
Abstract
Significance:
Cancer is a complex disease, which not only involves the tumor but its microenvironment comprising different immune cells as well. Nitric oxide (NO) plays specific roles within tumor cells and the microenvironment and determines the rate of cancer progression, therapy efficacy, and patient prognosis.
Recent Advances:
Key understanding of the processes leading to dysregulated NO flux within the tumor microenvironment over the past decade has provided better understanding of the dichotomous role of NO in cancer and its importance in shaping the immune landscape. It is becoming increasingly evident that nitric oxide synthase 2 (NOS2)-mediated NO/reactive nitrogen oxide species (RNS) are heavily involved in cancer progression and metastasis in different types of tumor. More recent studies have found that NO from NOS2+ macrophages is required for cancer immunotherapy to be effective.
Critical Issues:
NO/RNS, unlike other molecules, are unique in their ability to target a plethora of oncogenic pathways during cancer progression. In this review, we subcategorize the different levels of NO produced by cells and shed light on the context-dependent temporal effects on cancer signaling and metabolic shift in the tumor microenvironment.
Future Directions:
Understanding the source of NO and its spaciotemporal profile within the tumor microenvironment could help improve efficacy of cancer immunotherapies by improving tumor infiltration of immune cells for better tumor clearance.
Introduction
Cancer is a complex disease that has many stages and involves multiple mechanisms, the understanding of which requires examination of many components/features of the tumor: like the tumor cell itself as well as endothelial cells and angiogenesis, immune cell polarization, and fibroblast behavior. The complexity of the communications extends from the intracellular and intercellular communications within the cancer cell neighborhood to the entire physiology and can also involve the microflora of the body. It has been established over the years that reactive oxygen species (ROS) and nitric oxide (NO)/reactive nitrogen oxide species (RNS) play critical roles within these pathways as do eicosanoids (156). Even today, new players such as hydrogen sulfide and persulfides are emerging in this landscape (68, 76, 85, 182). Despite the complexity of these signaling pathways, there are logical and precise mechanisms that result in specific phenotypic behavior. The key is to have the schematic of this landscape that encompasses specific mechanisms within specific contexts, providing maps for therapeutic targeting. Following the intracellular/extracellular path of redox species such as NO provides a unique opportunity to reveal this mechanism because of their involvement in numerous aspects of cancer biology (77a).
NO in infection and inflammation
NO has emerged as one of the critical mediators of inflammation in humans as well as in murine models of cancer (159, 180). However, many of the mechanisms in rodents do not always correlate with humans and this has made direct comparison difficult (134). Throughout the history of NO, this disengagement between animal models and humans has often slowed down scientific progress. The biology of NO emerged out of two lines of research: one, through the cardiovascular effects of endothelium-derived relaxing factor (53, 77, 86, 119), and the second from studies on the role of NO in immune response to tumor and infections (58, 69, 96, 143, 144, 151). Before these discoveries, NO biology was investigated in association with studies on the toxicity of cigarette smoke and air pollution. One of the ironies of this research is that NO in cigarette smoke is known to induce lung and cardiovascular disease, whereas endogenously generated NO is essential for preventing these diseases. This illustrates the idea of location and context dependence in trying to decipher the specifics of these mechanisms. In cancer, Hibbs et al. first described a small molecule produced from macrophages, which was critical in the antipathogen response and was dependent on the availability of arginine (69). Two years before this, another group had shown that macrophages could produce nitrosamines, nitrite, and nitrate during infection (143). It was found that NO was produced by inducible nitric oxide synthase/nitric oxide synthase 2 (iNOS/NOS2) and that these levels could kill tumor cells (189). The variability in the rate of production of NO from different cell types provided the necessary perspective on how NO may be important in normal physiology while also playing roles in antipathogen response.
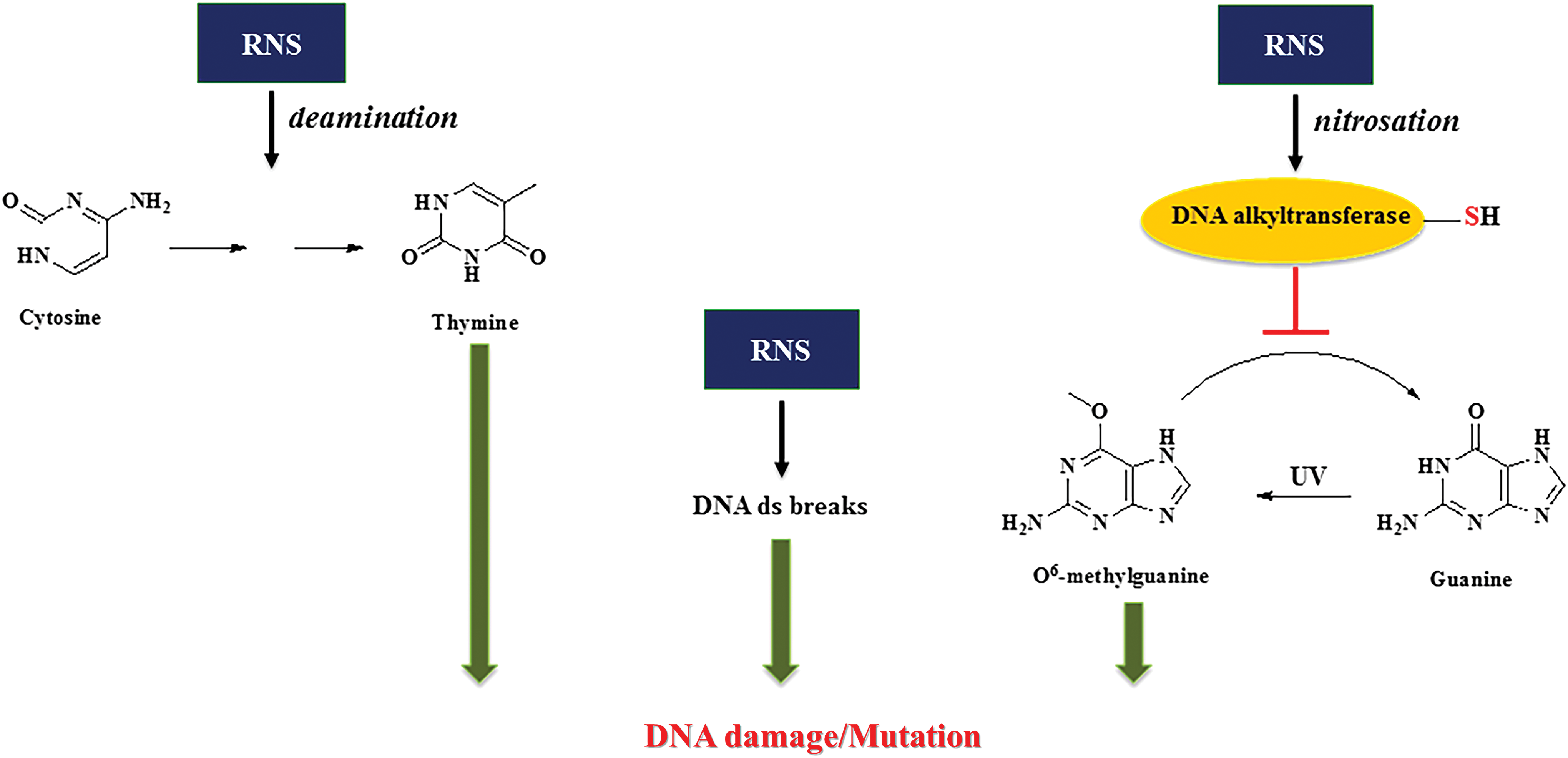
High NO flux causes genotoxicity and protein modification
At higher levels of NO such as those found to be produced by the immune system, there is the possibility of other types of chemical reactions involving novel targets: such as nitrosation of amines, particularly the primary amines in nucleic acids. It was shown that high levels of NO can also result in deamination, leading to C to T transition in DNA (115, 178). Further investigations show that these higher levels of NO could inhibit specific DNA repair systems, particularly those that are thiol dependent such as alkyl transferase and zinc finger proteins (95, 181). Oxidation of carcinogenic nitrosamines via cytochrome P450 (CYP450) formed DNA alkylating metabolites that cause DNA damage. This damage is repaired by alkyl transferases. Nitrosation of alkyl transferases inactivates the enzyme, which prevents DNA repair, thereby increasing potential mutations. It has recently been shown that alcohol dehydrogenase 5 (ADH5)/glutathione-dependent S-nitrosoglutathione reductase that, in part, controls S-nitrosation of proteins also controls NO effect on alkyl transferase, resulting in nitrosamine-induced carcinogenesis (170). In contrast, it has also been shown that NO could enhance the efficacy of chemotherapeutics such as the alkylating agent BCNU (1,3-bis (2-chloroethyl)-1-nitroso-urea), which is used to treat gliomas (174). Thus, high levels of NO through RNS can induce cytotoxic and genotoxic mechanisms, whereas lower doses can modulate normal physiology and improve chemotherapeutic efficacy (Fig. 1).
The NO/O2 reaction
In the early 1990s, the chemistry of NO and RNS could provide some insight into the biological dichotomy of both reactive species. One major question was how can NO in an aerobic environment be biologically important but also produce toxic RNS such as NO2 or N2O3? The answer lies in the chemical kinetics of the NO/O2 reaction wherein the reaction is second order with respect to NO, which makes the half-life in aerobic solutions proportional to NO concentration (175). The rate of RNS generation is also exponentially proportional to NO flux. Thus, low (≤100 nM) levels would only regulate NO/cGMP not resulting in appreciable RNS, whereas higher levels of NO produced by macrophage associated with tumor or pathogen response would mediate effects through NO/O2 reaction products. Diffusion away from this site results in exponential dilution of NO, thereby dramatically slowing the NO/O2 reaction. Thus, chemistry and diffusion play important roles in shaping the RNS profile, thereby making the location and geometry of NOS in the tissue or tumor critical.
