
Abstract
Aims:
Remote ischemic conditioning (RIC) protects against organ ischemia/reperfusion injury in experimental and clinical settings. We have demonstrated that RIC prevents liver and lung inflammation/injury after hemorrhagic shock/resuscitation (S/R). In this study, we used a murine model of S/R to investigate the role of nuclear factor (erythroid-derived 2)-like 2 (Nrf2) in mediating hepatoprotection.
Results:
The combination of RIC with S/R caused a synergistic rise in Nrf2 and its translocation to the nucleus in the liver. Increased activation of Nrf2 by RIC augmented heme oxygenase-1 (HO-1) and autophagy and exerted hepatoprotection, concurrent with reductions in S/R-induced TNF-α (tumor necrosis factor alpha) and IL-6 (interleukin-6). In Nrf2 knockout (KO) animals, RIC did not exert hepatoprotection, and it failed to upregulate HO-1 and autophagy. Further, resuscitating wildtype (WT) animals with blood from donor WT animals undergoing RIC was hepatoprotective, but not in Nrf2 KO recipient animals. Interestingly, RIC blood from Nrf2 KO donor animals was also not protective when used to resuscitate WT animals, suggesting a role for Nrf2 both in the afferent arm of RIC where protective factors are generated and also in the efferent arm where organ protection is exerted. Finally, RIC plasma prevented oxidant-induced zebrafish mortality, but not in Nrf2a morpholino knockdown fish.
Innovation:
Activation of Nrf2 is an essential mechanism underlying the hepatoprotective effects of RIC. Nrf2 appears to play a role in the afferent limb of RIC protection, as its absence precludes the generation of the protective humoral factors induced by RIC.
Conclusion:
Our studies demonstrate the critical role of Nrf2 in the ability of RIC to prevent organ injury after S/R.
Introduction
I
Remote ischemic conditioning (RIC) is a clinical intervention shown to lessen distant organ injury after ischemia/reperfusion. Our studies demonstrate an essential role for activation of nuclear factor (erythroid-derived 2)-like 2 (Nrf2) as an upstream effector of protection through its role as an inducer of the antioxidant response. In addition, the blood derived from animals that have undergone RIC is hepatoprotective when used to resuscitate animals undergoing shock/resuscitation. Interestingly, RIC blood from Nrf2 knockouts was not protective in wildtype animals, suggesting an added role for Nrf2 in the generation of protective factors by RIC. Transfusing ischemic conditioned blood may represent a novel approach to resuscitating trauma patients.
Using a murine model of S/R, we demonstrated that RIC exerted a protective effect on S/R-induced liver and lung injury (23), even when applied during the resuscitation phase of S/R. In a zebrafish model of neutrophilic inflammation, we also showed that plasma derived from mice after an RIC intervention reduced the number of neutrophils migrating toward the site of a tailfin cut injury, suggesting the transfer of a humoral factor that either directly or indirectly exerted anti-inflammatory effects in the recipient animal (23).
In various models of organ ischemia/reperfusion, the upregulation of antioxidative proteins, including superoxide dismutase, heme oxygenase-1 (HO-1), and NAD(P)H quinone dehydrogenase by RIC, has been associated with protection against organ injury (12, 21, 26). In liver ischemia/reperfusion, HO-1-induced autophagy after RIC has been shown to mediate hepatoprotection (38). The mechanisms of action of RIC have been less well studied in the context of S/R, despite S/R inducing ischemia/reperfusion in multiple organs. However, the report by Jan et al. complemented our studies by showing that HO-1 mediated the protective effects of RIC against S/R-induced lung injury (18).
A common feature of gene transcription in antioxidant genes is the presence of an antioxidant response element (ARE) in the cis-region of the promoter. The transcription factor, nuclear factor (erythroid-derived 2)-like 2 (Nrf2), binds to this ARE and is known to be important for the induction of a number antioxidant genes that are upregulated in response to oxidative stress (27). Nrf2 is controlled primarily at the level of protein stability. Under basal conditions, Nrf2 is sequestered in the cytosol by its interaction with Kelch-like ECH-associated protein 1 (Keap1), an adaptor for the Cullin 3 ubiquitin ligase, which together target Nrf2 for ubiquitination and proteasomal degradation (41). On exposure to oxidative stress, Nrf2 dissociates from Keap1, which allows Nrf2 to translocate to the nucleus, to bind to the ARE, and to promote transcription of antioxidant genes. In addition, Nrf2 contains multiple serine, threonine, and tyrosine residues that can be phosphorylated by protein kinase C, AKT, and ERK1/2, further promoting its stabilization and nuclear translocation (1). Supporting a potential physiological contribution of Nrf2, pharmacological induction of Nrf2 has been shown to diminish hepatic ischemia/reperfusion injury through upregulation of antioxidant molecules (20).
Given the importance of Nrf2 in promoting the antioxidant response, we hypothesized that RIC prevents S/R-induced organ injury and inflammation through induction/activation of Nrf2 and downstream antioxidant genes. In this article, we showed that RIC increased the nuclear translocation of Nrf2 in the liver after S/R and contributed to hepatoprotection through the induction of HO-1 expression and augmentation of autophagy. The key role of Nrf2 in this effect was further demonstrated by the finding that RIC blood, taken from donor animals undergoing the RIC intervention, was hepatoprotective when used to resuscitate shocked wildtype (WT) animals but not Nrf2 knockout (KO) animals. Interestingly, blood derived from Nrf2 KO animals undergoing RIC was unable to exert protection when used to resuscitate WT animals undergoing S/R, suggesting an additional role for Nrf2 in the elaboration of the protective humoral factor produced by RIC. These studies position Nrf2 as a central element of a positive feedback loop leading to the protective effect of RIC and also suggest alternative targets for improving organ protection after S/R.
