
Abstract
Significance:
Calpains (CAPNs) are a family of calcium-activated cysteine proteases. The ubiquitous isoforms CAPN1 and CAPN2 have been involved in the maintenance of vascular integrity, but uncontrolled CAPN activation plays a role in the pathogenesis of vascular diseases.
Recent Advances:
It is well accepted that chronic and acute overproduction of reactive oxygen species (ROS) is associated with the development of vascular diseases. There is increasing evidence that ROS can also affect the CAPN activity, suggesting CAPN as a potential link between oxidative stress and vascular disease.
Critical Issues:
The physiopathological relevance of ROS in regulating the CAPN activity is not fully understood but seems to involve direct effects on CAPNs, redox modifications of CAPN substrates, as well as indirect effect on CAPNs via changes in Ca2+ levels. Finally, CAPNs can also stimulate ROS production; however, data showing in which context ROS are the causes or the consequences of CAPN activation are missing.
Future Directions:
Detailed characterization of the molecular mechanisms underlying the regulation of the different members of the CAPN system by specific ROS would help understanding the pathophysiological role of CAPN in the modulation of the vascular function. Moreover, given that CAPNs have been found in different cellular compartments such as mitochondria and nucleus as well as in the extracellular space, identification of new CAPN targets as well as their functional consequences would add new insights in the function of these enigmatic proteases.
Introduction
The family of Ca2+-dependent cysteine proteases, or calpains (CAPNs), mediates the limited proteolytic cleavage of a spectrum of proteins that affect a wide range of physiological processes. Indeed, deleterious effects of CAPNs have been reported in a number of different diseases, including diabetes (35,108), Alzheimer's disease (124), and myocardial infarction (71,82), to name just a few, making CAPNs interesting therapeutic targets. CAPNs also appear to play important roles in the physiological regulation of the vascular system, and the dysregulation of CAPN activity has been associated with the development of cardiovascular diseases, including hypertension (71,74,81) and atherosclerosis (91). Many of the diseases linked with altered CAPN activity are also conditions where reactive oxygen species (ROS) levels are elevated and an imbalance toward an excess production of oxidants or an alteration of the antioxidant defense results in oxidative stress (32,83,150). The activation of these two pathways may not simply occur in parallel as there is increasing evidence that the CAPN activity can be