Another theory proposed that the toxic nature of NO was due to the interaction of superoxide and NO to form a reactive species, peroxynitrite (ONOO−), which like other RNS can modify numerous macromolecules in the cell (59). From a first approximation using synthetic peroxynitrite to treat cells, this seemed like a plausible hypothesis. However, the biochemistry and biology of the NO/O2 − reaction are more complex than one involving a single intermediate. Although synthetic bolus peroxynitrite (exogenous) can oxidize different biological substrates, the NO and O2 − flux results in a series of intermediates that lead to nitrite and nitrate and is in fact part of NO-mediated antioxidant mechanisms (82). The reason is that NO/O2 − formation of ONOO− leads to rapid secondary reactions that resulted in less reactive NO2/N2O3, which is readily hydrolyzed to nitrite. Also, research showed that physiological CO2 reacted with ONOO- within milliseconds to form ONOOCO2 −. Despite being a powerful oxidant, it rapidly isomerized (in <10 ms) to nitrite and nitrate. Therefore, the reaction of RNS generated from NO/O2 − occurs in specific and isolated locations of O2 − generation with limited diffusion capacity for the peroxynitrite species (158, 179). Furthermore, NO has other antioxidant properties wherein low concentration abated Fenton reaction and Haber Weiss chemistry, thus protecting against lipid peroxidation (176). This further confirms that, at high doses, NO/acidic nitrite through NO2 can act as an oxidant and NOS2 inhibition prevents lipid peroxidation (13). Thus, looking at the primary mechanism of NO within the context of its formation and reactivity, it becomes clear that concentration, temporal, and spatial considerations are important.
NO/RNS in cancer
Owing to the interesting chemical toxicology of these species, other aspects of NO and cancer emerged. With the advent of research focusing on the transfection of NOS2 into different murine and human cancer lines, two dramatic effects of NOS2 on cancer progression were revealed. Transfection of NOS2 into different cancer cells leads to decrease in viability and NO was determined to be an anticancer molecule (39, 186). However, NOS2 transfection into human cancer cells increased the aggressiveness of these cells in vivo into xenograft models (79, 80). In later studies, it was found that NOS2 in tumor cells promoted cancer, whereas NOS2 in immune mediators had antitumor/proinflammatory effects, suggesting that besides the NO flux, spatial distribution of NO within the cancer microenvironment was also vital to understanding the dichotomy of NO effects in cancer (55). This further emphasizes that localization and cell-specific production of NO can have profoundly different outcomes.
To better define the role of NO/RNS in cancer, an important discovery was the relationship between NOS2/NO and p53. Malfunction in the p53 system can account for >60% of cancers and the vast majority of carcinomas and adenocarcinomas (74). NO was shown to stabilize p53 through phosphorylation of key serine/threonine moieties (73). In the case of wt p53, this can result in anticancer, apoptotic mechanisms mediated by NO (98). In addition, it was found that NOS2 expression was negatively regulated by p53, thus leading to a crosstalk between p53 and NOS2 in a feedback-regulated loop where p53 controls NOS2 activity. This relationship has been found in a variety of cancers (5, 6, 48). Yet, dysregulation of p53 takes the system off the brakes, allowing increased and prolonged expression of NOS2 in many solid tumors. In animal studies, Tp53−/−/NOS2−/− double knockouts show rapid increase in lymphomas, thus supporting the in vitro studies (75). This relationship of NO and p53 is important in the context of cancer progression.
As research in the NO and cancer realm focused on tumor physiology, the importance of cyclic guanosine monophosphate (cGMP) in modification of blood flow and angiogenesis in the tumor vasculature became apparent. Aberrant angiogenesis, a hallmark of cancer, due to rapid proliferation of tumor cells leads to poor blood flow and an increase in hypoxia and ischemia reperfusion. Many angiogenic agents such as vascular endothelial growth factor (VEGF) require NOS3/cGMP to facilitate the vascular network (41, 52). This chronic hypoxic and inflammatory state induced by aberrant vasculature favors selection of more aggressive cancer stem-like cells that are highly metastatic and resistant to therapy (133, 137). To help cancer progression further, NO at these low levels prevents immune cells from entering the tumor (93, 94). Normal vasculature when stimulated by pathogens/lipopolysaccharide (LPS) results in adhesion, rolling, and extravasation of immune cells into neighboring tissue (183, 184). However, adhesion molecule expression in tumor vasculature is downregulated and this inhibits adhesion and homing of immune cells into the tumor (89, 132).
Until recently, NO was thought to be an inhibitor of T cell function and endothelial activation and generally immunosuppressive in tumor biology (14, 87, 139, 167). This is the case in the aberrant vasculature seen associated with tumors. However, local low-dose irradiation was found to normalize the vasculature and allow T cell infiltration and subsequent tumor rejection and prolonged survival in pancreatic cancers. These effects were attributed to the differentiation of NOS2+ macrophages in the tumor microenvironment (35, 89). Interestingly, another group found that NO induced the expression of adhesion molecules on endothelial cells and favored T cell infiltration at low doses, whereas high doses inhibited vascular normalization (132). This demonstrates dramatic differences in NO effects based on its flux in another facet of cancer that directly affects efficacy of the current cancer immunotherapy modality.
NOS2 and Patient Outcomes
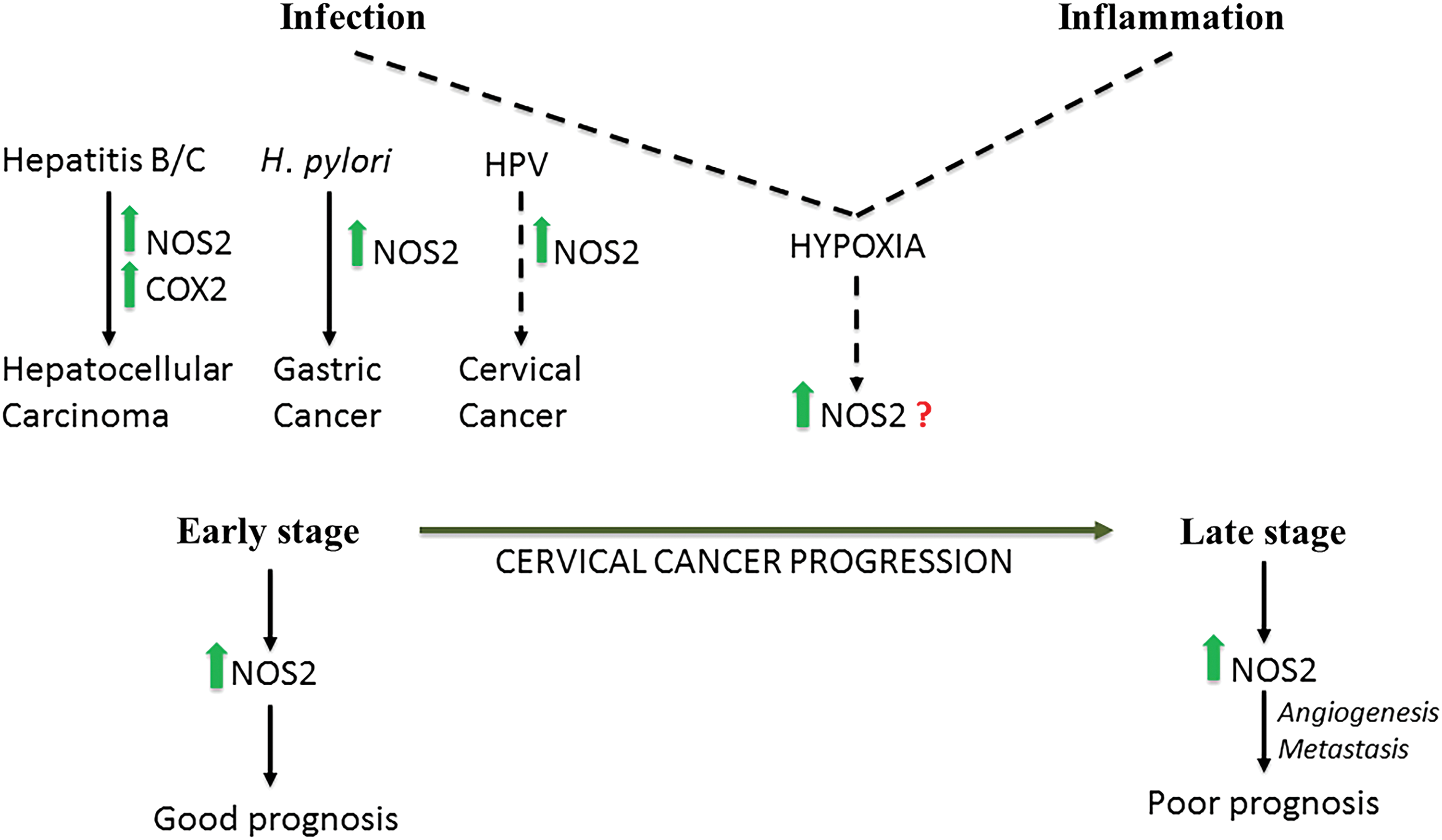
In the past decade, clinical trials have shown that NOS2 expression is seen in many cancers. Overexpression of NOS2 is present in >50% of glioma, melanoma, breast, prostate, pancreatic, liver, cervical, ovarian, nasopharyngeal, lung, stomach, colon, and esophageal cancer patients [summarized in (26)]. High NOS2 predicts poor patient outcome in numerous cancers such as estrogen receptor negative [ER (-)] breast, glioma, melanoma, pancreatic, stomach, liver, and colon cancers. In many cases, NOS2 is associated with increased angiogenic and metastatic potentials [summarized in (159)].