Results
Hepatoprotection and activation of the HO-1-autophagy pathway by RIC
S/R (protocol illustrated in Fig. 1A) resulted in hepatocellular injury, as indicated by a markedly elevated level of plasma alanine aminotransferase (ALT) (Fig. 2A), increased tissue levels of malondialdehyde as a measure of lipid peroxidation (Fig. 2B), and the presence of hepatocellular necrosis in histological sections (Fig. 2C). As previously reported, RIC reduced hepatocellular injury caused by S/R, as evidenced by reduced ALT release, lipid peroxidation, and necrosis (Fig. 2A–C, respectively).

Given the previously described role of the HO-1-autophagy pathway in exerting hepatoprotection in liver ischemia/reperfusion injury (38), we evaluated the levels of liver HO-1 protein expression in animals subjected to S/R ± RIC. As illustrated in Figure 2D, S/R alone caused a rise in HO-1 expression that was further significantly increased by antecedent RIC. This pattern mirrored that shown for HO-1 mRNA levels, suggesting that regulation occurred at the transcriptional level (Fig. 2E). We then explored changes in autophagy induced by RIC in animals undergoing S/R. As shown in Figure 2F, the ratio of microtubule-associated protein 1A/1B-light chain 3 (LC3)-II to LC3-I, an indicator of autophagic vesicle formation, was lower after S/R than in sham animals, although not reaching statistical significance. However, the LC3-II/LC3-I ratio was significantly increased in RIC plus S/R animals. Further, levels of the adaptor molecule p62, whose levels are reduced through the autophagic process, were significantly reduced in RIC plus S/R animals (Fig. 2G), consistent with the conclusion that RIC augments autophagy in S/R animals. As an additional measure of autophagy, we also performed transmission electron microscopy and counted autophagosomes. Figure 2H shows representative micrographs illustrating the double-membrane vesicle containing cellular contents, characteristic of autophagosome formation. Blinded quantitation of autophagosomes and mitophagosomes showed that the number of autophagosomes and mitophagosomes were significantly increased in S/R plus RIC animals compared with S/R alone (Fig. 2H). Autophagy and mitophagy were infrequently observed in sham or RIC-alone livers. Therefore, similar to liver ischemia/reperfusion, our studies indicate that activation of the HO-1-autophagy pathway is involved in the protective effect of RIC in the setting of S/R.
Activation of Nrf2 by RIC
We then explored the upstream pathways leading to HO-1 activation by RIC. The promoter region for HO-1 contains an ARE that binds the transcription factor Nrf2. We examined whether RIC might influence Nrf2 levels and distribution in the cells. As shown in Figure 3A, S/R significantly increased Nrf2 protein expression in whole-cell lysates of control S/R animals, a rise that was significantly potentiated by antecedent RIC. To investigate the cellular distribution of Nrf2, we performed nuclear and cytosolic fractionation. These studies revealed that RIC in S/R animals not only increased total Nrf2 but also profoundly increased its presence in the nucleus, consistent with nuclear translocation (Fig. 3B). Representative blots are shown in the upper panel, whereas quantifications of Nrf2 in the cytosolic and nuclear fractions are shown in the lower panels, respectively. Since dissociation of Nrf2 from Keap1 is required for Nrf2 stabilization, we immunoprecipitated Nrf2 and then immunoblotted for Keap1 to discern the effect of S/R ± RIC on its association. In a representative blot, Figure 3C illustrates that Nrf2-Keap1 association was lessened after S/R, an effect that was further augmented by RIC. Under basal conditions, the association of Nrf2-Keap1 in the complex with the Cul3 ubiquitin ligase results in Nrf2 ubiquitination, targeting its degradation. S/R plus RIC caused a marked reduction in ubiquitination, consistent with the dissociation of Nrf2 from Keap1, reduced degradation, and an increase in cellular Nrf2 (Fig. 3D). This effect was present, although less profound with S/R alone.

Because the transcription of HO-1 is also regulated by the transcriptional repressor Bach1 (31), we performed immunoblots of nuclear and cytosolic fractions to determine whether S/R or RIC influenced Bach1 levels. As shown in Supplementary Figure S1, Bach1 levels were not significantly different between groups after S/R and RIC plus S/R. Together, these studies demonstrate that the HO-1 antioxidant response induced by S/R is a result of Nrf2 stabilization and RIC augmented this effect through a post-translational modification process.
