
Abstract
Significance:
Eukaryotic cells execute various functions in subcellular compartments or organelles for which cellular redox homeostasis is of importance. Apart from mitochondria, hypoxia and stress-mediated formation of reactive oxygen species (ROS) were shown to modulate endoplasmic reticulum (ER) and Golgi apparatus (GA) functions.
Recent Advances:
Research during the last decade has improved our understanding of disulfide bond formation, protein glycosylation and secretion, as well as pH and redox homeostasis in the ER and GA. Thus, oxygen (O2) itself, NADPH oxidase (NOX) formed ROS, and pH changes appear to be of importance and indicate the intricate balance of intercompartmental communication.
Critical Issues:
Although the interplay between hypoxia, ER stress, and Golgi function is evident, the existence of more than 20 protein disulfide isomerase family members and the relative mild phenotypes of, for example, endoplasmic reticulum oxidoreductin 1 (ERO1)- and NOX4-knockout mice clearly suggest the existence of redundant and alternative pathways, which remain largely elusive.
Future Directions:
The identification of these pathways and the key players involved in intercompartmental communication needs suitable animal models, genome-wide association, as well as proteomic studies in humans. The results of those studies will be beneficial for the understanding of the etiology of diseases such as type 2 diabetes, Alzheimer's disease, and cancer, which are associated with ROS, protein aggregation, and glycosylation defects.
Introduction
A
However, nowadays, substances such as hydrogen peroxide (H2O2) are also considered ROS because they may constitute the basis for the formation of proper oxygen radicals. In most, if not all, cases, incomplete reduction of oxygen to superoxide anions (O2 •−) represents the first step in ROS formation (212). Thus, an insufficient O2 supply, which does not meet the energy demands of the cell (commonly called hypoxia) as well as an uncontrolled generation of ROS, can be life threatening and lead to oxidative damage of DNA, lipids, and proteins. To cope with periods of acute or chronic hypoxia and/or excessive ROS accumulation, aerobic living cells and organisms have evolved complex biochemical systems and highly efficient antioxidant strategies to allow adaption and survival.
Eukaryotic cells execute various functions in subcellular compartments, which contribute to overall ROS homeostasis. For example, it is well known that mitochondria consume about 80% of the oxygen in the cell and produce the vast majority of ROS due to electron leaks at the electron transport chain (ETC) complexes in the form of O2 •−. From the five ETC complexes, the NADH-ubiquinone oxidoreductase (complex I) and ubiquinone-cytochrome c oxidoreductase (complex III) are the best-known ROS-generating sites (72).
Much less ROS are generated in the endoplasmic reticulum (ER), the Golgi apparatus (GA), lysosomes, peroxisomes, the nucleus, the extracellular space, or at the cytoskeletal interfaces between these compartments. While the recently identified molecules interacting with CasL-CRK-associated substrate-related proteins were found to oxidize actin and to generate ROS by organizing the cytoskeletal interfaces (53), various enzyme systems such as NADPH oxidases (NOX1-5, dual oxidase [DUOX] 1-2), xanthine oxidases, monoamino oxidase(s), cyclooxygenase(s), lipoxygenase(s), uncoupled nitric oxide (NO) synthases, lysyl oxidase(s), the cytochrome P450s, and peroxidases can be found within the respective compartments [for review, see Samoylenko et al. (170)].
However, when the compartmental functions are disrupted due to an altered metabolism or the lack of oxygen and/or antioxidants, the adaptation and defense mechanisms may not work properly. As a consequence, many diseases, including inflammatory, cardiovascular, metabolic, and neurodegenerative diseases, as well as cancer, are associated with hypoxia and/or the increased availability of ROS, commonly termed oxidative stress (49).
Research during the last decade has shown that the ER and Golgi are critical for cellular communication and the physiological and pathological responses to cellular signals. In accordance with this, hypoxia and ROS were shown to modulate ER and Golgi function. Conversely, perturbed ER and Golgi functions were shown to affect the hypoxia response, ROS generation, and subsequently also diverse cellular functions (11, 142). Hence, it appears to be of special interest that the mutual interplay between the ER and the Golgi compartments via ROS may affect signaling pathways and disease pathology. In the current review, we summarize some recent aspects of the role of hypoxia and ROS on ER and Golgi functions as well as their links to pathological consequences such as cancer.
Basic Structure and Functions of the ER
The ER is best known for its main tasks in the synthesis and export of proteins and lipids and as a regulator of calcium (Ca2+) homeostasis. Advanced microscopical techniques, including three-dimensional electron tomography and high-resolution confocal microscopy, have revealed that the ER is a single membranous network that includes the nuclear envelope, which extends to the peripheral ER [for reviews, see Hu et al. (74) and Westrate et al. (211)]. The nuclear envelope is composed of the inner nuclear membrane and outer nuclear membrane; both contain nuclear pores allowing transport of molecules, including RNAs and proteins. Their permeation with proteins binding to chromatin, lamin, and the cytoskeleton maintains the shape of the nuclear membrane. The peripheral ER branches out from the outer nuclear membrane and forms a series of cisternal sheets and dynamic tubules with a common lumen (Fig. 1).
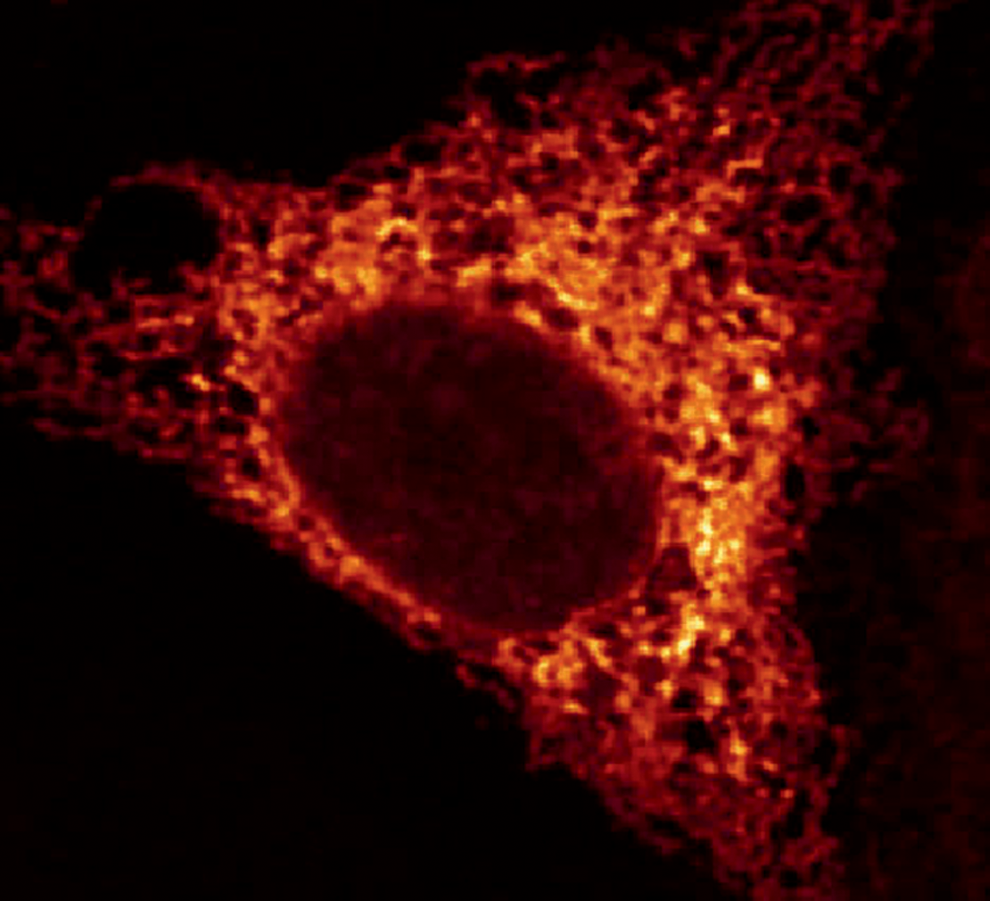
The stacked “rough” peripheral ER sheets are characterized by the high density of ribosomes on the cytosolic surface indicating the occurrence of synthesis, folding, and post-translational modification of membrane-bound or secreted proteins. By contrast, the ER tubules associate with fewer ribosomes, giving them their “smooth” character. Furthermore, the tubular network is curved and subject to continuous growth and rearrangement (74, 211).
All cells contain rough sheets and smooth tubules; however, their ratio varies in different cell types. Hence, there are specific cells where the one or the other is more abundant. Cells of the stomach and gastrointestinal tract (which secrete gastric acid and digestive enzymes, respectively), cells of the lung (which secrete surfactant), cells of salivary glands (which secrete saliva), or antibody producing plasma cells are just a few examples of cells enriched with rough sheets. By contrast, the ER with enriched smooth tubules is highly abundant in cells producing high amounts of fatty acids, cholesterol, phospholipids, and steroids; primarily these cells are found in the liver, testes, and ovaries, as well as in muscle, where the smooth ER (also known as sarcoplasmic reticulum, SR) has a major role in calcium sequestration.
Since the ER is involved in the synthesis of membrane and organelle proteins as well as of virtually all proteins destined for secretion, it represents the first compartment of the so-called secretory pathway. Within that pathway, the ER works in concert with the GA to process newly synthesized proteins that carry a signal peptide and/or a transmembrane domain that direct their insertion into the ER before their delivery to their final destinations in the cell. This involves, apart from folding of the nascent polypeptides and post-translational modifications such as disulfide bond formation or N-glycosylation, protein quality control to ensure that only properly folded proteins can be transported via COP II vesicular carriers to the GA and the plasma membrane.
In addition, recent emerging findings have shown that numerous proteins also reach these destinations in a different manner. This so-called unconventional protein secretion is not fully understood yet but comprises secretion of proteins without a signal peptide by mechanisms involving pore-mediated translocation, ABC transporter-based secretion, or autophagosome/endosome-based secretion. Alternatively, signal peptide/transmembrane domain containing proteins that enter the ER may be secreted via bypassing the GA by involving the Golgi reassembly stacking proteins [for details, see Rabouille et al. (151)].
Importantly, conventional and unconventional protein secretions are both sensitive to various types of stress such as glucose deprivation, exposure to inflammatory mediators, or mechanical stress, which are all linked to the use of oxygen and the generation of ROS (170). For example, a change in the luminal content of oxygen, ROS, or Ca2+ can affect protein synthesis and folding. Improper or defective protein folding has severe consequences, which are thought to contribute to pathological situations such as inflammation, lipid accumulation, oxidative stress, autophagy, and apoptosis (49). Vice versa, a buildup of unfolded proteins as seen with endoplasmic reticulum storage diseases (167) such as familial hypercholesterolemia, osteogenesis imperfecta, diabetes insipidus, or congenital hypothyroid goiter can alter ER redox or Ca2+ homeostasis (20, 112). These features reveal that the ER is a dynamic organelle where ROS are involved in signaling. In addition, the discovery of ER interactions with other cellular compartments via mitochondria-associated membranes (MAMs) and plasma membrane-associated membranes indicates that the ER contributes also to compartmental communication and cell fate via the passage and transport of ions and other small molecules such as ROS (61).
Protein Folding in the ER
To gain its biological function, a polypeptide that has cotranslationally entered the ER needs to be folded into its specific three-dimensional structure. Thereby, the correct spatial arrangement of intra- or intermolecular disulfide bonds is very important. If a protein is not properly folded, the protein will not attain its optimal functional conformation. In that case, the quality control system of the ER determines whether the protein becomes subject to either refolding/isomerization or retranslocation into the cytosol where it will be degraded (43).