Numerous studies have shown that elevated NOS2 is associated with bacterial or viral infection even in cancers such as cervical, prostate, gastric, and hepatocellular carcinoma. In hepatocellular carcinoma, NOS2 and cyclooxygenase 2 (COX2) coexpression was only associated with hepatitis C and hepatitis B virus-induced cancer and not those associated with noninfectious causes, for instance, cirrhosis (120, 123). Gastric cancer is an example wherein a pathogen, Helicobacter pylori, directly elevates NOS2 (25). Cervical cancers associated with human papillomavirus infection also have elevated NOS2 (138). Interestingly, NOS2 has a positive prognostic effect at early stages of cervical cancer, but in later stages, NOS2 is a predictor of poor prognosis and leads to more vascularization and metastasis like in other solid cancers (43, 138). An alternative route to NOS2 induction is the metabolic change in the tumor environment where increases in hypoxia lead to increased NOS2 expression, and these metabolic changes may be caused due to infection and inflammation (Fig. 2).
The Chemical Biology of NO in Cancer
To place in context the cellular mechanisms that can be dramatically different over NO concentration ranges from sub-nM to μM, (0.1 nM to 5 μM sustained (seen in subcellular compartments of murine macrophages), we and others have studied the chemical biology of NO and described specific chemical and molecular mechanisms in the cancer landscape. One of the most important tools used in vitro are the NONOates or diazeniumdiolates, which spontaneously generate NO at specific concentrations and also exhibit varied temporal profiles of NO release, thus providing NO mimetics that reflect NO fluxes derived from NOS2 (158). These compounds have provided an important tool to understand specific oncogenic mechanisms that can be extrapolated to in vivo conditions and human cohorts. Other NO donors have been used to probe these mechanisms but are often complicated by their involvement in other reactions, besides simple NO release. Studies using the NONOates such as DETA/NO have revealed a concentration dependency for different biological effects (157) (Fig. 3).
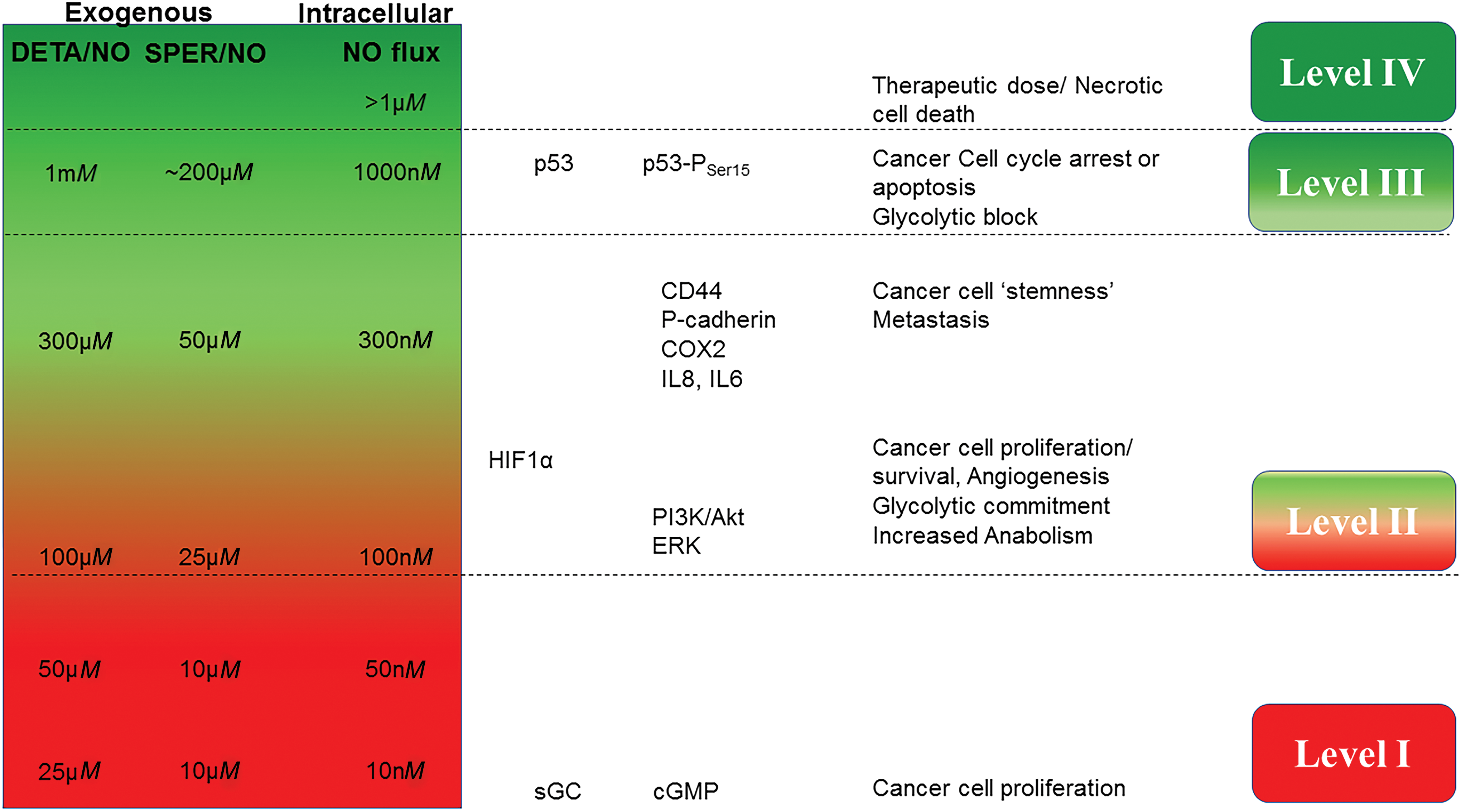
Using this technique, the oncogenic mechanisms that NO activates or inhibits have been examined with respect to time and flux. Two comprehensive reviews by Hanahan and Weinberg describe common signal transduction pathways that occur in most cancers (65, 66). Pathways regulated by NO such as DNA repair, phosphoinositide 3-kinase/protein kinase B (PI3K/Akt) survival signaling, extracellular signal-regulated kinase (ERK), β-catenin pathway, transforming growth factor beta (TGFβ) signaling, and hypoxia-inducible factor (HIF) and kruppel-like factor 4/5 networks among others are key players in promoting cancer (78, 108, 118). Although 500 nM intracellular NO flux leads to increase in p53 and nitrosative stress signaling, it is interesting that in a 100–400 nM range, NO is involved in protumorigenic mechanisms (156). This includes activation of RAS, HIF1α stabilization, epidermal growth factor receptor (EGFR)/proto-oncogene tyrosine-protein kinase Src (SRC) activation, and CD63/TIMP1 interaction that leads to PI3K activation and cancer cell survival and proliferation. Examining the fluxes of NO with these common oncogenic processes shows an important level that is above the physiological fluxes but below nitrosative stress levels of NO. This is the flux at which NO induces nitrosative signaling. In general, this level is associated with wound-healing responses but also is a key activator of primary oncogenic pathways, making this NO flux a determinant of poor prognostic phenotype. In this review, we discuss the mechanisms of NO action in terms of sustained NO fluxes such as those that would be expected in a chronic disease such as cancer (Fig. 3). These are level I, NO/cGMP level (<100 nM); level II, nitrosative signaling of oncogenic pathways (100–500 nM); level III, nitrosative stress (>500 nM), and level IV, ubiquitous RNS formation (>>1 μM).
Level I. Role of NO/cGMP in Progression of Cancer
At NO flux <100 nM, NO-cGMP/protein kinase G pathways are the primary signaling mechanisms. The level of NO that is relevant to normal physiology may be dysregulated by certain cancer-related cues. For example, it was found recently that endothelial NOS traffic inducer (NOSTRIN) inhibits disease progression and is downregulated in aggressive pancreatic cancer phenotypes (168). Within the tumor microenvironment, leukocyte interactions with the endothelium and regulation of blood flow can be attributed to the acute/short-term effects of the NO/cGMP pathway, whereas tumor progression and angiogenesis are chronic/long-term effects (52). Intracellular calcium flux controls the acute effects of NO from endothelial nitric oxide synthase/nitric oxide synthase 3 (NOS3) and neuronal nitric oxide synthase/nitric oxide synthase 1 (NOS1), whereas long-lived NO flux produced from phosphorylation of NOS3 and NOS2 is both calcium independent and generate sustained higher levels of NO (52). In an environment that is abundant in substrate and devoid of NO scavengers and chelators, the Km for O2 for NOS1, NOS2, and NOS3 is 350, 130, and 4, respectively; making oxygen tension in the tumor microenvironment an important determinant of NOS induction and NO production (145). This implies that in the hypoxic core of solid tumors, NOS3 would be the most likely source of NO.
