
Abstract
Aims:
Cardiac-specific overexpression of metallothionein (MT) has been shown to be beneficial in ischemic heart disease, but the detailed mechanisms through which MT protects against myocardial infarction (MI) remain unknown. This study assessed the involvement of the mTORC2/FoxO3a/Bim pathway in the cardioprotective effects of MT.
Results:
MI was induced in wild-type (FVB) mice and in cardiac-specific MT-overexpressing transgenic (MT-TG) mice by ligation of the left anterior descending (LAD) coronary artery. Cardiac function was better; infarct size and cardiomyocyte apoptosis were lower in MT-TG mice than in FVB mice after MI. Moreover, MT-TG mice exhibited better phenotypes after LAD ligation than FVB mice treated with Mn(III)tetrakis (1-methyl-4-pyridyl) porphyrin pentachloride (MnTMPyP; a reactive oxygen species [ROS] scavenger) and cardiac-specific catalase-overexpressing transgenic (CAT-TG) mice, which showed the same ROS levels as MT-TG mice after MI. Activation of mechanistic target of rapamycin complex 2 (mTORC2) was essential for the cardioprotective effects of MT against MI. In addition, MT attenuated the downregulation of phospho-FoxO3a after MI, inhibiting the expression of the apoptosis-associated gene Bim, located downstream of FoxO3a, and reducing the level of apoptosis after MI. To mimic ischemic-injured FVB and MT-TG mice in vitro, H9c2 and MT-overexpressing H9c2 (H9c2MT7) cardiomyocytes were subjected to oxygen and glucose deprivation, with the results being consistent with those obtained in vivo.
Innovation and Conclusion:
The cardioprotective effects of MT against MI are not entirely dependent upon its ability to eliminate ROS. Rather, MT overexpression mostly protects against MI through the mTORC2-FoxO3a-Bim pathway.
Introduction
Myocardial infarction (MI) is a common and lethal heart disease, with high incidence and high annual mortality rates (3, 24, 25, 32, 39, 45). Cardiac dysfunction during MI is due largely to ischemia-induced cardiomyocyte apoptosis, resulting from a decrease in oxygen supply and dysregulation of the mechanistic target of rapamycin (mTOR) pathway (1, 10, 26, 42, 46).
This study identified that metallothionein (MT) preserved cardiac function, lowered infarct size, and reduced cardiomyocyte apoptosis in mice after myocardial infarction (MI). More importantly, the functional importance of MT during MI is not limited to its reactive oxygen species scavenging ability. Rather, its novel role in the activation of mTORC2/FoxO3a/Bim pathway alleviates MI-induced cardiomyocyte apoptosis, thus protecting the heart. Therefore, MT-modulated mTORC2/FoxO3a/Bim pathway may play an important role in the treatment of MI.
mTOR, a master regulator of cell growth, proliferation, and survival, interacts with specific adaptor proteins, forming mTOR complexes 1 (mTORC1) and 2 (mTORC2), with each having specific biological functions (35). mTOR is important in cardiac physiology and disease. For example, cardiac dysfunction and heart failure can be induced in mice by inducible cardiac-specific mTOR and Raptor gene deletion (36, 53). By contrast, genetic or pharmacological partial inhibition of mTORC1 activity is cardioprotective in multiple pathological conditions, including pressure overload, hypertrophic cardiomyopathy, and MI (7, 23). Using models of chronic MI, mTORC1 was found to be activated in the remote myocardium and to contribute to ventricular remodeling (7, 47).
Although mTORC2 signaling is not as well characterized as mTORC1 signaling, mTORC2 signaling was recently found to be involved in cardiomyocyte growth and survival during pressure overload. Mice with a cardiac-specific deletion of the Rictor gene showed progressive development of cardiac dilation and dysfunction associated with massive apoptosis after MI (47).
mTORC2 was shown to phosphorylate Akt, which promotes survival and has antiapoptotic kinase activity, at serine 473, leading to Akt activation and enhancement of cell survival. Under conditions of acute stress, mTORC2 activates Akt, promotes regeneration, and prevents apoptosis (11). Among the important downstream targets of Akt are members of the FoxO subfamily of forkhead transcription factors, which can be inactivated by Akt-dependent phosphorylation (5, 37, 55). FoxO3a, one member of the FoxO subfamily, regulates cellular apoptosis by inducing expression of the proapoptotic gene Bim (12, 41). Activation of Akt by mTORC2 and the subsequent hyperphosphorylation and inactivation of FoxO3a were found to inhibit Bim expression, thereby preventing apoptosis (30).
Metallothionein (MT) is a cysteine-rich, low molecular weight protein. Its basal levels in biological systems are usually very low, although they may vary with age and type of tissue (16, 22). MT expression can be induced by heavy metals, starvation, heat, inflammation, and other stress conditions (17, 34). To date, however, the mechanisms underlying the cardioprotective effects of MT in MI remain unclear.
MT has been reported to protect against L-NAME-induced myocardial anomalies possibly through the restoration of autophagy (51). Moreover, MT was shown to protect the heart against ischemia/reperfusion by inhibiting apoptosis mediated by mitochondrial cytochrome c release and caspase-3 activation (21). Using cardiac-specific MT-overexpressing transgenic (MT-TG) mice, we found that MT was associated with high resistance to the development of diabetic cardiomyopathy (9) and age-associated cardiovascular diseases (8). Most of these studies found that MT acted as a scavenger of reactive oxygen species (ROS), suggesting that the cardioprotective effects of MT are due to its suppression of oxidative stress.
This study investigated the mechanism by which MT overexpression attenuated MI-induced cardiac damage and determined whether the underlying cardioprotective effects of MT are associated with mTORC2/FoxO3a/Bim-mediated apoptosis, both in vivo and in vitro.
Results
MT significantly attenuates MI-induced apoptosis, reduces infarct size, and preserves cardiac function
MI was induced in MT-TG mice and wild-type (FVB) control littermates by ligation of the left anterior descending (LAD) coronary artery for 1 week. Kaplan–Meier analysis showed that post-MI survival rate was significantly higher in MT-TG than in FVB mice (Fig. 1A).
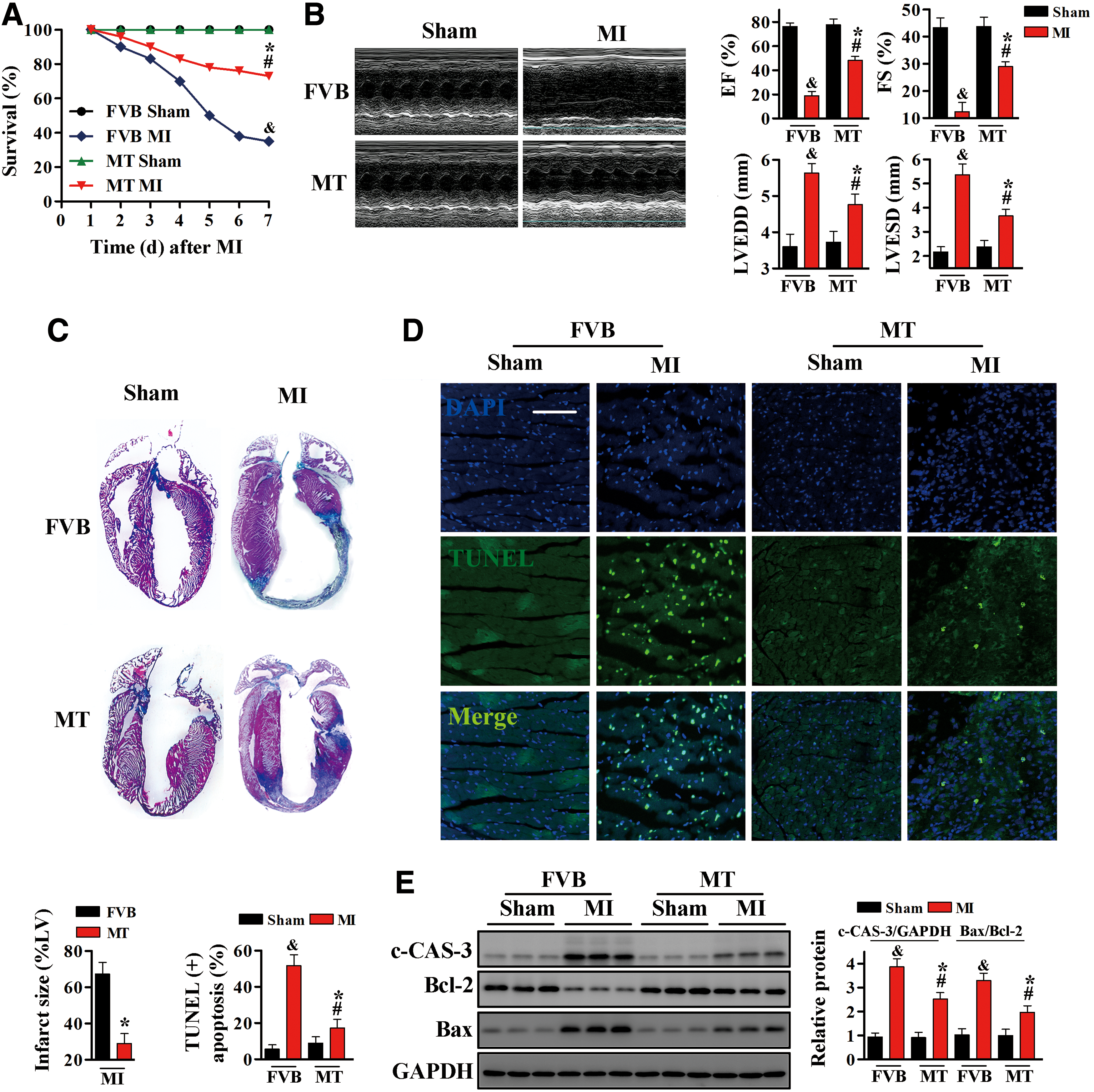
Consistent with the improved survival rate, MT-TG mice had better cardiac function than FVB mice, as determined by M-mode echocardiography. Before MI, left ventricular end-systolic diameter (LVESD), left ventricular end-diastolic diameter (LVEDD), left ventricular ejection fraction (EF %), and fractional shortening (FS %) did not differ significantly in FVB and MT-TG mice (Fig. 1B). One week after MI, however, LVESD and LVEDD were greater, EF (%) and FS (%) were lower, in FVB than in MT-TG mice. To evaluate actual infarction areas, mouse hearts were incubated with Masson's trichrome stain. MT was found to markedly reduce infarct size from 65% to 28% (Fig. 1C).