It is now commonly accepted that disulfide bond formation in the ER lumen requires, apart from an environment allowing oxidation, reduction, and isomerization of disulfide bonds, correctly folded enzymes that function as catalysts. Oxidoreductases, including members of the protein disulfide isomerase (PDI) family (44, 65), can carry out these tasks (Fig. 2). Interestingly, controlled disulfide bond formation exists also in the cytoplasm, in particular during virus biogenesis (169), and in the intermembrane space of mitochondria where PDI-like oxidoreductases were found (68), indicating that disulfide bond formation is not restricted to the ER.
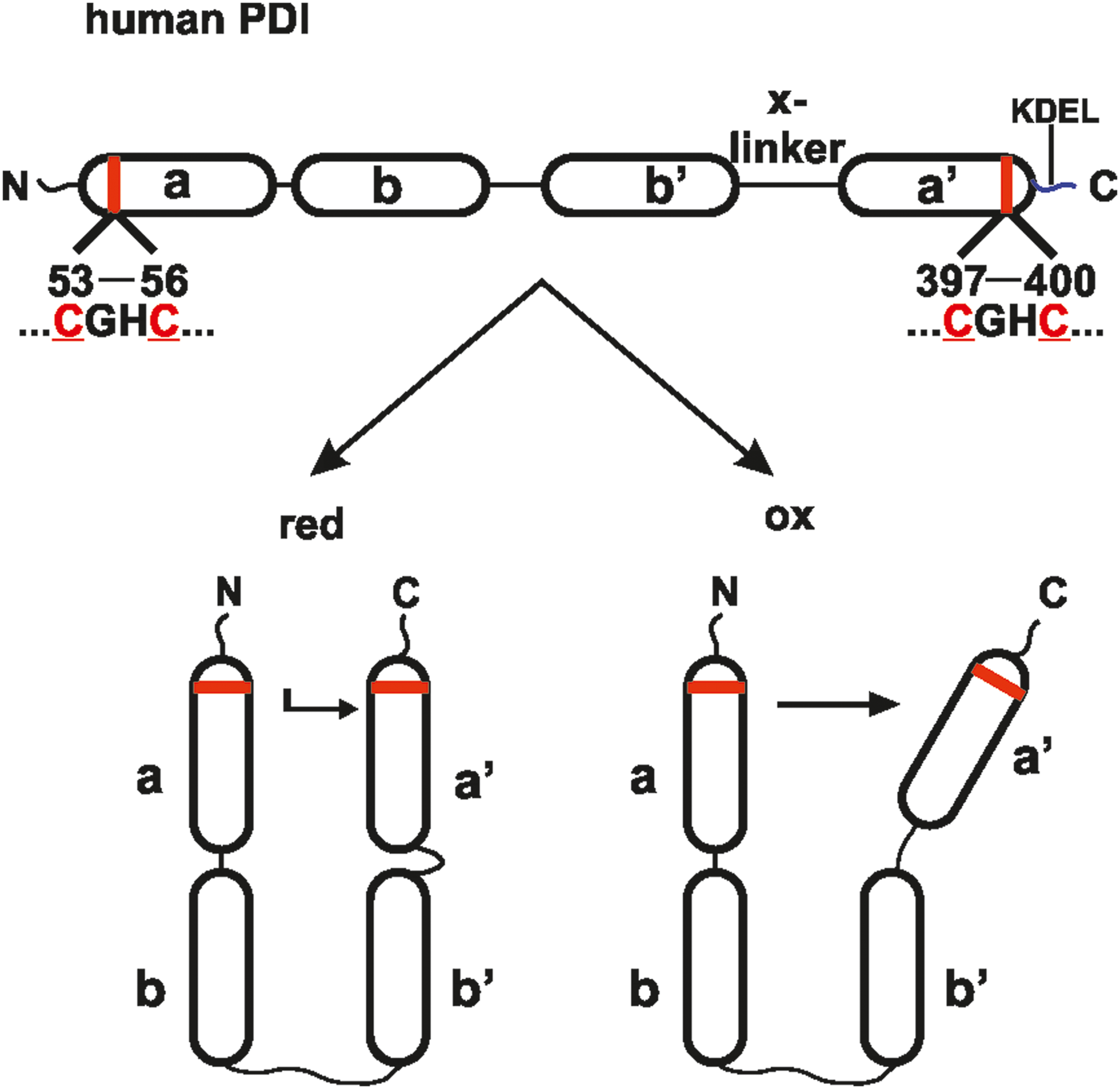
In humans, more than 20 PDI family members, which all possess at least one thioredoxin-like domain, have been characterized thus far (51). The archetype member PDIA1 (commonly and herein referred to as PDI) contains four thioredoxin-like domains in the order
By working as an oxidase, PDI folds nascent ribosomal translated proteins after their translocation into the ER. Under these conditions, the two cysteines in PDI's
As the result of the latter reactions, PDI converts disulfide bonds in the substrate into the respective thiols of the cysteines and becomes itself oxidized. It is important to emphasize that oxidation and reduction of disulfide bonds are linked to the transfer of two electrons, whereas isomerization is electroneutral.
In addition to PDI, other members of the PDI family such as ERp57, peptidyl-prolyl isomerases such as cyclophilin B (CypB), as well as the Ca2+-regulated lectin-type chaperones calreticulin (CRT) and calnexin (CNX), contribute to post-translational folding of many glycosylated, secreted, or integral membrane proteins synthesized within the ER (15, 16, 31). Thereby, complexes of CNX/CRT-ERp57 and/or CNX/CRT-CypB are parts of a cycle that determines whether the N-glycosylated proteins are correctly folded and can be released to the Golgi or need to be refolded or even terminally degraded by the ER-associated degradation (ERAD) system (15, 16, 31). In addition to efficient folding and assembly of glycoprotein substrates, CNX was recently shown to assist efficient inositol deacylation and proper maturation of glycosylphosphatidylinositol-anchored proteins (111).
Glycoproteins are formed cotranslationally in a process where an oligosaccharyltransferase (OST) complex transfers glycans en-bloc to Asn residues residing in the sequence Asn-X-Ser/Thr of the target protein (15, 16, 31). To acquire its native conformation, the glycoprotein needs to be bound to CNX/CRT. Binding is dependent on the presence of a specific monoglucosylated oligosaccharide chain (GlcMan9GlcNAc2). This monoglucosylated oligosaccharide chain is generated from a precursor glycan with three terminal glucose molecules (Glc3Man9GlcNAc2) due to the sequential action of glucosidases I and II. Once the monoglucosylated oligosaccharide chain is bound to CNX/CRT, the CNX/CRT-ERp57 complexes favor oxidative folding, while CNX/CRT-CypB complexes promote isomerization of peptidyl-prolyl bonds (15, 16, 31). During this step, glucosidase II removes the last glucose from the chain, thus releasing the protein and, if folding was correct, the protein is transported to Golgi via the ER-Golgi intermediate compartment (ERGIC) (4, 15, 31).
Misfolded proteins can be reglycosylated by the “folding sensor” UDP-glucose glycoprotein glucosyltransferase (UGGT) and re-enter the CNX/CRT cycle for another round of folding. Thereby, a member of the redox-sensitive selenoproteins, SELENOF (SEP15), has been shown to interact in a 1:1 complex with UGGT and is thought to function as a redox-sensitive PDI that contributes to reduction of disulfide bonds during refolding (97, 185).
If proper folding cannot be achieved, terminally misfolded proteins are taken out from the system by a number of processes commonly known as ERAD. The system starts with “mannose trimming,” which removes α1,2-mannose residues from the misfolded glycoprotein. Although all details are not completely resolved, it appears that ER mannosidase I and members of the EDEM (ER degradation-enhancing α-mannosidase-like protein) family (EDEM1–3) play an important role [for review, see Tannous et al. (194)]. Simultaneously, the PDI family member Erdj5 starts to reduce disulfide bonds in misfolded proteins. Mannose trimming also recruits BIP to keep the protein in a soluble state and targets the misfolded glycoproteins for ubiquitylation and retrotranslocation into the cytoplasm where they will be finally degraded by the 26S proteasome. These processes involve binding of the misfolded protein to the lectin-like proteins OS9 and XTP3-B as well as to the HRD1 adaptor protein SEL1 and the ubiquitin ligase HRD1 [for review, see Ruggiano et al. (166)]. Furthermore, the HRD1 complex is in contact with another multiprotein transmembrane complex involved in retrotranslocation of the ubiquitylated protein. In addition to HRD1, this complex consists of Derlin proteins (DERLIN1-3), the two selenoproteins SELENOS and SELENOK (184, 201), as well as the ATPase valosin-containing protein (VCP) together with NPL4 and UFD1. While VCP seems to transport substrates from the ER to the cytoplasm with its ATPase activity, the selenoproteins are thought to reduce particularly stable disulfides that were not reduced before retrotranslocation by ERdj5 [for review, see Ruggiano et al. (166) and Tannous et al. (194)].
Disulfide Formation Is Coupled with H2O2 Generation
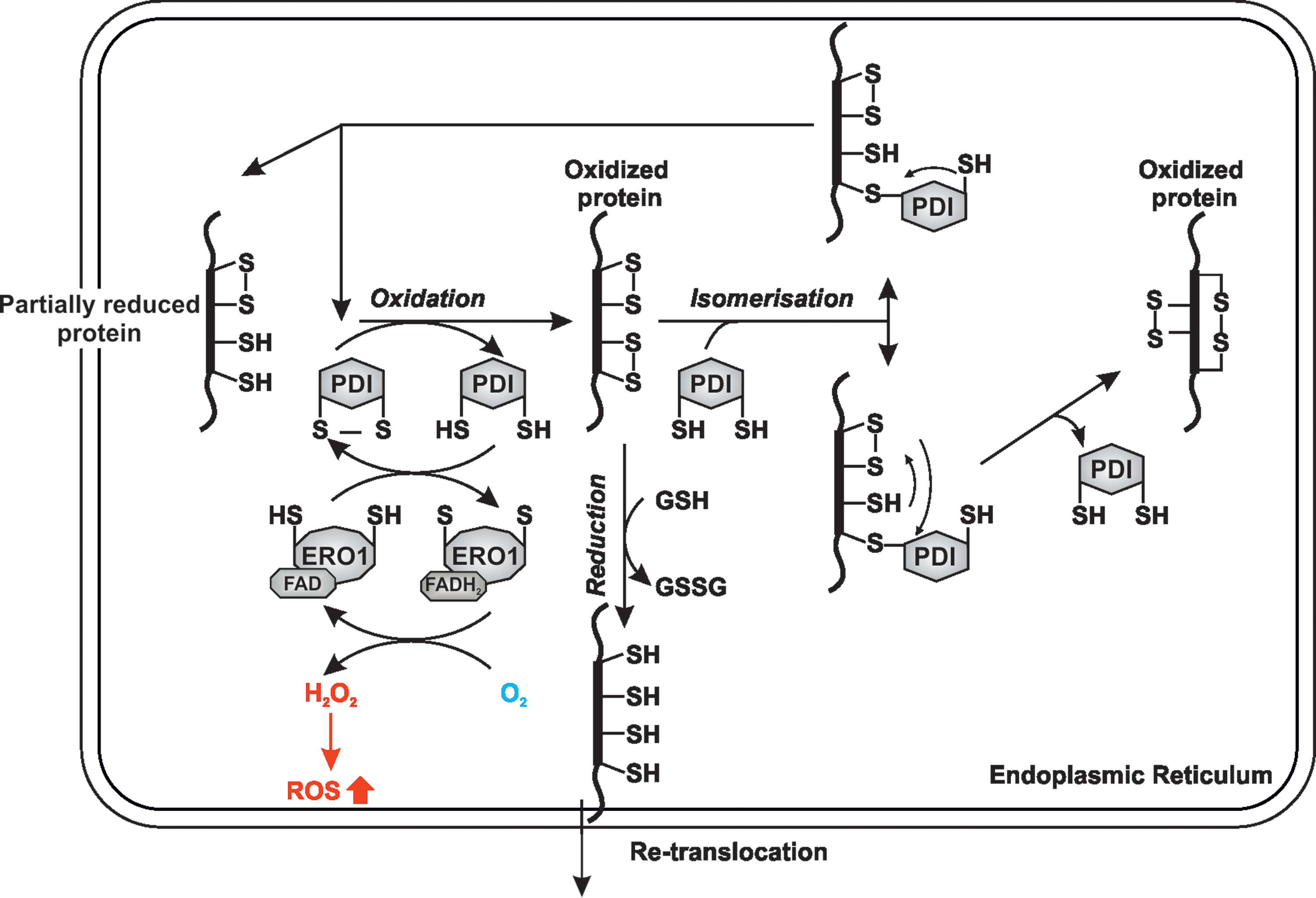
Overall, protein folding is based on an exchange of a disulfide between the substrate and the catalyzing enzyme where the active site motif of the latter shuttles between reduced and oxidized states. As mentioned above, when transferring a disulfide to a target protein, PDI's active site cysteines, in particular in the
The action of ERO1 leads to reoxidation of PDI through which the electrons from PDI are shuttled via two cysteine pairs in a flexible loop of ERO1 to a CXXC motif proximal to the cofactor FAD that is then reduced to FADH2 (199). Subsequently, ERO1's FAD is regained by transferring the electrons from FADH2 to molecular oxygen as a terminal electron acceptor, which consequently becomes reduced to hydrogen peroxide (199) (Fig. 3).