Although NOS2 expression is strongly related to prognostic status in various cancers, the roles of NOS1 and NOS3 have not been delineated. This is largely due to the wide range of roles played by NO/cGMP pathway in normal physiology. The role of NO/cGMP can be dichotomous, being pro- or antitumorigenic, in different contexts. NO produced from NOS3, through cGMP, is involved in tumor initiation, progression, and therapeutic response (Fig. 4). Specific polymorphisms in NOS3 can be predictors of treatment outcome (51, 163) and have been correlated to reduced angiogenesis (111, 113, 169). Although NOS1 has been associated with several tumors, it has not been implicated in determining patient outcome (152). This low flux of NO decreases leukocyte adhesion and infiltration while increasing vascular leakiness and blood flow (93, 94). It is to be noted that all isoforms of NOS can activate cGMP signaling pathways. NOS3 can directly activate RAF-1 as well as RAS in RAS-driven cancers (101). Activation of RAS and subsequently the PI3K/Akt pathway promotes NOS3 phosphorylation and increases nitrosation of cysteine 118 on RAS (101). This nitrosation event is associated with dramatic increase in activity (149). NOS3 knockout has been found to improve prognosis in pancreatic cancer that is RAS driven. Wnt/β-catenin signaling is a key player in progression of many cancers, and NOS3 could potentially inhibit this pathway as observed with sulindac, a nonsteroidal anti-inflammatory drug (160). In gastric cancer, Type II cGMP-dependent protein kinase (PKGII) can inhibit phosphorylation of EGFR and other receptor tyrosine kinases as well as the metastasis factor c-Met (81, 185). However, in hepatocytes, cGMP increases activity of tumor necrosis factor (TNF) α-converting enzyme (TACE/ADAM17) leading to increased NOTCH as well as TNFα and EGF signaling, this then leads to activation of ERK as well as NFκB, and PI3K resulting in cell proliferation/survival.
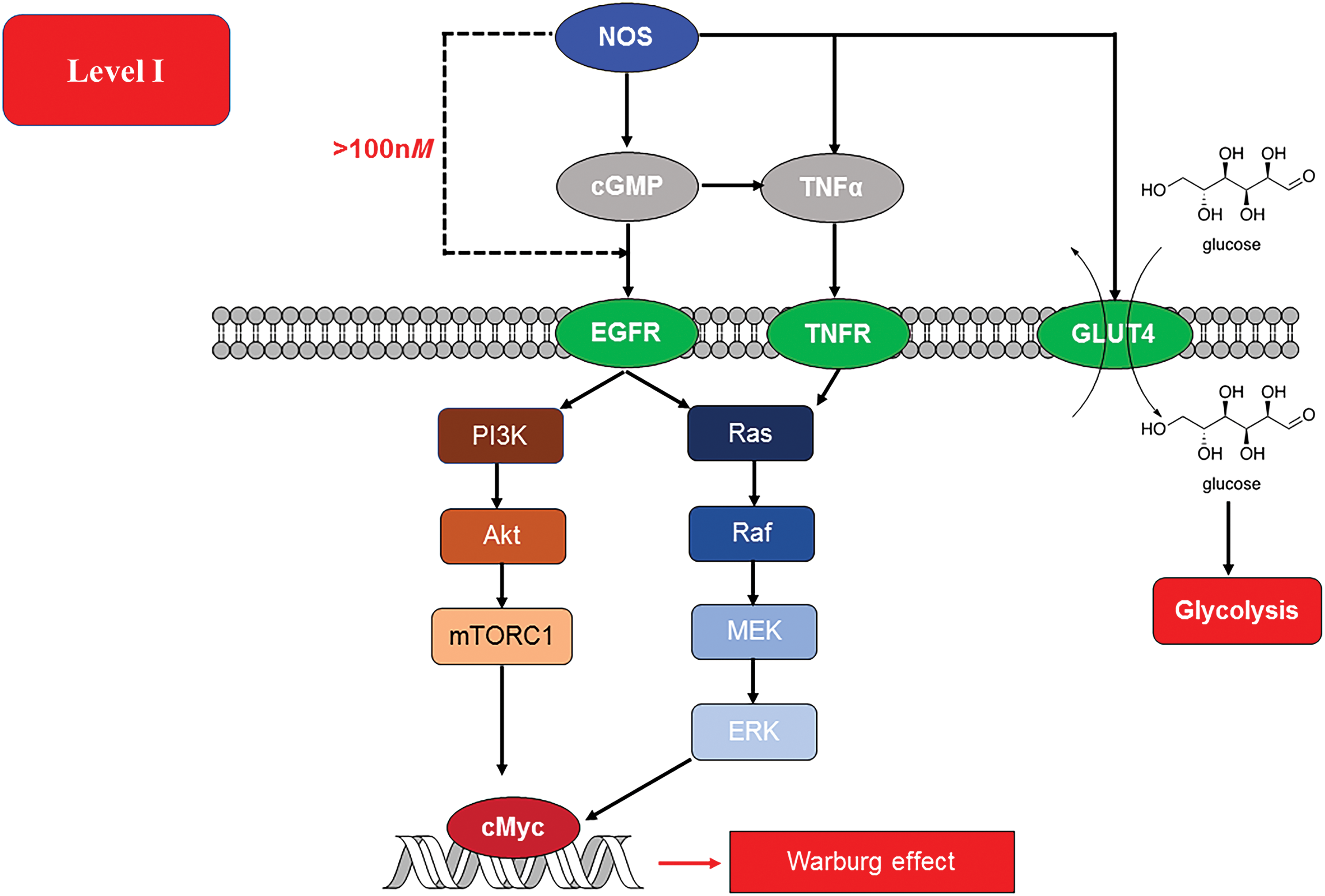
Effect on metabolism
This flux of NO, unlike the nitrosative signaling or stress levels, has little direct effect on the cellular respiration in cancer cells. NO and cGMP can increase RAF/ERK signaling, subsequently leading to increased c-Myc expression, which, in turn, can lead to increased glycolysis. Yet, another study shows that increased activity of soluble guanylyl cyclase (sGC) in breast cancer inhibits proliferation (172). Interestingly, low level of NO may provide an opportunity for signaling through ROS or hypoxia. Increase in cGMP downstream of NOS2 can induce TACE/ADAM17 via p38-mitogen-activated protein kinases (MAPK) pathways and ERK and glucose transporter 4 (GLUT4) and hence, increase glucose uptake (24, 45, 187). This pattern of pathways at this level predicts an outcome favorable to glycolysis. It should be noted that at these levels of NO, in vitro experiments do not show evidence for stabilization of HIF1α (155). Therefore, more detailed analysis is required into different types of cancers as well as different phenotypes with in specific diseases.
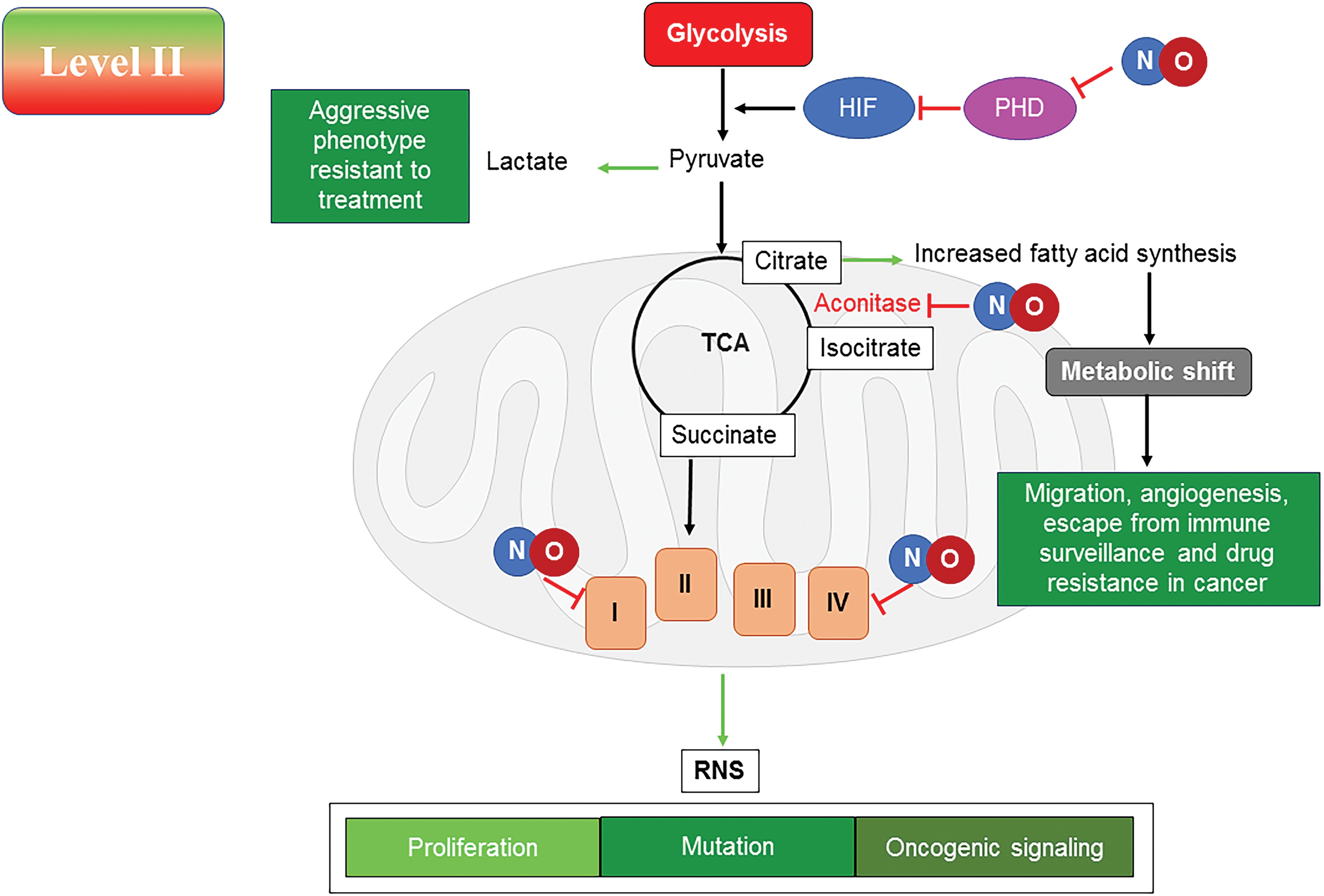
Level II. Nitrosative Signaling (cGMP Independent)
A total of 100–500 nM NO levels induce several classical pro-oncogenic pathways, including but not limited to HIF1α stabilization and subsequent signaling events and activation of PI3K/Akt, TGFβ, and ERK signaling pathways through ligand-independent mechanisms (156). At this NO flux, there is increase in genomic instability, cancer “stemness”, chemoresistance, angiogenesis, metastasis, and immunosuppression (156). This flux of NO generated in vitro with NO donors has been shown to correlate with NOS2 signaling that is associated with human patient samples and that seen in animal studies (56, 67). These in vitro systems using NO donors recapitulate the signaling events that drive poor patient prognosis and has become an important model to study this intermediate level of NO flux (56, 67). It should be noted that these pathways are also part of the natural wound healing process, thus the expression “Cancer is the wound that does not heal.”