Cardiomyocyte apoptosis induced by ischemic injury is a major cause of cardiac functional impairment (31). Terminal deoxynucleotidyl transferase dUTP-mediated nick-end labeling (TUNEL) assays showed that the rate of cardiac apoptosis was higher in FVB hearts after MI than Sham operation, but was significantly lower in MT-TG hearts after MI (Fig. 1D). These results were confirmed by Western blotting analysis of cleaved caspase-3 (c-CAS-3) and of the Bax/Bcl-2 ratio. Both of these markers were significantly increased in FVB mice, but alleviated in MT-TG mice, after the induction of MI (Fig. 1E and Supplementary Fig. S6).
To mimic ischemic-injured FVB and MT-TG mice in vitro, stable human MT-IIA-overexpressing H9c2 cardiomyocytes (H9c2MT7) and control H9c2 cardiomyocytes were subjected to oxygen and glucose deprivation (OGD). Exposure of H9c2 cells to OGD significantly increased the number of TUNEL-positive cells, the level of c-CAS-3, and the Bax/Bcl-2 ratio. By contrast, OGD-induced apoptosis of H9c2MT7 cardiomyocytes was effectively reduced by MT overexpression (Supplementary Figs. S1A, B, and S10).
Taken together, these results suggested that MT plays an important role in the maintenance of cardiac function and in reducing cell apoptosis in response to MI or OGD.
MT transduces cardioprotective signals in MI via pathways that are not entirely dependent on the elimination of ROS
MT is an intracellular antioxidative protein with marked ability to scavenge ROS, which are important pathogenic mediators of myocardial damage in MI. Therefore, the association between the ability of MT to remove ROS and its cardioprotective effects during MI was evaluated.
ROS accumulation was detected by Amplex Red reagent, which detects a specific ROS: hydrogen peroxide, and dichloro-dihydro-fluorescein diacetate (DCFH-DA) probe, which detects ROS nonspecifically.
ROS accumulation after MI was much greater in the hearts of FVB than those of MT-TG mice (Fig. 2A). To investigate the contribution of ROS elimination to the phenotype of MT-TG mice in response to MI, FVB mice received daily intraperitoneal (i.p.) injections of various doses of Mn(III)tetrakis (1-methyl-4-pyridyl) porphyrin pentachloride (MnTMPyP), a cell-permeable dismutase mimetic that acts as a ROS scavenger, for 1 week immediately after LAD ligation. As expected, intracellular ROS induced by MI decreased, especially in mice treated with 15 mg/kg MnTMPyP, with these mice having the same cardiac ROS level as MT-TG mice (Fig. 2A).
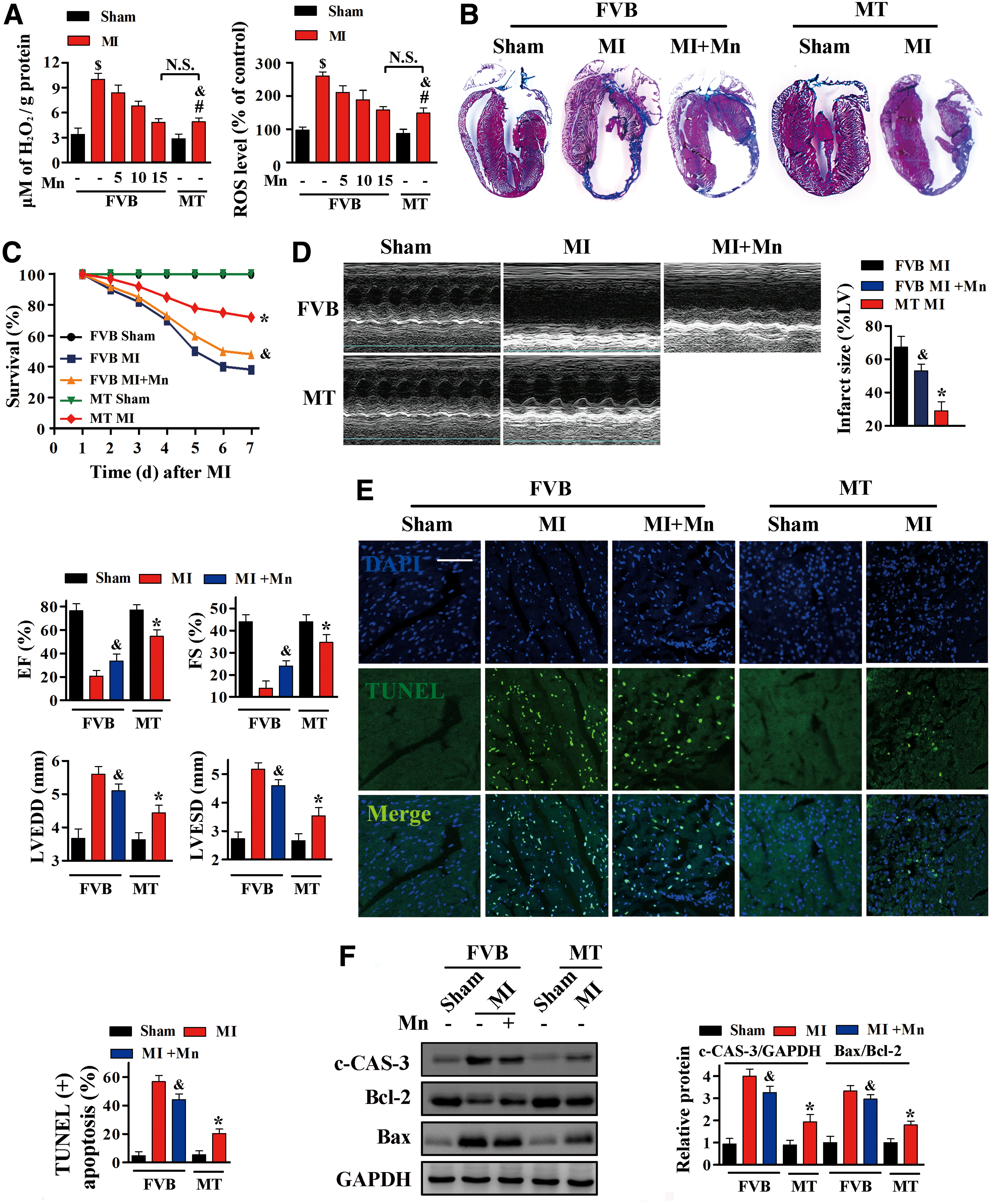
Interestingly, after MI, infarct size was much smaller (Fig. 2B), survival rates were higher (Fig. 2C), and cardiac function was better (Fig. 2D) in MT-TG mice than in FVB mice treated with MnTMPyP, which have the same cardiac ROS level as MT-TG mice after MI. Furthermore, TUNEL assays and Western blotting analysis indicated that cardiomyocyte apoptosis after MI was more extensive in FVB mice treated with 15 mg/kg MnTMPyP than in MT-TG mice (Fig. 2E, F and Supplementary Fig. S6).
Consistent with these in vivo results, ROS accumulation induced by OGD was effectively inhibited by treatment with MnTMPyP, especially at a concentration of 150 μM, similar to the effects of MT overexpression in H9c2MT7 cardiomyocytes (Supplementary Fig. S2A). However, the number of TUNEL-positive cells after OGD was lower in H9c2MT7 cells than in H9c2 cells treated with 150 μM MnTMPyP (Supplementary Fig. S2B).
Collectively, these results suggested that the mechanism underlying the cardioprotection of MT involved more than the elimination of ROS.