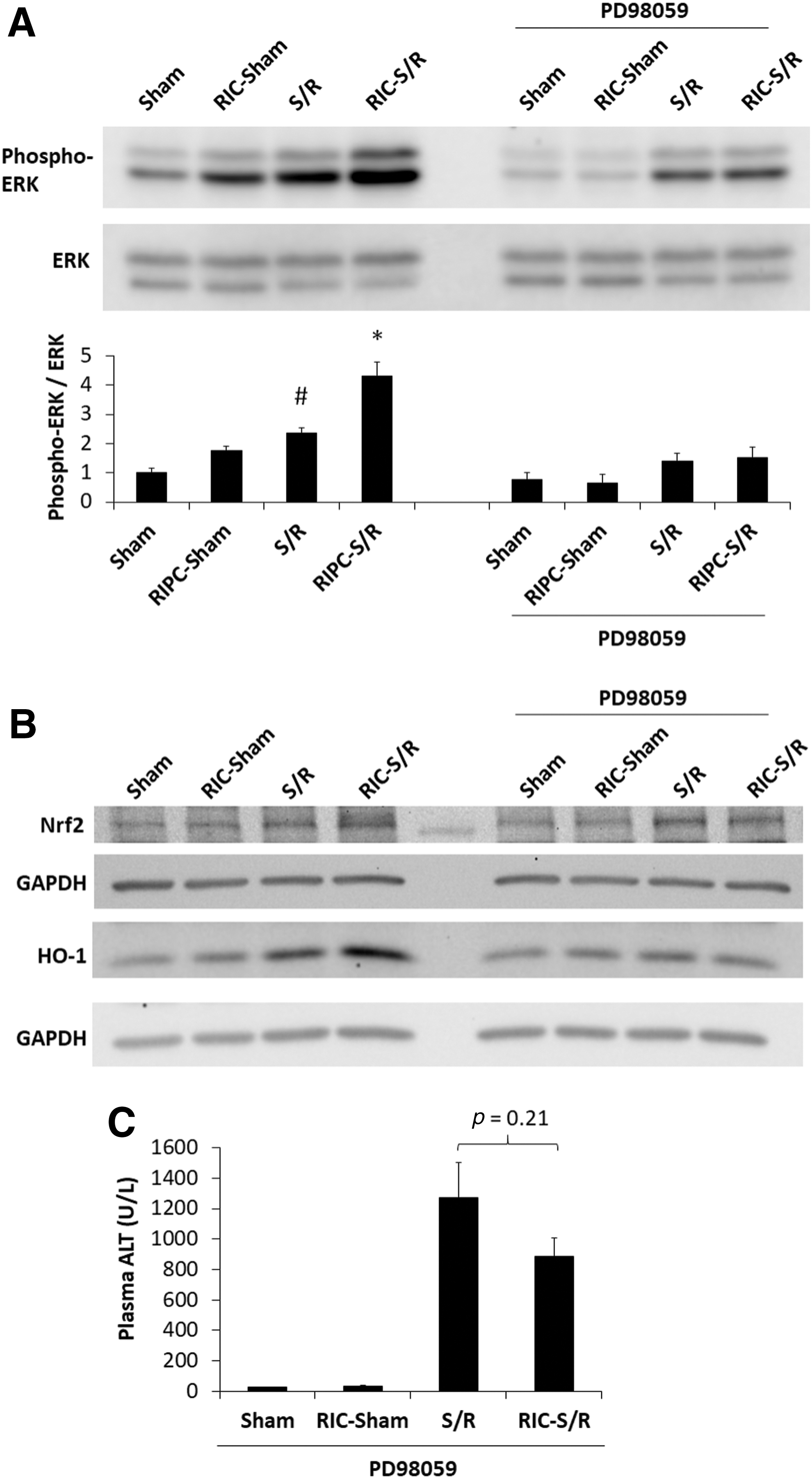
The mitogen-activated protein kinase ERK1/2-dependent phosphorylation of Nrf2 has been shown to contribute to its downstream effects on HO-1 expression (29, 40). We, therefore, evaluated the effect of RIC on ERK1/2 activation and its impact on Nrf2. As shown in Figure 4A, S/R increased ERK1/2 phosphorylation and this effect was augmented with RIC plus S/R. To examine the contribution of ERK1/2 phosphorylation on downstream events, the MEK inhibitor PD98059 was administered to animals. Figure 4A shows that PD98059 administration before RIC precluded the additional increase in phospho-ERK1/2 in RIC plus S/R animals compared with S/R animals. In the presence of the ERK1/2 inhibitor, the rise in Nrf2 and HO-1 levels observed with RIC plus S/R was prevented (Fig. 4B). Further, this agent reversed the hepatocellular protection exerted by RIC (Fig. 4C, S/R vs. RIC-S/R: not significantly different) compared with untreated animals, as shown in Figure 2A.
RIC attenuates liver injury and inflammation: role of Nrf2
The results cited earlier suggest a potential role for the Nrf2-HO-1 axis in the hepatoprotective effect of RIC on S/R-induced liver injury. To evaluate the role of Nrf2 in this pathway, we studied the impact of RIC on Nrf2 KO animals. As illustrated in Figure 5A, S/R alone caused comparable liver injury in both WT animals and Nrf2 KO animals (compare left histogram with right histogram). However, RIC again significantly lessened S/R-induced liver injury in WT animals, whereas the hepatoprotection exerted by RIC after S/R was abolished in Nrf2 KO animals. Rather, injury was exacerbated in Nrf2 KO animals by the addition of the RIC intervention to S/R. Similarly, systemic inflammation associated with S/R, as shown by a rise in the plasma levels of tumor necrosis factor alpha (TNF-α) (Fig. 5B) and interleukin (IL)-6 (Fig. 5C) in WT and Nrf2 KO animals, was prevented by RIC in WT animals, but not in Nrf2 KO animals. Conversely, the levels of these cytokines were augmented by the addition of RIC to S/R in Nrf2 KO animals. The rise in pro-inflammatory cytokines after S/R was mirrored by a significant increase in the plasma levels of the anti-inflammatory cytokine IL-10 (Fig. 5D). This increase in IL-10 in S/R animals, however, was prevented in WT animals receiving RIC but not in Nrf2 KO animals. As TNF-α can directly stimulate the production of IL-10 in monocytes (39), the reduction in IL-10 levels observed in WT RIC animals is likely a result of decreased production due to lower TNF-α levels.

Finally, the induction of the HO-1-autophagy pathway by RIC did not occur in Nrf2 KO animals. As shown in Figure 5E, in Nrf2 KO animals, RIC plus S/R did not augment the expression of HO-1 above that observed in S/R alone. This is in contrast to that observed in WT animals. Similarly, autophagy was not augmented by RIC in Nrf2 KO animals undergoing S/R. Unlike the increase in the LC3-II/LC3-I ratio induced by RIC plus S/R in WT animals, this ratio was not increased by RIC in Nrf2 KO animals sustaining S/R (Fig. 5F). Further, the reduction in p62 by RIC that was observed in WT animals was not evident in Nrf2 KO animals (Fig. 5G). Taken together, these data indicate a central role for Nrf2 and downstream HO-1-autophagy in mediating hepatoprotection exerted by RIC after S/R.
Resuscitation with RIC whole blood attenuates liver injury
Our previous work showed that plasma from animals undergoing the RIC protocol exerted an anti-inflammatory effect on neutrophil function when injected into zebrafish embryos, suggesting that RIC elaborates a protective humoral factor (23). We sought to investigate whether there was also transferrable protection when blood derived from animals undergoing RIC was used to resuscitate animals subjected to S/R. As shown in Figure 6A, transfusion with RIC blood derived from WT RIC animals into WT animals undergoing S/R significantly reduced plasma ALT levels compared with S/R animals resuscitated with Control Blood derived from WT control animals. In addition, resuscitation with RIC blood significantly attenuated the S/R-induced rise in IL-6 levels (Fig. 6B) and, to a lesser extent, reduced plasma TNF-α levels compared with Control Blood resuscitated animals (Fig. 6C). The significant rise in plasma IL-10 levels induced by S/R was also prevented by resuscitation with RIC blood (Fig. 6D).
