
Abstract
Aims:
Pulmonary fibrosis (PF) is characterized by myofibroblast activation through oxidative stress. However, the precise regulation of myofibroblast transdifferentiation remains largely uncharacterized.
Results:
In this study, we found that tanshinone IIA (Tan-IIA), an active component in the root of Salvia miltiorrhiza Bunge, can suppress reactive oxygen species (ROS)-mediated activation of myofibroblast and reduce extracellular matrix deposition in bleomycin (BLM)-challenged mice through the regulation of nuclear factor-erythroid 2-related factor 2 (Nrf2). Additionally, Tan-IIA restored redox homeostasis by upregulating Nrf2 with NADPH oxidase 4 suppression and effectively prevented myofibroblast activation by blocking ROS-mediated protein kinase C delta (PKCδ)/Smad3 signaling. Nrf2 knockdown in the fibroblasts and the lungs of BLM-treated mice reduced the inhibitory effects of Tan-IIA, indicating the essential role of Nrf2 in the Tan-IIA activity. Tan-IIA impaired the binding of kelch-like ECH-associated protein 1 (Keap1) to Nrf2 by promoting the degradation of Keap1 and thereby increasing Nrf2 induction by protecting Nrf2 stability against ubiquitination and proteasomal degradation. Importantly, we also found that the glutamate anaplerotic pathway was involved in energy generation and biosynthesis in activated myofibroblasts and their proliferation. Tan-IIA shunted glutaminolysis into glutathione (GSH) production by activating Nrf2, resulting in the reduction of glutamate availability for tricarboxylic acid cycle. Ultimately, myofibroblast activation was prevented by impairing cell proliferation.
Innovation and Conclusion:
In addition to the regulation of redox homeostasis, our work showed that Tan-IIA activated Nrf2/GSH signaling pathway to limit glutaminolysis in myofibroblast proliferation, which provided further insight into the critical function of Nrf2 in PF.
Introduction
Pulmonary fibrosis (PF) is characterized by a progressive and irreversible decline in the lung function due to the excessive deposition of collagen in the pulmonary interstitium (26). While inflammation is considered the root cause for initiating lung tissue scarring (9), myofibroblast activation is critical in the pulmonary fibrogenic responses (15, 21). Under pathological conditions, fibroblasts accumulate in the areas of damage and differentiate into activated myofibroblasts, capable of secreting collagen and other proteins, which are involved in the connective tissue synthesis and remodeling (31).
Several lines of evidence demonstrate that the association of oxidative stress with inflammation is a driving force of myofibroblast activation (12, 33). Excessive reactive oxygen species (ROS) generation and persistent redox imbalance can promote sustained fibroblast activation and increase resistance to apoptosis (11). As the primary enzymatic source of extracellular ROS production, only NADPH oxidase 4 (Nox4) is shown to modulate myofibroblast activation in response to the transforming growth factor beta 1 (TGF-β1) (12, 14, 27). Accumulated evidence indicates that the Nox-dependent redox signaling in the profibrotic responses is largely mediated through TGF-β1/Smad3 signaling (14, 13).
Nuclear factor-erythroid 2-related factor 2 (Nrf2) and NADPH oxidase 4 (Nox4) imbalance contributes to fibrotic responses in pulmonary fibrosis (PF). Tanshinone IIA (Tan-IIA) inhibited myofibroblast activation by protecting Nrf2 stability against degradation and suppressing Nox4 via interfering with transforming growth factor beta 1 (TGF-β1)/Smad3 signaling. Tan-IIA shunted glutaminolysis into glutathione production by activating Nrf2, resulting in the reduction of glutamate availability for the tricarboxylic acid cycle. Ultimately, myofibroblast activation was prevented by impairing cell proliferation. These results presented another mechanism for restraining myofibroblast transdifferentiation and suggested that Nrf2 pharmacological agonist combined with glutaminase inhibitor might be a potential therapeutic strategy for combating PF.
As a downstream mediator in the TGF-β1-induced profibrotic responses, Nox4-induced ROS formation elicits the conversion of the latent form of TGF-β1 to its active form, which emerges in a feed-forward manner (17). Therefore, an increasing amount of alpha smooth muscle actin (α-SMA), fibronectin, and other extracellular matrix (ECM) proteins induced by TGF-β1 eventually give rise to PF in the process (23, 24).
In response to oxidative stress, the antioxidant system initiates a series of programs to restrain excessive ROS generation. The role of nuclear factor-erythroid 2-related factor 2 (Nrf2) has been characterized as a crucial transcription factor that protects cells against oxidants (18). Nrf2 binds to antioxidant response elements and stimulates the transcription of the antioxidant proteins, as well as proteins involved in glutathione (GSH) biosynthesis and regeneration (20, 22). Recent studies showed that Nrf2 increases mitochondrial biogenesis (4), and the loss of redox homeostasis promotes profibrotic myofibroblast phenotypes in the lung of aged mice (11), indicative of the role of redox balance in control of cell growth. In fact, myofibroblast activation is an initial cause of fibrotic response, and the proliferation and differentiation are required for myofibroblast activation.
Furthermore, the critical role of oxidative stress-related glutaminolysis in organismal homeostasis and myofibroblast differentiation has been recently recognized (3). As a source of carbon and nitrogen for biomass accumulation, more glutamine is required in rapidly growing cells to support energy generation and biosynthesis (1). For instance, Nrf2 activation is shown to promote GSH synthesis in cancer cells, and this process increases the consumption of glutaminolysis-derived glutamate, thereby limiting glutamate availability for tricarboxylic acid (TCA) cycle and other biosynthetic reactions (1, 6, 28). It is likely that rapidly growing fibroblasts have a high demand for nutrients to support the enhanced proliferation in response to TGF-β1 stimulation.
Although the dependence on glutamine metabolism is mainly observed in cancer cells and activated lymphocytes (1), it is possible that Nrf2 activation-increased GSH production can inhibit myofibroblast activation by limiting glutamate availability for cell growth.
Tanshinone IIA (Tan-IIA) is an active component from Chinese herbal medicine Danshen, the dried root of Salvia miltiorrhiza Bunge, with the ability to suppress fibrotic responses (10, 36) and inhibit TGF-β1-dependent epithelial to mesenchymal transition in PF (30). Tan-IIA activated Nrf2 with reducing ROS production in aortic smooth muscle cells (41) and protected cardiomyocytes and neurons from apoptosis by Nrf2-dependent antioxidant effects (5, 39). Tan-IIA upregulates Nrf2 induction, and therefore, it is tempting to know if it could prevent myofibroblast activation via improving redox homeostasis and metabolic regulation.
For these, in the current study, we elucidated the potential influence of Tan-IIA on myofibroblast activation from the aspect of Nrf2-Nox4 balance and metabolic regulation. We showed that in addition to the amelioration of PF by Nrf2-Nox4 balance restoration, Tan-IIA activated Nrf2 to consume glutamate for GSH synthesis and thereby restraining myofibroblast activation due to the limited glutamate supply for proliferation. These findings indicate that more than regulation of antioxidant program, Nrf2 activation can suppress fibrotic response via the regulation of glutamine metabolism.
Results
Tan-IIA attenuated PF in mice
The effect of Tan-IIA on PF was investigated in mice challenged with bleomycin (BLM). Hematoxylin–eosin (H&E) staining revealed the occurrence of diffuse alveolar collapse and wall thickening in the lung tissue of BLM-treated mice (Fig. 1A). Additionally, the enhanced Masson trichrome staining for peribronchial and interstitial fibrosis showed that collagen deposition increased in the lungs of BLM-treated mice (Fig. 1A). However, these pathological changes were dose dependently attenuated by oral administration of Tan-IIA at doses ranging from 5 to 20 mg/kg (Fig. 1A).