Nitrosative signaling results in specific chemical species that mediate signaling in cancer cells, leading to a more proliferative and metastatic phenotype (156). At this level of NO, specific chemical reactions such as nitrosation and activation of nonheme iron occur and this activates discrete pathways. Like with growth factor ligands that bind to specific receptors, thiols and metals are two main targets of NO- and RNS-mediated signaling. Kinetics of NO reactivity with different metal ligands allows tuning of the affinity of the targets to react at specific NO concentrations. The metal ligand environment and oxidation state dictate this selectivity. This allows kinetically determined targets to form precise initiators of specific signal transduction pathways. RNS-mediated activation of thiols is accelerated by hydrophobic environments as well as the availability of low pK a thiols. Thus, tuning of NO reactivity by specific molecular motifs allows for unique signaling events.
In contrast to effects seen with level I NO fluxes, RAS and EGFR can also be activated by RNS-mediated nitrosation and modification of thiols in a cGMP-independent mechanism, leading to activation of RAF/MEK/ERK signaling (149). NO at this level also regulates RAS activity by causing S-glutathiolation in a redox-sensitive manner (3, 72). Nitrosation and subsequent activation of ERK correlates with poor prognosis in ER (-) breast cancer (56). This ERK activation further induces c-Myc while inhibiting RAS and leading to suppression of PI3K/Akt and MAPK signaling pathways (148).
Growth factors activate receptor tyrosine kinases and subsequently turn on PI3K/Akt signaling. Mutations in PIK3CA and PTEN that are prevalent in many cancer types increase the PI3K/Akt pathway, indicating that these are critical pathways in promotion of poor outcome. At this level of NO, ligand-independent activation of SRC and EGFR occurs through nitrosation of thiol motifs. The presence of detectable S-NO moieties indicates that cysteine-rich domains are prone to thiol modification (7, 121). Sulfenic acid formed from ROS can activate these pathways possibly through the formation of disulfide bonds via specific redox electrophile intermediates (162). Interestingly, O2 − has been found to attenuate PI3K signaling, indicating that superoxide can antagonize NO-mediated pathways and NO can, in turn, inhibit superoxide-mediated molecular pathways (157). At this level of NO, the NO/O2 reaction and not the NO/ROS reaction causes the nitration of specific tyrosine residues of the tissue inhibitor of metalloproteinase 1 (TIMP1) enzyme in MDA MB 231, a triple negative, mesenchymal stem-like breast cancer line (125). Although this tampers its ability to inhibit matrix metalloproteinase (MMP), nitrated TIMP1 has a higher affinity to induce CD63-mediated prosurvival signaling via the phosphorylation of PI3K/Akt and BAD proteins (125). Elevated TIMP1 levels have been shown to cause the accumulation of cancer-associated fibroblasts that then promote progression of many types of cancers.
One of the major downstream targets of PI3K, namely Akt, is a critical node in many protumorigenic pathways, leading to the activation of Wnt/β-catenin and mTOR signaling pathways. At the nitrosative signaling level, Akt phosphorylation, activation of β-catenin, and mTOR signaling are observed even in the absence of growth factors or other associated ligand–receptor interactions (105, 125, 149). PI3K activation from SRC also induces the activation of critical DNA repair family proteins, such as DNA-PK, ataxia-telangiectasia and Rad3-related kinase, and ataxia telangiectasia mutated kinase (12). Although it has been suggested that many proteins such as RAS, SRC, and EGFR are directly activated by NO through R-SNO formation, the downstream kinase inhibitors effectively inhibit these pathways exactly as is seen with growth factor stimulation (ligand-dependent pathway), suggesting that RNS formation may initiate these pathways in the membrane but do not influence the downstream signaling events. The kinetics of these events are determined by the higher lipid solubility of NO and O2 and would preferentially occur in the membrane as opposed to the cytoplasm where the kinetics are less favorable and RNS scavengers such as reduced glutathione (GSH) are present. Growth factor receptors are predominantly membrane bound and hence, the precision of pathway activation is determined by several factors that work concurrently.
High, medium, and low levels of NO stabilize HIF1α by inhibiting prolyl hydroxylase (PHD) and activate hypoxia responsive elements (HREs) (18). Activation of the HRE binding proteins increases expression of various proteins involved in metabolic processes. Hence, NO interestingly provides a simulated hypoxia state where there is inhibition of mitochondrial respiration while sufficient oxygen is still present. This is recapitulated using NO donors that stabilize the HIF1α in a dose-dependent manner independent of O2 level.
Effects on metabolism
Nitrosative signaling can influence the carbon flow and fuel utilization in cancer and stromal cells through direct interactions with key enzymes or by eliciting specific pro-oncogenic pathways. Although NO/RNS can directly interact with specific sites in the respiratory chain and the tricarboxylic acid (TCA) cycle, these species can also induce several oncogenic pathways that shift metabolism from utilization of fatty acids to glycolysis (10, 40, 193). This shift to an aerobic glycolytic process, or Warburg effect, may require both the direct reaction with NO/RNS and downstream signaling pathways, leading to this shift in metabolism. At this level of NO flux, the anabolic kinase mTOR is active and can drive cancer cell proliferation. However, it has been found that in nutrient-deprived macrophages, this level of NO (upper range ∼500 nM) can lead to activation of the ATP sensor AMP kinase, which, in turn, can also activate the catabolic processes such as fatty acid oxidation (10, 36).
NO can affect mitochondrial function at complexes 1 and 4 (32, 130, 131) of the electron transport chain (ETC). In cancer cells, lactate production is observed as a result of aerobic glycolysis or Warburg effect, which has been proposed as a hallmark of a more aggressive phenotype (84). It is still unclear how NO directly or indirectly may play a role in mitochondrial function in cancer cells and induction of aerobic glycolysis. Further in this review, we discuss the various pieces of evidence and predictions about the effects of NO on the bioenergetics of cancer cells.
At the level of NO that correlates with nitrosative signaling, it was reported that there was loss of oxidative phosphorylation with the immediate application of the NO donor in breast cancer cells, indicating that NO inhibits cellular respiration (37). Concordantly, removal of NO restored the oxidative phosphorylation, thus confirming that the acute effect of NO was through direct interaction with the respiratory machinery and not through indirect signaling events. However, prolonged NO exposure induces a number of oncogenic pathways, in particular, HIF1α, PI3K, and ERK signaling already discussed, which are known to change the metabolic function and induce glycolysis and increase fatty acid synthesis in large part from cytoplasmic citrate (36). From these observations, it can be concluded that induction of specific signaling pathways such as with growth factors and other oncogenic stimuli may lead to specific effects on metabolic pathways.
Stabilization of HIF1α induces metabolic genes that are involved in increasing glycolysis, and induction of NFκB increases glucose uptake through induction of the GLUTs (36, 92). PI3K through Akt activates mTORC1, which leads to an increase in fatty acid and protein synthesis (36). Pyruvate kinase M2 (PKM2), a part of the “glycolytic enzyme complex”, unlike PKM1, leads to Warburg effect by controlling the flow of pyruvate (192). The tetrameric form of PKM2 leads to pyruvate production, whereas the dimeric form is less active. In addition, HIF1α and PI3K conspire to increase PKM2 expression, which preferentially directs pyruvate to lactate. Although tyrosine phosphorylation inhibits PKM2, serine phosphorylation activates it. PKM2 can regulate HIF1α, signal transducer and activator of transcription (STAT) 3, and c-Myc signaling (192). NO has been shown to increase NFκB in tumor cells. PKM2 is overexpressed in cancer cells and can regulate HIF1α through NFκB (9). PKM2 phosphorylation by ERK leads to formation of a complex with β-catenin, which, in turn, induces lactate dehydrogenase A (LDHA) favoring lactate formation. PKM2 is a gatekeeper and has a redox active cysteine, which when oxidized or modified diverts glucose from glycolysis to pentose phosphate shunt. Thus, increase in PKM2 through signaling will favor the Warburg effect and increase numerous anabolic pathways for protein, fatty acid, and ribose synthesis, as well as increase NADPH.