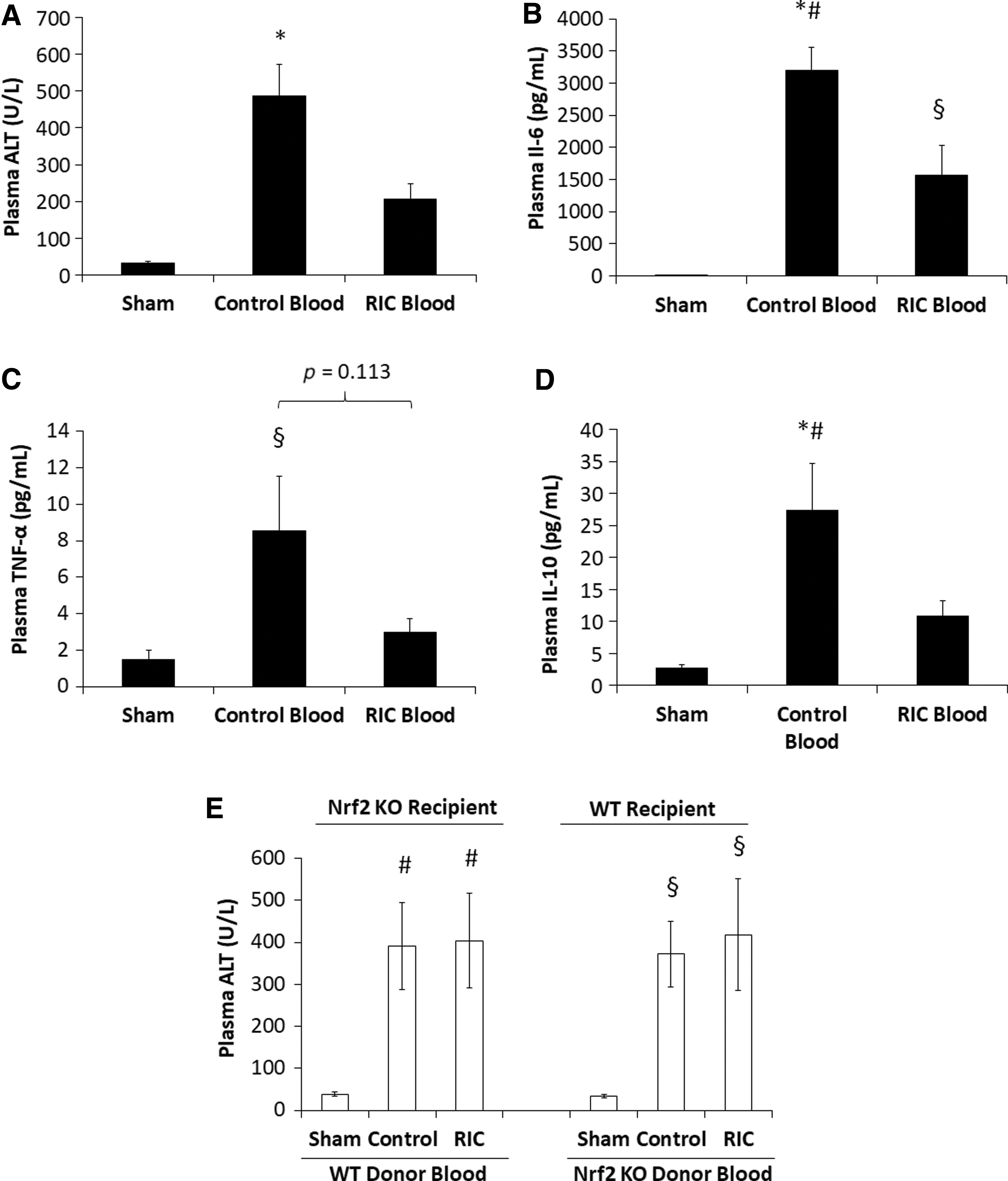
To determine whether Nrf2 is required in the recipient animal to induce transfer of hepatoprotection, Nrf2 KO animals subjected to S/R were resuscitated with blood taken from WT RIC donor animals. As shown in Figure 6E (left histogram), the protection exerted by WT RIC blood was not evident when used to resuscitate Nrf2 KO animals subjected to S/R. Considered in aggregate, these studies suggest that a transferrable humoral factor generated by the RIC protocol is able to induce protection in S/R animals in an Nrf2-dependent manner. We also asked whether Nrf2 was necessary to induce the protective propensity of RIC blood. To do so, the RIC protocol was performed in Nrf2 KO animals and the blood from these animals was used to resuscitate WT animals subjected to S/R. Interestingly, hepatoprotection was not observed by using this protocol (Fig. 6E, right histogram). These studies, therefore, suggest that Nrf2 is required both to generate the protective factor(s) by RIC and to exert the protective effect at the remote organ after transfusion of S/R animals.
RIC plasma improves survival in H2O2-treated zebrafish
In zebrafish, oxidant-mediated inflammation leading to mortality can be studied by exposing animals to exogenous H2O2. To evaluate whether plasma derived from RIC blood protects against oxidant-induced mortality, 3-day-old zebrafish larvae were microinjected into the common cardinal vein with control or RIC plasma and subjected to 2 mM H2O2 treatment. WT zebrafish pretreated with RIC plasma demonstrated significantly improved survival by day 8 (35%) as compared with zebrafish pretreated with control plasma (0%), vehicle (3%), or noninjected control (0%) (Fig. 7A, p < 0.01 by Log-Rank test). To study the role of Nrf2 in this protection, we knocked down Nrf2a in zebrafish by morpholino. As shown in Figure 7B, zebrafish injected with scrambled morpholinos were again protected by an injection with RIC plasma (solid lines—p < 0.01 by Log-Rank test). Knockdown of Nrf2a, in itself, resulted in earlier mortality compared with scrambled morpholino zebrafish exposed to exogenous H2O2 (dotted lines). As observed in the murine model, the protective effect of RIC Plasma was abolished in Nrf2a knockdown recipients. Finally, RIC plasma derived from Nrf2 KO animals did not exert protection in WT zebrafish against oxidant stress (Fig. 7C).