In addition to the role of ERO1 in oxidizing PDI, it has been shown that PDI family members such as PDIA2, PDIA3, PDIA4, and PDIA6 can exchange disulfides among each other (5, 133), which indicates the existence of additional reoxidation mechanisms (51). This was supported by studies in mice showing that knockout of both Ero1Lα and Ero1Lβ, alone or in combination, was not lethal, unlike the knockout of the single ERO1 gene in yeast. Only milder phenotypes such as selective cardiac conduction abnormalities in ERO1Lα-deleted mice and compromised folding of proinsulin in ERO1Lβ-deficient mice were observed (227).
Interestingly, the H2O2 produced during ERO1's action can itself oxidize PDI yielding two molecules of water (89) or it can be used by peroxiredoxin 4 (PRDX4) (195, 227), glutathione peroxidase 7 (GPX7), and glutathione peroxidase 8 (GPX8) (128) to form disulfides and to oxidize PDI and several other family members (Fig. 4). Interestingly, hypoxia has been shown to induce GPX8 expression and to suppress growth factor signaling in HeLa cells (13). Although not directly shown, these data imply that scavenging of H2O2 suppresses proliferative signaling via a reduction in protein folding, since growth factors usually pass the ER-GA secretory pathway to function.

In addition to PRDX4 and GPX7/8, the transmembrane vitamin K epoxide reductase (VKOR) and the enzyme called quiescin sulfhydryl oxidase 1 (QSOX1) are good candidates that may play roles in disulfide bond formation. Oxidized VKOR appears to be able to donate a disulfide to the PDI family members TMX and TMX4 (176) and was proposed to be a folding backup system (168). Similar to ERO1, QSOX1 couples disulfide bond formation with H2O2 generation, however, at a much higher rate (1). Interestingly, this reaction appears to occur outside the cell since QSOX1 is processed within the GA and secreted (165). This is in line with the findings that show QSOX1 contributes to the incorporation of laminin into the extracellular matrix (ECM) (77).
While the action of PRDX4 and GPX7/8 may limit the potential for the generation of toxic ROS within the ER, they appear to use H2O2 from different sources. In particular, knockdown experiments with GPX8 and PRDX4 indicate that only GPX8 reduces ERO1-derived H2O2 (152), a process fostered by the specific interaction of GPX8 with the cys208/cys241 region of ERO1α (153). By contrast, PRDX4 appears to act also in an ERO1-independent manner since it can transfer electrons from reduced PDI to H2O2 in ERO1-deficient cells and tissues (227). Hence, it can sustain disulfide bond formation by using H2O2 from a different source than ERO1.
Since the abovementioned findings indicate a role for H2O2, GPX7, and GPX8 in protein folding, one would expect a similar clear role for reduced glutathione (GSH) and GSSG. Although in vitro the rate-limiting step for PDI-catalyzed disulfide bond formation is the reoxidation of PDI, which is commonly achieved through the addition of a glutathione redox buffer, the role of the GSH/GSSG ratio for disulfide formation in cells is less clear [for review, see Delaunay-Moisan et al. (35)]. Recent evidence pointed out that depletion of GSH synthase or GSH-degrading enzymes did not prevent disulfide formation (23, 198). Instead, the involvement of cytosolic thioredoxin reductase was proposed to act as a system providing the reducing equivalents for correct disulfide formation in the ER (147). Although these findings argue against a predominant role of GSH in the folding process, GSH oxidation to GSSG was shown to activate ERO1, suggesting that GSH may have a yet not well-explored function for ER homeostasis [for review, see Delaunay-Moisan et al. (35)]. A recent study investigating GSH transport in yeast showed that GSH enters the ER via the Sec61 channel and that the oxidized yeast homologue of BIP, Kar2, inhibits this transport (148). Since BIP is a crucial player in the unfolded protein response, these findings suggest a model in which thioredoxin reductase may maintain oxidative protein folding under normal conditions, whereas GSH becomes important in coping with ER stress.
NADPH Oxidases as H2O2 Generating Folding Assistants
The above findings indicate that H2O2, in addition to its recently attributed role as a second messenger, appears to be an important cofactor for disulfide bond formation. NADPH oxidases of the NOX family may generate H2O2 that could potentially feed PRDX4-mediated disulfide bond formation (Fig. 4). In mammals, seven members have been identified, which partially differ in their tissue-specific expression and subcellular localization, as well as in their mode of activation (55). Most NOXes, including the two DUOX (Dual oxidase 1 and 2) members, require interactions with cytosolic proteins for activation. Among those are the proteins NOXO1 and NOXA1, which activate NOX1 and NOX3, the NOX2 activators p40phox, p47phox, p67phox, and Rac1 (8), and the proteins DUOXA1 and DUOXA2, which regulate DUOX1 and -2, respectively (55, 144).
Interestingly, recent findings indicate that overexpression of PDI can increase NOX1 expression and a concomitant increase of ROS production in vascular smooth muscle cells (46). Although these effects were not dependent on the catalytic function of PDI, they indicate a functional interplay between the folding machinery in the ER and cytosolic ROS formation. This interrelationship was further substantiated by findings from neutrophils and vascular cells, which indicate that PDI can coimmunoprecipitate with the cytosolic activators of the neutrophil and vascular NOX2, namely p47phox, p22phox (173), and Rac1 (143). Moreover, the thiol-mediated interaction between PDI and p47phox appeared to be a switch for ROS production where oxidized PDI stimulates ROS production, while reduced PDI inhibits it (34).
The ER itself harbors NOX4, NOX5, and DUOX1/2, which are differentially regulated. NOX5 activation depends on calcium and calmodulin (99), whereas DUOX1/2 requires, in addition to the abovementioned cytosolic regulators, only calcium (197) (Fig. 4). NOX4 appears to be unique (118) since it was shown to generate H2O2 independent of regulatory subunits (129). Thereby it seems to be mainly regulated via its expression (181), which can be induced by several stimuli such as hypoxia or transforming growth factor β1 (75). However, a recent study has shown that NOX4 can interact with CNX in the ER. This interaction appears to include a feedback mechanism since the absence of CNX also reduced NOX4 expression and ROS formation (150).
The interplay between NOX4 and CNX may also be important for disulfide bond formation in N-linked glycoproteins where CNX and its partner lectin, CRT, interact preferentially with the PDI family member ERp57, which has a higher affinity to N-glycosylated proteins than PDI in vitro (221). Thereby, NOX4 and/or H2O2 could contribute to the CNX/CRT system that determines whether the N-glycosylated proteins are correctly folded and can be released to the Golgi or need to be refolded or terminally degraded by the ERAD system (see the Protein Folding in the ER section). Although direct evidence is still lacking, these findings suggest that NOX proteins and NOX activators may contribute to H2O2 generation for protein folding or further signal transduction.
ROS, ER Calcium Signaling, and MAMs
Calcium pumps, called SERCA (sarco/endoplasmic reticulum calcium ATPase), transport calcium in an energy-dependent manner from the cytoplasm into the ER. In mammals, three distinct SERCA genes, SERCA 1, 2, and 3 exist. This and cell- and/or tissue-type-dependent alternative splicing make it possible that more than 10 different isoforms have been identified (42). Two of these SERCA isoforms, SERCA1 and SERCA2, exist in fast-twitch and slow-twitch muscle, respectively. Both isoforms exhibit a significant oxidation of Cys residues during aging, indicating involvement of ROS (207). While high-performance liquid chromatography/mass spectrometry experiments with native SR vesicles revealed that oxidative modification of Cys349 of SERCA1 is enough to modulate enzyme activity (207), knockdown of NOX4 or NOX2 in endothelial cells showed that NOX-derived ROS can modulate SERCA1 activity via S-glutathiolation of Cys674 (42).
Conversely, two ER calcium release channels, the ryanodine receptor (RyR) and the inositol 1,4,5-trisphosphate receptor (IP3R), promote the release of calcium from the ER. Several RyR tissue-specific isoforms with RyR1 residing in skeletal muscle, RyR2 in cardiac muscle, and RyR3 as the brain-specific isoform are known to exist. Experiments with single RyR channels incorporated into lipid bilayers have shown that H2O2 can directly modulate RyR activity by oxidizing redox-sensing thiol groups (225). Oxidation of RyR thiols enhances channel activity, augments SR Ca2+ leak (196), and increases Ca2+ spark frequency (218). In primary cultures of rat skeletal muscle, inhibitor experiments showed that RyRs can be regulated by NOX (41, 225). Specifically, in skeletal muscle cells, NOX4 coimmunoprecipitated with RyR1, thereby modulating its Ca2+ release activity. Furthermore, RyR1 contains a redox-sensitive Cys at position 3635, which is supposed to be involved in Ca2+ homeostasis (190).
In addition to the direct effects on RyR1, the interaction of NOX4 with CNX/CRT may also be of relevance for Ca2+ homeostasis. CNX itself is a Ca2+-binding protein that regulates Ca2+ levels, depending on its palmitoylation status via interactions with SERCA2b [for review, see Pitts and Hoffmann (146)]. Recent evidence pointed to a role of selenoprotein M (SELENOM) as being a candidate who potentially could counteract the action of NOX4. Although the details by which SELENOM regulates Ca2+ homeostasis need to be shown, it presumably acts by reducing disulfide bonds in its partner proteins such as RyRs, IP3Rs, and SERCA2b (146). In addition to SELENOM, two other selenoproteins, SELENON and SELENOT, contribute to Ca2+ homeostasis in the ER (146). SELENON was shown to bind Ca2+ via its EF hands and to counteract hyperoxidation of SERCA2 (116), whereas absence of SELENON was found to increase susceptibility of cells to H2O2-induced oxidative stress [for review, see Pitts and Hoffmann (146)]. SELENON's importance is further underlined by findings showing that mutations in SELENON lead to myopathies in skeletal muscle (116).
Although the molecular aspects how SELENOT contributes to Ca2+ homeostasis are not resolved to the last detail, SELENOT was found to be essential for life and to potentially protect the heart against ischemia/reperfusion injury through inhibition of apoptosis and oxidative stress (14, 160).