Considering that cardiac-specific catalase-overexpressing transgenic (CAT-TG) mice exhibited strong ROS eliminating ability in the heart (19, 56), the above experiments were also performed in CAT-TG mice to confirm our observations. Catalase-overexpressing levels in those mice were reflected as the elevation of catalase activities in the heart (19), for the CAT-TG mice we chose in this study, the cardiac catalase activities were ∼60-fold higher than those for the control mice, and the cardiac ROS levels after MI did not differ significantly in CAT-TG mice and age-matched MT-TG mice (Supplementary Fig. S3A).
As expected, infarct size was greater (Supplementary Fig. S3B) and survival rates were lower (Supplementary Fig. S3C) in CAT-TG than in MT-TG mice. Furthermore, echocardiography showed no significant differences in cardiac systolic and diastolic functions between CAT-TG and MT-TG mice before ischemic injury. One week after MI, however, EF (%) and FS (%) were much lower, while LVEDD and LVESD were higher in CAT-TG than in MT-TG mice (Supplementary Fig. S3D). TUNEL assays and Western blotting analysis showed that cardiomyocyte apoptosis after MI was much lower in MT-TG than in CAT-TG mice (Supplementary Figs. S3E, F, and S11). These findings confirmed that the cardioprotective effects of MT against MI were not due simply to the elimination of ROS, but that other mechanisms are likely involved.
MT activates the mTORC1 and mTORC2 signaling pathways in vivo and in vitro
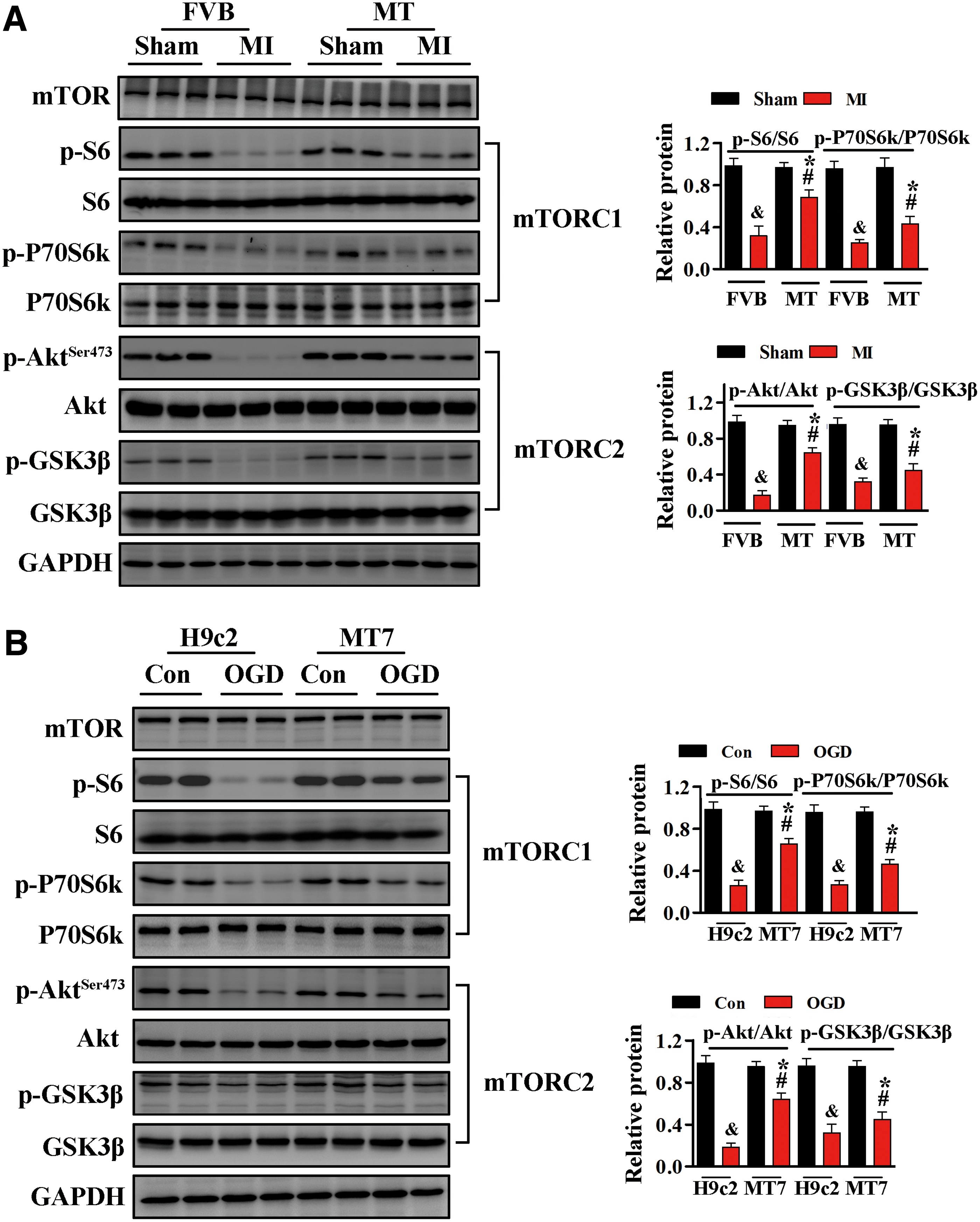
Dysregulation of mTOR signaling in cardiac diseases contributes to cardiomyocyte apoptosis, with associated impairment of cardiac function (7, 23, 36, 47, 53). Accordingly, we evaluated the effects of mTOR signaling on MT-mediated cardioprotection. mTORC1 signaling was assessed by evaluating the phosphorylation levels of S6 and P70S6k (53), whereas mTORC2 signaling was assessed by evaluating the phosphorylation levels of Akt and GSK3β (47).
After MI, the levels of phosphorylation of both sets of proteins were markedly decreased in the hearts of FVB mice (Fig. 3A and Supplementary Fig. S7). However, cardiac-specific MT overexpression increased the levels of p-S6 and p-Akt Ser473 in myocardial tissues from ischemic hearts (Fig. 3A). Exposure of H9c2 cells to OGD resulted in markedly reduced phosphorylation levels of S6 and Akt. By contrast, the reductions in p-S6 and p-Akt Ser473 after OGD were effectively alleviated in H9c2MT7 cells (Fig. 3B and Supplementary Fig. S7).
mTORC2 inhibition abrogates the cardioprotective effects of MT in vivo and in vitro
Although our results indicated that MT activates both mTORC1 and mTORC2 after MI or OGD, these experiments could not determine which one plays a dominant role in the cardioprotective effects of MT. Therefore, immediately after MI, MT-TG mice were treated with 20 mg/kg Torin1 or 8 mg/kg rapamycin daily for 1 week to inhibit mTOR. The effects of different inhibitors were assessed by several key molecular signaling.
Treatment of MT-TG mice with Torin1 markedly reduced mTORC1 and mTORC2 activities, as indicated by the reductions in p-S6 and p-Akt Ser473 (Supplementary Figs. S4A and S11). By contrast, rapamycin treatment inhibited only mTORC1 activity, as indicated by reductions in p-S6 (Supplementary Figs. S4B and S11). More importantly, Torin1 treatment induced an additional increase of infarct size after MI in MT-TG mice compared with vehicle-treated mice (Fig. 4A), as well as significantly impairing cardiac functions (Fig. 4B). In addition, Torin1 administration significantly increased cell apoptosis, contributing to increases in infarct size and cardiac dysfunction (Fig. 4C–E and Supplementary Fig. S8). By contrast, rapamycin did not alter the protective effects of MT on infarct size, cardiac function, and apoptosis (Fig. 4A–E).

Similar to our in vivo findings, in vitro exposure of H9c2MT7 cells to Torin1 and rapamycin during OGD showed that Torin1 suppressed the activities of both mTORC1 and mTORC2, whereas rapamycin suppressed only mTORC1 activity (Supplementary Figs. S5A and S11). TUNEL assays and Western blotting analysis suggested that Torin1 exacerbated OGD-induced apoptosis in H9c2MT7 cells (Supplementary Fig. S5B, C), whereas rapamycin did not (Supplementary Fig. S5B, C).