Discussion
This study provides significant new insights into understanding how RIC exerts its protective effect in a murine model of S/R. The central finding is that RIC exerts hepatoprotection through the activation of the transcription factor Nrf2. Specifically, RIC increases the expression of the transcription factor Nrf2 in the liver and in addition, promotes its nuclear translocation where it induces the upregulation of the cytoprotective molecule HO-1 and autophagy. ERK1/2-dependent phosphorylation also appears to contribute to this effect. In the absence of Nrf2, this cascade of events does not occur and the effect of RIC on downstream mediators and on hepatoprotection is lost. Using blood transfer experiments, we further demonstrate that Nrf2 is necessary for the hepatoprotection in the recipient animal, such that protection occurs in WT but not in Nrf2 KO animals. In support of these findings, we show that plasma derived from RIC animals is able to prevent zebrafish mortality in an oxidative stress model in animals that are replete in Nrf2, but not in morpholino Nrf2a knockdown animals. Although others have shown that RIC exerts protection against ischemia/reperfusion injury by increasing HO-1 and autophagy, our findings clearly define the upstream signaling molecule Nrf2 as being essential to this protection. Importantly, these are likely relevant to other ischemia/reperfusion models where RIC has been shown to be protective and may be targets for intervention.
The role of Nrf2 in ischemic conditioning has received limited study, demonstrating only an association with protection in models of renal ischemia/reperfusion (17), retinal ischemia/reperfusion injury (42), and cerebral ischemia/reperfusion injury (26), but no causal relationship. Our studies are unique in that we use Nrf2 KO animals and zebrafish to clearly demonstrate that activation of Nrf2 is a critical element in exerting the protection by RIC. We also show that, in conditioned animals, Nrf2 dissociates from Keap1, resulting in reduced ubiquitination/degradation, allowing it to translocate to the nucleus to exert its effects. In addition, RIC-induced activation of ERK1/2 appears to participate in Nrf2 stabilization. Phosphorylation of Nrf2 by ERK1/2 has also been shown to contribute to Nrf2 translocation to the nucleus (44). Our studies support the notion that HO-1 as being a downstream mediator of Nrf2 as Nrf2 KO animals exhibited reduced activation of HO-1 and hepatoprotection. Wang et al. have implicated HO-1-induced autophagy as being causally related to organ protection after RIC (38), although the specific type of autophagy was not defined.
One attractive possibility is that mitochondrial autophagy (mitophagy) is augmented by RIC. Cellular ischemia/reperfusion depolarizes mitochondria, causing mitochondrial dysfunction and augmented mitochondrial reactive oxygen species generation. Mitophagic clearance of these damaged organelles reduces reactive oxygen species production and promotes restoration of cellular homeostasis. In support of this hypothesis, a recent study has shown a potential role for mitophagy in mediating the protective effect of RIC in a model of cerebral ischemia/reperfusion injury (43). Further, Sun et al. demonstrated that mitophagy is involved in hepatoprotection against hemorrhagic shock at 4.5 h postresuscitation (34). We observed that RIC plus S/R induced a significant increase in the number of mitophagosomes at 2 h postresuscitation, suggesting that early induction of mitophagy by RIC may participate in organ protection against S/R. The precise role of mitophagy in RIC-induced protection requires further investigation.
The protective paradigm of RIC against ischemia/reperfusion injury can be considered, in aggregate, a hormesis effect whereby a small dose of reactive oxygen species generated from the limb undergoing RIC systemically protects against subsequent lethal ischemia/reperfusion injury in a remote organ. The upregulation of hepatic HO-1 in our RIC and S/R model suggests the involvement of an emerging hormetic system known as Vitagenes (4), which involves the induction of stress response proteins composed of heat shock proteins, including (HSP32 [HO-1], HSP60, and HSP70) thioredoxin, and sirtuins regulated by Nrf2 and heat shock factors to maintain redox homeostasis. These cytoprotective stress response proteins in concert protect the cell against redox damage under numerous biological processes, including aging (8) and neuroinflammation (5). We measured the expression of HSP60 and HSP70 in WT and Nrf2 KO animals and did not observe significant differences after S/R or RIC (Supplementary Fig. S2), suggesting that the Nrf2-HO-1 axis but not Heat Shock Factor/HSP60/HSP70 is involved in organ protection against S/R. The absence of RIC-induced hepatoprotection in Nrf2 KO animals supports the critical role of Nrf2 in the hormetic effect of RIC.
The present studies demonstrate that a humoral factor(s) exerts protection when blood/plasma is derived from animals undergoing the RIC protocol, further validating the hormetic effect of RIC. Dickson et al. presented the first evidence that whole blood taken after ischemic preconditioning in rabbits could be transfused to precondition another rabbit against myocardial ischemia/reperfusion injury (11). Since then, several candidate humoral factors elicited by RIC have been identified in the context of preventing myocardial ischemia/reperfusion injury. These include autocoids such as opioids (10), adenosine (24), and nitric oxide (30), cytokines including stromal-derived factor (9) and IL-10 (3), apolipoprotein A-1 (19), microRNA-144 (25), and metabolites including glycine (6) and kynurenic acid (6, 28). Changes in the plasma proteome after the RIC protocol have also been previously reported (14, 16). Although these papers revealed a broad range of potential effector molecules in the plasma, this study demonstrating that Nrf2 activation is required for the RIC-mediated hepatoprotection has suggested some interesting potential candidates.