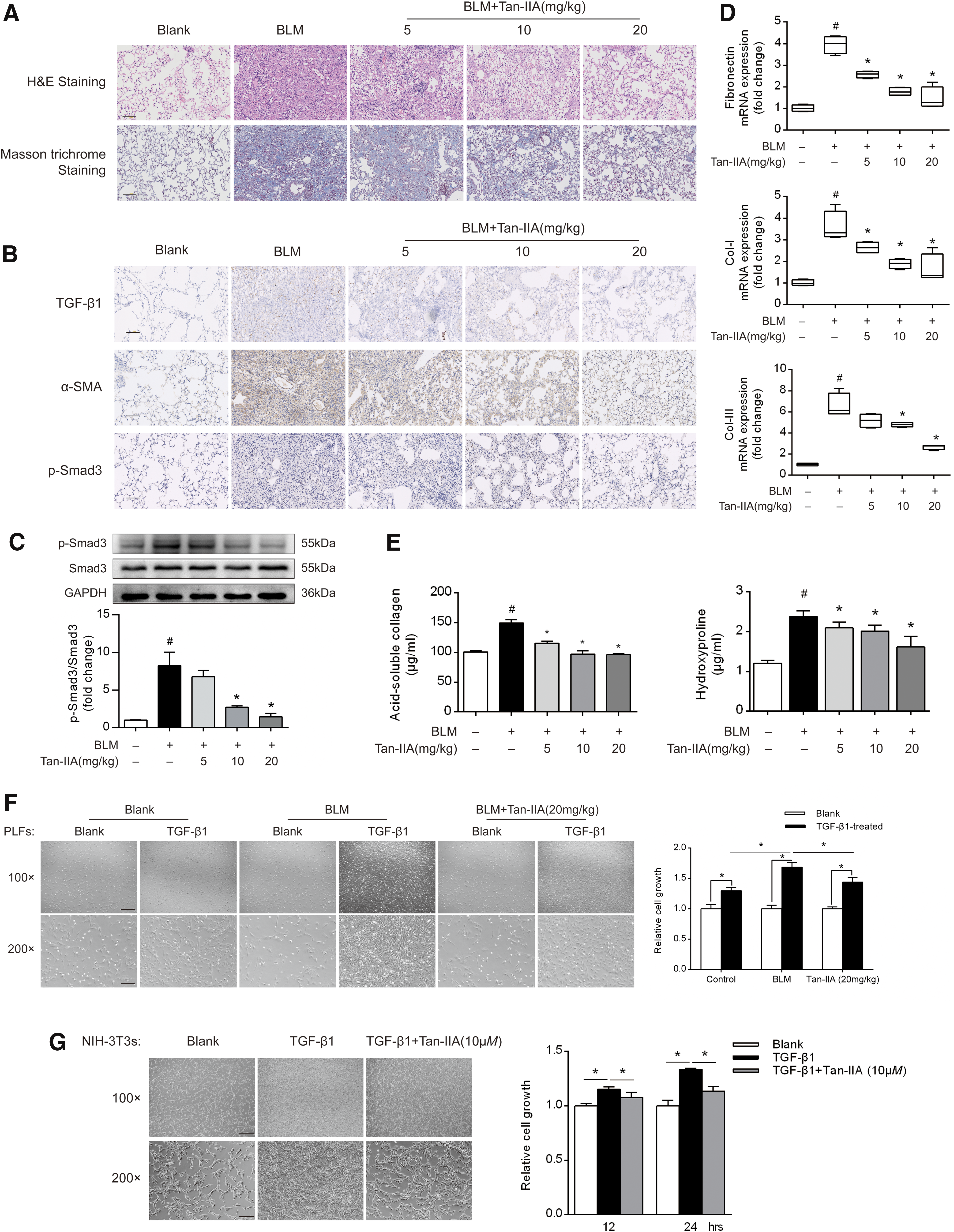
Immunohistochemical examination showed that Tan-IIA suppressed TGF-β1 and α-SMA expression with reduced Smad3 phosphorylation (p-Smad3) (Fig. 1B). Western blot showed that Tan-IIA inactivated Smad3 by dephosphorylation while total protein was not affected (Fig. 1C and Supplementary Fig. S7). Activated fibroblasts secreted fibronectin to bind the ECM components. Concurrently, Tan-IIA inhibited gene expression of fibronectin, type I collagen (Col-I), and type III collagen (Col-III) with reduced acid-soluble collagen and hydroxyproline accumulation, indicating the reduced ECM deposition in the lung tissue (Fig. 1D, E).
In addition, Tan-IIA effectively downregulated mRNA and protein expression of α-SMA, Col-I, and Col-III in response to TGF-β1 in cultured NIH-3T3 fibroblasts and MRC-5 cells (a human cell culture line composed of fibroblasts derived from the lung tissue) (Supplementary Fig. S1A–F). Sulforaphane is a dietary isothiocyanate with potent antioxidative effects via the regulation of Nrf2 signaling (8). As a positive control, sulforaphane also effectively inhibited TGF-β1-induced collagen induction in fibroblasts (Supplementary Fig. S1A–F).
Since cell proliferation is essential for fibroblast activation, we isolated primary lung fibroblasts (PLFs) from BLM-challenged mice to determine whether Tan-IIA administration could restrain fibroblast proliferation in response to TGF-β1. Although TGF-β1 stimulation effectively increased fibroblast growth, the effect was more potent in pulmonary fibroblast isolated from BLM-treated mice, indicative of the increased sensibility to fibrotic response. However, TGF-β1-induced cell growth was restrained in PLFs isolated from Tan-IIA-treated mice (Fig. 1F).
These results demonstrated that Tan-IIA administration increased the resistance of lung fibroblasts to TGF-β1-induced fibrogenic responses. Similarly, in cultured NIH-3T3 cells, Tan-IIA (10 μM) significantly inhibited the TGF-β1-induced cell growth (Fig. 1G). In resting cells, Tan-IIA showed no toxicity at the concentrations ranging from 0.01 to 500 μM, excluding the possibility that Tan-IIA inhibited fibroblast activation via cytotoxicity (Supplementary Fig. S1G). Together, these results suggested that Tan-IIA attenuated PF by inhibiting myofibroblast activation.
Tan-IIA regulated Nrf2 and Nox4 expression in fibroblasts
ROS are the driving force for myofibroblast activation, and Nrf2 is a key orchestrator of the cell responses to oxidative stress and exerts a protective effect against oxidative damage (12). Although BLM treatment mildly increased Nrf2 protein expression in the lung tissue, an effect likely due to the compensatory response to oxidative stress, a dose-dependent increase in total Nrf2 expression was observed in the lung from Tan-IIA-treated mice (Fig. 2A and Supplementary Fig. S8). Similar upregulation was detected in fibroblasts exposed to TGF-β1 (Fig. 2B and Supplementary Fig. S8). We detected Nrf2 protein expression at 68 kDa, in consistent with the published studies (37). Interestingly, it is proposed that the molecular weight of Nrf2 should be at 95–100 kDa (19). The abundance of amino acid residues and/or polyubiquitination of Nrf2 might be an acceptable explanation for the difference.
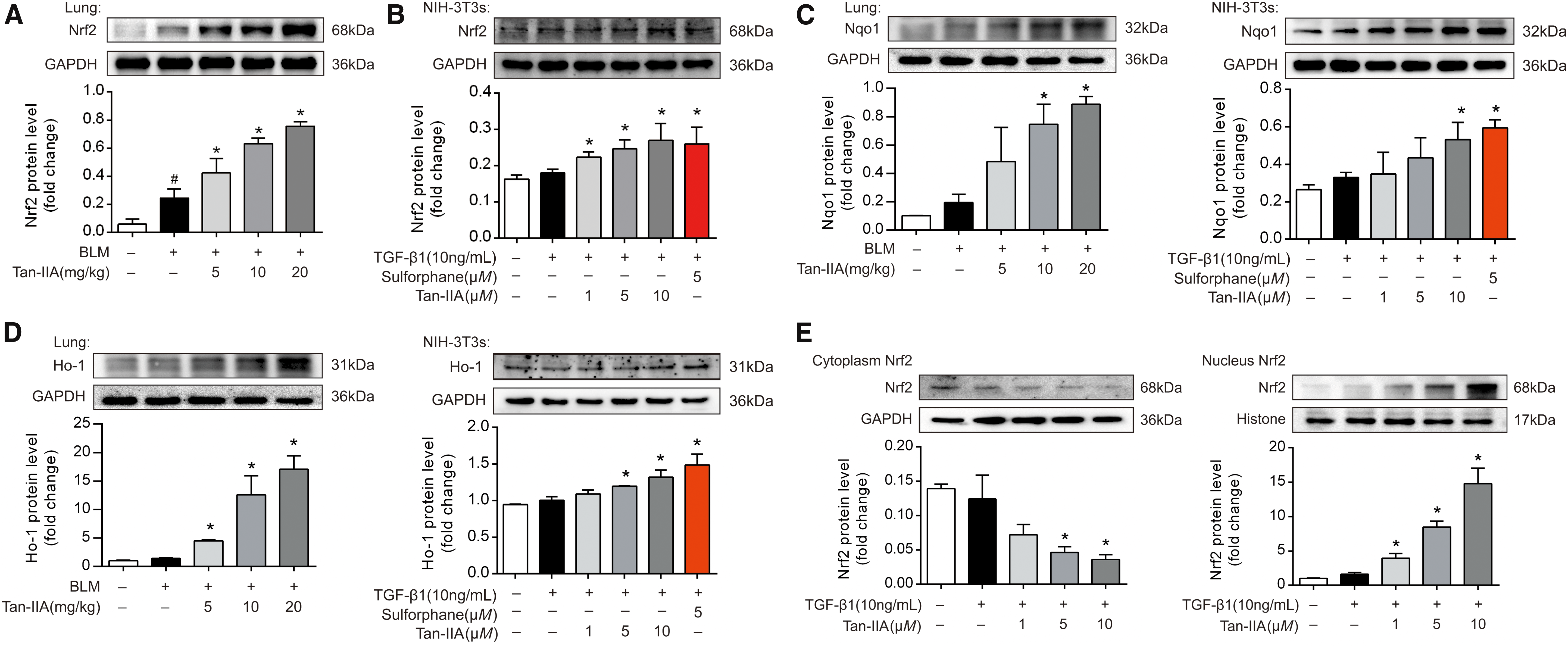
NAD(P)H quinone dehydrogenase 1 (Nqo1) and heme oxygenase1 (Ho-1) are the downstream targets of Nrf2. Tan-IIA increased the protein expression of Nqo1 and Ho-1 in mice lung and fibroblasts, indicative of Nrf2 activation (Fig. 2C, D, and Supplementary Fig. S8). Meanwhile, Tan-IIA treatment also increased the mRNA expression of Nqo1 and Ho-1 in the lung of BLM-challenged mice and NIH-3T3s (Supplementary Fig. S2A, B).