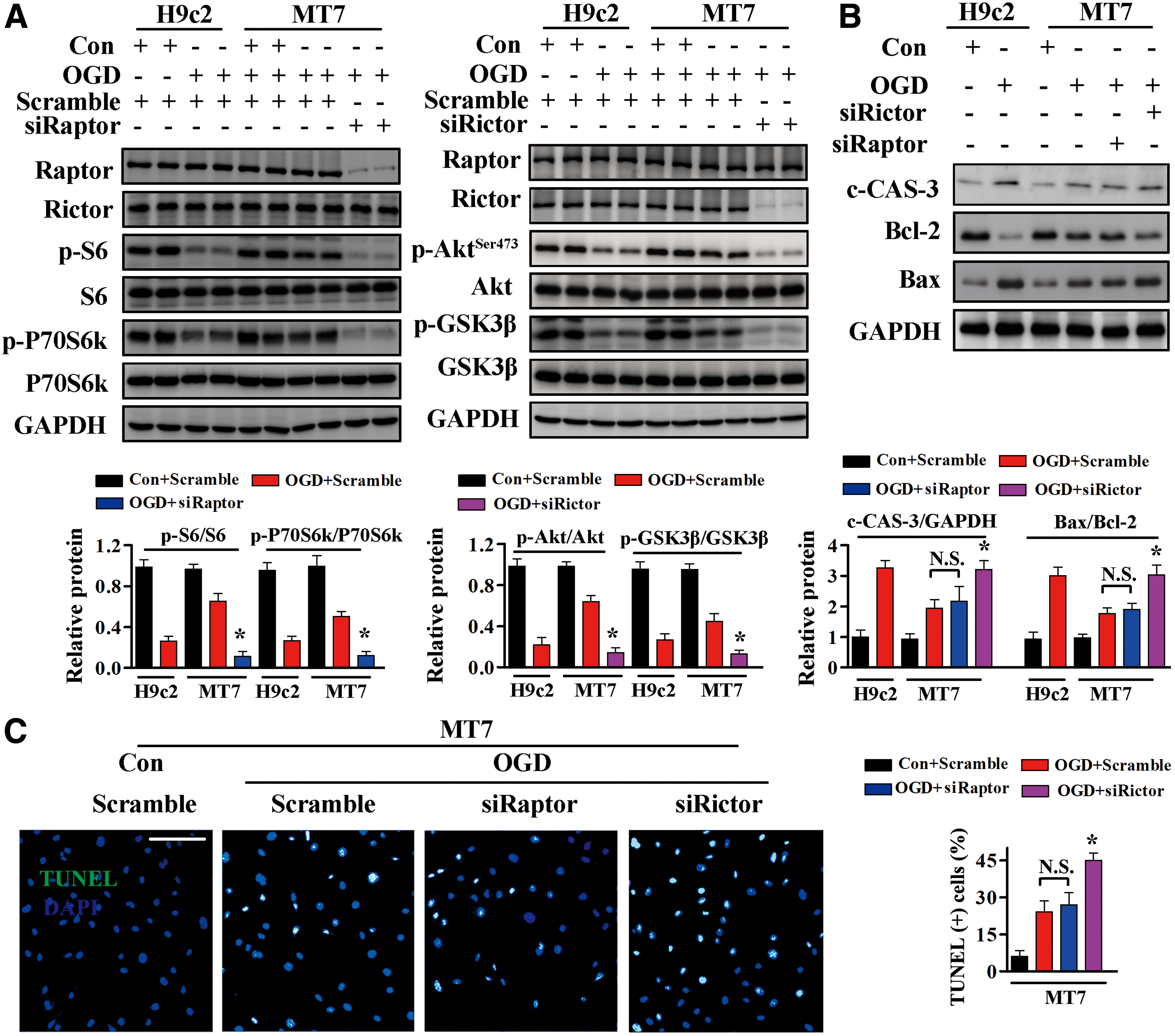
To further assess whether the mTORC2 activation is involved in the cardioprotective effects of MT, H9c2MT7 cells were transfected with small interfering RNAs (siRNAs) specific for Raptor or Rictor and then subjected to OGD. Transfection with Raptor siRNA dramatically suppressed mTORC1 activity, whereas transfection with Rictor siRNA markedly reduced mTORC2 activity, in H9c2MT7 cardiomyocytes (Fig. 5A and Supplementary Fig. S8). H9c2MT7 cell apoptosis did not differ significantly after transfection of Raptor or scramble siRNA (Fig. 5B, C and Supplementary Fig. S8), whereas siRNA-mediated knockdown of Rictor abolished the protective effects of MT against OGD-induced cell apoptosis, as evidenced by increased levels of TUNEL-positive cells (Fig. 5C) and c-CAS-3 (Fig. 5B), and increases in the Bax/Bcl-2 ratio (Fig. 5B).
The functional activities of mTORC1 and mTORC2 were selectively inhibited in vivo by targeted downregulation of Rictor or Raptor expression using adenoviruses carrying Raptor short hairpin RNA (shRNA), Rictor shRNA, or control scramble sequences. These adenoviruses were injected into the myocardia of MT-TG mice, followed by LAD ligation after 1 week, with successful transfection confirmed by the presence of GFP in the myocardia (Fig. 6A). Transfection of Ad-sh-Rictor or Ad-sh-Raptor into MT-TG hearts dramatically reduced Rictor or Raptor expression, respectively (Fig. 6B and Supplementary Fig. S9).
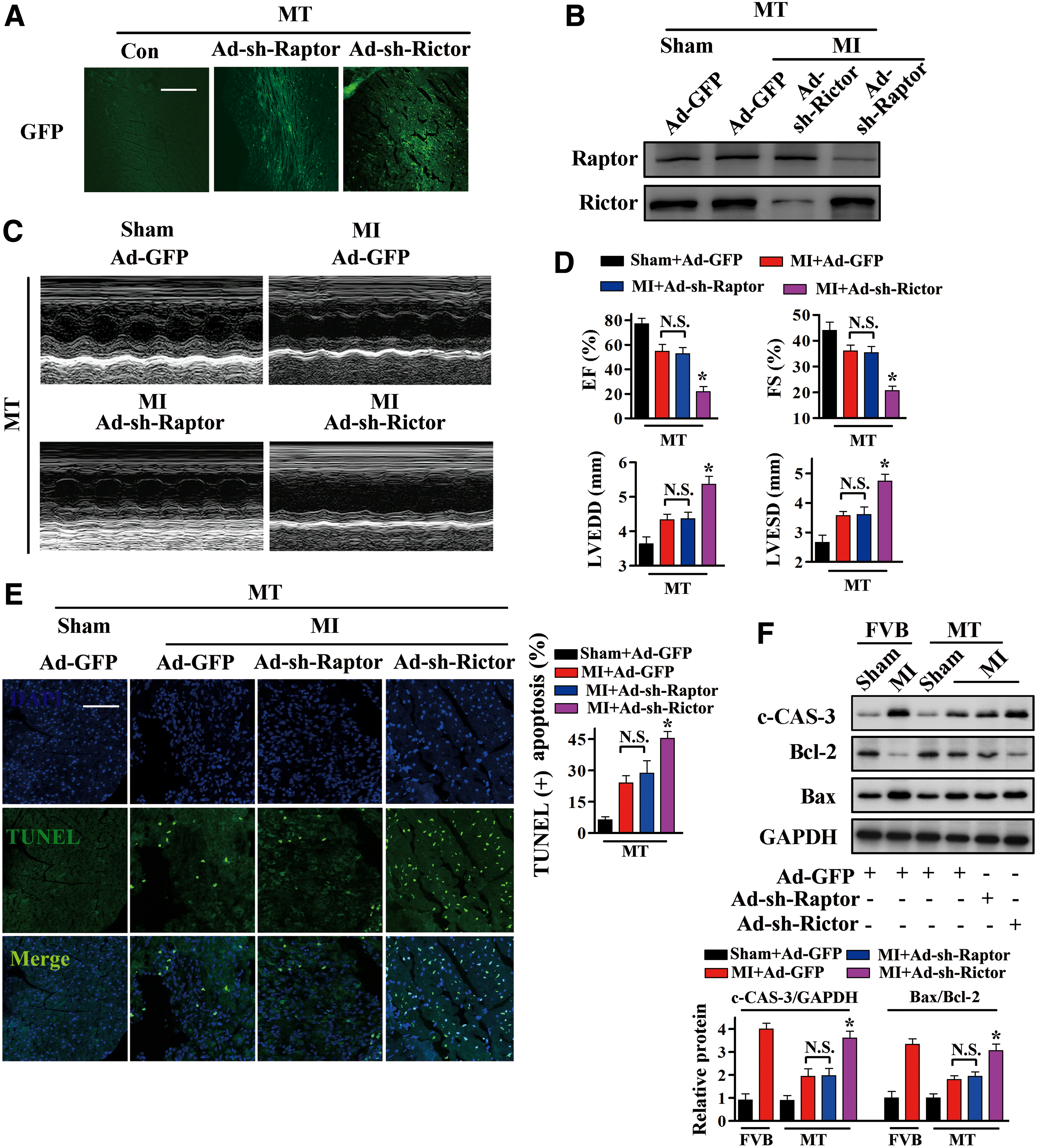
After MI, cardiac function was significantly impaired in MT-TG hearts transfected with Ad-sh-Rictor compared with hearts transfected with Ad-GFP, as indicated by increases in LVESD and LVEDD, and reductions in EF (%) and FS (%) (Fig. 6C, D). In addition, transfection with Ad-sh-Rictor significantly increased myocardial apoptosis, as evidenced by increased levels of TUNEL-positive cells (Fig. 6E) and c-CAS-3 (Fig. 6F and Supplementary Fig. S9), and increases in the Bax/Bcl-2 ratio (Fig. 6F). By contrast, transfection of Ad-sh-Raptor into MT-TG mouse hearts did not significantly alter heart function or cell apoptosis compared with Ad-GFP transfected hearts after MI (Fig. 6C–F). These results indicated that inactivation of mTORC2 abolished the cardioprotective effects of MT and that mTORC2, but not mTORC1, is essential for the cardioprotection of MT against MI.