Chen et al. recently reported that RIC induced the release of the myokine irisin into the blood from skeletal muscle of the treated limb and that irisin exerted protection to lungs subjected to ischemia/reperfusion injury (7). Interestingly, irisin has been shown to stimulate Nrf2 expression in A549 cells (33). We measured irisin in the plasma after RIC but did not observe any changes in its levels (Supplementary Fig. S3). Another potential candidate is nitrite, derived from the oxidation of the nitric oxide induced by limb ischemic conditioning. Rassaf et al. (30) reported that nitrite circulating to the heart after ischemic conditioning was reduced to nitric oxide in the myocardium where it exerted protection. Notably, administration of the nitric oxide donor has been demonstrated to induce the expression and translocation of Nrf2 and downstream HO-1 gene expression in vascular endothelium cells through ERK1/2 activation (2). In addition, nitrosative modification of Keap1 with nitric oxide donor allowed Nrf2 to stabilize (13). Our finding that RIC plasma derived from mice provides cross-species protection in the zebrafish suggests that these are highly conserved factors. Our findings are consistent with a study by Shimizu et al. (32) that demonstrated that RIC plasma taken from humans exerts cross-species protection in rabbit cardiomyocytes against ischemia/reperfusion injury.
A limitation from our zebrafish model is that Nrf2a knockdown resulted in higher sensitivity to oxidative stress and resulted in earlier mortality, which may preclude the beneficial effect of RIC. Nevertheless, this experiment is consistent with our murine model from which Nrf2 expression is required in the recipient animal to exert transfer of protection. Further studies are warranted to determine the molecules involved in activating the vitagene system and in the transferrable protection of RIC plasma. Using activation of the Nrf2-HO-1 axis as a readout for protection should help to elucidate potentially important effector molecules induced by RIC.
One unexpected observation was that plasma derived from Nrf2 KO animals undergoing RIC was not protective in either the murine S/R model or the zebrafish oxidative stress model. We have not investigated the mechanism underlying failed protection in this setting. Nrf2 regulates the expression of antioxidant proteins through its interaction with the ARE in the promoter of their genes. Since the RIC technique induces repeated cycles of ischemia/reperfusion in skeletal muscle in the hindlimb, it is likely that this stress may induce an antioxidant response in the muscles. In the absence of Nrf2, the antioxidant response would likely be reduced and the constituents of the RIC plasma would be altered. In support of this hypothesis, we observed that superoxide dismutase activity in the plasma is significantly increased after RIC but not in Nrf2 KO animals (Supplementary Fig. S4). Whether this alters the elaboration of the protective humoral factor or results in the release of molecule(s) that counteract the protective effect is unclear. For example, Vega et al. showed that rat hind limb ischemia/reperfusion caused release of xanthine oxidase into the plasma (37). Enhanced release might augment oxidative stress in the liver and preclude hepatoprotection by RIC. This unexpected yet intriguing finding warrants further study. In any case, our observations suggest that both the afferent and the efferent arms of RIC require Nrf2. Further, comparing RIC plasma derived from Nrf2-replete and Nrf2-deficient animals may represent an opportunity to better determine the key molecules elaborated by this intervention.
Our studies identify Nrf2 as a major participant in RIC-induced hepatoprotection after S/R. Given the demonstrated role of various antioxidants in mediating the beneficial effect of RIC in other models of ischemia/reperfusion injury, it is probable that this mechanism may be relevant to other diseases where RIC has been shown to be effective. It also identifies a potential target for activation, which may alone recapitulate the effects of RIC or when given in combination with RIC, may enhance its protective effects. Finally, although RIC is a clinically relevant and safe intervention, the protective effect of resuscitation with remote ischemic conditioned blood suggests a potentially novel approach to delivering this intervention.