affected by ROS, suggesting that CAPNs act as a molecular link between oxidative stress and the development of cardiovascular diseases. Indeed, ROS can initiate calpain activation indirectly by increasing intracellular Ca2+ or directly by inducing the oxidative modification of the proteases themselves (52,53,88,118). Moreover, ROS-mediated modifications can affect the susceptibility of different proteins to CAPN-mediated cleavage (105,138).
Calpains
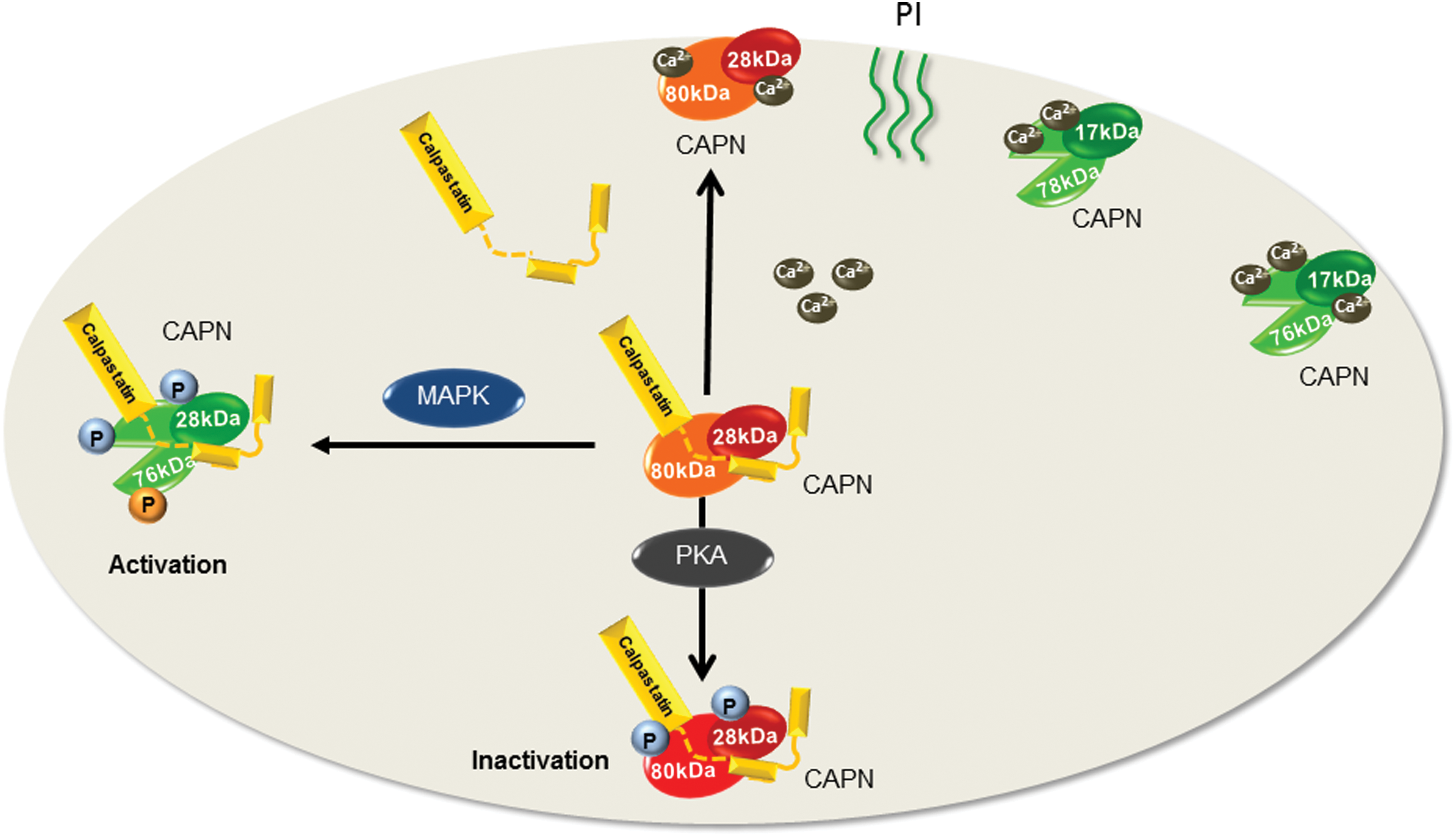
CAPNs are a large family of Ca2+-activated cysteine proteases that are the products of at least 15 genes in mammals. The regulation of calpain activity is tightly controlled by the generally low intracellular concentration of Ca2+ as well as by the presence of calpastatin (CAST), which is an endogenous calpain inhibitor. One important characteristic of CAPNs is that the cleavage of their target proteins more frequently elicits a change in their function rather than their proteolytic degradation (49,144). The ubiquitously expressed “conventional” or “typical” CAPNs, CAPN1 and CAPN2, were originally named μ-calpain and m-calpain, respectively, to reflect the micromolar or millimolar concentrations of Ca2+ required for their in vitro activation (49,141). Both CAPN1 and CAPN2 are heterodimers composed of a common 28 kDa regulatory subunit known as CAPN4 and distinct 80 kDa catalytic subunits. The enzyme heterodimers are present as a proenzyme complex in the cytosol and maintained in an inactive state by virtue of their binding to CAST. Following an increase in intracellular Ca2+, CAST dissociates from CAPNs to allow their membrane translocation. Once associated with the membrane, CAPNs undergo autoproteolysis, which cleaves the N-terminal of both the 80 kDa catalytic subunit and the 28 kDa regulatory subunit to generate the fully active 76 kDa protein and the 17 kDa small subunit (94,112). Although Ca2+ is the main stimulus for CAPN activation, other interacting proteins such as the heat shock protein (HSP) 90 (8) as well as membrane lipids and phosphoinositides are also involved in the regulation of CAPN activity (27,123). For example, the association of HSP90 with CAPN1 prevents its complete inhibition by CAST and therefore acts to increase the proteolytic activity (8), whereas phospholipids enhance the sensitivity of CAPN2 to Ca2+ (111). Proteolytic activity can also be regulated by post-translational modification, and the phosphorylation of CAPN2 on Ser369 and Thr370 by protein kinase (PK) A negatively regulates its activity in vitro and in vivo (132,136), whereas CAPN phosphorylation by mitogen-activated protein kinases (MAPKs) enhances the proteolytic activity (157,156) (Fig. 1).