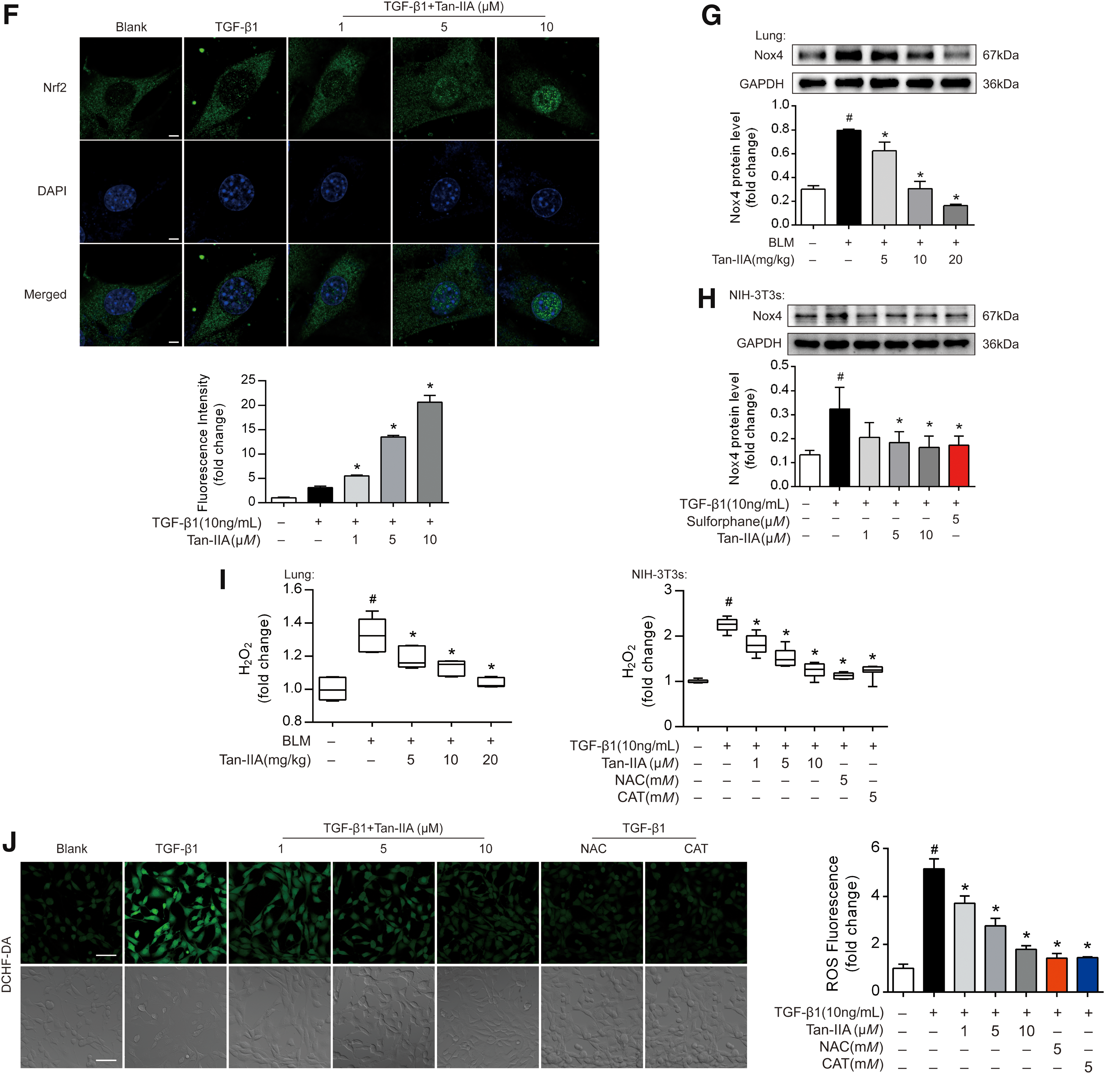
Nrf2 is mainly located in the cytoplasm in resting cells. In response to the activation, it translocates to the nucleus to counteract oxidative stress through transcriptional regulation (20). Tan-IIA increased Nrf2 expression in nucleus, whereas Nrf2 protein in the cytoplasm was concordantly reduced, indicative of Nrf2 translocation into nucleus (Fig. 2E and Supplementary Fig. S9). The visualization by confocal microscopy and the quantified data confirmed the functional role of Tan-IIA in Nrf2 activation (Fig. 2F).
The Nox4-dependent generation of ROS is required for TGF-β1-induced myofibroblast differentiation, ECM production, and contractility (33). In line with this, Nox4 expression increased in BLM-instilled mice and TGF-β1-stimulated fibroblasts, whereas these effects were reversed by Tan-IIA in a dose-dependent manner (Fig. 2G, H and Supplementary Fig. S9). TGF-β1 stimulation induced gene and protein expression of Nox4 but not Nox2 in NIH-3T3 fibroblasts (Supplementary Fig. S2C).
Furthermore, we evaluated the effects of Tan-IIA on ROS production by determining the hydrogen peroxide levels in vivo and in vitro using commercial assay kit (Fig. 2I). As anticipated, Tan-IIA effectively reduced ROS generation in the lung tissue and fibroblasts, demonstrating its oxidative stress suppressing action. The view of DCFH-DA staining confirmed this suppression of Tan-IIA in TGF-β1-stimulated fibroblasts (Fig. 2J). Consistently, quantitative result demonstrated that Tan-IIA concentration dependently reduced cellular hydrogen peroxide production in NIH-3T3s (Fig. 2K). As positive controls, ROS scavengers N-acetyl-
The regulation of Nrf2 and Nox4 by Tan-IIA was reproduced in the human cell line of MRC-5 (Supplementary Fig. S2E, F). These data revealed that Tan-IIA regulates redox homeostasis by upregulating Nrf2 and downregulating Nox4, possibly contributing to the prevention of myofibroblast activation.
Nrf2 and Nox4 reciprocally regulated myofibroblasts activation
Tan-IIA reciprocally regulates Nrf2 and Nox4 induction, and it was tempting to elucidate the potential implications to myofibroblast activation. In NIH-3T3 cells, Nrf2 overexpression, Nox4 knockdown, or vehicle treatments were individually prelabeled with their own distinct CellTracker fluorescence dyes, seeded at equivalent densities into the same cell culture dish, and then treated with TGF-β1 for 24 h. Fluorescence microscopy analysis with the quantified illustration revealed that in response to TGF-β1 treatment, the red-labeled untreated cells preferentially grew relative to their green-labeled Nrf2 overexpressing counterparts (Fig. 3A and Supplementary Fig. S10). Similar results were also observed in Nox4 knockdown cells (Fig. 3A and Supplementary Fig. S10).