While RyRs are the Ca2+ channels in excitable cells, IP3Rs are the primary Ca2+ release channels in the ER of both nonexcitable and excitable cells [for review, see Reddish et al. (156)]. ER Ca2+ release via IP3R is initiated by binding of inositol 1,4,5-trisphosphate (IP3). As with RyRs, three different IP3R isoforms have been identified (87).
Interestingly, RyRs, IP3R, ERO1α, GPX8, ERp44, immunoglobin heavy chain binding protein/glucose-regulated protein of 78kDa (BIP/GRP78), and the sigma-1 receptor (SIGMAR1) were found to be enriched in MAMs. MAMs are physical connections consisting of membrane fragments from both the ER and the outer mitochondrial membrane. They are highly dynamic structures and are, apart from ROS production and Ca2+ homeostasis, involved in metabolic cell functions, including lipid biosynthesis and transfer, as well as in inflammatory processes (155). Thereby, it might be envisaged that a common regulation of the IP3Rs in MAMs and the Ca2+ uniporter activity in the inner mitochondrial membrane could direct ER-derived Ca2+ fluxes preferentially to mitochondria (138). Ca2+ in the mitochondria can stimulate the Krebs cycle and oxidative phosphorylation, which lead to higher respiratory rates and ROS production. Consequently, ROS production due to Ca2+ overload in mitochondria can lead to apoptosis via permeability transition pore opening, cytochrome c, GSH, and adenosine triphosphate (ATP) release (47, 155).
IP3Rs in MAMs interact with BIP, SIGMAR1 (66), ERp44 (70), and ERO1α (2). ERp44 cycles between the ER and the GA to retrieve ER proteins with reduced cysteines (171) and inhibits IP3R1 activity by competing with BIP for IP3R1 binding (70). On the contrary, ERO1α appears to counteract ERp44 binding to IP3R1, which results in IP3R-mediated Ca2+ efflux from the ER and apoptosis. These processes appear to be especially important under conditions such as ER stress where ERO1 is transcriptionally induced (104). These links between Ca2+ regulation and ROS formation are also supported by a recent finding indicating that GPX8, which was found to control ERO1α-generated ROS (152), is an MAM-enriched protein that reduces ER Ca2+ levels concomitant with a reduced activity of SERCA2b, and increased Ca2+ leakage from the ER (219). Although these findings suggest that ERO1 has a proapoptotic role in addition to its function in protein folding, ERO1 was found to be associated with a poor prognosis in breast and gastric cancer patients (180, 192); the underlying mechanisms remain elusive but may involve a hypoxic response (192).
Together, ROS production and ER-Ca2+ homeostasis are closely linked and MAMs constitute an important physical link between the ER and mitochondria. Although it remains so far open, it is attractive to speculate that MAMs could also integrate a feedback response from mitochondria to the ER.
ROS and PDI Family Members in ER Stress and Cell Signaling
The generation of H2O2 within the ER by processes involving ERO1, NOX4, NOX5, or DUOX1/2 appears to be relevant in a context where the ER participates in intercompartment signaling. Data from yeast, worms, and mammals support this view and thus ER-triggered conversion of H2O2 into other ROS, such as the formation of •OH via a Fenton reaction (110), may play a role. In this case, H2O2 was shown to contribute to epidermal growth factor, platelet-derived growth factor (191), and granulocyte colony-stimulating factor signaling (135). H2O2 generation in the ER also participates in the activation of survival pathways (174), and in the regulation of ER-luminal peroxidases in various settings of cytosolic signal transduction (13, 140). The permeability of the ER membrane for H2O2 is facilitated by the presence of aquaporin channels, in particular aquaporin 8, and potentially by aquaporin 11 [for review, see Appenzeller-Herzog et al. (3)] (Fig. 4). Although the role of aquaporin 11 is rather indirect, homology modeling as applied for seven vertebrate aquaporin 8 channels with the recently solved structure of a plant aquaporin (AtTIP2;1 from Arabidopsis thaliana) (93) may help to gain more insight into the involvement of aquaporin 11 in H2O2 transport.
While these findings suggest that under normal circumstances protein folding in the ER is quite balanced, the ER may be challenged under conditions where inhibition of N-glycan synthesis, disturbance of calcium homeostasis, or oxidative stress will create an imbalance in protein folding. This is commonly referred to as ER stress.
To deal with ER stress, cells activate the unfolded protein response (UPR), which decreases folding load and increases folding capacity (163). The folding load is reduced by an increase in ERAD and by downregulating transcription and translation of almost all genes encoding secretory proteins (56, 163). By contrast, the folding capacity is enhanced via synthesis of chaperones and folding enzymes. Thereby, the UPR relays its effects via three distinct sensors, inositol requiring kinase 1 (IRE1), double-stranded RNA-activated protein kinase (PKR)-like endoplasmic reticulum kinase (PERK), also called pancreatic eukaryotic initiation factor 2α (eIF2α) kinase, and the activating transcription factor 6 (ATF6). Under unstressed conditions, each sensor is largely inactive due to its high rate of association with the ER luminal chaperone BIP (also called GRP78). On induction of the UPR, BIP dissociation from the sensors stimulates their activity. Although accumulation of unfolded proteins appears to be the major UPR inducer, it was reported that additional mechanisms, for example, hypoxia, could initiate and modulate the activity of each individual arm of the UPR (12) (Fig. 5).

In mammalian cells, there are two homologues of IRE1, IRE1α and IRE1β. IRE1α is ubiquitous, whereas IRE1β is restricted to the intestinal epithelium and to the respiratory epithelium (117). IRE1 is a serine/threonine protein kinase with endoribonuclease activity. Accordingly, activated IRE1 catalyzes splicing of the messenger RNA (mRNA) encoding full-length X-box binding protein 1 [XBP1(U)]; the loss of a 26-base RNA fragment generates the active transcription factor [XBP1(S)] (19, 102). IRE1 also catalyzes the IRE1-regulated mRNA decay (RIDD) (71). Furthermore, IRE1 can bind to tumor necrosis factor receptor-associated factor 2 (TRAF2), a RING finger protein, and activates apoptosis signal-regulating kinase 1 (ASK1), which activates JNK (c-Jun N-terminal kinase) and p38 to subsequently trigger apoptosis (204) (Fig. 6).
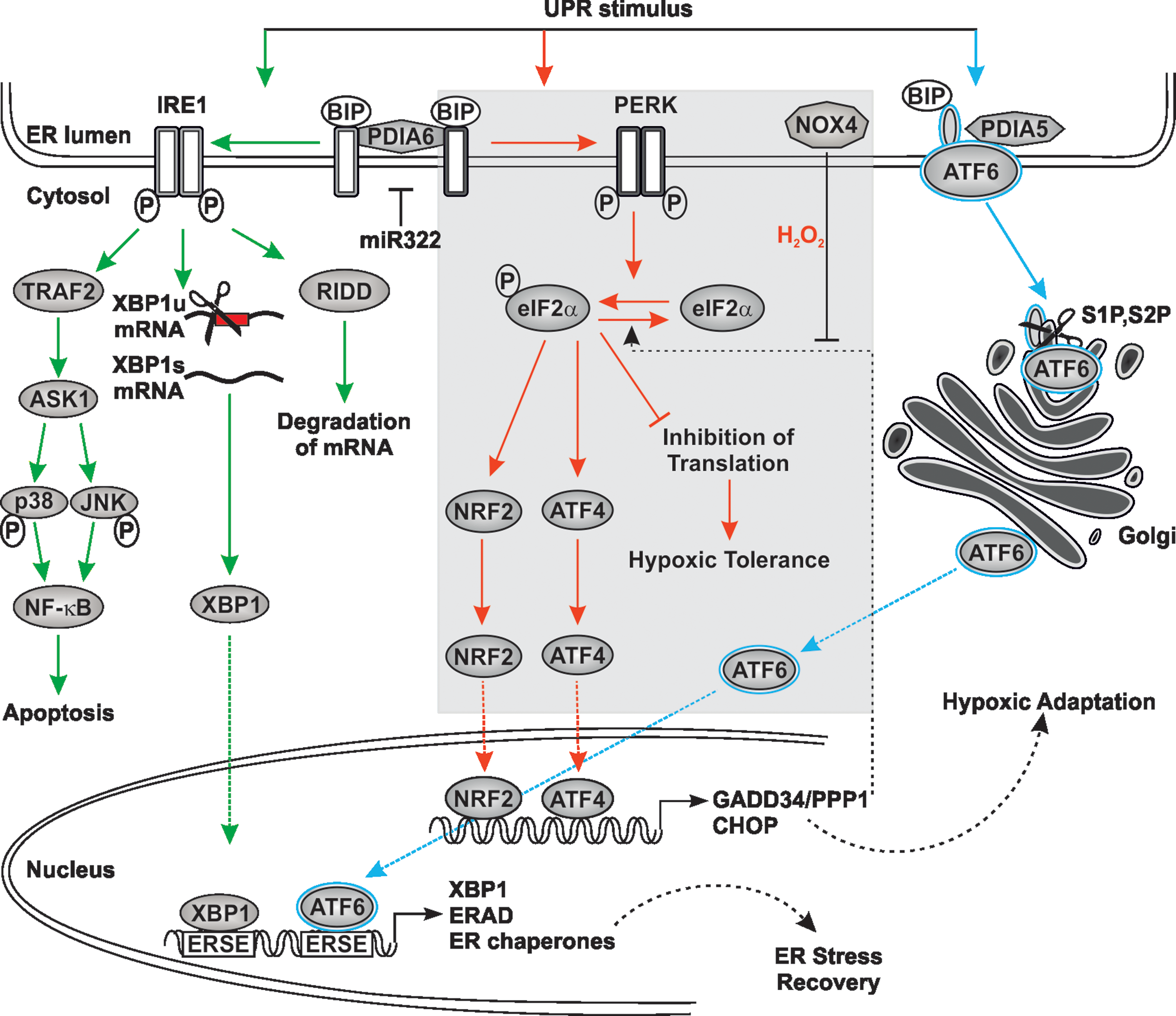
PDIA6 interacts with IRE1 (39) and appears to be able to modulate and fine-tune several aspects of IRE1 signaling. Silencing of PDIA6 also reduced insulin production and insulin secretion in response to glucose in insulin-secreting cells (38). These effects were mediated by IRE1 via its RIDD activity and not by its XBP1 splicing activity. This suggests that, in addition to BIP dissociation, oxidations/reductions of disulfide bonds during insulin synthesis might, via PDIA6, be involved in IRE1 activation/inactivation. This would, in a more general view, provide another level of IRE1 signaling where one or the other activity of IRE1 can be switched on or off to restrict its signaling within a physiological range (38).
Furthermore, PDIA6 also appears to fine-tune IRE1 activation by using Ca2+ ions. A recent study using a small interfering RNA (siRNA) screen revealed that miR-322 can target PDIA6 (Fig. 6). This appears to happen in a Ca2+-dependent manner where Ca2+-depletion reduces miR-322 levels, leading to increased PDIA6 levels and, consequently, IRE1α activity (59). Conversely, silencing of PDIA6 reduced ER Ca2+-depletion-dependent activation of IRE1α. Together, these findings further substantiate the intricate interplay between ROS-regulated compounds such as PDI members, ER Ca2+, and the UPR, which are reviewed elsewhere in much more detail (56, 60).