There are also a number of atypical tissue-specific calpain isoforms, for example, CAPN3 or p94 is expressed in skeletal muscle (139), CAPN8 (nCL-2) and CAPN9 (nCL-4) are found in the gastrointestinal tract (140), CAPN11 is expressed in the testis (37), and CAPN12 is specific for hair follicles (36). Eye tissues have been reported to express different splice variants of the muscle-specific p94, that is, Lp82 and Lp85, which are present in the lens (79,80), whereas Rt88 is expressed in the retina (9) and Cn94 is expressed in the cornea (99). Two additional splice variants of CAPN3, namely hMp78 and hMp84, which contain an atypical initiation exon and a putative nuclear localization signal, have been identified in melanoma cells (97). CAPN14 is one of the newly characterized isoforms and has been shown to be specifically expressed in the esophagus (68,135) (Table 1).
Classification of Calpains
CAPN, calpain.
Activated CAPNs cleave a broad spectrum of substrates and are implicated in a wide variety of Ca2+-regulated cellular processes, such as signal transduction, cell proliferation and migration, cell cycle progression, differentiation, apoptosis, membrane fusion, and platelet activation (49) (Fig. 2). However, while these physiological processes are usually triggered by transient CAPN activation, the sustained activation of CAPNs is associated with its pathological actions. Indeed, the overactivation of CAPNs has been linked with cell death in several neurodegenerative diseases, such as glaucoma, retinal degeneration, and Alzheimer's disease (124,151) as well as in myocardial infarction (54,59,101). Moreover, CAPNs have been implicated in the pathogenesis of diabetes. Certainly, CAPN2 activation is involved in beta cell apoptosis (58) and different aspects of the vascular complications of diabetes, including increased vascular permeability and endothelial dysfunction (126,159). Although most of the diseases described result from CAPN overactivation, loss-of-function mutations in the Capn3 gene (exons 1, 4, 5, 8, 10, 11, 21, and 22) are known to account for the limb girdle muscular dystrophy 2A, which is, so far, the only disease associated with an alteration of CAPN3 levels (120,121). Mechanistically, the mutations have been linked to an increased rate of autoproteolytic degradation of the enzyme (48). Finally, polymorphism of Capn genes has been linked with disease development. For example, there is clear evidence that polymorphism of the Capn10 gene, in particular the single polymorphism UCSNP-43, is involved in the etiology of type 2 diabetes (35,108) as well as in the severity of polycystic ovary syndrome (4,129).

How do CAPNs recognize its substrate? Unfortunately, there is no easy way to predict a potential calpain cleavage site and the PEST sequence, that is, regions rich in proline (P), glutamate (E), serine (S), and threonine (T), which was proposed as an intramolecular signal for CAPN (38), has proved to be unreliable. In fact, most of the known CAPN substrates do not possess a PEST sequence. It is highly likely that there are other determinants of protein susceptibility to calpain cleavage. For example, the binding of calmodulin has been suggested to play a role in the recognition process as several calmodulin-binding proteins are calpain substrates (152). However, this interaction is controversial as although the presence of a calmodulin-binding region is essential for the proteolysis of the Ca2+ pump by CAPN1 (93), other proteins appear to be protected from CAPN-mediated cleavage by binding to calmodulin (60,134,158). It is most probably that the 3D structure of a protein determines whether or not it can be cleaved by calpains, and different algorithms have been developed as prediction tools for CAPN cleavage sites (39,78,147).