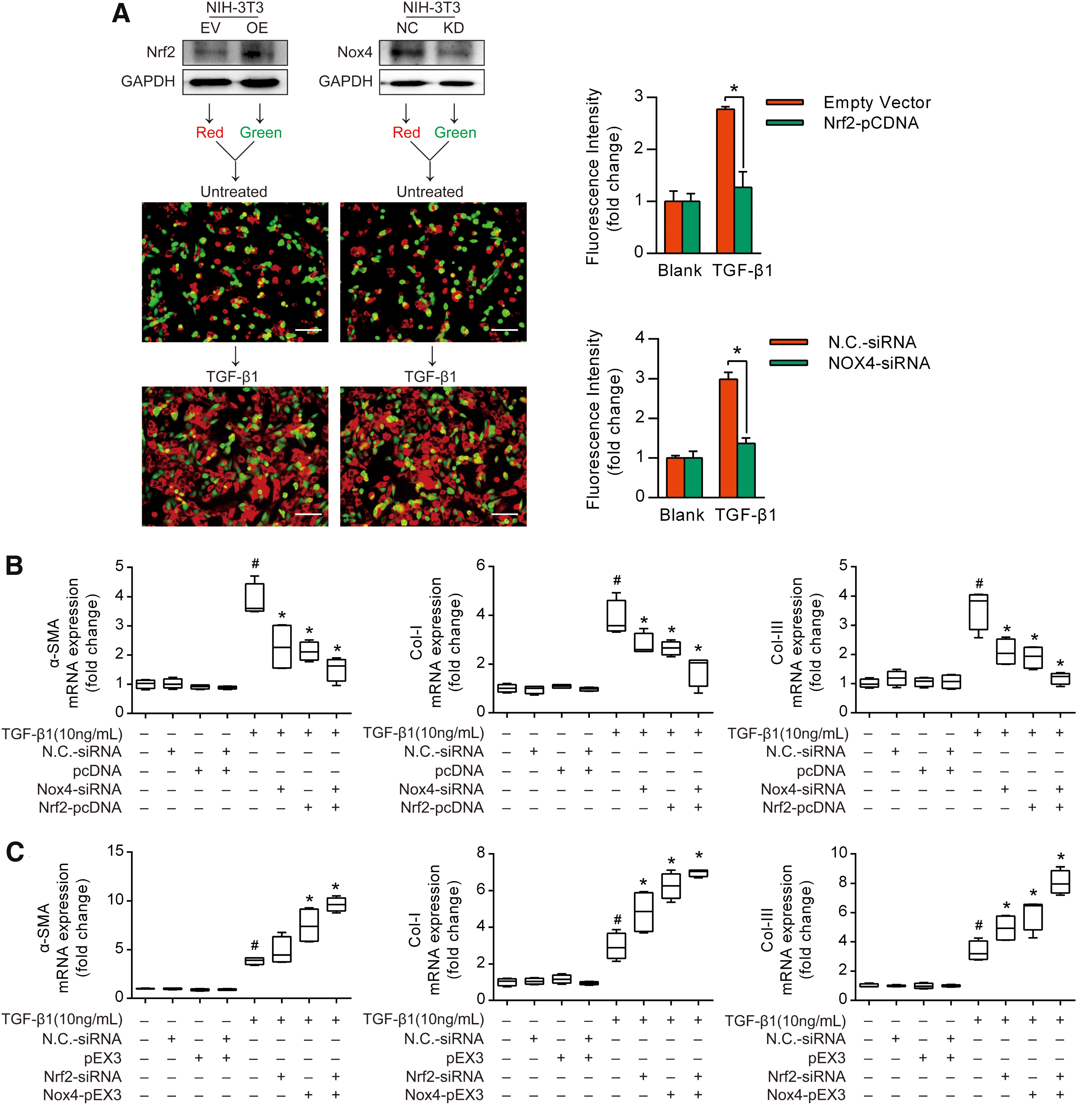
To further examine the reciprocal regulation of fibroblast activation by Nrf2 and Nox4, we overexpressed Nrf2 and knocked down Nox4 to establish an enabling environment against fibroblast activation. We found that Nrf2 overexpression or Nox4 knockdown attenuated the TGF-β1-induced gene expression of α-SMA, Col-I, and Col-III, respectively; this effect was further potentiated by cotreatment with Nrf2 overexpression and Nox4 knockdown (Fig. 3B). In contrast, Nrf2 knockdown and Nox4 overexpression collaborated to enhance the TGF-β1-induced myofibroblast activation (Fig. 3C). These data suggested the involvement of redox imbalance in fibroblast proliferation and indicated that Nrf2 and Nox4 reciprocally regulated myofibroblast activation.
Tan-IIA suppressed myofibroblast activation via the regulation of Nrf2 and Nox4
Tan-IIA promoted Nrf2 induction coupled with Nox4 suppression, and it was interesting to find out whether this regulation was involved in the suppression of the fibrogenic response. In NIH-3T3 fibroblasts, Tan-IIA suppressed the TGF-β1-induced gene and protein expression of α-SMA, Col-I, and Col-III, but this alternation was reversed by Nrf2 knockdown (Fig. 4A and Supplementary Fig. S11). The suppression of ROS production by Tan-IIA was also attenuated by Nrf2 knockdown, whereas the effects of antioxidative agents NAC and CAT were not affected in the presence of Nrf2-siRNA (Supplementary Fig. S3A). In contrast, overexpression of Nrf2 by transfection with Nrf2-pCDNA further potentiated the inhibitory effects of Tan-IIA on the fibrogenic response (Supplementary Fig. S3B).

Similarly, Nox4 knockdown tended to potentiate the inhibitory effect of Tan-IIA on the TGF-β1-induced myofibroblast activation (Supplementary Fig. S4A). It is noteworthy that in resting fibroblasts without TGF-β1 stimulation, overexpression of Nox4 independently increased gene and protein expression of α-SMA, Col-I, and Col-III, and this effect was also blocked by cotreatment with Tan-IIA (Fig. 4B and Supplementary Fig. S11). Similar regulation was also observed in ROS production (Supplementary Fig. S4B). These results suggested that the ROS-induced oxidative stress is a driving force for the fibrotic response, and Tan-IIA can regulate myofibroblast activation in an Nrf2-dependent manner.
It is proposed that ROS is involved in TGF-β1/Smad3 signaling (13, 14). We hypothesized that Nox4-derived ROS activated protein kinase C delta (PKCδ), which mediated the TGF-β1-induced myofibroblast activation via Smad3 activation. GKT137831, a bioavailable dual inhibitor of the NADPH oxidase isoforms Nox4 and Nox1, was used to elucidate the role of Tan-IIA in PKCδ/Smad3 signaling. The Western blot assay showed that Tan-IIA and GKT137831 inhibited Smad3 activation in response to TGF-β1 by dephosphorylation (Fig. 4C and Supplementary Fig. S12). Moreover, overexpression of Nox4 attenuated the inhibitory effect of Tan-IIA on p-Smad3 (Fig. 4D and Supplementary Fig. S12), providing evidence that the suppression of Nox4 was involved in Smad3 inactivation by Tan-IIA.
The confocal microscopy analysis showed that the treatment with Tan-IIA and GKT137831 prevented TGF-β1-induced PKCδ membrane translocation in fibroblasts (Fig. 4E). Similar to Tan-IIA, the selective PKCδ inhibitor rottlerin also inactivated Smad3 by dephosphorylation, indicative of the involvement of PKCδ in Smad3 activation (Fig. 4F and Supplementary Fig. S12). These data suggested that the suppression of PKCδ/Smad3 activation was involved in the action of Tan-IIA.
Tan-IIA protected Nrf2 from protein ubiquitination
We investigated the effect of Tan-IIA on Nrf2 induction in fibroblasts without TGF-β1 treatment and found that Nrf2 concentration dependently increased Nrf2 mRNA and protein expression (Fig. 5A and Supplementary Fig. S13). Concordantly, Tan-IIA upregulated gene expression of Nqo1, Ho-1, Gclc, and Gclm, indicative of Nrf2 activation by Tan-IIA (Supplementary Fig. S5).