MT promotes FoxO3a translocation to the cytoplasm after MI or OGD
Because FoxO3a is a substrate of Akt that at least partly facilitates its proapoptotic effects, we assessed whether FoxO3a is involved in the cardioprotective effects of MT. FoxO3a phosphorylation was significantly lower in FVB mice after MI than after Sham operation, but this reduction was blocked by MT overexpression (Fig. 7A and Supplementary Fig. S9). Western blotting analysis of cytoplasmic and nuclear fractions showed that the nuclear translocation of FoxO3a was increased after MI in FVB mice, and that this translocation was suppressed by MT overexpression (Fig. 7B and Supplementary Fig. S9).
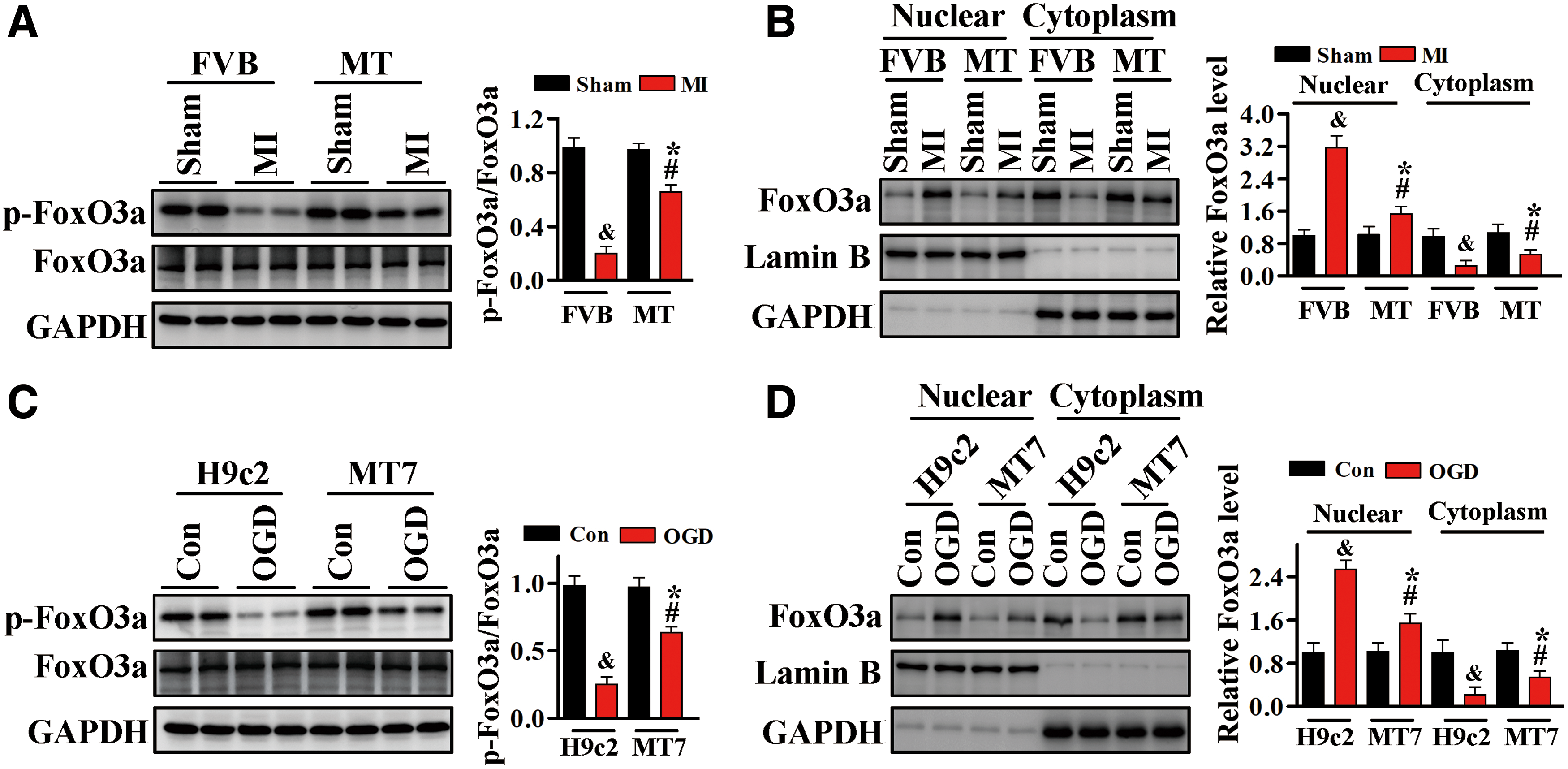
Consistent with these in vivo results, we found that FoxO3a phosphorylation in H9c2 cardiomyocytes was significantly decreased after OGD, whereas FoxO3a phosphorylation was higher in H9c2MT7 cells, which overexpress MT (Fig. 7C and Supplementary Fig. S9). Measurement of FoxO3a protein levels in the nuclear and cytoplasmic fractions of H9c2 and H9c2MT7 cells showed that OGD markedly increased the nuclear level of FoxO3a protein in H9c2 cells, but that this increase was blocked in H9c2MT7 cells (Fig. 7D and Supplementary Fig. S9).
MT inhibits the nuclear translocation of FoxO3a thus affects its transcriptional activity to exert the cardioprotective effects
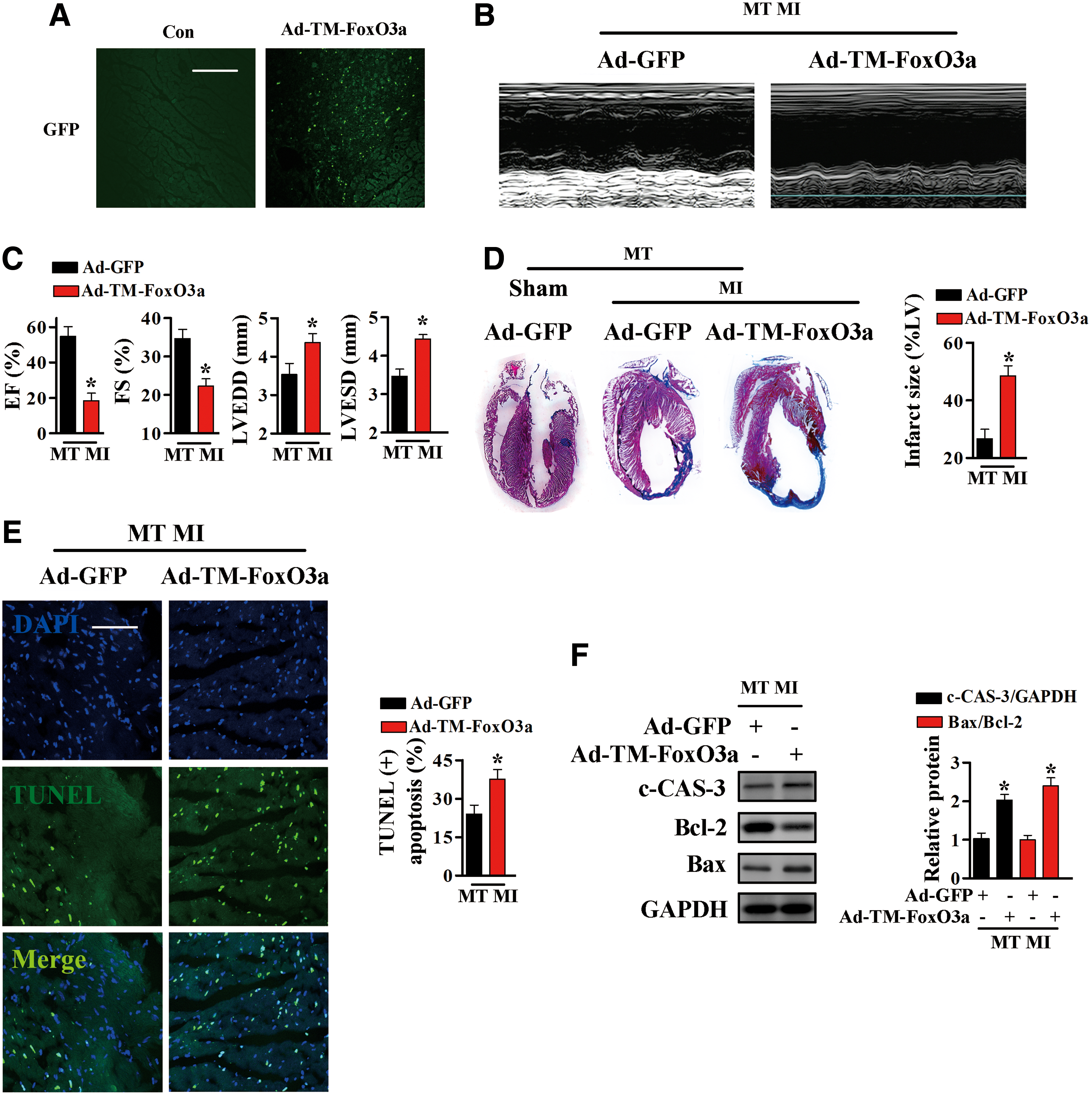
To test whether FoxO3a transcriptional activity is involved in the cardioprotective effects of MT, an adenoviral vector expressing constitutively active FoxO3a (TM-FoxO3a) and a vector expressing GFP were injected into the myocardia of MT-TG mice. In TM-FoxO3a, the serine/threonine residues at positions 32, 253, and 315 were replaced by alanine residues; TM-FoxO3a is constitutively active because it cannot be inactivated by phosphorylation (5, 40).