PERK is also a serine/threonine kinase, and on activation its major target is the α-subunit of eIF2α. This causes a general stop in translation to decrease protein influx into the ER lumen, except for that of ATF4 (12) (Fig. 6). Consequently, ATF4 promotes expression of ER chaperones, genes involved in amino acid metabolism, and glutathione biosynthesis. Impaired signaling in this UPR part due to loss of PERK or ATF4 function was shown to raise ROS levels in ER-stressed cells (64). In addition, ATF4 induces another downstream transcription factor called CHOP (CAAT/Enhancer binding protein [C/EBP] homologous protein or DNA damage-inducible transcript 3). CHOP is involved in the regulation of a gene expression program in response to a variety of cell stresses, including oxidative and ER stress (202). Interestingly, ERO1α is a transcriptional target of CHOP (115). Hence, CHOP could exacerbate ER stress via ERO1 and make the ER lumen more oxidative. Conversely, CHOP deficiency in cells and mice reduced oxidative and ER stress (119, 226) (Fig. 6). For example, in a mouse model of Charcot–Marie–Tooth 1B neuropathy, deletion of CHOP largely rescued the neurological phenotype (141) and in diabetes type 2 models, a lack of CHOP improved beta-cell function and survival (187). ATF4 is also involved in a negative feedback mechanism to PERK since it induces expression of growth arrest and DNA damage-34 (GADD34)/protein phosphatase 1 (PPP1R15A) that reverses PERK's action on eIF2α and is correlated with apoptosis (172). Importantly, NOX4 was also shown to be an ATF4 target (174), but this was shown to prolong eIF2α phosphorylation (Fig. 6). This effect was achieved by the ability of NOX4 to form a complex with GADD34/PPP1R15A and to oxidize it with the consequence that GADD34/PPP1R15A was inhibited and eIF2α phosphorylation was prolonged. Although GADD34/PPP1R15A contains redox- sensitive cysteines, which were oxidized by NOX4-derived H2O2, it was found by mutation of amino acids involved in metal ion coordination (N124D or D64N) that the metal center was the site of oxidative inhibition (174). Consequently, NOX4 appears to be in the center of a fine tuning mechanism between ATF4 and eIF2α where its action is considered to promote survival, at least in cardiomyocytes (174).
PERK has another important target that integrates a variety of responses to oxidative stress, the basic leucine zipper (bZIP) Cap “n” Collar transcription factor nuclear factor erythroid-2 (NF-E2)-related factor-2 (Nrf2) (Fig. 6). Nrf2 is important to counteract oxidative stress since it controls the expression of a number of “antioxidant” genes, among them those for γ-glutamylcysteine synthetase, glutathione S-transferase A1 and A2, NAD(P)H:quinone oxidoreductase, UDP-glucuronosyl transferase, and heme oxygenase 1 [for review, see Levonen et al. (103)]. Nrf2 −/− cells are very sensitive to ER stress (32), and importantly, glutathione production is increased in Ero1Lα overexpressing cells, indicating a link between the altered redox state within the ER and Nrf2-mediated transcription (123).
Similar to IRE1, PERK can also interact with PDIA6 (39, 59). Although PDIA6 also seems to act negatively on PERK activity, there appear to be differences in the PDIA6 actions with respect to the stimulus. While PDIA6 seems to attenuate IRE1 and PERK signaling in response to chemical ER-stress such as thapsigargin in a similar manner, it inhibits glucose-stimulated insulin secretion primarily only via the RIDD activity of IRE1, but not its XBP1 splicing activity. This also occurred independent of PERK as indicated by the use of PERK inhibitors (38).
The so-called ATF6 signaling is another contributor to the UPR. ATF6 itself is a bZIP transcription factor, which is confined to the ER membrane under unstimulated conditions. Two isoforms, ATF6α and ATF6β/cAMP-response-element-binding protein-related protein/G13, exist. Both are ubiquitously expressed in all tissues. In response to ER stress, ATF6 migrates from the ER to the GA where it is sequentially cleaved by site-1 protease (S1P) and site-2 protease (S2P). As a consequence, activated ATF6 translocates to the nucleus where it can bind ATF/cAMP response elements and ER stress response elements in genes encoding ER chaperones, including CRT, GRP94, BiP, or transcription factors such as XBP1 [for review, see Haze et al. (67), Lee et al. (102), Yamamoto et al. (217)] (Fig. 6).
Importantly, in the inactive state, ATF6 forms monomers, dimers, or oligomers, which are held together via disulfide bonds (with C467 and C618 being most critical), and only reduced monomeric ATF6 reaches the GA (124). This implies that redox changes and redox-sensitive PDI members could control ATF6 activation in addition to dissociation of BIP. Indeed, PDIA5 was found to contribute to disulfide bond rearrangement and packaging of ATF6α into coat protein complex II (COPII) vesicles to allow its exit from the ER as well as to promote subsequent activation of ATF6α target genes (69). Thus, BIP dissociation and PDIA5-induced reduction of ATF6 are required to allow full activation of ATF6 under ER stress.
Overall, these findings indicate the existence of intricate feed-forward and feedback loops between the redox/PDI and UPR systems that are involved in fine tuning of cellular responses to different stimuli.
Hypoxia, PDI Members, and ER Stress
Protein synthesis and folding appear to be O2-demanding redox-regulated processes, and consequently, a drop in the ambient O2 pressure or the absence of O2 (anoxia) would hamper proper protein folding and cause ER stress. Indeed, severe hypoxia or anoxia can rapidly activate PERK, which induces ATF4 via eIF2α phosphorylation (12). This activation could be abrogated by the expression of catalase and GPX, pointing to the involvement of ROS in this response (109). In addition to the PERK-eIF2α-ATF4 axis, hypoxia acts via the transcription factors from the hypoxia-inducible factor family (HIFs). Three HIFs, from which HIF-1α and HIF-2α were shown to be responsive to ROS (92), are so far known and partially linked with ER function (110). Although both, the PERK-eIF2α-ATF4 and the HIF axis, are responsive to hypoxia and ROS, they are activated independently from each other (178).
The ROS involved in the regulation of these responses may be derived from ERO1, NOX4, NOX5, or DUOX1/2; however, so far ERO1 appears to be the most studied candidate in this context. Of note, ERO1Lα but not ERO1Lβ was shown to be a HIF-1 target (52), which would enable a feedback cycle allowing thiol oxidation in the ER via hypoxic adaptation of ERO1Lα transcription.
The latter features may be important in tissues with a high proliferation rate and tissue hypoxia, features that are common in solid tumors. Both the PERK-eIF2α-ATF4 and HIF responses appear to cooperate in tumors since cells with compromised PERK-eIF2α-ATF4 signaling are more sensitive to hypoxia and form slower growing tumors (45). Conversely, hypoxia and ER stress seem to promote cell survival autophagic flux induction via ATF4-mediated ULK1, LC3B, and ATG5 expression (145, 164) as well as via augmentation of HIF-1α-dependent VEGF expression and angiogenesis (142).
Moreover, analysis of patient cohorts with triple-negative breast cancer revealed a specific gene expression signature that was highly correlated with expression of XBP1 and hypoxia-inducible genes (26), whereas loss of XBP1 inhibited tumor growth (188, 214). As with PERK, hypoxia/anoxia can activate XBP1 and ATF6 expression in an HIF-1α-independent manner (36, 162). While the further contribution of ATF6 to the HIF system is so far unknown, XBP1 contributes to the assembly of a transcriptional complex, which augments HIF-1α activity and enhances tumorigenicity of mammospheres under hypoxia (26).
In addition, PDI expression itself was found to be upregulated by hypoxia/anoxia in endothelial cells (57) and to promote survival in a model of cardiomyopathy in ischemic mice (182). In line with these observations, a number of cancers, including breast, prostate, lung, ovarian, and brain tumors, also display an increased expression of PDI members (216). Increased PDI expression in breast cancer or glioblastoma patients is associated with chemotherapy resistance (54), and lower survival rate (183). In addition, increased PDIA4 and PDIA6 appeared to be involved in cisplatin resistance of lung adenocarcinomas since knockdown of PDIA4 and PDIA6 regained cisplatin sensitivity (200).
These findings are to a certain extent counterintuitive since they indicate that PDI family members, which are thought to act as potential folding catalysts or chaperones, are primarily expressed under conditions where O2, the terminal electron acceptor for disulfide bond formation, is limited or even absent. However, a recent study has shown that disulfide bond formation is a process with two major phases. In the first, very early phase (within 3 min of protein synthesis), disulfide bonds are formed in the absence of O2. The subsequent second post-translational disulfide bond and/or isomerization phase are O2 dependent (95). These findings point to the role of different PDI members and an alternative electron acceptor with H2O2 being a good candidate and oxidases such as NOX proteins as its source. Furthermore, it indicates that the differential requirement for O2 affects the transit of proteins through the secretory pathway from the ER to the Golgi and beyond (16), which suggests that it also has profound effects on Golgi physiology.
The GA, Hypoxia, and ROS
In addition to protein folding, hypoxia and ROS affect many other cellular pathways, including transcription, translation, vesicular trafficking, pH homeostasis, energy metabolism, autophagy, proliferation, and cell death, as well as tumor cell invasion and metastasis (11, 27, 139). Several recent studies have also shown that ROS and hypoxia not only impair Golgi-associated vesicular trafficking and protein sorting steps but also alter glycosylation (10, 40, 186). Therefore, the Golgi is by no means different from any other organelle known to be affected by ROS and hypoxia. Yet, amazingly little is known about the Golgi-resident proteins that are affected by hypoxia or regulate redox state, for example, by counteracting the ROS load. In addition, the mechanistic details underlying hypoxia or ROS-driven changes in glycosylation remain much less clear than their effects on the functioning of the ER. In the next paragraphs, we discuss some effects of hypoxia and ROS on glycosylation processes within the Golgi and maintenance of its ion homeostasis.
Golgi Structure and Its Basic Functions
The GA is recognized as an independent secretory pathway organelle, and it is physically separated from the ER by the ERGIC (4). In higher eukaryotes, the GA consists of 30–80 distinct Golgi stacks assembled together into a compact peri- or juxtanuclear Golgi structure (Fig. 7, top left), while in many cancer cell lines, the Golgi remains mostly fragmented for reasons that are not completely understood (91) (Fig. 7, top right).