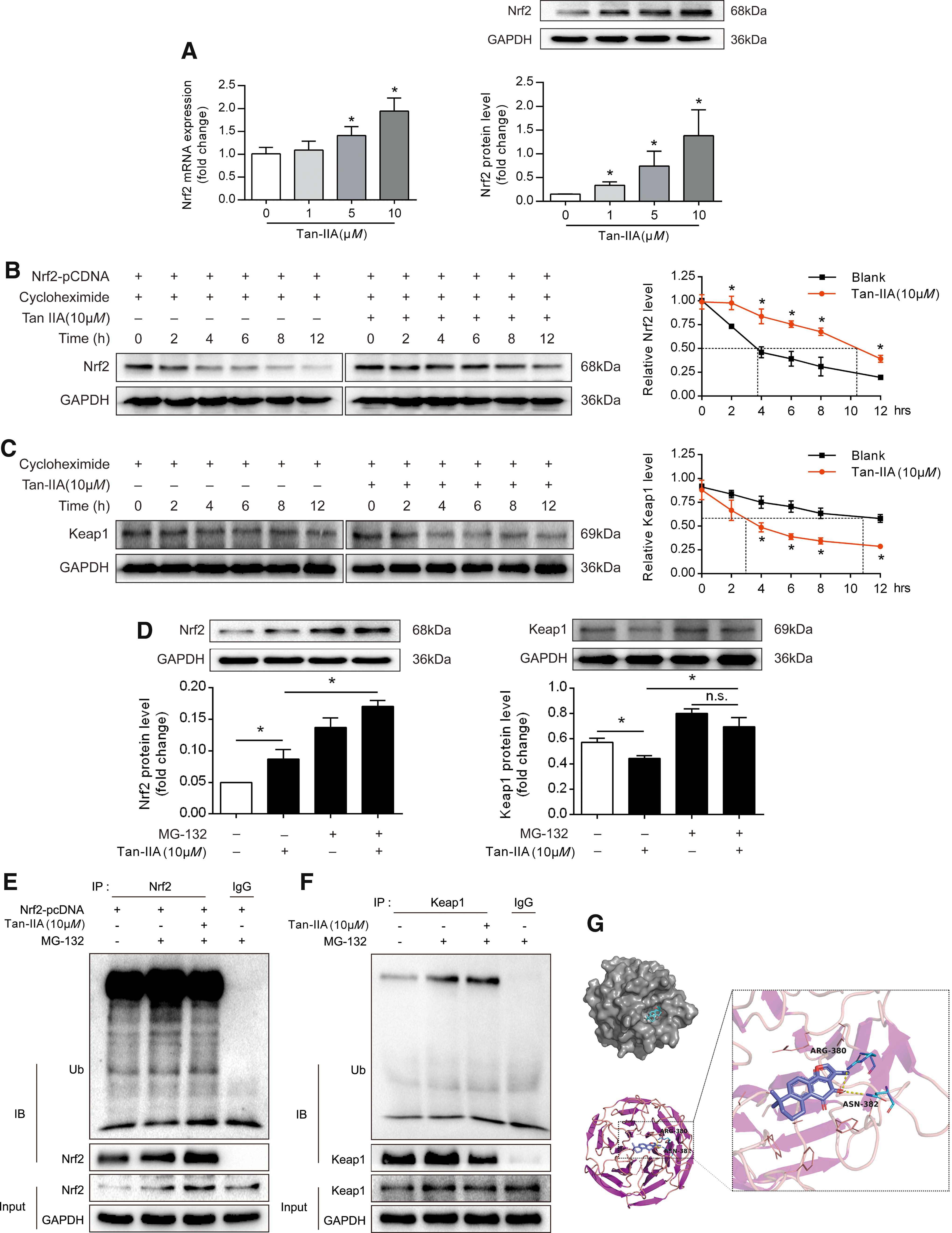
In resting cells, the cytoplasmic protein kelch-like ECH-associated protein 1 (Keap1) has been shown to bind Nrf2 and promote its degradation through protein ubiquitination (16). Accordingly, we became interested in establishing whether Tan-IIA preserves Nrf2 protein expression by preventing its degradation through the regulation of Keap1. To this end, we used eukaryotic inhibitor cycloheximide to inhibit protein synthesis in NIH-3T3 cells to study the degradation of Nrf2 with or without Tan-IIA. We found that exogenous Nrf2 protein was continuously degraded from 2 to 12 h, whereas Tan-IIA treatment effectively preserved Nrf2 protein expression by improving its stability (Fig. 5B and Supplementary Fig. S13). In contrast, Keap1 was expressed in a steady state, whereas Tan-IIA treatment impaired the stability of Keap1 and the significant effect remained from 4 to 12 h (Fig. 5C and Supplementary Fig. S13).
In addition, Tan-IIA increased the endogenous induction of Nrf2 induction, and this effect was further enhanced by cotreatment with the proteasome inhibitor MG-132 (Fig. 5D and Supplementary Fig. S13). We also noted that MG-132 increased Keap1 protein expression and attenuated the inhibitory effect of Tan-IIA on Keap1 induction (Fig. 5D). These results indicated that Tan-IIA promoted Keap1 degradation through the proteasome pathway, thus improving the stability of Nrf2.
Keap1 functions as an adaptor protein for the Cul3 E3 ubiquitin ligase, responsible for the continuous ubiquitylation and degradation of Nrf2 (17, 32). We transfected a plasmid-encoding Nrf2 in the presence and absence of MG-132 and Tan-IIA. Ubiquitin was analyzed by immunoblotting to reveal ubiquitinated Nrf2. As shown in Fig. 5E, ubiquitinated Nrf2 was evident in the presence of MG-132 (lane 2), but its abundance was decreased by Tan-IIA (lane 3), suggesting that Tan-IIA interfered with the ubiquitination of Nrf2 (Fig. 5E and Supplementary Fig. S14). Furthermore, we found that Tan-IIA can promote Keap1 ubiquitination (Fig. 5F and Supplementary Fig. S14).
The molecular docking result showed that Tan-IIA docks into the cavity of the Keap1 protein with a reasonable fit, and the binding energy between Keap1 and Tan-IIA was −6.57 kJ/mol, indicating a strong bond between them (Fig. 5G). However, the molecular weight of Nrf2 was too small to bind with Tan-IIA (data not shown). Together, these results suggested the possibility that Tan-IIA promoted Keap1 degradation through direct interaction, thereby protecting Nrf2 induction from the effect of protein ubiquitination.
Tan-IIA increased the sensitivity of fibroblasts to the loss of GSH
Activation of Nrf2 can impair tumor cell growth due to the reduced glutamate availability for anaplerosis through the TCA cycle (28). Accordingly, we pondered whether Nrf2 activation can restrain myofibroblast activation via the regulation of glutaminolysis. We found that Tan-IIA treatment increased GSH content in PLFs, likely due to Nrf2 activation (Fig. 6A). Glutaminase catalyzes the conversion of glutamine to glutamate, which is an important source for the generation of the antioxidant GSH (38). The glutaminase inhibitor CB-839 blocked the Tan-IIA-induced GSH production without influencing untreated cells, indicating that GSH production was mainly through glutaminolysis and was sensitive to glutaminase inhibition in the case of Tan-IIA (Fig. 6B).
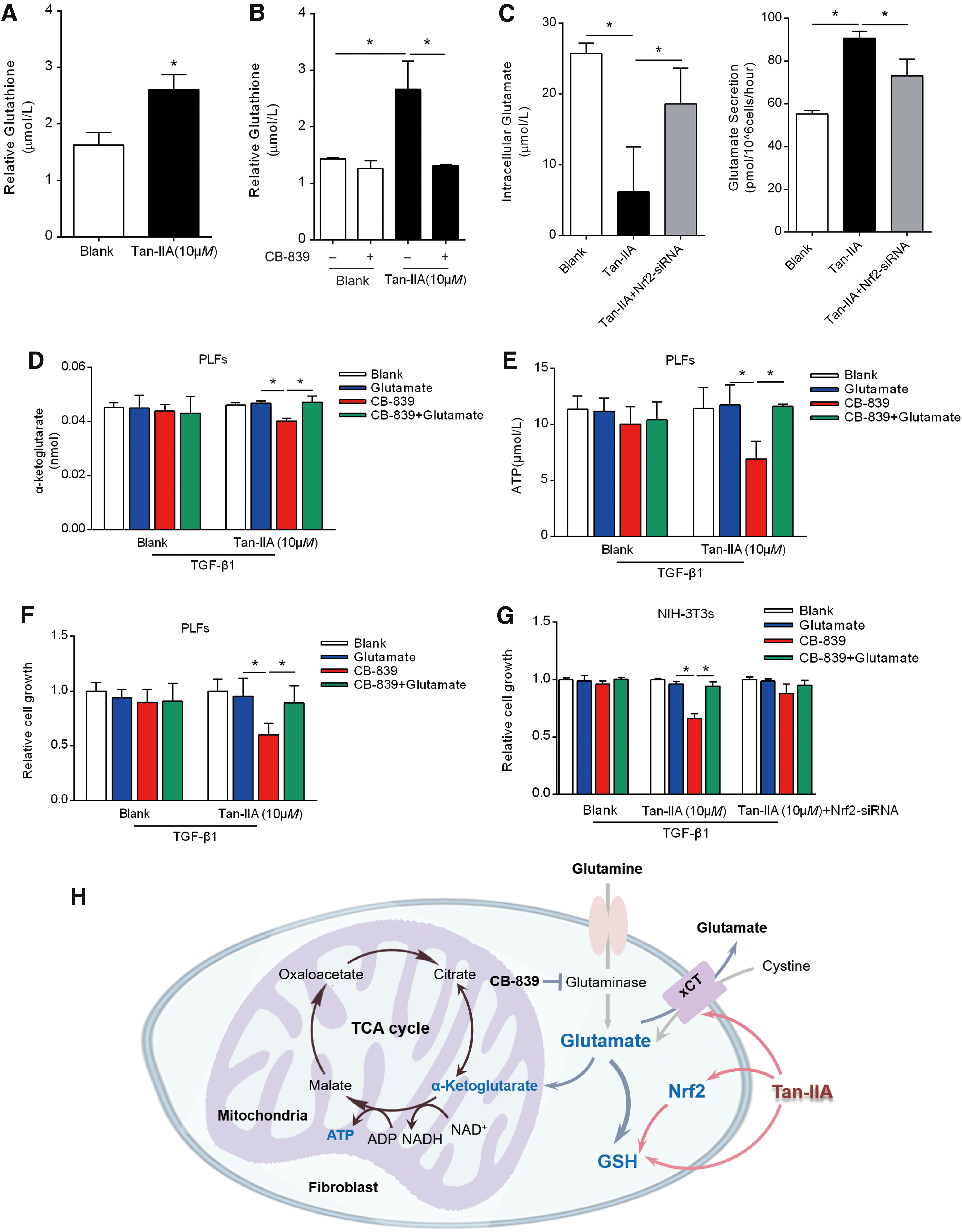
Remarkably, Tan-IIA-treated fibroblasts had lower intracellular but higher extracellular levels of glutamate compared with untreated cells, but these alternations were reversed by Nrf2 knockdown, indicating that Tan-IIA influenced intracellular glutamate pool in a manner dependent on Nrf2 induction (Fig. 6C).