One week after injection of the adenoviral vectors into the myocardia of MT-TG mice, these mice were subjected to LAD ligation. Immunofluorescence was performed to confirm the presence of the TM-FoxO3a-expressing adenoviral vectors (Fig. 8A). Nuclear TM-FoxO3a in MT-TG mice completely blocked the improvements in post-MI heart function (Fig. 8B, C) and reductions in infarct size resulting from MT overexpression (Fig. 8D). Furthermore, Ad-TM-FoxO3a transfection induced higher level of apoptosis in MT-TG hearts after MI, as demonstrated by increased levels of TUNEL-positive cells (Fig. 8E) and c-CAS-3, and increases in the Bax/Bcl-2 ratio (Fig. 8F and Supplementary Fig. S9).
In vitro evidence supporting the role of FoxO3a in MT-associated cardioprotection was obtained by transfecting H9c2MT7 cells with adenovirus expressing TM-FoxO3a or GFP and subjecting these cells to OGD. Transgene expression was assessed by the detection of GFP immunofluorescence or by nuclear/cytosolic fractionation (Fig. 9A, B and Supplementary Fig. S10). In agreement with our in vivo results, Ad-TM-FoxO3a transfection significantly increased the proportion of TUNEL-positive cells (Fig. 9C), the levels of c-CAS-3, and the Bax/Bcl-2 ratios (Fig. 9D and Supplementary Fig. S10).
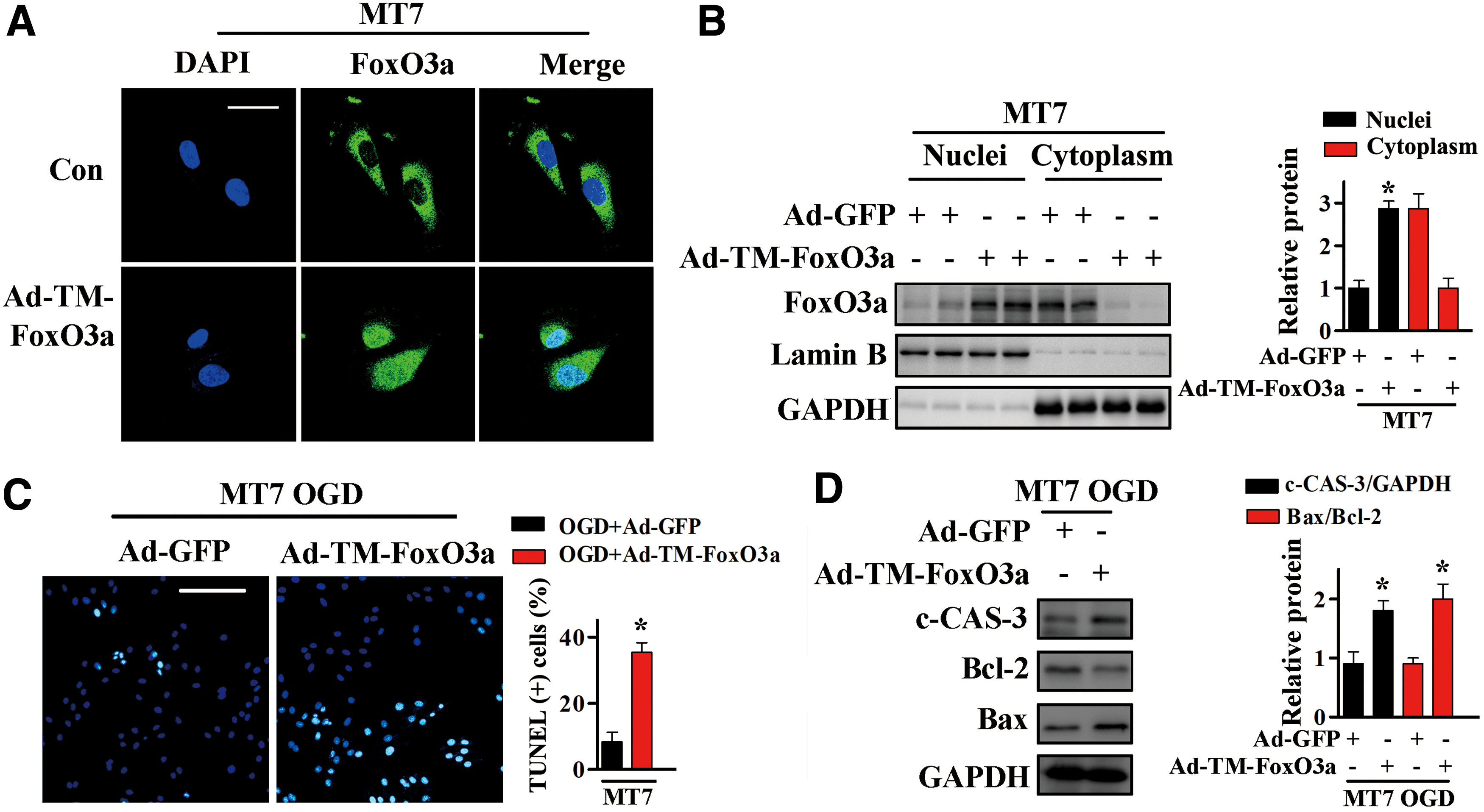
Collectively, these results demonstrated that the cardioprotective activity of MT during MI involves the downregulation of FoxO3a transcriptional activity.
MT downregulates Bim expression in a FoxO3a-dependent manner
Bim, a member of the Bcl-2 family and a downstream target of FoxO3a, is an important mediator of cell apoptosis in MI (50, 54). The involvement of Bim in MT-associated cardioprotection was assessed by Western blotting analysis. Bim expression was significantly higher in FVB than in MT-TG mice after MI (Fig. 10A and Supplementary Fig. S10). These changes were also confirmed by reverse transcription polymerase chain reaction (RT-PCR), which showed that Bim messenger RNA (mRNA) levels were higher in FVB than in MT-TG mice after MI (Fig. 10A). Compared with MT-TG mice transfected with Ad-GFP, transfection with Ad-TM-FoxO3a abolished the ability of MT to inhibit MI-stimulated upregulation of Bim (Fig. 10B and Supplementary Fig. S10).
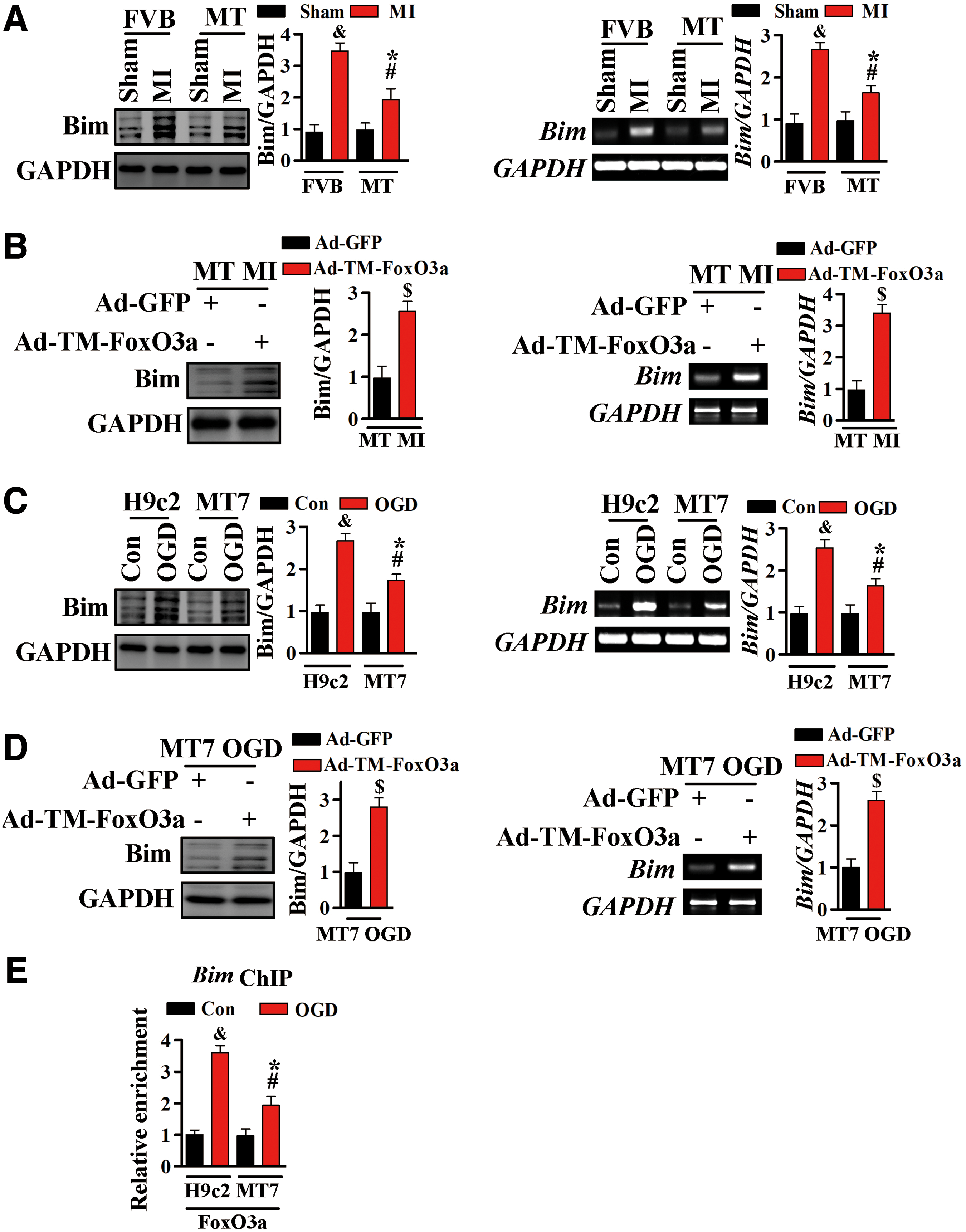
Consistent with our in vivo results, Bim protein and mRNA levels were higher in H9c2 cells after exposure to OGD, but were significantly inhibited by MT overexpression (Fig. 10C and Supplementary Fig. S10). Moreover, constitutively active FoxO3a markedly reduced the ability of MT to inhibit OGD-stimulated Bim upregulation (Fig. 10D and Supplementary Fig. S10).