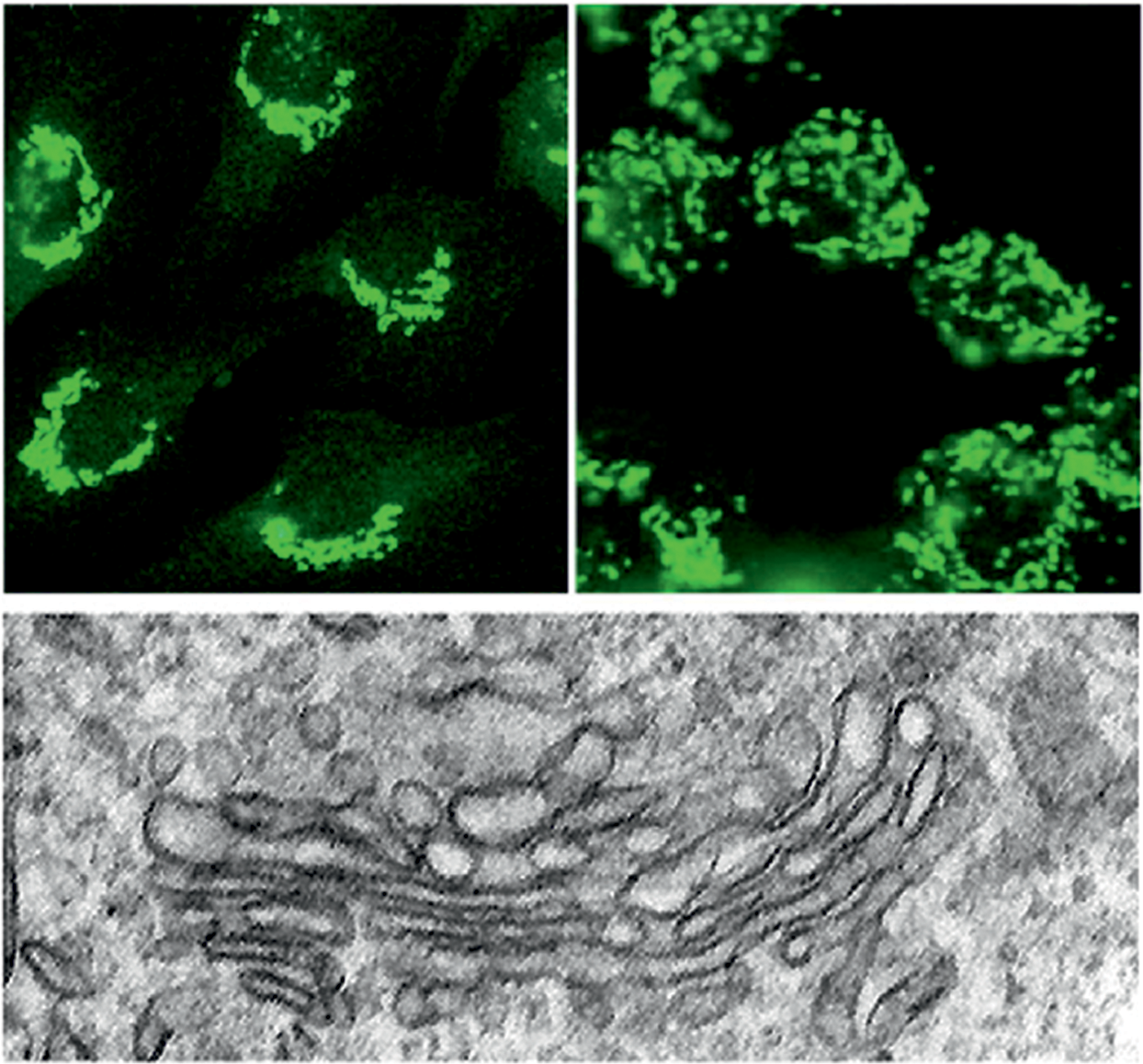
Each individual Golgi stack is a single functional unit studded with dozens of glycosidases and glycosyltransferases as well as nucleotide sugar transporters. All these proteins are needed for glycosylation of proteins and lipids, which are the main post-translational modifications taking place in this organelle (113). This also involves further processing and maturation of ER-derived N-glycans as well as the synthesis of O-glycans, proteoglycans, or glycolipids. Although the cis-Golgi network (CGN) and the trans-Golgi network (TGN), which closely associate with both sides of the Golgi stack, also participate in the glycosylation processes (189, 223), they primarily function as sorting stations in the secretory pathway. Thereby, mainly the TGN enables delivery of mature glycoproteins and glycolipids into post-Golgi transport vesicles, whereas both facilitate recycling of ER-resident chaperones and folding catalysts as well as Golgi-resident glycosylation enzymes. This basic Golgi architecture is highly dynamic as the GA sits at the crossroads of secretory and endocytic compartments; thereby it receives and sends vesicular carriers back and forth between these compartments.
ROS, Hypoxia, and the Golgi Redox State in Glycosylation
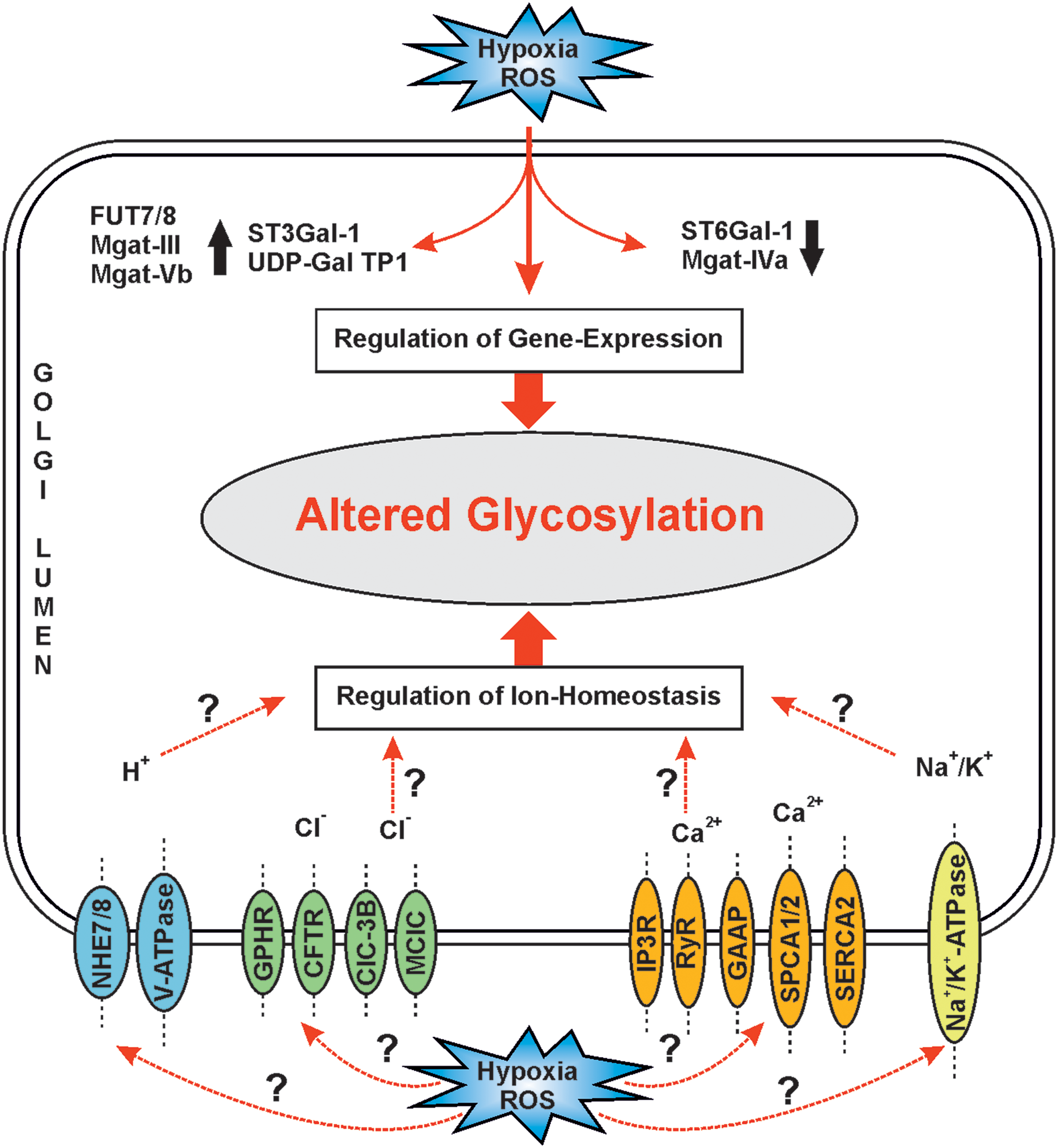
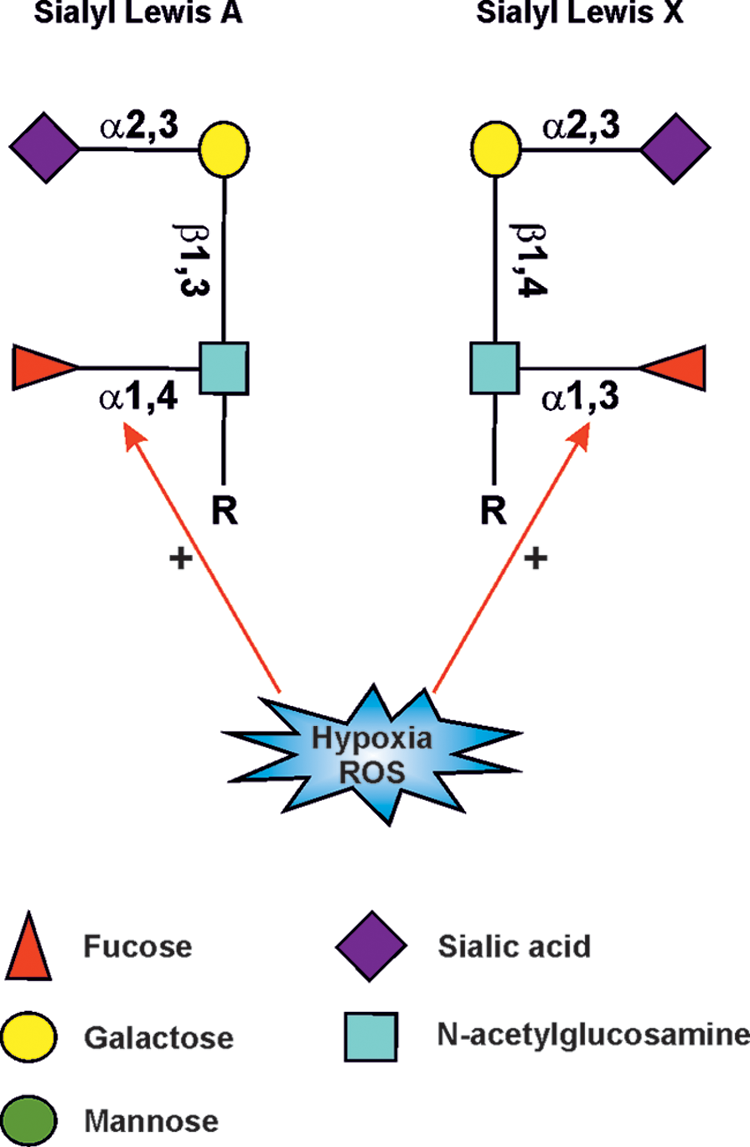
While hypoxia-mediated changes in the other subcellular organelles have been well defined, the way they alter glycosylation in the GA is still incompletely understood. However, HIF-1, -2, and -3, which are known to regulate the expression of hundreds of genes required to sustain normal and malignant cell growth (179), have been found to regulate only a handful of about 250 genes needed to make all glycan structures in mammalian cells. These include down- or upregulation of enzymes that make nucleotide sugars in the cytoplasm (186). Hypoxia/HIFs modulate the expression of medial-Golgi enzymes Mgat-II, Mgat-III, and Mgat-V and Mgat-Vb, and the trans-Golgi-localized ST6Gal-1 (193) (Fig. 8). Oxidative stress was shown to increase the transcription of fucosyltransferase VII (FUT7), sialyltransferase ST3Gal-I and transporters for UDP-Galactose, sialic acid (Sialin), and N-acetylglycosamine (UGT1) (94) (Fig. 8). Some of these are involved in the synthesis of cancer-associated sialyl Lewis A/X carbohydrate epitopes for selectins (33, 186) (Fig. 9) found typically on O-linked glycans and glycolipids. Other findings indicate that HIF-1α suppressed the expression of α1,2-fucosyltransferase genes (FUT1 and FUT2) in the pancreatic cancer cell lines Pa-Tu-8988S and Pa-Tu-8988T (10). Similar attenuation has been detected at the mRNA level with UDP-glucuronosyltransferases (90), O-GlcNAc transferase (OGT) (108), and glucosylceramide synthase (GCS) (224). Based on these observations, it is clear that hypoxia and HIFs can modulate many glycosylation pathways via altering the availability of nucleotide sugars, the level of their transporters, and the repertoire of transferases expressed by the cells. These changes, which likely vary depending on the cell type, and the type and duration of hypoxia treatment, however, do not directly correspond to the altered glycan structures produced by the cells (88). Other defects are therefore likely to be involved.