As the export of glutamate in exchange for the import of cysteine through the antiporter system is required for the GSH generation (26), the increased glutamate release in Tan-IIA-treated cells supports the existence of intracellular cysteine pools for GSH production. Consequently, Tan-IIA treatment reduced the cellular α-ketoglutarate contents and adenosine triphosphate (ATP) generation in the presence of the glutaminase inhibitor CB-839, but these alterations were reversed by adding glutamate (Fig. 6D, E), indicating that Tan-IIA reduced the TCA intermediators and energy generation.
To determine the dependency on glutamate, we observed cell growth in TGF-β1-treated fibroblasts when the glutamate generation was inhibited by CB-839. Consistently, glutamate was able to rescue fibroblast growth from Tan-IIA treatment (Fig. 6F). In NIH-3T3 cells, Nrf2 knockdown rescued cell growth from Tan-IIA treatment in the presence of CB-839 (Fig. 6G), strongly supporting the notion that Nrf2 activation is responsible for the increased cell sensitivity to glutamate deprivation. Taken together, these results indicated that Tan-IIA-induced Nrf2 activation shifts the glutamine/glutamate pathway for antioxidant GSH production and thereby limiting glutamate availability for biosynthesis to support fibroblast growth (Fig. 6H).
Nrf2 knockdown attenuated inhibitory effects of Tan-IIA on PF
To ascertain the essential role of Nrf2 in suppressing PF, we instilled Nrf2 siRNA into the mice trachea at the time of the BLM injury and confirmed the efficiency on Nrf2 knockdown by examining protein and gene expression in the lung (Supplementary Fig. S6). H&E staining and Masson trichrome staining showed that Tan-IIA treatment attenuated alveolar architecture damage and reduced collagen deposition, but the protective effects were attenuated by Nrf2 knockdown (Fig. 7A). The immunohistochemistry analysis also demonstrated that Nrf2 knockdown blocked the suppressive effect of Tan-IIA on Smad3 activation by dephosphorylation (Fig. 7A).
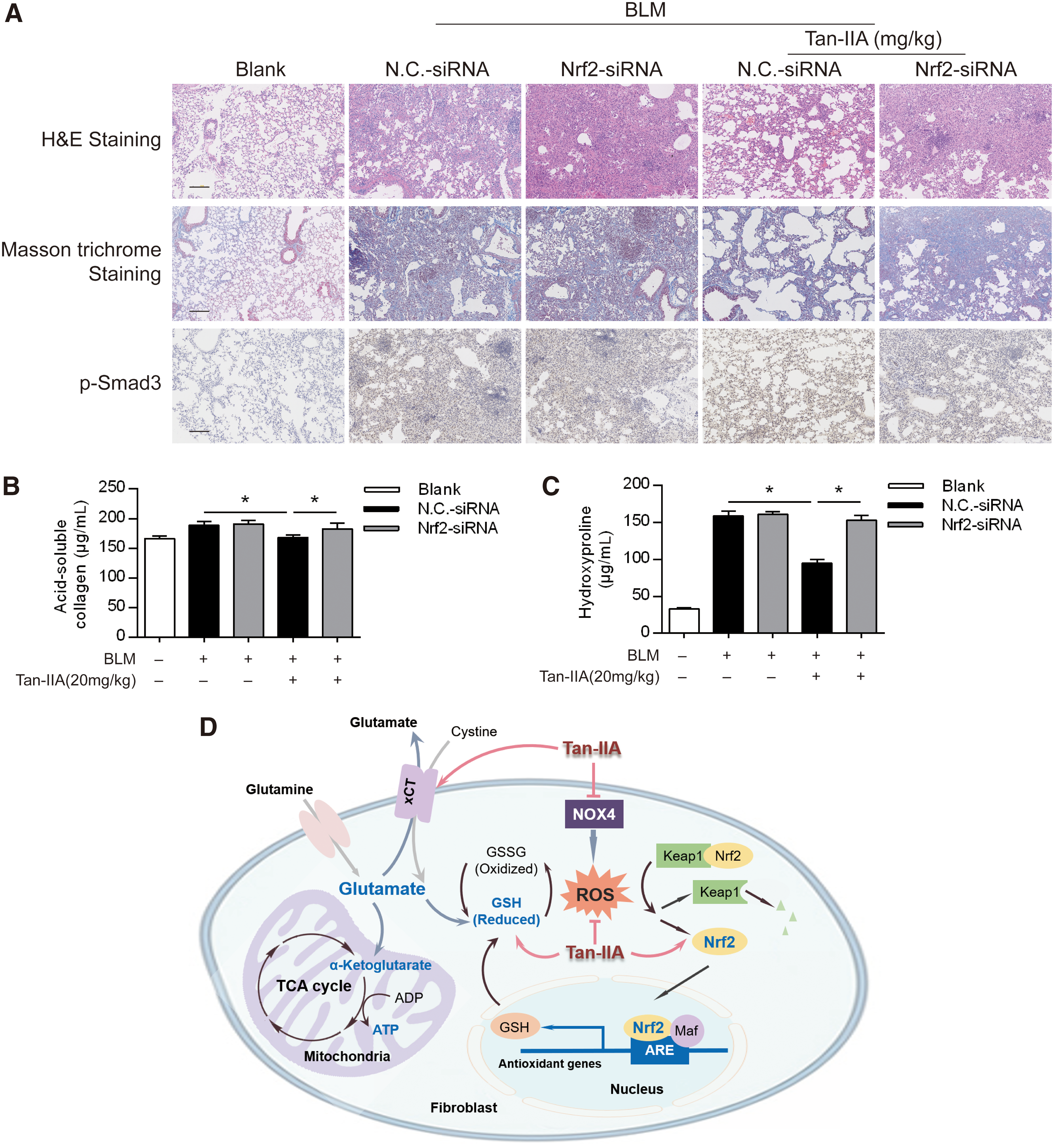
Furthermore, Nrf2 knockdown diminished the inhibitory effect of Tan-IIA on BLM-induced collagen and hydroxyproline depositions (Fig. 7B, C). These results provided evidence in vivo that Nrf2 activation is essential for Tan-IIA to suppress PF. Based on the presented in vitro and in vivo results, we concluded that Tan-IIA inhibited myofibroblast activation through the regulation of glutaminolysis by restoring redox homeostasis, resultantly suppressing the fibrotic response in PF (Fig. 7D).
Discussion
In this work, we found that Tan-IIA activated Nrf2 to restore redox homeostasis, thus contributing to inhibit myofibroblast activation in PF. Consistent with previous study, NADPH oxidases generate ROS to enhance fibrotic responses, whereas Nrf2 suppresses oxidative stress and attenuates fibrosis by transcriptionally upregulating antioxidant system in response to TGF-β1 induction (11, 14). Interestingly, we noted that Tan-IIA-induced Nrf2 activation led to the regulation of glutaminolysis and shunted glutamate into areas devoted to synthesizing GSH, which restricted the supply of glutamate availability for TGF-β1-stimulated myofibroblast proliferation. Although the antifibrotic role of Tan-IIA has been documented (10, 30, 36), these findings provide a novel strategy to restrain the fibrotic response from the perspective of fibroblast metabolism.
Pulmonary fibrogenic responses are characterized by the differentiation of fibroblast to myofibroblast, and excessive ECM deposition is a major consequence of myofibroblast activation (15). Oral administration of Tan-IIA suppressed myofibroblast activation and reduced collagen deposition in the lung of BLM-treated mice. Additionally, the therapeutic benefits of Tan-IIA were also confirmed in cultured fibroblasts. Activated myofibroblasts are derived from mesenchymal fibroblasts through differentiation and proliferation (31). In Fig. 1E, we isolated PLFs from Tan-IIA-treated mice that had been challenged with BLM and found that the ability of TGF-β1 to promote proliferation was impaired. Given the critical role of TGF-β1 in myofibroblast proliferation and activation (7, 29), these results indicated that Tan-IIA restrained PF by inhibiting fibroblast activation, an effect likely resulting from the interfering with cellular metabolism.