To further confirm that these changes in Bim were mediated by FoxO3a, ChIP assays using FoxO3a-specific antibodies were performed to assess the physical interaction between endogenous FoxO3a and Bim promoter after ischemic injury. Exposure to OGD markedly increased the association of FoxO3a with the Bim promoter in H9c2 cells, an association significantly inhibited by MT overexpression (Fig. 10E). This and the preceding results suggested that, under conditions of OGD, the restrained transcriptional activity of FoxO3 (mainly manifested as reduced recruiting to the Bim promoter) contributed by MT was mediated by a mechanism involving mTORC2/Akt activation.
Discussion
During the pathogenesis of MI, a host of insults collectively inflicts damage on cardiomyocytes, contributing to the loss of heart function, acute cardiac arrest, and death. This study showed that MT has cardioprotective effects against MI, and that this protection involves the activation of the mTORC2/FoxO3a/Bim-mediated antiapoptotic pathway.
Most previous studies of MT focused on its ability to scavenge ROS. By contrast, our results are the first to indicate that MT-associated cardioprotection was not entirely dependent on the ability of MT to eliminate ROS after MI. MnTMPyP and CAT-TG mice were utilized to verify whether the ability to remove ROS is key to the cardioprotective effects of MT. We found that infarct size was smaller, the level of apoptosis was lower, and cardiac function was better in MT-TG mice after MI than in MnTMPyP-treated mice or CAT-TG mice, suggesting that the cardioprotection conferred by MT after MI was due at least in part to a mechanism other than scavenging ROS.
The mTOR pathway is likely responsible for cardiomyocyte apoptosis induced by MI, with this apoptosis being considered the primary cause of cardiac dysfunction and heart failure. Although we found that MT activates both mTORC1 and mTORC2 after MI or OGD, the effects of mTOR activation on MT-mediated cardioprotection remain unclear. Because mTORC1 and mTORC2 have different functions, their possible roles in MT-mediated cardioprotection were assayed.
Pharmacological inhibition of mTORC1 and mTORC2 with Torin1 blocked MT-associated cardioprotection in vitro and in vivo, whereas rapamycin did not alter the ability of MT to protect the heart from MI. Genetic deletion of mTORC2 by silencing of Rictor abolished the ability of MT to protect the heart from ischemic injury, both in vitro and in vivo. By contrast, genetic deletion of mTORC1 by silencing of Raptor did not affect MT-associated cardioprotection. Taken together, these results indicated that mTORC2, but not mTORC1, is essential for the cardioprotection of MT.
Nevertheless, MT overexpression and its associated mTORC2 activation may be complicated. MT is a transition metal-binding protein associated with metal homeostasis in living organisms (18). In mammals, MT binds predominantly to zinc, which is involved in the metabolism of nucleic acids, proteins, lipids, and carbohydrates, as well as in the control of gene expression and cell growth, differentiation, and proliferation (18, 20). Overexpression of MT can result in Zn sequestration (18), thus inhibiting the activation of p53, another zinc-binding protein and its key biochemical activity depends on the incorporation of zinc (15), and subsequently leading to the activation of mTORC2 (6). This regulatory loop suggests a possible link between MT and mTORC2, but other mechanisms may also be present.
In addition, previous studies have demonstrated that high-zinc diet or zinc sulfate injection could also induce MT expression in the heart, thus protecting against diabetic cardiomyopathy (13, 48). These findings indicated that MT and its associated metal homeostasis change play important roles in the treatment of various diseases.
mTORC2 is associated with Akt activation, and activated Akt (phospho-Akt) can block apoptosis by inactivating several targets, including Bad, forkhead transcription factors, and caspase-9. Reduced Akt Ser473 phosphorylation after Rictor silencing may reduce the phosphorylation of its downstream targets, including FoxO, and contribute to cell death (29, 33, 55). FoxO3a, a downstream target of Akt, is involved in regulating various cellular functions, including differentiation, metabolism, proliferation, and apoptosis (2, 38, 49). Overexpression of constitutively active FoxO3a could markedly enhance the expression of its target gene Bim, resulting in apoptosis (44).
Because mTORC2 is a primary Akt Ser473 kinase, which is functionally connected to FoxO3a-linked cardiomyocyte apoptosis (5, 12, 44), we assessed the role of FoxO3a in MT-induced cardioprotection after MI. We found that MT induced the phosphorylation (inactivation) of FoxO3a in the hearts of mice after MI and in cardiomyocytes subjected to OGD. FoxO3a inactivation markedly reduced the expression of its transcriptional target, the proapoptotic gene Bim. Activated Bim can directly interact with proapoptotic factors, such as Bax, to form a complex, which can translocate into the mitochondrial membrane, promoting the release of cytochrome c and activating caspase-dependent apoptosis (27). Transfection of adenoviral vectors expressing constitutively active FoxO3a (TM-FoxO3a) inhibited MT-mediated cardioprotection after MI or OGD.
Thus, the possible mechanism underlying the protective effects of MT against MI might include the activation of the mTORC2/FoxO3a/Bim signaling pathway, via four steps: (i) MT activates Akt by inducing mTORC2 activity, (ii) p-Akt Ser473 inactivates FoxO3a by phosphorylating FoxO3a, (iii) phosphorylation of FoxO3a results in its nuclear exclusion and transcriptional inactivation, and (iv) the expression of Bim, a transcriptional target of FoxO3a, is reduced, downregulating apoptosis and protecting the heart.
The most important finding of this study is that MT has significant cardioprotective effects, reducing infarct size, improving cardiac function, and inhibiting apoptosis, through the mTORC2/FoxO3a/Bim pathway.
Materials and Methods
Animals
Wild-type FVB mice, cardiac-specific MT-TG mice, and cardiac-specific CAT-TG mice with 8–10 weeks of age were used in our study. MT-TG mice and CAT-TG mice are all in FVB genetic background, and are generated as previously described (19, 20). For the MT-TG mice, human MT-IIA is overexpressed ∼10-fold in the cardiomyocytes, and for the CAT-TG mice, cardiac catalase activities are ∼60-fold higher than those for the control mice. All mice were housed in temperature-controlled environment under a 12-h light/dark cycle, and offered free access to food and water. All experimental procedures for animal studies were carried out conforming to the guide for the care and use of laboratory animals and were approved by the Animal Policy and Welfare Committee of Wenzhou Medical University, Wenzhou, Zhejiang Province, China.
For MI, the LAD branch of the coronary artery was ligated in male mice. In brief, a rodent ventilator was used with 65% oxygen during the surgical procedure. After mice were anesthetized by isoflurane inhalation, the chest was opened by a horizontal incision via the space between the fourth and fifth ribs. Ischemia was achieved by ligating the LAD using a 7-0 nylon suture. Regional ischemia was confirmed by the cyanosis on the myocardial surface and dyskinesis of the ischemic region. The Sham-operated animals underwent the same surgical procedures, while the ligature around the LAD was not fastened. No differences in perioperative mortality rates (within 24 h) were observed among the FVB, MT-TG, and CAT-TG mice. At least six mice were contained in each group, including Sham-operated group and MI group.
For assessment of post-MI survival, mice that had survived the first 24 h after surgery were followed for 1 week and were inspected daily.