In addition to effects on glucose metabolism, NO can also modulate glutamine metabolism. Activation of the ERK pathway, which increases c-Myc as well as PI3K/Akt/mTOR, increases conversion of glutamine to glutamate that can feed into the TCA cycle via α-ketoglutarate (62). This can lead to what is referred to as “glutamine addiction” seen in many cancer cells. Besides being used as a fuel source, glutamate is required for GSH, which is elevated in many cancer cells. Glutamate can also enter the TCA through anaplerosis; another prosurvival metabolic program seen in cancer cells. Deamination of the glutamine pool leads to a buildup of amines in the cells. These amine groups are used for amino acid synthesis and are particularly relevant to upregulation of arginine pathways. Deamination also leads to increased ammonia and this is sometimes referred to as “ammonia stress” in cancer cells as excess ammonia can induce autophagy (27).
It may be predicted that at nitrosative levels of NO, glutamine would be utilized to fuel the TCA and ETC (Fig. 5). Although fatty acid oxidation is reduced under these conditions and glucose is diverted to lactate, glutamine would be expected to be the predominant fuel. However, in the presence of NO at this level, aconitase is inhibited, preventing the conversion of citrate to isocitrate and α-ketoglutarate. In macrophages, this may lead to export of citrate and increased fatty acid accumulation (10). In cancer cells, the increased signaling through mTORC1 and ERK suggests a metabolic diversion in NO-rich conditions to generate fat from exported citrate (109). Thus, glucose and glutamine can be utilized as carbon sources to generate building blocks for proliferation at the nitrosative stress level. Interestingly, during epithelial–mesenchymal transition (EMT), these cells exhibit elevated glutaminase (GLS) expression as well as a tendency to be more committed to glycolysis (97, 106). Since NO will induce EMT, there may be a shift away from glutamine utilization and proliferation to glucose-dependent migration and cancer stem cell-like behavior. If this is the case, then targeting of glycolytic enzymes may prevent metastasis.
Effect on arginine and amine metabolism
As already discussed, NOS2 activity is arginine dependent and is determined by arginine metabolism. Arginine is critical in numerous pathways and its concentration is largely determined by arginase and NOS2 in many cells (154). Arginine can be formed from recycling citrulline through arginosuccinate synthase 1 (ASS1) and arginosuccinate lyase (ASL) or by addition of amine groups to ornithine to generate citrulline that then recycles arginine. The increased lysis of glutamine to ammonia due to increase in GLS (resulting from nitrosative signaling) would, in some tumors, increase carbamoyl phosphate, which can be an amine donor for many reactions like the conversion of ornithine to citrulline. NOS2 generates NO, that inhibits ornithine decarboxylase (ODC) through thiol modification, and generates α-amino acid Nω-hydroxy-nor-L-arginine (NOHA), an intermediate in the NOS2 reaction as well as an inhibitor of arginase. Hence, under the conditions of nitrosative signaling, there are numerous pathways that coordinate to favor increase in NOS2 activity and increased aerobic glycolysis.
Methyl and acetyl metabolism
Methyl and acetyl metabolisms are not bioenergetic reactions but are interesting with regard to their epigenetic effects. Herein, we review the possible roles for NO in methyl and acetyl metabolism. Inhibition of aconitase 2 by NO increases citrate (10). The cytosolic citrate can then be converted to acetyl CoA that is used in fatty acid synthesis and may result in an increase in acetyl groups that can subsequently be used for protein modification as well as amino acid synthesis (64). Diversion of metabolism away from arginase and polyamine synthesis (via ODC) in presence of high NO flux could increase the methyl equivalence available for a variety of processes such as CpG- and histone-methylation. NO directly inhibits the activity of the Jumonji C domain containing demethylase, KDM3A, by forming a nitrosyl–iron complex in the catalytic pocket (70). Ten-eleven translocation methlycytosine (TET) dioxygenase enzymes use similar cofactors (α-ketoglutarate) and substrate as the Jumonji C domain demethylases. TETs epigenetically increase gene expression by inducing formation of hydroxymethyl cysteine (70). NO also inactivates histone deacetylases (sirtuins) by cysteine S-nitrosation, whereas HDACs, in turn, influence NO production (83). Hence, NO directly regulates acetylation and methylation events and subsequently affects epigenetic changes and could be important especially with regard to changes observed between epithelial and mesenchymal states of the cancer cell.
NO and iron metabolism
An important aspect of this level of NO is that it interacts with numerous nonheme iron complexes. Nitrosative signaling levels of NO lead to an increase in iron uptake and decrease in storage due to the regulation of iron responsive elements through iron binding proteins, such as cytosolic aconitase (IRP1) (103, 146). Interestingly, IRP1 is downregulated by NO via STAT5 proteins (141). This may suggest a feedback mechanism controlling NO uptake and storage. NO can activate Nrf2A through nitrosation of KEAP1, leading to increase in GSTπ (2, 50, 104, 164). When nonheme iron forms a nitrosyl complex, it is through GSTπ that these are converted to iron–dinitrosyl glutathione products that are exported through the multidrug resistance proteins (104). This balance of iron uptake and iron export is an important regulatory mechanism. These effects of import and export of iron–dinitrosyl complexes may be an important aspect of controlling NO flux and its reaction products (104, 147).
PHD at the hub of NO oncogenic behavior
Another important aspect of NO at this level is its interaction directly with the iron of PHDs. This class of enzymes have an iron(II) or ferrous metal that is dependent on O2, and the substrate can be α-ketoglutarate or ascorbic acid that makes it a unique intersection of different metabolic functions (191). In addition to facilitating the degradation of HIF1α, PHD-like proteins regulate several processes. Jumonji C domain containing demethylases and TET dioxygenase enzymes, for example, are both PHDs that can inhibit IKKβ, thereby inducing the canonical NFκB pathway (33). PHDs are controlled by the reductant α-ketoglutarate and inhibited by the mutant isocitrate dehydrogenase 2 product D-2-hydroxyglutarate (D2HG) (190). Glioma cells (largely high NOS2) can have a mutation that leads to increased D2HG and inhibition of HIF1α. PHD influences many processes including changes in the TCA (production of αKG), decreased demethylation, and increase in NFκB and HIF1α activity. Thus, this fortifies the idea that NO transforms cells to a more aggressive phenotype with the ability to thrive even in hypoxic conditions.
Level III. Nitrosative Stress Signaling
The next higher level of NO activates pathways that increase the protective mechanisms against both nitrosative and oxidative stress. This level, defined by a sustained NO flux between 500 and 1000 nM, results in the induction and stabilization of several proteins that are involved in cell cycle arrest as well as antistress mechanisms (156). One of the hallmarks of this NO level is the activation of p53, which occurs at 500 nM in many cell types, resulting in post-translational modification (phosphorylation and acetylation) of this critical protein (73, 155). In addition, MKP-1, an ERK phosphatase, is also activated, subsequently preventing cell proliferation (122, 127). Many of the processes mediated by NO at this flux are proapoptotic. However, if viewed from a temporal perspective, many of the proteins activated concurrently with p53 including but not limited to binding partners of antioxidant response elements could potentially be protective in some cells (8). Nitrosation of caspases is an acute/short-term effect of nitrosative stress that prevents apoptosis. However, chronic/long-term nitrosative stress signaling itself activates the caspase cascade (88). Thus, there is a critical balance that is maintained between prosurvival and cytotoxic/antitumor mechanisms that determine the mode of NO action.
In mouse models, infection, inflammation, and wound response mechanisms can induce an initial Th1 immune response that upregulates NOS2 activity, subsequently stabilizing p53 (17), a critical step in cancer immune surveillance. Research has found that this level of NO downregulates the Th1 response through reduction of NF-κB signaling (21, 107) and activation of TGFβ (166). Furthermore, NOS2 and NO lead to upregulation of interleukin (IL)-10 in regulatory T cells (116). If placed in a temporal context, increase in NOS2 possibly occurs 4–8 h after the Th1 induction in macrophage and this then leads to the downregulation of Th1 inflammatory system. Thus, this level of NO is critical for switching the immune response away from Th1.
Effect of nitrosative stress level of NO on intermediate metabolism
NO levels that induce nitrosative stress can have the opposite effect on many of the carbon pathways already described. Although nitrosative signaling induces tumor progression, higher levels that stabilize p53 will reverse this and are important to understanding cancer treatment response and eradication. In murine models, NO has been shown to be a critical component in tumor clearance as well as for immune surveillance. In the case of wild-type p53, there is a dramatic change of the carbon flow through intermediate metabolism from the nitrosative signaling level. P53 reduces glucose uptake by inhibiting GLUT expression, and increases glutamine fueling of TCA by upregulating glutaminase (GLS) that converts glutamine to glutamate that then feeds into the TCA through α-KG (32, 36, 92). P53 blocks pyruvate dehydrogenase kinase 2 (PDK2) and, in turn, activates pyruvate dehydrogenase (PDH), thus increasing acetyl CoA preferentially over lactate formation, which counteracts aerobic glycolysis. P53 also inhibits the pentose phosphate pathway by inhibiting glucose 6-phosphate dehydrogenase (G6PD) (92). Also, p53 increases fatty acid oxidation instead of synthesis, thus forming acetyl CoA. In conclusion, this favors a change of the carbon flow from nitrosative signaling compared with that induced by activation of the oncogenic pathways (Fig. 6). One difference is that many direct targets such as mitochondrial aconitase as well as cytosolic aconitase are inhibited and the mitochondria are still depolarized. In this case, there would be a prediction for increased production of citrate from glutamine. However, in the case of mutated p53 (observed in 50% cancers), the nitrosative level signaling would be allowed to continue unabated, providing another reason why p53 and NOS2 are critically linked in cancer.