Persistent fibrosis in the lungs of aged mice is characterized by impaired redox homeostasis (11). Nox4 overexpression and the impaired ability to induce the Nrf2-mediated antioxidant response indicated the involvement of metabolism-associated oxidative stress in PF (11). Indeed, Nrf2 is shown to induce myofibroblastic dedifferentiation in PF (2). Consistent with the upregulation of Nrf2 in BLM-challenged mice, Tan-IIA activated Nrf2 with the suppression of Nox4 overexpression in TGF-β1-stimulated fibroblasts, indicating the ability to restore redox homeostasis.
Nox4-dependent ROS generation is required for myofibroblast differentiation (12), and as shown in Fig. 3A, the inhibition of cell growth by Nrf2 overexpression in TGF-β1-stimulated fibroblasts could be a result related to the suppression of ROS production. Nrf2 and Nox4 reciprocally regulated the fibrotic response in TGF-β1-stimulated fibroblasts, and therefore, it is logical to believe that Tan-IIA inhibited the myofibroblast activation by regulation of the balance between Nrf2 and Nox4. The inhibitory effects of Tan-IIA on the fibrotic response were attenuated by the Nrf2 knockdown or Nox4 overexpression, providing evidence to support the conclusion.
Protein kinase C is a family of protein kinase enzymes that regulate the function of other proteins through the phosphorylation of Ser/Thr residues. Upon activation, PKCδ can translocate to multiple subcellular localizations and the location in subcellular compartments is a key determinant of its biological effects. Tan-IIA prevented the TGF-β1-mediated PKCδ translocation to the membrane and thus inhibited myofibroblast activation by dephosphorylation of Smad3 activation. The previous study also showed that PKCδ and Smad2/3 signaling mediates the Nox4-associated myofibroblast activation (25).
The published studies demonstrated that Tan-IIA attenuated pulmonary and hepatic fibrosis with the suppression of TGF-β1 and inflammation, but the possible implication in oxidative stress is not known (10, 30, 36). Our work demonstrated that the improved redox homeostasis contributed to the suppression of myofibroblast activation, although protein abundance is not always consistent.
Next, we investigated the regulation of Keap1 to elucidate the molecular target by which Tan-IIA activated Nrf2. In resting cells, Keap1 functions as an adaptor of the Cul3-based E3 ligase, which conjugates ubiquitin to Nrf2 and promotes rapid degradation of Nrf2 by proteasome (16). Tan-IIA impaired the binding of Keap1 to Nrf2 by promoting Keap1 degradation and thus protected Nrf2 from E3 ligase-mediated ubiquitination and proteasomal degradation. This finding provided solid evidence that Tan-IIA counteracted oxidative stress to prevent myofibroblast transdifferentiation by activating Nrf2. Almost concurrently, Tan-IIA was reported to modulate Nrf2/Keap1 signaling in cardiomyocytes to inhibit apoptosis (39), further confirming the regulation of Nrf2 by Tan-IIA in fibroblasts.
Because Nrf2 is a transcriptional factor that combats oxidative stress by upregulating antioxidant response elements, we reasoned that the suppression of Nox4 by Tan-IIA should be a downstream result from the antioxidant effects. Interestingly, we found that Tan-IIA also increased Nrf2 mRNA expression in resting fibroblasts, and this regulation is unable to be explained by Keap1 degradation. The mechanism by which Tan-IIA transcriptionally regulates Nrf2 expression remains to be elucidated.
It is well established that the metabolism of cancer cells mainly depends on aerobic glycolysis, whereas emerging evidence indicates that glutaminolysis is required by tumorigenesis for energy generation and biosynthesis (3). Glutaminolysis-derived glutamate can be converted to α-ketoglutarate, which enters the TCA cycle to generate ATP or to serve as an alternative source of carbon for protein and nucleotide synthesis in proliferating cells (6, 35).
Nrf2 transcriptionally increases GSH biosynthesis and regeneration (20). When glutaminolysis-derived glutamate is consumed in GSH production, reduced glutamate availability impairs the TCA cycle anaplerosis. In tumorigenesis, this impaired TCA cycle anaplerosis leads to defective glutamate-dependent fueling of central carbon metabolism (28). Similar to cancer cells, the metabolism of myofibroblasts is vigorous to sustain uncontrolled cellular proliferation. Accordingly, we hypothesized that this regulation also likely occurs in activated myofibroblasts.
GSH is synthesized from the amino acids cysteine, glutamate, and glycine, and cysteine and glutamate are mainly derived from glutaminolysis (28). In TGF-β1-stimulated myofibroblasts, Tan-IIA reduced intracellular glutamate due to GSH synthesis and thus limited glutamate availability for the TCA cycle to support energy generation and biosynthesis. Tan-IIA reduced the TCA cycle anaplerosis and impaired cell growth in a similar glutamate-dependent manner. The data presented here supported our hypothesis that Nrf2/GSH signaling can impair the cell growth via the regulation of glutaminolysis in proliferating myofibroblasts.
In summary, this study confirmed that Nrf2 and Nox4 reciprocally regulate myofibroblast activation, and the dynamic equilibrium between Nrf2 and Nox4 is essential for the amelioration of PF. Tan-IIA activated Nrf2 by interfering with Keap1 and counteracted the Nox4-elicited oxidative stress to suppress myofibroblast activation. More importantly, Tan-IIA could activate Nrf2/GSH signaling to restrain myofibroblast proliferation by limiting glutamate availability to support cell growth. Thus, these findings provided new insight into the relationship between glutaminolysis and myofibroblast activation from the aspect of metabolism.
Materials and Methods
Materials
Tan-IIA (purity 98%) was purchased from Shanghai YuanYe Biotechnology Co., Ltd (Shanghai, China) and dissolved in dimethyl sulfoxide (DMSO, with the final concentration 0.1%, v/v). BLM was obtained from Nippon Kayaku Co., Ltd (Tokyo, Japan). Recombinant human TGF-β1 (HEK293 derived) (100-21) was provided by PeproTech (Rocky Hill, NJ). Sulforaphane,
Antibodies against ubiquitin (CY5520), Nrf2 (CY1851), TGF-β1 (CY1115), Nox4 (CY5255), p-Smad3 (S423+S425) (CY5140), Nqo1 (CY6710), and α-SMA (CY5295) were procured from Abways Technology (Shanghai, China). Anti-GAPDH (AP0063), anti-Col1A2 (S3) (BS1530), anti-Col3A1 (P104) (BS1531), Goat Anti-Rabbit IgG (H+L) HRP (BS13278), and Goat Anti-mouse IgG (H+L) HRP (BS12478) were obtained from Bioworld Technology (St. Paul, MN). Antibody against Ho-1 (sc-136960) was from Santa Cruz Biotechnology (Santa Cruz, CA) and anti-Keap1 (H436) was from Cell Signaling Technology (Beverly, MA).
Cycloheximide (HY-12320), CB-839 (HY-12248), GKT137831 (HY-12298), and MG-132 (HY-13259) were from MedChemExpress (Brea, CA). Mouse IgG (A7028), RIPA lysis buffer (P0013), Protein A + G agarose (Fast Flow), CAT, NAC, DCFH-DA assay kit, DHE assay kit, DAPI staining solution, DiI assay kit, hydrogen peroxide assay kit, and Alexa Fluor 488-labeled goat anti-rabbit IgG (H+L) antibody were purchased from Beyotime Institute of Biotechnology (Shanghai, China).
Murine model of BLM lung injury
Female C57BL/6 mice (20 ± 2 g), purchased from the Laboratory Animal Center of the Yangzhou University (Yangzhou, China), were housed in colony cages 12-h light/dark cycles with free access to food and water. The animal care and experimental procedures were approved by the Animal Ethics Committee of the China Pharmaceutical University. Mice were anesthetized with intraperitoneal injection of 4% chloride hydrate (10 mL/kg), and the trachea was exposed by a cervical incision. BLM (0.025 U) was dissolved in saline and then intratracheally instilled using a 27-gauge needle.
Animals in the sham groups received equivalent volume of saline in the same way. After BLM infusion, mice were administrated with Tan-IIA (5, 10, and 20 mg/kg, i.g.) or 0.5% CMC-Na solution as vehicle for 21 consecutive days. The mice were euthanized for collection of lung samples on day 21 after BLM treatment. For acid-soluble collagen and hydroxyproline assays, lung tissues were detected with commercial kits. For Western blot and quantitative real-time polymerase chain reaction (Q-PCR) analysis, right lobes were quickly frozen in liquid nitrogen followed by storing at −80°C. For histological examination and immunofluorescent assay, left lobes were directly fixed in 4% paraformaldehyde.
Cell preparation and culture
PLFs isolated from the lungs of C57BL/6 mice as described (34). Tissue sections were washed twice in sterile phosphate-buffered saline (PBS), resuspended in Dulbecco's modified Eagle's medium (DMEM) (Keygen Biotech, Jiangsu, China) with 4.5 g/L glucose supplemented with 10% fetal bovine serum (Thermo Scientific, MA), transferred to a tissue culture dish, and incubated for 1–3 weeks to allow fibroblasts to migrate out of tissue sections. Fibroblasts were purified by repeated trypsinization and passaging to achieve a homogenous population of spindle cells. PLFs at three and five passages were used in experiments.