For intramyocardial gene delivery, MT-TG mice were anesthetized with isoflurane inhalation and intubated, the chest was opened through a midline sternotomy, mice received an intramyocardial injection with Ad-GFP or Ad-sh-Raptor or Ad-sh-Rictor or Ad-TM-FoxO3a. Each mouse received a 4-point injection in a total volume of 100 μL. One week after intramyocardial injection, mice were subjected to MI surgery.
Cell culture
H9c2 and H9c2MT7 (stable overexpression of human MT-IIA) rat cardiomyocytes were purchased from American Type Cell Collection (ATCC) and cultured at 37°C under 5% (v/v) CO2 in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (Gibco, Thermo Fisher Scientific) and 1% penicillin/streptomycin.
OGD model and drug treatment
The cells were subjected to OGD to mimic myocardial ischemia. OGD was initiated as previously described (14). In brief, the complete growth medium was replaced by serum-free, glucose-free modified Eagle's medium, and the cells were incubated at 37°C with 5% CO2 and 95% N2 (v/v) for 3 h in a modular incubator chamber. The control cells were kept in Neurobasal Medium in a normal environment.
During hypoxia, the following chemicals were applied as indicated in the figure legends: MnTMPyP (0–150 μM), rapamycin (50 nM), and Torin1 (100 nM).
For the adenovirus infection, H9c2MT7 cells were washed with phosphate buffered saline (PBS), then serum-free DMEM medium containing diluted viral stock was added to wells (multiplicity of infection = 20).
Echocardiography measurement
At the end of 1 week after the coronary occlusion, six mice from each group were used for cardiac function determination by using a Sonos-5500 echocardiograph (Agilent Technologies) with a 15-MHz linear transducer. Mice during echocardiographic analysis were anesthetized with an i.p. injection of 2,2,2-tribromoethanol (240 mg/kg; Wako Pure Chemical Industries). LVEDD and LVESD were measured to calculate left ventricle (LV) FS % and LV EF %.
Measurement of the infarct size
Masson's trichrome-stained slides were examined by light microscopy scanning instrument, digitized and analyzed using ImageJ software. The infarct size was assessed morphologically and calculated as the ratio of the total infarct area to the total endocardial circumference.
Oxidative stress measurement
For the detection of ROS accumulation, Amplex Red reagent (Invitrogen) was used to detect hydrogen peroxide levels in mouse hearts and H9c2 cells according to the manufacturer's protocol and a previous report (28). For mouse hearts, fresh frozen tissue (30–60 μg) was incubated in Amplex Red working solution (containing 100 μM Amplex Red reagent and 1 U/mL horseradish peroxidase) in HEPES buffer for 30 min at 37°C. The supernatant was then collected, and the fluorescence was measured at 530 nm excitation wavelength and 590 emission wavelength, using a Mithras LB 940 reader (Berthold Technologies). All procedures were protected from light, and a standard curve using fresh H2O2 was generated for comparison. The tissue was then dried and weighed, and the final results were normalized to dry tissue weight.
For the detection of hydrogen peroxide levels in H9c2 cells, equal amounts of cells seeded on 96-well plates were incubated with Amplex Red working solution (containing 50 μM Amplex Red reagent and 0.1 U/mL horseradish peroxidase) for 30 min at 37°C. Fluorescence was measured at 530 nm excitation wavelength and 590 emission wavelength, using a Mithras LB 940 reader (Berthold Technologies). All procedures were protected from light, and a standard curve using fresh H2O2 was generated for comparison.
The production of ROS was also measured by the DCFH-DA probe (Sigma). For the in vivo study, 5 μg protein (homogenate) was incubated with 4 μM DCFH-DA for 10 min at 37°C, and for the in vitro study, equal amounts of cells cultured in 96-well plates were incubated with 25 μM DCFH-DA for 15 min at 37°C, all protected from light. The fluorescence was then measured at 485 nm excitation wavelength and 535 emission wavelength using a Mithras LB 940 reader; the data were analyzed with MikroWin 2010 software (Berthold Technologies).
TUNEL staining
The TUNEL staining was performed using a commercially available in situ apoptosis detection kit (Roche Molecular Biochemicals) according to the manufacturer's protocol for apoptotic cell nuclei and 4′6-diamidino-2-phenylindole (DAPI; Sigma) for all cell nuclei, as previously described (43).
The apoptotic index was determined as the number of TUNEL-positive nuclei divided by the total number of nuclei stained with DAPI. For heart tissues, specimens were fixed by 4% paraformaldehyde in PBS overnight at 4°C, permeabilized with 0.1% Triton X-100, and then incubated with TUNEL reagent containing terminal deoxynucleotidyl transferase and fluorescent isothiocyanate-dUTP. For cells, equal numbers of cells were exposed to OGD injury, and then treated as described above with the 4% paraformaldehyde fixation for 15 min at room temperature. DAPI was used for nuclear staining. TUNEL-positive cardiomyocytes were counted in randomly selected three fields of the slide. Images were visualized and captured with a Leica SP8 confocal microscopy.
RNA isolation and semiquantitative RT-PCR
Total RNA was extracted from frozen heart or cultured cells by using TRIzol reagent (Invitrogen) and reverse-transcribed into complementary DNA (cDNA) by using the GoScript Reverse Transcription Kit (Promega). The cDNA was then subjected to semiquantitative RT-PCR analysis, and gene expression was quantified as previously described (22). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was tested as an internal control.
Primer sequences are shown as follows: Bim (rat): sense 5′-GTCTTCCGCCTCTCGGTAAT-3′, antisense 5′-AGAGATACGGATCGCACAGG-3′; Bim (mouse): sense 5′-CGGATCGGAGACGAGTTCA-3′, antisense 5′-TTCAGCCTCGCGGTAATCA-3′; GAPDH (rat): sense 5′-TGGCGCTGAGTACGTCGTG-3′, antisense 5′-ATGGCATGGACTGTGGTCAT-3′; GAPDH (mouse): sense 5′-AGGTCGGTGTGAACGGATTTG-3′, antisense 5′-TGTAGACCATGTAGTTGAGGTCA-3′.
Nuclear/cytoplasmic fractionation
The heart tissues and cardiomyocytes were subjected to nuclear and cytosolic fractionation using Nuclear/Cytosolic Fractionation Kit (BioVision, Milpitas, CA), following the protocol recommended by the manufacturer.
RNA interference
Cells were transfected with Raptor small interfering RNA (siRaptor; sc-270140; Santa Cruz) or Rictor small interfering RNA (siRictor; sc-270141; Santa Cruz) or siRNA scrambled (negative) control (siScramble; sc-37007; Santa Cruz) by using Lipofectamine 2000 for 12 h in Opti-MEM. Cells were then placed in full-growth medium for 24 h before treatment with OGD.
Immunoblotting assays
Immunoblotting assays were used to detect cleaved caspase-3 (Cell Signaling Technology), Bcl-2 (Abcam), Bax (Abcam), mTOR (Cell Signaling Technology), FoxO3a (Cell Signaling Technology), p-FoxO3a (Cell Signaling Technology), Akt (Cell Signaling Technology), p-Akt (Cell Signaling Technology), p-P70S6k (Cell Signaling Technology), P70S6k (Cell Signaling Technology), p-S6 (Cell Signaling Technology), S6 (Cell Signaling Technology), GSK3β (Cell Signaling Technology), p-GSK3β (Cell Signaling Technology), and Lamin B (Cell Signaling Technology). Total protein from heart tissues or cells was extracted with lysis buffer. The protein concentration was measured by Bradford assay.
Twenty to fifty micrograms of protein were then subjected to electrophoresis on 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis gel at 120 V and transferred to a PVDF membrane. Membranes were blocked with 5% bovine serum albumin in Tris-buffered saline containing 0.1% Tween 20 and incubated with primary antibodies overnight at 4°C. Subsequently, proteins were detected after incubation with horseradish peroxidase-conjugated secondary antibodies (Dako Cytomation) and visualized with enhanced chemiluminescence reagent ECL (GE Healthcare). The expression of specific antigens was quantified using Image Quant 5.2 software (Molecular Dynamics, Inc.), and the expression of GAPDH (Abcam) was used as a loading control, except where indicated.
ChIP assay
ChIP was performed as described previously (52). The primer sequences of Bim promoter were as follows: forward, 5′-TGCCACCAAAGATCTCTACC-3′; reverse, 5′-GCATTTCCTCACAGAGTTGG-3′.
Statistical analysis
Data were analyzed by GraphPad Prism 5.0, and results are expressed as mean ± standard error of the mean. Differences in each sample were evaluated using unpaired Student's two-tailed t-test or analysis of variance. A value of p < 0.05 was considered significant. All experiments were repeated at least three times.
Footnotes
Acknowledgments
This work was supported by the National Nature Science Foundation of China under Grants 81570368, 81573069, 81600304, 81770498, and 81773346, the Zhejiang Provincial Natural Science Foundation (LY19H020005 and LYY18H310009), and the Technology Program of Wenzhou (Y20160001 and Y20160004).
Author Disclosure Statement
No competing financial interests exist.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Figure S7
Supplementary Figure S8
Supplementary Figure S9
Supplementary Figure S10
Supplementary Figure S11
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.