Subcellular localization of CAPNs
Although CAPNs were long assumed to be exclusively cytosolic proteins, it is now clear that these proteases are also present in the nucleus and mitochondria. CAPN10 was the first isoform to be detected in the mitochondria and has been found in the outer membrane, the intermembrane space, as well as in the inner membrane and matrix fractions (6,66). Other studies reported the presence of CAPN1- and CAPN2-like isoforms in mitochondria. These isoforms demonstrated a similar Ca2+ dependency as the cytosolic CAPNs but differed in their susceptibility to CAPN inhibitors. To differentiate between the classical cytosolic CAPNs, the newly identified CAPNs were named mitochondrial CAPN1 and CAPN2 (102,106,107). CAST is also present in the mitochondria where its association with CAPN1 prevents the uncontrolled activation of the protease (65,66). What is the role of mitochondrial CAPNs? In rat liver, the mitochondrial CAPNs were reported to be involved in the truncation of apoptosis-inducing factor (AIF) and the cleavage of the voltage-dependent anion channel, which accounts for their involvement in mitochondrial apoptotic signaling (106,107). The role of mitochondrial CAPNs in the release of AIF has also been reported in neuronal cell death after cerebral ischemia (25). In the mitochondria, CAPN10 cleaves the complex I core subunit V2, and ND6 leading to the activation of mitochondrial permeability transition pore and mitochondrial dysfunction (6). Although it was initially unclear whether CAPN1 and CAPN2 were resident to the mitochondria or imported from the cytosol, it seems that CAPN1, but not CAPN2, can be imported into mitochondria. Indeed, similar to CAPN10, CAPN1 possess a mitochondrial targeting sequence at its N-terminal, and the removal of the N-terminal 22 amino acids of CAPN1 prevents its mitochondrial import. The addition of this N-terminal region to either CAPN2 or green fluorescent protein, however, enabled mitochondrial import (10). Interestingly, CAPN4 can also be imported into the mitochondria, but only in the presence of CAPN1 (10).
CAPNs, in particular CAPN2, are also able to translocate from the cytosol to the nucleus upon activation to play a role in both DNA repair and apoptosis. Indeed, CAPN2 was found to translocate to the nucleus and to cleave Ku80, a subunit of the Ku protein complex, which is an initiator of the nonhomologous, end-joining, double-strand DNA break repair pathway in mammary epithelial cells (11). Moreover, in cardiomyocytes, the nuclear translocation of CAPN2 is secondary to an increase in ROS production and is involved in apoptosis (28). In the latter study, CAPN2 was shown to bind to Ca2+/calmodulin-dependent protein kinase II (CaMKII) δB in the nucleus leading to CaMKIIδB degradation. This in turn led to the downregulation of Bcl-2 mRNA and a reduction in the ratio of Bcl-2 to Bax proteins to initiate the mitochondrial apoptosis pathway. Interestingly, CAPNs can also be found extracellularly where they can target extracellular proteins. Certainly, CAPNs have been detected in extracellular fluids either as a result of their secretion from inflammatory cells (47) or as a cargo carried by microparticles, particularly platelet-derived microparticles (46,109,116).
Redox regulation of CAPNs
ROS-regulated Ca2+-dependent CAPN activation
CAPNs are targets of redox modification (52,53,88,118), but most studies investigating the redox regulation of CAPNs have focused on an indirect role of ROS in regulating the enzymatic activity. Certainly, ROS can interfere with intracellular Ca2+ levels to affect calpain regulation. One example is the sustained activation of CAPNs in platelets from diabetic patients, which was found to be the consequence of the redox-mediated dysregulation of the intracellular Ca2+ homeostasis (118). One factor known to contribute to the disturbed platelet Ca2+ homeostasis in diabetic subjects was a marked reduction in the Ca2+-ATPase activity (122). Among the multiple Ca2+ATPases expressed in human platelets, the activity of the sarco-endoplasmic reticulum Ca2+ATPase (SERCA)-2 has been reported to be sensitive to oxidative stress. In particular, peroxynitrite (ONOO−) can differently affect the SERCA-2 activity such that low concentrations result in the S-glutathionylation and activation of SERCA-2 (2,3), whereas high concentrations of ONOO− induce its tyrosine nitration and inactivation. Certainly, the tyrosine nitration of SERCA-2 was significantly enhanced in platelets from diabetic patients compared with those from healthy individuals, and the consequences of diabetes could be mimicked by the in vitro stimulation with high concentrations of ONOO−. The latter led to the hyperactivation of CAPNs and resulted in the degradation of a spectrum of platelet proteins and consequently to the alteration of the platelet proteome. In particular, the platelet and endothelial cell adhesion molecule CD31, septin-5, integrin-linked kinase, and the chemokine CCL5 were all identified as CAPN targets in diabetic platelets (116). As is typical of calpain activation, the proteolytic cleavage of all the latter proteins did not immediately trigger their degradation but rather a change in function that contributed to platelet activation (116). Some of the effects observed were synergistic as the CAPN-dependent cleavage of septin-5 led to its dissociation from the soluble N-ethyl-maleimide-sensitive protein attachment protein receptor (SNARE) protein, syntaxin-4, and promoted the secretion of α-granule contents, including that of CCL5 (116). Moreover, the CAPN-mediated cleavage of CCL5 led to the generation of an isoform with enhanced chemotactic activity and thus presumably higher atherogenic potential (116). Given that CAPNs can be detected in platelet releasate, the CAPN-mediated cleavage of CCL5 may take place in α-granules or in the extracellular space following platelet degranulation.