NIH-3T3 cells (a cell line of embryo fibroblasts from mice; Cell Bank of the Chinese Academy of Sciences, Shanghai, China) were cultured in DMEM supplemented with 10% calf serum (ExcellBio, Shanghai, China), 100 U/mL penicillin, and 100 μg/mL streptomycin. Cells were incubated at 37°C in 5% CO2, 95% air. After reaching confluence, cells were washed with PBS for twice and switched to the serum-free medium before proceeding with further experiments.
Transfection
For in vivo RNAi studies, specific Nrf2 or nonspecific control siRNA (GenePharma, Suzhou, China) was administrated by intratracheal injection to the BLM-treated lungs at a dose of 50 μg per mouse. NIH-3T3 cells were transfected with siRNA targeting Nrf2 or Nox4 using Lipofectamine 2000 reagent (Invitrogen, CA) for knockdown. For in vitro overexpression studies, we employed Lipofectamine 2000 reagent to transfect plasmid targeting Nrf2 or Nox4 and empty vector (pCDNA, pEX-3) (GenePharma) in NIH-3T3s. After 24 h of transfection, cells were cultured in fresh medium for further experiments.
The assay for acid-soluble collagen and hydroxyproline contents
The lungs were homogenized in the ice-cold normal saline, and the contents of acid-soluble collagen and hydroxyproline were, respectively, assayed with the Sircol Collagen Assay Kit (Biocolor, Northern Ireland, United Kingdom) and Hydroxyproline Assay Kit (Jiancheng Bioengineering Institute, Nanjing, China) according to the manufacturer's instructions.
Histological and immunohistochemical examination
Following sacrifice, the lung left lobe of mice was removed and fixed in 4% paraformaldehyde, dehydrated, and embedded in paraffin. The paraffin-embedded tissue samples were sectioned into 5-μm slices and then stained with H&E or Masson trichrome for collagen and examined under a digital scanner (NanoZoomer 2.0, Hamamatsu, Japan). Meanwhile, the paraffin-embedded tissues for all groups were processed for immunohistochemical staining of α-SMA, TGF-β1, and the p-Smad3 for TGF-β1/Smad3 activation. Images were viewed by a section digital scanner (NanoZoomer 2.0).
Assay of ROS production
Cultured NIH-3T3 cells were treated with indicated agents for 24 h, and the intracellular ROS generation was determined by measuring the oxidative conversion of cell permeable DCFH-DA to fluorescent dichlorofluorescein and utilizing commercial assay kit for hydrogen peroxide detection. To visualize ROS production in the lung, the tissues were homogenized for hydrogen peroxide measurement or stained with the superoxide oxidizes dihydroethidium. Images were viewed by a confocal scanning microscopy (Zeiss LSM 700, Jena, Germany) at an excitation wavelength 488 nm and an emission wavelength 525 nm.
Immunofluorescence assays
NIH-3T3 cells were transfected with Nrf2 overexpressed plasmid or Nox4 siRNA for 24 h. Cells of the same background were prelabeled with 25 μM of their own distinct red or green CellTracker dyes (Thermo Scientific) and incubated at 37°C for 30 min. Cells were then washed three times with PBS and seeded at equal densities into the same culture dishes. After 4 h, the cells were stimulated with 10 ng/mL TGF-β1 for 24 h, and the images were viewed with a fluorescent microscope (Olympus IX53; Nikon, Japan). For the view of Nrf2 nucleus trafficking and PKGδ membrane translocation, NIH-3T3s were treated with indicated agents for 24 h and visualized under a confocal scanning microscope (Zeiss LSM 700).
Molecular docking study
To evaluate the binding potential of Tan-IIA to Keap1 protein, in silico protein-ligand docking experiment was carried out using Autodock program (version 4.2) (Olson Laboratories, CO). The structure of Keap1 was downloaded from the protein data bank (PDB ID: 3wdz), and the crystal water molecules and other small molecules were removed. The Autodock program (Olson Laboratories) was then applied to generate the conformational ensemble for Tan-IIA to Keap1 protein. We used the genetic algorithm for conformational search. To extensively explore the conformational space of Tan-IIA, we performed 100 individual genetic algorithm runs to generate 100 docked conformations. The size of the docking box was properly set to enclose the possible binding pocket. The protein structure was kept fixed during molecular docking.
Cell proliferation assay
PLFs isolated from BLM-challenged mice or NIH-3T3 cells were seeded at equal densities and then stimulated with 10 ng/mL TGF-β1 for 24 h. Cell growth was observed under an optical microscope (Olympus IX53; Nikon) and detected by CCK-8 assay kit (Dojindo Laboratories, Kyushu Island, Japan). For glutamine deprivation experiments, cells were seeded in DMEM containing 10 ng/mL TGF-β1 at the time of plating. After cell attachment, cells were incubated with indicated agents for 3 days, and cell proliferation was determined by CCK-8 assay kit.
Measurement of GSH, glutamate, ATP, and α-ketoglutarate content
PLFs were treated with indicated agents in the presence or absence of 2.5 μM CB-839. After treatment, the supernatant was harvested for the assay of extracellular glutamate using a commercial kit (Jiancheng Bioengineering Institute). Treated cells were collected, washed with PBS twice, and lysed for measuring the intracellular levels of GSH, glutamate, ATP, and α-ketoglutarate with commercial kits (Abcam, MA).
Cellular viability detection
Cellular viability was detected with CCK-8 assay kit (Dojindo Laboratories). Cells were grown in 96-well culture plates and were treated with Tan-IIA at the concentrations ranging from 0.01 to 500 μM for 48 h. Then, CCK-8 solution (10 μL) was added to each well. After 4 h of incubation, the absorbance was measured using a microplate reader (Thermo Varioskan LUX, MA) at 450 nm.
Quantitative real-time polymerase chain reaction
TRIzol reagent (Invitrogen) was used according to the manufacturer's protocol to obtain the total RNA from the lungs or cells. A 2 ng section of RNA was employed for the synthesis of first strand of cDNA by using Hifair II 1st Strand cDNA Synthesis SuperMix (Yeasen, Shanghai, China). We performed real-time PCRs for each cDNA sample in triplicate using Hieff qPCR SYBR Green Master Mix (Yeasen) and gene-specific primer pairs for GAPDH, fibronectin, α-SMA, Col-I, Col-III, Ho-1, and Nqo1 (Table 1).
Nucleotide Sequences of Gene-Specific Primers Used for Quantitative Real-Time Polymerase Chain Reaction
α-SMA, alpha smooth muscle actin; Col-I, type I collagen; Col-III, type III collagen; Ho-1, heme oxygenase1; Nox4, NADPH oxidase 4; Nqo1, NAD(P)H quinone dehydrogenase 1; Nrf2, nuclear factor erythroid 2-related factor 2.
A CFX96 real-time system (BioRad, CA) was used for PCR amplification by the following steps: enzyme activation at 95°C for 5 min, 95°C for 10 s of denaturing, annealing, and extending for 30 s at 60°C followed by 40 cycles. We expressed semiquantitative real-time PCR data for each target gene as 2−ΔΔCt relative quantitation versus endogenous GAPDH, with error bars representing the standard error of the mean of triplicate reactions.
Western blot analysis and immunoprecipitation
We prepared cell and tissue lysates in RIPA buffer and performed Western immunoblotting as described (40). Equal amounts of protein were immunoblotted with specific primary antibodies. The values of band intensities were developed with ECL (enhanced chemiluminescence; Yeasen) and quantized by Image-ProPlus 6.0 software (Rockville, MD).
For immunoprecipitation, cell lysates were then centrifuged at 12,000 g for 20 min, and the soluble fraction was collected. Next, the antibody was immunoprecipitated overnight at 4°C and then with protein A + G agarose beads for another 2 h. After that, the protein A + G agarose beads were washed four times with the lysis buffer. The beads were then boiled in 1% SDS loading buffer for Western blotting with the indicated antibodies.
Statistical analysis
Data are expressed as the mean ± SD (standard deviation) of at least three independent experiments. Statistical analysis was performed using GraphPad Prism software, version 6.0 (San Diego, CA). Single-factor ANOVA and two-tailed Student's t-test were used to compare the statistical significance of differences between the samples and their respective controls. A p-value of <0.05 was considered to be statistically significant.
Footnotes
Acknowledgments
This study was supported in part by the National Natural Science Foundation of China (No. 81573705 and No. 81421005) and “Double-First Class” Development (CPU2018GF07). We thank the Cellular and Molecular Biology Center of the China Pharmaceutical University for technical assistance.
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.