It is intriguing that ROS or hypoxia-mediated glycosylation changes have not yet been detected in the initial synthesis of N-glycans in the ER. However, the recent finding that the redox-sensitive selenoprotein T was shown to function as a new OST and to be indispensable for ER homeostasis as well as for maturation and secretion of glycohormones from endocrine glands (62) suggests that hypoxia and/or ROS could affect this process. However, further studies are needed to clarify whether and how hypoxia and/or ROS may affect ER-associated glycosylation events.
Hypoxia-mimicking agents or ROS have been shown to cleave cell surface-localized glycosaminoglycans (i.e., proteoglycans) and N-glycans (i.e., N-linked oligosaccharides) in an N-acetyl-amino-sugar-specific manner by involving a transition metal (37). These changes affect cell/cell or cell/ECM interactions, and coincide with inflammatory responses leading to rheumatoid arthritis, atherosclerosis, and cancers (193). In addition, oxidative stress contributes to the onset of type II diabetes, a disease that is characterized by hyperglycemia and insulin resistance, and by its association with a high-fat diet (131). In this particular case, oxidative stress is caused by an excess of both glucose and free fatty acids together with high overproduction of ROS in mitochondria. Of these, high amounts of free fatty acids have been shown to inhibit the activity of two transcription factors termed FOXA2 and Hnf1a, which normally positively regulate the expression of MGAT-IVa, a glycosyltransferase needed for β1,4-GlcNAc branching of N-glycans. This modification is crucial for the normal transport of the glucose transporter 1 (GLUT-1) to the cell surface on insulin stimulation, through which it is able to bind and import glucose into cells for further use. In its absence, however, the GLUT-1 transporter is unable to move to the cell surface, leading to markedly diminished glucose transport into pancreatic β cells. Chronic elevation of free fatty acids and increased production of ROS in mitochondria also attenuate insulin secretion in pancreatic β cells and thereby trigger the onset of type-2 diabetes. Based on existing evidence, the term “Glyco-redox” was recently introduced to describe a tight link between structural and functional changes of glycans and oxidative stress-generated ROS or reactive nitrogen species (193). ROS-induced glycan alterations are important, as they closely associate with many devastating diseases such as cancers, diabetes, Parkinson disease, Alzheimer's disease, amyotrophic lateral sclerosis (ALS), and chronic obstructive pulmonary disease.
Genetically encoded targeted redox probes such as roGFP and HyPer can be used to determine the existing redox state of any subcellular organelle (122). The use of surface-enhanced Raman spectrum-sensitive monitoring of the redox potential of the cytoplasm in both normoxic and pseudohypoxic cells (CoCl2 treated) has shown that the cytosolic redox potential becomes progressively more reductive on hypoxia. This appears likely to be due to a metabolic switch-induced increase in the production of NADH and NADPH (86). In line with this, it was also shown that hypoxia-induced redox changes differ among mitochondria and the cytosol in pulmonary and systemic smooth muscle cells (210). By expressing a redox-sensitive, ratiometric fluorescent roGFP in the cytosol (Cyto-roGFP), the mitochondrial matrix (Mito-roGFP), or the mitochondrial intermembrane space (IMS-roGFP), it was shown that hypoxia increases the oxidation potential of the cytosol and mitochondrial intermembrane space, whereas it decreases that of the mitochondrial matrix. Hypoxia-induced oxidation of the cytoplasm was attenuated through the overexpression of cytosolic catalase, suggesting that ROS were involved.
Although these findings left out to what extent hypoxia modulates the redox state of the Golgi lumen or other compartments, we have recently compared the redox status of mitochondria, ER, and GA under normoxia. By using roGFP2, we found that mitochondria have a less oxidizing environment than the ER and the Golgi (170). Intriguingly, we also observed that the lumen of the GA is even more oxidizing than the ER (170) despite the fact that the ER is generally considered the most oxidizing organelle within the secretory pathway due to its ability to promote oxidative disulfide bond formation during folding of nascent polypeptide chains. The reason for having a high oxidizing potential in the Golgi is currently unclear. One possibility is that it facilitates disulfide bond formation or reorganization. In support of this view, assembly of von Willebrand factor into ER-derived oligomers to multimers requires tail-to-tail disulfide bond formation in the Golgi (208). Such a sequential assembly into oligomers and multimers in different secretory compartments is likely to be assisted by the QSOX family, although the precise physiological roles remain uncertain (30). These enzymes represent an ancient fusion between PDI-like thioredoxin (Trx) domains and ERV-like oxidase domains, which allows QSOX to efficiently couple the generation of protein disulfide bonds with the reduction of molecular oxygen to hydrogen peroxide. QSOX1 is found in the ER, in the Golgi, at the cell surface, and in the extracellular space (24). Of the two splice variants, QSOX1a retains a C-terminal transmembrane helix and may remain membrane associated, while the shorter QSOX1b form transits from the ER and at least some fraction of it is secreted from the cells. Human QSOX1a is a long variant accumulating in the Golgi (24), suggesting that the predominant role of this long form is in late-stage disulfide bond formation, possibly in the maturation of ECM components and in the formation of higher order structures.
In contrast to the ER, only a few proteins that regulate the redox state or protect the Golgi from ROS load are known. The glutaredoxins, GRX6 and GRX7, represent members of a ubiquitous family of proteins that catalyze the reduction of disulfide bonds in their substrate proteins by the use of GSH. GRX6 and GRX7 represent closely related monothiol glutaredoxins that are located in the cis-Golgi in baker's yeast, thereby representing the first glutaredoxins found in this compartment (121). Both of these glutaredoxin-like proteins show a high glutaredoxin activity in vitro, and yeast cells lacking both proteins show growth defects and a strongly increased sensitivity toward oxidizing agents such as hydrogen peroxide or diamide. These observations suggest that GRX6 and GRX7 may play a general role by counteracting the oxidation of specific thiol groups in substrate proteins in the Golgi.
Hypoxia, ROS, and Golgi pH Homeostasis
Unlike the ER, other secretory pathway compartments, that is, the ERGIC, GA, and secretory vesicles, are all known to be mildly acidic (137, 215). Each of them also has its own unique pH set point that is important for efficient membrane trafficking, protein sorting, or glycosylation (7, 91). In general, the pH decreases from the ER toward the plasma membrane, and in the case of GA, also along the cis-trans axis of the Golgi stack, in which the pH varies from 6.7 in the CGN (entry face) to 6.0 in the TGN (exit face). Previously, it was suggested that the decrease in pH between successive secretory compartments is likely due to decreased proton leakage (efflux) or increased density of the V-ATPase [vacuolar H+ ATPase (215)].
To function properly, the GA requires constant remodeling and strict control of luminal ion and pH homeostasis. Given that the GA is mildly acidic, has higher calcium and manganese levels than the cytoplasm, and is highly permeable to K+ (175), it is no wonder why so many ion transporters and channels have been shown to be present in the Golgi membranes. These include the V-ATPase, the Golgi pH regulator (GPHR, a chloride channel), the cystic fibrosis transmembrane conductance regulator (CFTR, a chloride channel) (9), a mid-1-related chloride channel (125), and voltage-gated chloride channels (ClC-3B in mammal cells and Gef-1 in yeast) (85). Golgi membranes also contain the Na+/K+-ATPase (149) as well as two different isoforms of the Na+/H+ exchanger (NHE) (127, 130) (Fig. 8). These transporters contribute either directly or indirectly to the preservation of the resting membrane potential, regulation of luminal pH, as well as vesicular trafficking and protein sorting in the organelle. In line with this, monensin, a potent Na+/H+ ionophore, has been shown to block intra-Golgi transport between the medial- and trans-Golgi cisternae (58).
Given the strict dependence of correct glycosylation on Golgi luminal pH (159), an obvious question is whether hypoxia directly affects Golgi pH homeostasis. It is known to be regulated by three main ion transport systems: ATP-mediated proton pumping, counter ion conductance, and intrinsic proton leakage across organellar membranes (137, 215). Of these, the V-ATPase uses ATP to pump protons into the Golgi lumen and thus is responsible for Golgi acidification (82). Counter ion conductance is needed to prevent the buildup of excess positive charge due to continuous ATP-mediated proton import that otherwise would increase the resting membrane potential and block proton import by affecting the V-ATPase activity. It involves either passive influx of anions through the GPHR chloride channel (114) or efflux of cations through relevant cation (e.g., potassium, calcium) transporters. An example of this is a chloride channel protein 5 (CLC-5) mutant in the kidney (63); its presence is associated with defective acidification of the endosomes in kidney cells (85). In the Golgi, counter ion conductivity is considered to be high enough to support continuous proton pumping (175). Therefore, it serves as an indispensable “background component” that enables normal proton pumping and Golgi acidification well below the pH set point of the organelle. The last, but not the least, important pH regulating component is the intrinsic and ill-defined “proton leakage” pathway that serves to counteract proton import and thus to prevent overacidification of the Golgi lumen (215). It is likely that this component dictates the pH set point of the Golgi lumen, as the rate of proton leakage (in contrast to proton import) varies between different organelles (215), decreasing from the ER toward the plasma membrane. Proton export was also found to be voltage sensitive and inhibited by Zn2+, suggesting the involvement of a regulated channel (175). The protein channel responsible for this proton “leakage” still awaits identification, but as suggested previously (159), other pathways such as anion exchanger-mediated bicarbonate import may also play a role.
In all eukaryotes, the V-ATPase is a multisubunit protein complex that consists of a peripheral cytosolic domain V1 and an integral membrane domain Vo. Under normal conditions, the activity of the V-ATPase is known to be under strict control. It is regulated by glucose (affecting VoV1 assembly and disassembly), by reshuffling of disulfide bonds within the ATP binding A subunit, by changing the coupling between ATP hydrolysis and proton pumping, and by some regulatory proteins, such as the V-ATPase accessory subunit Ac45 [for review, see Jansen and Martens (81)]. The physiological meaning of such a regulation remains unclear (137).
Currently, there are no direct data showing whether hypoxia and ROS indeed affect Golgi luminal pH homeostasis. However, many hypoxia-mediated cellular changes may indirectly modulate Golgi luminal pH. For example, they may reduce cellular ATP levels needed for proton pumping, via redirecting energy production from oxidative phosphorylation to glycolysis, and thereby reduce proton pumping and proper acidification of the Golgi lumen. Alternatively, they may modulate V-ATPase activity via affecting disulfide bond formation, given that hypoxia will inevitably result in the loss of oxygen, which in turn likely will affect the oxidative potential of the Golgi lumen. Recall that under normoxic conditions, it is higher than that of the ER (170).
A hypoxia-induced metabolic shift from oxidative phosphorylation to glycolysis is also known to be associated with increased expression of CAIV, a membrane-associated carbonic anhydrase and Na-dependent bicarbonate exchanger, to allow increased import of bicarbonate into the cytosol, and to overcome glycolysis-derived acid load due to lactate production. Increased intracellular bicarbonate may also increase its import into the Golgi lumen, through which Golgi luminal pH would increase and alter glycosylation. It is of note that only a 0.2 pH unit increase in Golgi luminal pH is sufficient to interfere with N- and O-linked glycosylation in the Golgi (158). Two ubiquitously expressed and Golgi-localized NHE isoforms, NHE7 and NHE8 (127, 130), may also increase proton export (Fig. 8). The Na+/H+ exchange is driven by ion gradients and can serve to export protons (in exchange for Na+ import) when the H+ concentration is as high as in the Golgi lumen. In support of this, overexpression of both NHE7 and NHE8 was found to increase Na+ and K+ influx to the Golgi (130). Both also significantly raised luminal Golgi pH (127). All these phenomena, that is, reduced V-ATPase activity (due to the lack of ATP or altered disulfide bond formation), increased import of bicarbonate, or increased proton export, are potential targets that can be affected by hypoxia. These examples may not be the only ones, but they offer the starting point for studies aimed to define in detail to what extent and how hypoxia may affect Golgi pH homeostasis and its glycosylation potential in mammalian cells.