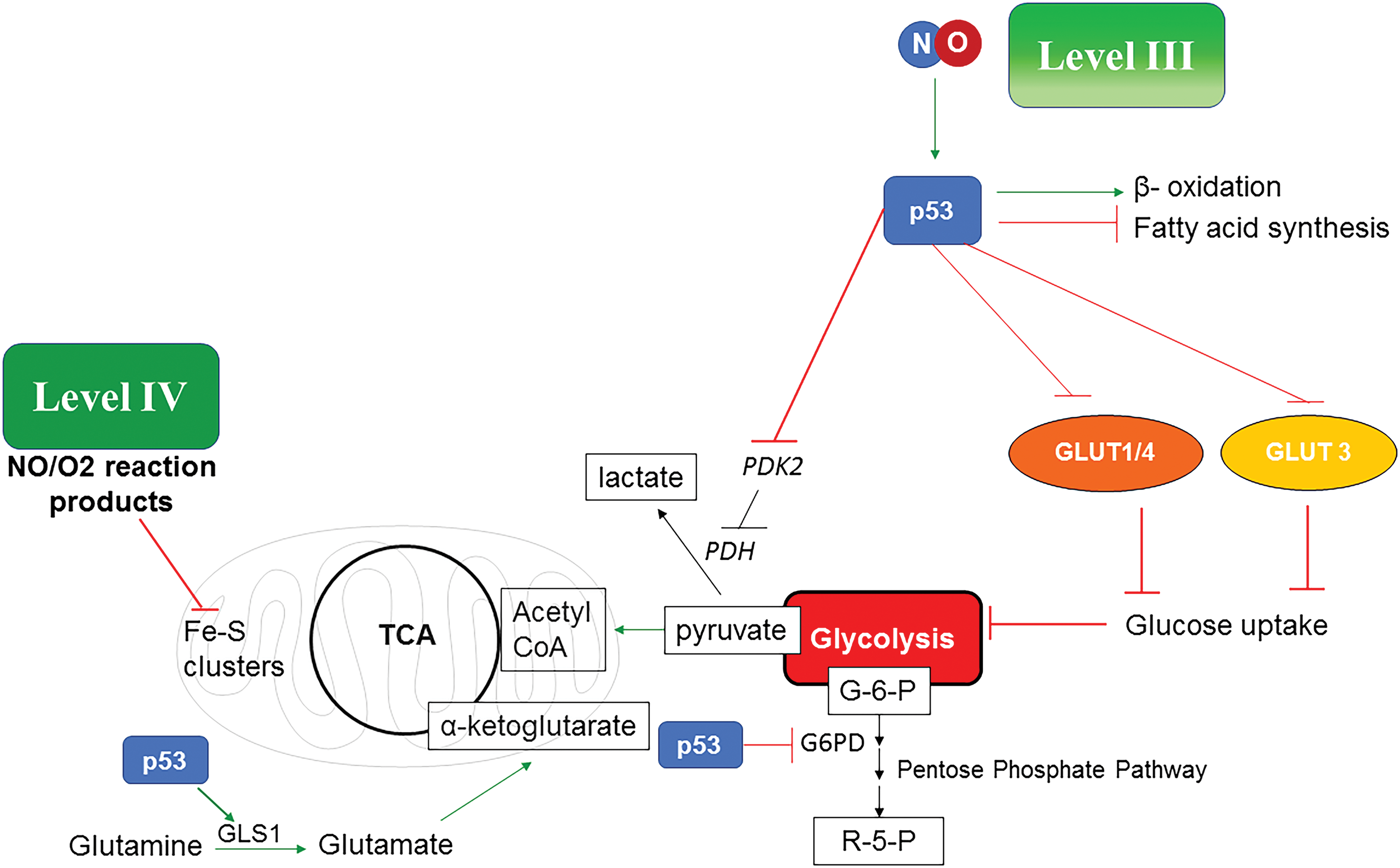
At these higher levels of NO, there is increased mitochondrial stress resulting in decreased NADH utilization due to inhibition of targets in complex 1 (32, 57, 140). Also, at higher levels of NO, there is suppression of both the TCA and ETC, a potentially catastrophic event. This may in fact be why NOS2-positive cells correlate strongly with p53 mutation. This interference with metabolism that reduces availability of glucose and fatty acids as energy sources, thereby limiting fueling to glutamine may be a mechanism of how NO, through Tp53 induction, is a potential anticancer pathway.
Level IV. Role of RNS Formation and Pervasive Nitrosative Stress
This flux of NO was historically the level that was studied first with respect to cancer. Activation of Nrf1/2 (a part of the antioxidant system), H2AX phosphorylation (γH2AX, an element of the DNA damage response mechanism) are two important events that occur at this NO flux (117). At these high levels estimated at 1–5 μM flux, NO generates locally high RNS that modify proteins and other macromolecules. These levels are physiologically relevant in local regions of an active murine macrophage and persist within the macrophage microenvironment (19, 73, 150, 155) and can lead to apoptosis and mitochondrial dysfunction.
One subcellular location in the macrophage where there are high levels of RNS is the phagosome. Rac1 translocation of NOS2 and NADPH oxidase results in the concentrated production and the interaction of ROS and NO, giving locally high concentrations of nitrite (177). In the acidic environment of the phagosome, the H+/NO2 − and increased iron and peroxide form a cocktail of ROS and RNS to fight different pathogens. Moreover, nitrosative stress involving high levels of NO could mediate tumor clearance, particularly in leukemias harboring wild-type Tp53 (75, 98, 144). Interferon gamma (IFNγ) and TNFα secreted by CD8+ T cells lead to NOS2 induction in macrophages in the tumor microenvironment. This, in turn, is critical for tumor clearance and may decide patient response to different immunotherapy regimen (89, 132). However, NOS2 in human macrophages does not produce as much NO as murine macrophages.
Implication of level IV on metabolism and cell death
The effects on metabolism at this level are quite profound. The direct chemical reactions of RNS modify numerous processes. Sustained high NO level would lead to chronic mitochondrial depolarization through direct chemical interaction of NO with the Fe–S clusters of the respiratory complex proteins (16, 31, 136). In addition, there are changes in DNA and other macromolecules. Although ubiquitous production from NOS2 may be tempered by consumptive mechanisms and effects on NOS2 activity through loss of substrate and electron flow, NO donors may be important to delineate these effects.
The antitumor activity of p53 via cell cycle arrest and apoptosis occurs at level III NO fluxes (Fig. 6). Recent research has shown that oxidative stress can induce p53-mediated necrosis in ischemia reperfusion model (165). This biology could also be applicable to tumor models at level IV NO flux (high RNS), which may lead to necrosis and subsequent immune activation.
Control of NOS2 Enzymatic Activity
The level of NO is ultimately tuned by the balance between the production from NOS and its consumption. This shapes the NO profile in specific cellular neighborhoods. Some have quoted that the NOS enzyme activity is among the most tightly regulated mechanisms, hence corroborating the importance of NO (14). If the activity of the enzyme from the biochemical point of view is considered, there are several factors that influence its kinetics. The factors that are required for enzyme activity are NADPH, FMN, O2, tetrahydrobiopterin (THBP), and the substrate L-arginine. In addition to this, iron metabolism and heme synthesis are important factors in the assembly of an active protein. GAPDH has been shown to be critical to the insertion of the heme to form active enzyme (23). Finally, the splice variants present may be important in determining the flux of NO.
The induction of NOS2 transcription
The proximal promoters of the human and mouse NOS2 have a common structure, with a TATA element ∼24 bp upstream of the transcription start site, preceded by consensus NF-kB, C/EBP, and OCT sites. In the murine NOS2 gene, an enhancer containing NF-kB, IRF, and STAT sites is located approximately 900 bp upstream of the transcription start site (TSS), and induction of NOS2 transcription by IFN-γ and/or LPS has been associated with the binding of STAT1 to the enhancer (54). The human NOS2 gene does not have an enhancer element in the 1 kb region upstream of the TSS. However, an enhancer element containing NF-kB, STAT, and AP-1 sites is located approximately 5 kb upstream of the human NOS2 promoter, and it has been associated with IFN-γ induction of NOS2 (188). NF-kB, AP-1, and p38 have also been shown to be required for the full transcriptional activation of the human gene (4, 42, 61). Although there are several common factors involved in NOS2 transcriptional activation in humans and rodents, the level of induction of NOS2 in human immune cells is considerably less than that in rat/mouse cells. Significant induction of NOS2 expression by IFNs, cytokines, and LPS can be seen in murine macrophages. However, in human tissue, high levels of NOS2 are found primarily in cancer cells rather than monocytes. Human cells such as epithelial cells, cancer cells, and astrocytes can induce NOS2 expression. This dynamic may be important for understanding differences observed between murine models and cancer patients.