ROS have also been implicated in CAPN activation in nonvascular cells. For example, increased mitochondrial ROS production and CAPN1 activation have been linked with doxorubicin-mediated myopathy in both cardiac and skeletal muscles (89). Although the authors did not describe the exact molecular mechanism(s) linking increased mitochondrial ROS with CAPN, they postulated that Ca2+ increased as a consequence of the O2 −-induced release of Ca2+ from the sarcoplasmic reticulum together with the attenuated Ca2+ removal from the cytosol as a result of oxidative damage to Ca2+ transporters located in the plasma membrane (89). Intermittent hypoxia has been found to increase ROS levels in SH-SY5Y neuroblastoma cells leading to Ca2+-induced CAPN activation (153). The same mechanism was also involved in the CAPN-mediated cleavage of the transcription factor Myc to generate the cytosolic cleavage product Myc-nick. The latter plays a role in cell differentiation and cancer cell survival through alpha-tubilin acetylation and autophagy (33,34). Finally, in glomerular mesangial cells, ROS generated by nicotinamide adenine dinucleotide phosphate (NADPH) oxidases result in CAPN activation and cell death (131).
S-nitrosation
Nitric oxide (NO) is synthesized from the amino acid
The possible regulatory role of S-nitrosation on the CAPN activity was first investigated over 20 years ago. Indeed, CAPN2 isolated from either skeletal muscle or neutrophils could be S-nitrosated and inactivated by sodium nitroprusside, an effect reversed by dithiothreitol suggesting the involvement of a modification on a cysteine residue. Interestingly, CAPN1 was less sensitive to modification by NO at neutral pH, and inactivation could be only observed in acidic conditions (88). Based on these observations, it was suggested that NO may regulate the CAPN activity depending on fluctuations in pH in contracting cells or pathological conditions. A role for NO in the regulation of CAPN activity has since been confirmed, and the age-related decrease in the expression of neuronal NOS was correlated with a decrease in CAPN S-nitrosation, which in turn led to CAPN activation and myofibril degradation (125). The link between NO and CAPN was also supported by the fact that NOS inhibition promoted CAPN1 autolysis, resulting in the degradation of titin and nebulin during postmortem aging (76). However, it is not known whether the latter effects can be attributed to the S-nitrosation of CAPN. More recently, in vitro studies confirmed the effect of S-nitrosation on the autolysis and activity of CAPN1 and identified the S-nitrosated cysteine residues as Cys49, Cys351, Cys384, and Cys592 in CAPN1 and Cys142 in CAPN4 (77). Functionally, the S-nitrosation of CAPNs was associated with protection against myocardial ischemia/reperfusion injury (148). The inactivation of CAPNs by NO-dependent S-nitrosation seems to be a highly conserved process in most cysteine proteases as it has also been reported in small organisms, such as parasites (7,16). Certainly, the NO donor-mediated life cycle arrest in Plasmodium, Trypanosoma, and Leishmania has been attributed to the inactivation of cysteine proteases via the S-nitrosation of a cysteine residing in the catalytic site.