ROS, Hypoxia, and Golgi Ca2+ Homeostasis
The GA has all the components needed to act as a Ca2+ store. It contains pumps for Ca2+ uptake (SERCA2, SPCA1/2 [secretory pathway calcium ATPase]), channels for Ca2+ release (IP3R, RyR), and luminal proteins that bind Ca2+ with high affinity (Fig. 8). So it is not a surprise that its lumen has high concentrations of Ca2+, Mn2+, and Mg2+ (205). These three cations have important roles in vesicular trafficking, glycosylation, proteolysis, or sorting of various cargo proteins from the TGN to their final destinations in the cell [for review, see Zampese and Pizzo (220)]. Lowering GA luminal Ca2+ and Mn2+ via downregulation of SPCAs has also been shown to alter the morphology of the GA (107). Mn2+ ions serve as important cofactors for Golgi-resident glycosyltransferases, but also seem to act as scavengers for hypoxia-induced ROS (29).
SERCA and SPCA pumps help to establish and maintain low cytosolic and high luminal-free Ca2+ concentrations in the ER and the other organelles of the secretory pathway. Failure to maintain this Ca2+ gradient results in ER and Golgi stress, and sometimes in cell death. In humans, allelic mutations in SPCA1 cause the Hailey–Hailey disease, in which keratinocytes show increased cytosolic Ca2+ concentration, and defects in protein sorting and Ca2+ signaling (154, 206). The patients show an increased propensity for the formation of erosive skin lesions in the flexural areas, and in some cases, skin lesions may evolve into squamous carcinomas (73). Whether these phenotypic changes are associated with altered glycosylation, one of the hallmarks of cancers, deserves further investigation. In contrast to SERCAs, SPCAs are also able to transport Mn2+ and thus supply this essential trace metal to the Golgi luminal glycosyltransferases (17, 206). The two Ca2+ pumps (SERCA2, SPCA1/2) also contribute differentially to the total uptake of Ca2+ into the Golgi lumen. It is now agreed that the cis- and trans-Golgi can differentially regulate their Ca2+ homeostasis (213). Partly, this appears to be due to the differential localization of these pumps and the Ca2+ release channels (see this section for further details) in the Golgi. SERCA2 is enriched in the cis-Golgi, while SPCA1 is mainly present in the trans-Golgi membranes. The Golgi-associated Ca2+ release channels, that is, IP3Rs and RyRs, similarly seem to have a differential localization, as IP3 did not release Ca2+ in the trans-Golgi, while caffeine did (107, 213).
The GA lumen harbors several Ca2+-binding proteins, including Cab45, CALNUC, p54/NEFA, and calumenin, all of which are distinct from their ER counterpart except CAB45. Of these, the most abundant is CALNUC, an EF-hand, Ca2+-binding protein resident of the CGN and cis-Golgi cisternae. It plays a major role in Ca2+ buffering and secretion through the Golgi (106).
In many situations, hypoxia and/or hypoxia-induced ROS have been shown to modulate Ca2+ homeostasis and cell signaling by increasing intracellular Ca2+ (and Mn2+) concentrations. These changes generally involve opening Ca2+ release channels via stimulation by, for example, inositol-1,3,5-trisphosphate, and inhibition of Ca2+/ATPase activity by ROS [see Görlach et al. (56)]. Moreover, it has been shown that intermittent hypoxia upregulates the expression of both SPCAs in HCT116 cells (84), which may indicate that Mn2+ transport especially via SPCA2 from the cytosol to the Golgi lumen coincides with increased ROS production, and thus suggests its detoxification via increasing cytosolic Mn2+. The upregulation of rat brain SPCA1, following Mn2+ exposure (222), is also in accord with this view.
The presence of similar Ca2+ pumps and release channels in the ER and Golgi suggests that the Golgi also contributes to a cytoplasmic Ca2+ increase induced by hypoxia, even though no direct evidence currently exists that shows to what extent the Golgi Ca2+ release contributes to the overall increase in cytosolic Ca2+. However, Ca2+ release will also result in a much reduced Ca2+ concentration inside the Golgi lumen. This could be even more important in terms of Golgi functioning, and alone could explain the multiple effects of Ca2+ depletion on vesicular trafficking, proteolysis, glycosylation, or protein sorting in the Golgi. As an example, it has been shown that high Ca2+ and low pH in the Golgi are needed to concentrate secretory cargo proteins to regulated post-Golgi vesicular carriers (25). Calcium depletion was also found to inhibit selectively the cleavage of pro-somatostatin without affecting the formation of secretory vesicles even at the optimal Golgi pH of 6.1, when the luminal free Ca2+ concentration was less than 15 μM. Similarly, depletion of luminal Ca2+ inhibited the endoproteolytic cleavage of proinsulin, but not the sorting of secretogranin II into immature granules in semi-intact PC12 cells (6). On the contrary, Ca2+ depletion was found to inhibit Brefeldin A-induced retrograde transport from the GA back to the ER in HeLa cells (79). These few examples, despite being somewhat controversial, also relate to cell-type differences with respect to the responses to changes in the Golgi luminal Ca2+ concentration.
Excess ROS are deleterious to the survival and proliferation of cancer cells. Hence, endogenous antioxidants are also upregulated to detoxify the cell and maintain a delicate balance of elevated intracellular ROS that is beneficial to malignant cells (136). In support of this, dysregulation of protein expression is associated with many cancer types and constitutes an important hallmark of malignancy. Recently, the transmembrane B cell lymphoma-associated factor (Bax) inhibitor-1 motif-containing (TMBIM) protein family has received increasing attention due to its expected role in tumorigenesis. (161). The Golgi antiapoptotic protein (GAAP) is a member of the TMBIM family and is projected by phylogenetic analyses to have originated before the divergence of plants and protozoa about 2000 million years ago. Orthologues of the human GAAP are remarkably conserved at the protein level (e.g., amino acid sequence, length, and hydrophobicity profile) throughout eukaryotes, prokaryotes, and some poxviruses, in agreement with a highly conserved ancestral structure and function [for review, see Carrara et al. (21)]. Since the first discovery of this gene in 2002 in camelpox virus, several cellular functions and structural properties of GAAPs from various origins have been described. Within eukaryotes, GAAPs regulate Ca2+ levels and fluxes from the principal intracellular stores (Golgi and ER), confer resistance to a broad range of apoptotic stimuli, and promote cell adhesion and migration via the activation of store-operated Ca2+ entry (22). Importantly, these multi-transmembrane proteins were shown recently to form cation-selective ion channels, potentially forming the basis for the modulation of diverse functions of GAAPs (22). In view of these functions and cancer, the effects of human GAAP on ROS homeostasis in the context of cancer cells were investigated. Significantly higher basal ROS levels, and more specifically basal H2O2, were detected intracellularly in cells overexpressing GAAP. In addition, cells overexpressing GAAP displayed stronger invasive capabilities in tissue culture, which was confirmed by the opposite effect on GAAP knockdown by siRNA. Furthermore, the activities of secreted matrix metalloproteinases 2 and 9, which are sensitive to regulation by ROS and play a key role in migration and invasion, were dysregulated in these cells. Although both the mechanistic links between these observations and the contribution of Ca2+ remain to be established, ROS appear to be a common factor of importance and relevance in understanding the contribution of GAAP in cancer development. Furthermore, the diverse multifunctional properties of GAAP provide a useful common starting point from which the complex interplay between ROS homeostasis and other important hallmarks of cancer such as cell invasion, migration, and resistance to apoptosis can be studied.
NO Homeostasis in the ER and Golgi
Nitric oxide (NO) is an important signaling molecule that also contributes to hypoxia response. For example, it induces vasodilation and thereby increases blood flow and oxygen supply (203). At the cellular level, hypoxia-induced NO inhibits the mitochondrial respiratory system by modifying thiol groups of specific cysteine residues in target proteins via S-nitrosylation. NO also contributes to the stability of the components of the HIF pathway, including HIF-1α (50). In addition to being a key substrate for the generation of NO, O2 regulates directly the production of NO by affecting the enzymatic activity and expression of various nitric oxide synthase isoforms, known as neuronal nitric oxide synthase, inducible nitric oxide synthase, and endothelial nitric oxide synthase (eNOS) (83). Of these, eNOS localizes mainly in the GA (80) where it enhances locally overall Golgi protein S-nitrosylation, and especially that of the N-ethylmaleimide-sensitive factor (NSF). Since NSF contributes to vesicular trafficking (membrane fusion), this modification reduces the speed of protein transport from the Golgi to the plasma membrane in a reversible manner. This finding at least partially explains why hypoxia inhibits ER-Golgi vesicular trafficking.
The GA is disassembled and shows a fragmented phenotype during apoptosis in neurodegenerative disorders such as ALS and Alzheimer's disease. These two phenomena precede neuronal cell death triggered by some toxins or oxidative damage, which suggests that the Golgi acts as a sensor of stress signals leading to cell death (126). Intriguingly, compounds that scavenge NO activity such as c-PTIO, methylene blue, N-acetylcysteine, and hemoglobin, induce Golgi fragmentation in several human endothelial cell lines (100). Golgi fragmentation by NO scavengers was inhibited by diethylamine NONOate and knockout of eNOS. It was also accompanied by depletion of the soluble NSF acceptor protein from the Golgi membranes, suggesting that NO may modulate global protein trafficking in vascular cells. Later on, it was shown that NO scavenging indeed prolongs secretion of soluble proteins without a complete block of vesicular trafficking between the ER and the Golgi (101). Such an effect could also impair Golgi glycosylation, even though no direct data currently exist to support this view.
Concluding Remarks
A number of important findings during the last decade have improved our understanding of disulfide bond formation, protein glycosylation, and secretion, as well as redox homeostasis. Thereby O2 itself, ROS, in particular H2O2, and pH changes appear to be of importance. An emerging concept is redox control of protein folding where several details in disulfide bond formation and redox regulation in the ER especially with respect to PDI and ERO1, as well as their interplay with NOX4, have been revealed, and they left surprises and new questions. For example, the relatively mild phenotypes of single ERO1-, NOX4-, and PRDX4-knockout mice (28, 78, 157) were unexpected and clearly suggest the existence of redundance and alternative pathways, which remain to be identified. In addition, the existence of more than 20 PDI family members from which the majority has an unknown function represents a key challenge. It will be interesting to learn how and whether the less-studied PDI family members, whether redox active or not, contribute to disulfide formation, redox regulation, and overall cellular homeostasis.
Findings indicating the interplay between hypoxia, ER stress, and Golgi function clearly indicate the intricate balance and importance of intercompartmental communication. The identification of MAMs indicates that these are just beginning to be understood with respect to ER and mitochondria. However, knowledge about the GA function in response to hypoxia and ROS is scarce as it is with respect to the other compartments with which the GA may communicate under these conditions. For example, hypoxia can reduce the amount of ATP via induction of a metabolic shift in mitochondria, but the mechanisms and key players by which this alters Golgi homeostasis and glycosylation remain so far elusive.
Resolution of these and other issues will not only help to better understand the etiology of diseases such as type 2 diabetes, Alzheimer's disease, and cancer, which are associated with ROS, protein aggregation, and glycosylation defects, but also to find new therapeutic clues. To achieve this, suitable animal models, genome-wide association and sequencing, as well as proteomic studies in humans, are needed.
Footnotes
Acknowledgments
We apologize to all researchers who excellently contributed to the field and whose work was not cited due to space limitations. This work was supported by the Finnish Academy of Science to S.K., T.K., the Jane and Aatos Erkko Foundation to T.K. as well as by the Biocenter Oulu, and the University of Oulu.