concept by designing ThiolQuant-Green 2 years ago (67), several groups including our own have followed up and designed probes with improved reaction kinetics (16, 66, 101, 151). All these GSH probes have reaction kinetics in the seconds to minutes range, and they have been demonstrated to quantitatively follow GSH dynamics in living cells.
It should be noted that these probes are claimed to be selective for GSH based on the fact that GSH is the most abundant small-molecule thiol in eukaryotic cells. However, some organisms use trypanothione (45), mycothiol (118), or bacillithiol (119) instead of glutathione. In these cases, the GSH probes could be repurposed to quantify these other small-molecule thiols assuming their concentrations are in the mM range.
Despite using similar Michael addition reactions, some GSH probes with suboptimal equilibrium constants and/or sluggish reaction kinetics cannot be used for time-lapsed experiments (93, 175). When coupling two addition reactions for cyclization, several folds of selectivity between different biothiols have been achieved. However, since GSH is the dominant free thiol in cells, it is difficult to achieve selective signal detection under physiological conditions (143). The nucleophilicity of thiols has also been utilized to design substitution reaction-based thiol probes (135). Similarly, these probes respond primarily to intracellular GSH under physiological conditions. Most of these probes are administered to cells at low micromolar concentrations. The available common properties for glutathione probes are summarized in Table 5.
Probes for other redox metabolites
Biochemical redox reactions involve more than just the molecules stated earlier. Continuous efforts have been made to discover other important redox signaling molecules involved in cellular metabolic; some examples include intermediates such as sulfenic acid, sulfane sulfur, sulfite, bisulfite, and peroxidized lipids. We have summarized some of the recently developed probes for these metabolic intermediates (Table 6).
When thiols are biologically modified in cells, they could go through several intermediates such as sulfenic acid, sulfane sulfur, sulfite, or bisulfite. These intermediates serve different roles in cells and are important in redox biology (57). The Carroll group has developed a series of probes targeting mostly sulfenic acid modifications on protein thiols (57 –59, 83, 121, 132). These sensing reactions are mostly based on selective addition of sulfenic acid onto a meta-cyclohexanedione. They have been used for labeling of sulfenic acid modified proteins from cell lysate but have relatively limited applications as live cell probes.
The thiol derivatization agent, 4-chloro-7-nitrobenzofurazan, widely used in HPLC is also used as a sulfenic acid probe (43). Due to the lack of cross-reactivity tests and relatively slow kinetics, all the sensing reactions mentioned earlier are geared toward lysate-based measurements.
There are also a number of sensors developed for polysulfides, including sulfane sulfur initially used by the Xian group. They first utilized a biotin switch assay to distinguish hydrodisulfides from the predominant RSH in cell lysates (186). The same group has also developed several probes for detecting different polysulfides in live cells (20, 95, 96).
A few probes for sulfite and bisulfite have also been developed based on the reductive addition of sulfite/bisulfite onto ketones or aldehydes (29, 30, 146, 165, 178). The role of such metabolites in redox biology is not yet well established, and the physiologically relevant concentration is unknown. Therefore, it is currently difficult to predict which attributes are important in designing probes for sulfite and bisulfite.
Other important redox intermediates include oxidized lipids, oxidized protein/peptide thiols, and oxidized sugar. However, there are no small-molecule probes that can selectively and reliably measure these peroxides potentially due to difficulty in sensing reaction design. There are a few reported probes for lipid peroxidation, but their cross-reactivity and bioavailability is not well studied (40, 117). More work needs to be done to demonstrate their applicability for studying redox biology.
Conclusions
Currently, the development of redox-responsive small-molecule fluorescent probes is primarily driven by chemists who sometimes fail to sufficiently test their probes in complex biological systems. Although many probes with new reaction centers have been reported, only a few have been tested and applied by redox biologists. There is an urgent need to strengthen the communication between probe developers, mostly chemists, and redox biologists.
The most challenging questions for developing new redox probes are: (i) how to develop new sensing reactions that target different analytes, and (ii) how to optimize these probes for real-time quantitation in biological systems with sub-cellular resolution. In this contribution, we have discussed several key factors that are important in assessing the quality of probes for both chemists and biologists, including selectivity, kinetics, and reversibility of sensing reactions, quantum yield, wavelength, and Stokes shifts of fluorophores, solubility, membrane permeability, protein interference, turnover, and organelle specificity of probe molecules. We specifically highlighted the advantages of reversibility in sensing reactions and its application in ratiometric probe design.
In the future, we hope that probe developers will focus not only on exploring new chemistries for sensing reactions but also on fine-tuning probes for biological applications. We believe that with further development in this field, small-molecule fluorescent probes will become useful complements to protein-based probes in redox biology studies.
Footnotes
Acknowledgments
The research was supported in part by the National Institutes of Health (R01-GM115622, R01-CA207701, R21-CA213535 to J.W., R01-AG045183, R01-AT009050, R21-EB022302, and DP1-DK113644 to M.C.W.) and the Welch Foundation (Q-1912 to M.C.W.).
Author Disclosure Statement
X.J., J.C., and J.W. are co-inventors of a patent application related to the RealThiol probe.