Oxidation
CAPN activity can be regulated by oxidants, and the treatment of human neuroblastoma cells with oxidants such as doxorubicin or 2-mercaptopyridin N-oxide in vitro resulted in the inactivation of CAPN, suggesting a direct modification of CAPN by oxidation. Given that the effect could be reversed by antioxidants, the oxidation-induced CAPN inactivation seems to be reversible (53). Similarly, the incubation of CAPN1 with oxidants such as H2O2 or sodium hypochlorite leads to the inhibition of its proteolytic activity without affecting its autoproteolysis, again an effect that was sensitive to dithiothreitol (52). H2O2 was also shown to decrease the ability of CAST to inhibit CAPNs, and it has been suggested that at high pH, CAST may limit the oxidation-induced inactivation of CAPN1 (26).
ROS-mediated regulation of CAPN expression
Oxidative stress has been shown to not only modulate the CAPN activity but also its expression. Indeed, exposing rat neuronal PC12 cells to H2O2 enhanced CAPN1 and CAPN2 mRNA levels and induced apoptosis (119). Other studies have reported the involvement of microRNAs, in particular miR-124 in the ROS-dependent regulation of CAPN expression (23,57,64). Indeed, in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson's disease and in dopaminergic neurons treated with methyl phenyl pyridinium iodide, the expression of the CAPN1 and CAPN2 was sensitive to miR-124. On one hand, the knockdown of miR-124 led to the increased production of ROS, suggesting the involvement of oxidative stress in the regulation of CAPN expression. On the other hand, the authors reported that miR-124 can directly interact with CAPN1 (64). MiR-124 was also reported to suppress the migration and invasion of glioma cells in vitro by targeting CAPN4 (23) and can suppress the proliferation and invasion of nasopharyngeal carcinoma cells through the Wnt/β-catenin signaling pathway (57).
Role of redox modification in the regulation of CAPN-mediated target protein cleavage
In addition to directly targeting CAPNs, ROS can induce the post-translational modification of target proteins and thus affect their sensitivity to CAPN-mediated cleavage. Carbonylation, for example, has been shown to enhance the sensitivity of the mitochondrial AIF to CAPN and account for AIF-mediated apoptosis. Accordingly, antioxidants blocked AIF carbonylation, as well as its subsequent cleavage and release from the mitochondria (105). Moreover, the oxidation of myofibrillar proteins was shown to enhance their susceptibility to proteolytic degradation by CAPN (138), which, in turn, was suggested to be a mechanistic link connecting oxidative stress with accelerated myofibrillar proteolysis during disuse muscle atrophy. However, oxidative modification of the muscle intermediate filament desmin decreased its susceptibility to CAPN at the same time as increasing its caspase-3- and caspase-6-mediated degradation (30). Although these observations were made in nonvascular cells, similar mechanisms may account for the degradation of proteins of the vascular system by CAPN.
Several kinases, including protein kinase C (PKC) (51,142) and tyrosine kinases (100), are sensitive to oxidative modification, and as phosphorylation can alter the susceptibility of a protein to CAPN-mediated cleavage, thus ROS-dependent PK activation can indirectly affect CAPN targets. One good example is the stress-induced phosphorylation of the nuclear ribonuclease III enzyme Drosha as a trigger of its CAPN-mediated degradation and cell death. Oxidative stress was found to enhance the activation of p38 MAPK, leading to the phosphorylation of Drosha and its dissociation from the cofactor DiGeorge syndrome critical region gene 8. The phosphorylated Drosha was exported from the nucleus to be degraded by CAPN and account for the stress-induced inhibition of Drosha-mediated cellular survival (155). Inhibitors of nuclear factor-κB (IκB) and ATP-binding cassette transporter A1 (ABCA1) are protein substrates reported to be regulated by phosphorylation on their serine or threonine residues (84,128). Indeed, phosphorylation of serine and threonine residues in the PEST domain of IκB by the casein kinase 2 has been shown to promote its CAPN-mediated cleavage in IgM(+) B cells (128). Such an enhanced IκB turnover was suggested to account for the increased basal nuclear factor-κB levels in these cells. ABCA1, which mediates the apolipoprotein A1-dependent efflux of excess cholesterol from cells, is known to be constitutively phosphorylated on Thr1286 and Thr1305. The phosphorylation of these residues enhanced its cleavage by CAPN. Mechanistically, it has been shown that the interaction of ABCA1 with apolipoprotein A1 leads to the dephosphorylation of the ABCA1 PEST sequence and thereby inhibits CAPN-mediated degradation resulting to an increase of ABCA1 cell surface expression (84).