Materials and Methods
Model of hemorrhagic shock/resuscitation
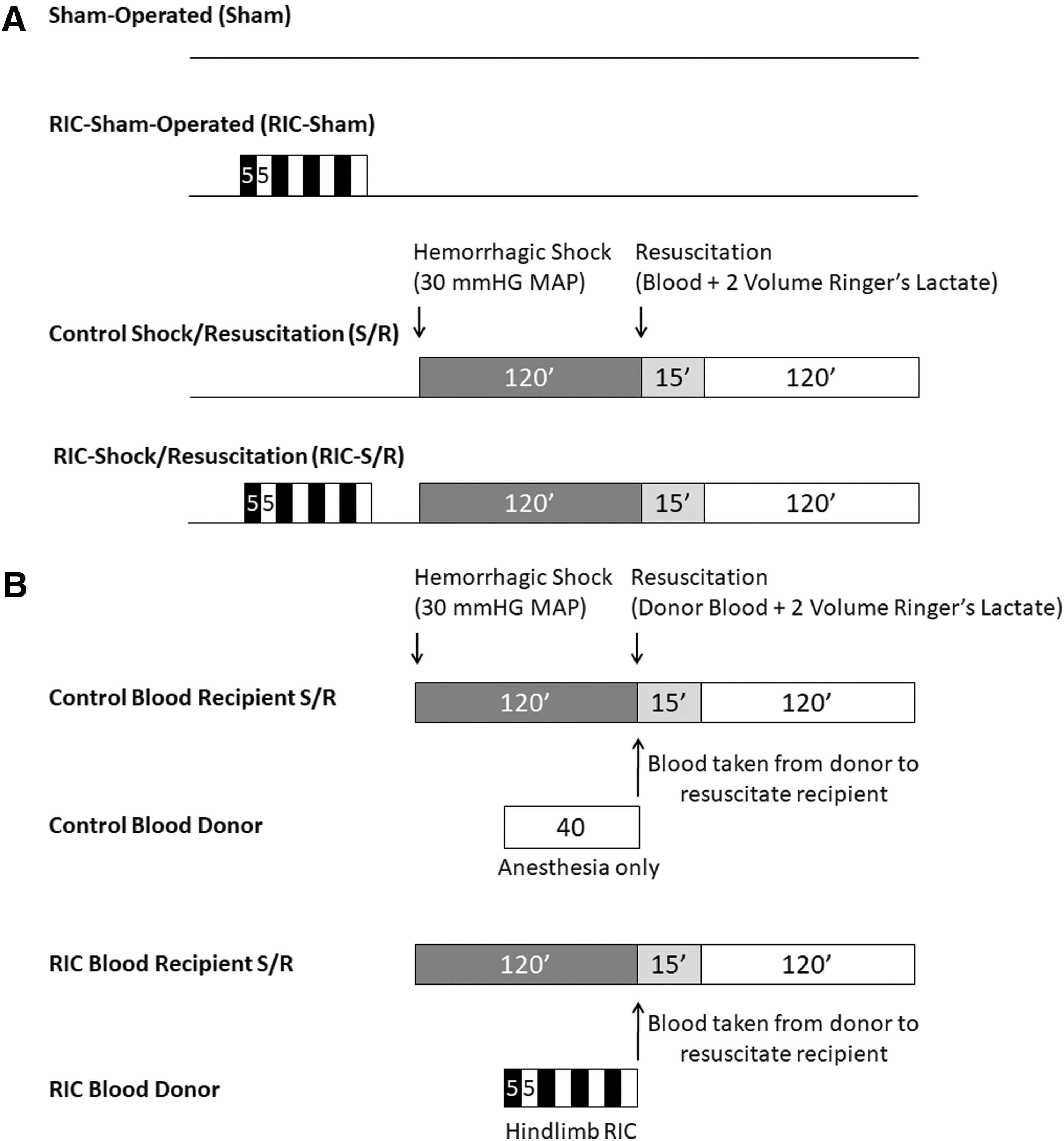
All animal studies including those in mice and zebrafish were approved by the Animal Care Committee of St. Michael's Hospital. Male WT C57BL/6J mice (Jackson Laboratories, Bar Harbor, ME) and Nrf2 KO mice (017009; Jackson Laboratories), 9–11 weeks old, were anesthetized with sodium pentobarbital (70 mg/kg), subjected to hemorrhagic shock at 30 mmHg mean arterial blood pressure for 2 h by controlled blood withdrawal through a 30-min period from the left femoral artery, and finally resuscitated with shed blood and Ringer's Lactate equal to two volumes of shed blood over 15 min. Mean arterial blood pressure was continuously monitored through the right carotid artery over this period. At 2 h after resuscitation, liver tissues were flash frozen in liquid nitrogen and blood samples were collected for analysis. Where indicated, RIC was performed before hemorrhage by occlusion of the left hindlimb by tightening a tourniquet for four cycles of 5-min ischemia and 5-min reperfusion. Control RIC animals underwent sham tightening of the tourniquet before hemorrhage. Sham-operated animals undwent anesthesia and operation but without S/R. In studies examining the role of Phospho-ERK1/2, MEK inhibitor (PD98059, 1 mg/kg) was administered by an intraperitoneal injection 20 min before initiation of RIC.
In studies evaluating the effect of resuscitation with remote ischemic conditioned blood on hepatoprotection (Fig. 1B), recipient mice (WT or Nrf2 KO) were subjected to the hemorrhagic shock protocol as described earlier and resuscitated with blood in an equal volume to shed blood taken from a donor mouse (either WT or Nrf2 KO) that had been subjected to either the protocol for RIC (RIC Blood) or sham conditioning (Control Blood), plus Ringer's Lactate equal to two volumes of shed blood over a 15-min period.
Plasma ALT
ALT levels as a marker of hepatocellular injury were measured in plasma samples at 2 h after resuscitation by the Diagnostic Laboratory at St. Michael's Hospital.
Thiobarbituric acid reactive substances assay
Liver tissue levels of malondialdehyde as a marker of lipid peroxidation were measured by using commercially available TBARS (thiobarbituric acid reactive substances) Assay kit (Cat No. KGE013; R&D Systems, Minneapolis, MN) and normalized to the total amount of protein assayed.
Liver histology
Liver tissues were fixed by immersion in 4% formaldehyde, embedded in paraffin wax, and cut into 5-μm slices. Sections were stained with hematoxylin and eosin and evaluated by light microscopy. The area of necrosis per five 10 × fields was quantified for each specimen in ImageJ.
Western blot analysis
Liver tissues were homogenized in lysis buffer containing: 10 mM NaCl, 30 mM HEPES, 20 mM NaF, 1 mM EGTA, 1% Triton X, 1 mM sodium orthovanadate, and complete protease inhibitor cocktail (Roche Diagnostics, Mannheim, Germany), and the supernatant was collected after centrifugation at 10,000 g for 10 min. Protein concentrations were determined by Bio-Rad DC Protein Assay (Bio-Rad, Hercules, CA). Forty micrograms of liver protein homogenates were separated via 8%, 10%, or 15% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to nitrocellulose or polyvinylidene fluoride membranes (Bio-Rad). The membranes were blocked at room temperature for 1 h and probed with primary antibody over night at 4°C followed by appropriate secondary antibodies conjugated with horseradish peroxidase. Protein loading was assessed and normalized with probing for glyceraldehyde 3-phosphate dehydrogenase (GAPDH) or corresponding nonphosphorylated protein. The blots were exposed to film or ChemiDoc Touch Imaging System (Bio-Rad) after enhanced chemiluminescence (Amersham Biosciences, Piscataway, NJ). Densitometry of the protein signals on film was analyzed by Quantity One software (Bio-Rad), and ChemiDoc Touch signals were analyzed with Image Lab software (Bio-Rad). Specificity of the Nrf2 band was confirmed in full length blot (Supplementary Fig. S5), demonstrating the absence of the Nrf2 band at ∼100 kDa in Nrf2 KO animals, consistent with previous validation by Lau et al. (22). To minimize nonspecific binding and antibody exhaustion, immunoblots were therefore cut above the 75 kDa molecular weight marker before probing with Nrf2 antibody.