Post-transcriptional regulation of human NOS2
The cellular level of human NOS2 mRNA is controlled not only by variation in the rate of gene transcription but also by the rate of mRNA degradation. The KH-type splicing regulatory protein (KSRP), poly(A)-binding protein (PABP), and hnRNP-E1 RNA-binding proteins have been found to interact with elements in the 3′ UTR of the human NOS2 mRNA (102). Cytokine stimulation with IFN-γ, IL-1β, and TNF-α reduced KSRP binding to AU-rich elements in the 3′ UTR of the NOS2 mRNA, thereby increasing its stability. PABP has been shown to bind both the 5′ and 3′ UTRs of human NOS2 mRNA. In contrast to KSRP, PABP binding was shown to stabilize NOS2 message (22). The hnRNP-E1 protein binds to 3′-UTR pyrimidine-rich elements to form stabilizing complexes. Binding of hnRNP-E1 to NOS3 mRNA increases its stability by protecting it from the inhibitory effects of micro-RNAs and antisense transcripts (71), implying that it may have the same effect on NOS2 mRNA. Micro-RNAs such as miR-146a can have an indirect effect on NOS2 expression by modulating cytokine expression (100). There is, however, one miRNA (miRNA-939) that has been shown to directly block human NOS2 expression by binding to the 3′ UTR of NOS2 mRNA (63). The same cytokines that induce NOS2 transcription (IFN-γ, IL-1β, and TNF-α) also increase the level of miRNA-939, which binds to two specific miR-939 binding sites in the human NOS2 3′-UTR, leading to translational inhibition of cytokine-induced NOS2 protein expression. This likely represents a feedback mechanism that limits the duration of NOS2 induction.
Epigenetic and post-translational regulation
There is very little literature about the human NOS2 regulation and its activity. NOS2 is hypermethylated in humans, suggesting a much lower level of induction (60). In human chondrocytes, hypomethylation is responsible for activation of NOS2 (34). However, it has been pointed out that under specific conditions in the lung alveolar macrophages, in astrocytes rather than microglia in the brain and in hepatocytes, NOS2 levels are much higher (29, 38, 129). This hints at the possibility of NOS2 expression being selective to certain cell types. Although there are many similarities between inflammation processes between species, NOS2 regulation is fundamentally different. It was shown that human NOS2 stability was proteasome dependent (112). An article by Foster et al. described an interactome of NOS2 in A549 cells (49). They demonstrated that cytokine stimulation resulted in a multiprotein network with allosteric activators and increase in ubiquitin through ubiquitin ligase FBX045. Notably, they did not observe important chaperone proteins such as HSP70, and others described in murine macrophages suggesting that the NOS2 interactome is different between mice and humans. Another important aspect of NOS2 in humans is the identification of different splice variants (44, 161), although several of these have been found to be nonfunctional.
Assembly of NOS2
The assembly of NOS2 to an active enzyme requires many processes that influence activity beyond the assembly of protein. It has been shown that miR939 controls human NOS2 expression by binding to the 3-UTR (63). Human NOS2 mRNA stability is also post-transcriptionally regulated through TTP, KSRP, and HuR (RNA binding proteins) (102). In the assembly of NOS2, first the iron is inserted into heme by the mitochondrial aconitase-like protein ferro-chetolase. After this, the heme moiety is inserted into the NOS2 protein to complete its assembly. Although heme assembly and insertion are important, iron status is critical; heme consumption by heme oxygenase 1 (HO-1) to make bilirubin, iron, and CO can be important too. HO-1 can be induced by a variety of pathways such as Nrf2A and IL-10. High levels of NO can induce both IL-10 and Nrf2A. This would, in turn, cause a disruption of the activity of NOS2. GAPDH has also been found to be important for heme insertion into the protein (23). This suggests a link between glycolysis activity and NOS2 active protein. Another important aspect is that the turnover in human cells is 1.6 half-life (90). Several articles suggest that RNS intermediates such as HNO can damage the protein and this requires a quick turnover (91, 124). Thus, sustained NOS2 production can be controlled by turnover and thus has a relatively quick response.
Enzymatic activity
The enzyme activity is critical in determining the rate of reaction, and proper dimerization is a major factor in the control of activity. The formation of the dimer tail to head configuration allows each monomer to provide the electron to the heme domain through the protein. Modification of this structure dramatically alters the activity of the enzyme (99). Arginine, the substrate, and THBP both contribute to the structure (153). It has been shown that deficiency of either leads to ROS production rather than NO similar to other P450 reductases (142). This is an interesting paradigm in that NO and ROS reflect the status of one another. Also, lower O2 has an effect and can even generate low ROS rather NO with NOS3. The Km for O2 is 70 μM and thus provides a range activity that is linear in the physiological O2 range (145). This also suggests that location in hydrophobic versus hydrophilic environments will have effects on NOS2 activity.
There are several factors that influence the NO levels beyond NOS2. These include cellular fueling as well as consumption by iron complexes and ROS. This tight control of NO from formation to scavenging leads to a tight spatial and temporal control on its activity and its ability to rapidly respond to its environment. One regulatory system that influences the fuel is arginine availability, which is maintained by uptake through the heavily regulated cationic amino acid transporters and metabolism by arginase (30, 110). Like other substrates, arginine uptake is limited through this. Arginine can be recycled from citrulline through ASS1 and ASL, which limit NOS activity. However, the largest regulator of arginine availability is arginase that converts arginine to ornithine, which is then committed to polyamine synthesis upon conversion to putrescine by ODC. Putrescine is subsequently converted to spermidine and spermine. ROS is formed in the conversion of spermidine to spermine through polyamine oxidase and this ROS could, in turn, reduce NO fluxes. Thus, arginase/polyamine synthesis limits the NOS2 activity and NO levels. In contrast, NOHA, a product of NOS2, leads to inhibition of arginase (20). Higher levels of NO lead to ODC inhibition through modification of its thiol moieties (11). At these levels of NO, mitochondrial TCA is also inhibited. Therefore, there is an important relationship between arginase and NOS2. Notably, low arginine leads to increased ROS rather than NO from NOS2 and this may present a novel switch in signaling (145).
Examination of these pathways indicates that they represent two different states of the cell. On one hand, increased arginase and polyamines are associated with increased proliferation, whereas on the other hand, NO, through signaling, leads to increased cancer “stemness”, immunosuppression, angiogenesis, and drug resistance. This may suggest that NO and arginase/polyamine pathway are important targets for two distinct populations of cancer cells within the tumor, one population that is more proliferative (arginase high/ODC high) and the other that is more quiescent, immunosuppressive, and harboring an increased metastatic potential (NO high).
NO and Immune Modulation
The communication between the tumor and the immune system is important in all stages of cancer from angiogenesis to drug resistance and radiation resistance and a broad range of NO flux plays cardinal roles in this crosstalk. The dichotomy in NO effects that is seen in cancer cells also applies to the immune system. In conventional radiation therapy, NOS2 induction produces NO that then activates IL10, leading to formation of an immunosuppressed microenvironment (126). Nevertheless, antitumor effects of tumor-associated macrophages (TAMs) are also largely regulated by NOS2 expression and associated NO fluxes (46, 114). Hence, there is a need to delineate this dichotomy. Several immunotherapies such as those using IL12 and IL2 or IL2 and anti-CD40 antibody act by inducing NOS2 expression in TAMs and converting them into more proinflammatory/antitumorigenic macrophages (171, 173). IL2/anti-CD40 therapy activates another arm of the immune system by activating IFNγ, which, in turn, modulates TIMP1 activity to reduce MMPs and control metastatic spread in an NO-dependent mechanism (128). NOS2 expression in tumor cells and associated cells in the microenvironment has been found to be the source of NO in murine models. However, the source NO in humans is still unclear.
Involvement of NO/cGMP-dependent immunogenic effects through increased VEGF results in neoangiogenesis. NO/cGMP can directly affect the tumor-associated immune cells favoring an anti-inflammatory phenotype that inhibits tumor clearance. While combating infections, mice produce high NO fluxes due to continuous NOS2 activation in myeloid cells. However, this chronic expression of NOS2 can induce immune suppressive effects. Research over the years has tied L-arginine metabolism in the myeloid cells to immune competence of T cells (15, 47). This hints at a direct, NO-mediated crosstalk between macrophages and T cells that could potentially regulate cancer therapy response. A mechanism that has been described for this involves sGC-mediated increase in tumor-infiltrating myeloid-derived suppressor cells that activate immune escape pathways within the tumor by inhibiting the infiltration and activation of cytotoxic T cells (135).
A major component of immune response to therapy is efficient recruitment of leukocytes to the tumor. In the early 1990s, it was found that leukocyte recruitment via the interaction with endothelial cells (of the aberrant vasculature) was absent in cancerous conditions, which, in part, can account for the immune escape of cancer (89, 183, 184). Radiation therapy releases inflammatory mediators that, in turn, favor the binding of neutrophils to endothelial cells, whereas low-dose γ irradiation in pancreatic cancer induces the differentiation of NOS2+ macrophages in the tumor microenvironment, which subsequently activate the endothelium and cause cytotoxic T cell infiltration (89, 184). A later study showed that endothelial activation is tightly regulated by the local intracellular NO flux (132).
Conclusions
NOS induction has multiple consequences ranging from induction of cell proliferation and Warburg effect at low doses (level I) to cell death and prolonged mitochondrial depolarization at high doses (levels III and IV). We have put the effects on signaling and metabolism into context with regard to flux and duration of NO persistence to better understand how current immunotherapies can be made more effective.
Footnotes
Acknowledgments
This research was supported, in part, by the Intramural Research Program of the NIH, Cancer and Inflammation Program. This project was funded in whole or, in part, with Federal funds from the National Cancer Institute, NIH, under Contract HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.