Immunoprecipitation
One milligram of liver protein homogenates as prepared earlier was immunoprecipitated with anti-Nrf2 antibody (1:100 dilution) overnight at 4°C followed by incubation with 10 μL of Pierce Protein A/G Plus UltraLink Resin beads (Thermo Fisher Scientific, MA) for 3 h at 4°C, and it was washed five times in lysis buffer. A negative control consisting of liver protein homogenate immunoprecipitated with isotype-matched unimmunized Rabbit IgG was included to confirm the specificity of immunoprecipitation. The immunoprecipitated proteins were eluted by boiling in SDS sample buffer for 5 min at 95°C and subjected to immunoblot as described earlier. The membranes were probed with anti-Keap1 antibody (followed by light chain specific horseradish peroxidase coupled secondary antibody) or anti-ubiquitin antibody. Membranes were then probed with anti-Nrf2 antibody to examine the total amount of Nrf2 protein immunoprecipitated. Five percent of input controls were immunoblotted and probed with Nrf2, Keap1, and ubiquitin antibodies.
Nuclear/cytosolic fractionation
Nuclear and cytosolic fractions from frozen liver tissue were isolated by using CelLytic NuCLEAR Extraction Kit (Sigma-Aldrich, Oakville, Ontario, Canada) according to the manufacturer's protocol and subjected to immunoblot as described earlier. Purity and loading of the cytosolic and nuclear fractions were determined by probing for nuclear marker Lamin B and cytosolic marker β-Tubulin, respectively.
Real-time quantitative reverse transcription polymerase chain reaction
Total RNA from flash frozen liver tissue was extracted by using RNeasy Mini Kit (Qiagen, Valencia, CA). One microgram of RNA was then subjected to DNAse treatment and reverse transcription (Bio-Rad). The relative abundance of mRNA for HO-1 and GAPDH was quantified by using SYBR green-based quantitative polymerase chain reaction (PCR; Applied Biosystems, Foster City, CA). Each reaction was performed in triplicate. Real-time PCR conditions were as follows: initial denaturation at 95°C for 10 min, followed by 40 cycles of 95°C for 15 s, and 60°C for 60 s. Purity of the PCR reaction was verified by the meltcurve analysis at the completion of the PCR. Relative abundance of gene expression was calculated by the 2ΔCT method, using GAPDH as the endogenous control. All primers were purchased from Sigma-Aldrich.
Plasma TNF-α, IL-6, IL-10, and irisin enzyme-linked immunosorbent assay
Plasma levels of TNF-α, IL-6, and IL-10 were quantitated in plasma samples taken 2 h after resuscitation by using commercially available enzyme-linked immunosorbent assay kits according to the manufacturer's instructions (Cat. No. M6000B, SMTA00B, and M1000B; R&D Systems). Irisin levels were measured in plasma samples taken after sham or RIC procedure according to the manufacturer's instructions (Cat. No. EK-067-29; Phoenix Pharmaceuticals, Burlingame, CA).
Transmission electron microscopy
Liver tissues were cut into 2 mm3 and submerged in primary fixative containing 4% paraformaldehyde and 1% glutaraldehyde. The tissues were then postfixed in 1% Osmium Tetraoxide, dehydrated in graded ethanol series, and embedded in Epon resin. Ultrathin sections (80–100 nm) were cut and contrasted with 5% uranyl acetate and Reynold's lead citrate. Specimens were examined by using Hitachi H-7000 transmission electron microscope at 75 kV, and digital images were captured on an Advanced Microscopy Technology XR60 CCD camera. The images were evaluated by a blinded observer. The number of autophagosomes and mitophagosomes per five 100 μM 2 fields per specimen was measured.
Zebrafish H2O2 survival study
Control or RIC plasma was acquired from WT or Nrf2 KO mice subjected to anesthesia only or four cycles of 5-min hindlimb ischemia and 5-min reperfusion. WT or Nrf2a knockdown 3-days-postfertilization zebrafish larvae were microinjected with 2 nL of 5% Control Plasma, 5% RIC Plasma, or vehicle (saline) into the common cardinal vein and subjected to treatment with 2 mM H2O2 at 1 h postinjection. Mortality was assessed by ventricular asystole. Knockdown of Nrf2a in zebrafish embryos was performed by Nrf2a morpholino (5′-CATTTCAATCTCCATCATGTCTCAG-3′) injection at the one-cell stage as described (35). Control morpholino (5′-CCTCTTACCTCAGTTACAATTTATA-3′) injection was also performed. Noninjected controls were included to control for positional and plate effects.
Reagents and antibodies
Chemicals and reagents were purchased from Sigma-Aldrich. Antibodies used for immunoblot and immunoprecipitation were purchased from Cell Signaling Technology (Danvers, MA): Phospho-Erk1/2 (#9101), Erk1/2 (#9102), GAPDH (#5174), Keap1 (#4617), Nrf2 (#12721), β-Tubulin (#2128), p62 (#5114), Abcam (Cambridge, MA): HO-1 (ab13243), Bach1 (ab124919), nanoTools (Teningen, Germany): LC3 (0260-100), Enzo Life Sciences (Farmingdale, NY): HSP60 (ADI-SPA-806), Thermo Fisher Scientific (Waltham, MA): HSP70 (MA3-007) and Santa Cruz (Dallas, TX): Lamin B (SC-374015), ubiquitin (SC-8017). Horseradish peroxidase-conjugated secondary antibodies were purchased from Jackson ImmunoResearch Laboratories (West Grove, PA): Donkey Anti-Mouse IgG (715-035-150), Donkey Anti-Rabbit IgG (711-035-152), and Light Chain Specific Mouse Anti-Rabbit IgG (211-032-171).
Statistics
Data are expressed as mean ± standard error of n independent studies. Pairwise comparisons were performed by t-test. Comparisons between multiple groups were performed by ANOVA followed by Tukey's post hoc test for pairwise comparisons. p-Value of <0.05 was considered significant. Comparisons between groups in zebrafish survival studies were performed by log-rank Mantel-Cox test.
Footnotes
Acknowledgments
This work was supported by the Keenan Chair to O.D.R., the Robert Salter Chair in Surgery to C.A.C., the Physicians' Services Incorporated Foundation, and the Canadian Institutes for Health Research and by funds from MITACS.
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.