While the phosphorylation of target proteins on serine or threonine residues has generally been associated with increased sensitivity to CAPNs, the tyrosine phosphorylation of target proteins tends to protect against CAPN cleavage. One CAPN substrate known to be regulated by tyrosine phosphorylation is the N-methyl-d-aspartate (NMDA) receptor. Truncation of the NMDA receptor NR2 subunits by CAPN is known to alter the structure and function of the receptor (15) and was suggested to be involved in the regulation of receptor turnover or in their relocation in synaptic membranes. Interestingly, the Src-mediated tyrosine phosphorylation of NR2 has long been recognized to increase channel conductance (163). It now seems that the phosphorylation of the receptor prevents the CAPN-mediated truncation of its C-terminal domain to account for the stabilization of the receptors in postsynaptic structures (14). A second example of protein protected from CAPN-mediated cleavage through its tyrosine phosphorylation is alpha II-spectrin. In this case, the Src-mediated phosphorylation of Tyr1176 located close to the SH3 domain in the CAPN cleavage site of alpha II-spectrin reduced its susceptibility to cleavage. This is of significance given the important role of spectrin in cross-linking actin filaments to the plasma membrane. It is therefore expected that the modulation of spectrin proteolysis by phosphorylation plays an important role in the stabilization and reorganization of the cytoskeleton and consequently in the regulation of spectrin-based cytoskeleton functions (104).
Redox Control of the Vascular System
Under physiological conditions, cells generate low concentrations of ROS, which function as signaling molecules involved in different physiological processes. However, the excessive production of ROS is involved in the pathogenesis of a number of diseases, including vascular diseases. In the vascular system, NADPH oxidases, represented by four Nox isoforms, that is, Nox1, Nox2, Nox4, and Nox5, are generally assumed to be the main cellular source of superoxide anions (O2 −) (73). Despite similarities in their core structures, Nox isoforms have different mechanisms of activation [reviewed in Ref. (18)]. Indeed, while Nox1 and Nox2 require the association with cytosolic components for activation, Nox5 is activated by Ca2+ and Nox4 is constitutively active. Superoxide anions can also be generated by cytochrome P450 enzymes (44,45), uncoupled NOS (154), and by the mitochondria (22). Other enzymes such as xanthine oxidase, myeloperoxidase (103), and cyclooxygenase are known to also participate in vascular O2 − generation, although to a lesser extent. After its generation, O2 − is quickly converted to H2O2 either spontaneously or by superoxide dismutases. H2O2 can further react with O2 in the presence of iron to generate a hydroxyl radical or catalyzed to H2O and O2 by catalases. Therefore, physiological levels of ROS are held in check by a tight balance between the activity of ROS-producing and ROS-degrading enzymes.
In addition to its reaction with H2O to form H2O2, O2 − can bind to NO to form ONOO− (114). This process leads to the reduction of NO bioavailability and endothelial dysfunction. Moreover, ONOO− can cause direct oxidative damage by nitrating tyrosine residues of proteins (12). In the vasculature, ONOO− has been shown to cause the tyrosine nitration and inactivation of the prostacyclin synthase (164,165) and to mediate angiotensin II-induced tyrosine nitration and activation of Erk (110). Moreover, tyrosine nitration is also associated with the high-glucose-induced oxidative injury of vascular smooth muscle cells (VSMCs) and endothelial cells (161,162). More recently, tyrosine nitration of phosphatidylinositol 3-kinase has been reported to mediate high-glucose-induced endothelial cell apoptosis (41). Although Nox1 and Nox2 were considered to be the main sources of vascular O2 −, the contribution of Nox5 has been recently reported. Nox5 was shown to enhance the vascular contraction by increasing VSMC Ca2+ and ROS levels and by enhancing the phosphorylation of the myosin light chain 20 (95). While these are just few examples of how ROS can affect the vasculature, the redox-mediated regulation of the vascular system involves much more complicated mechanisms, which have been described in detail by several reviews (19,145).
Role of CAPNs in ROS Production
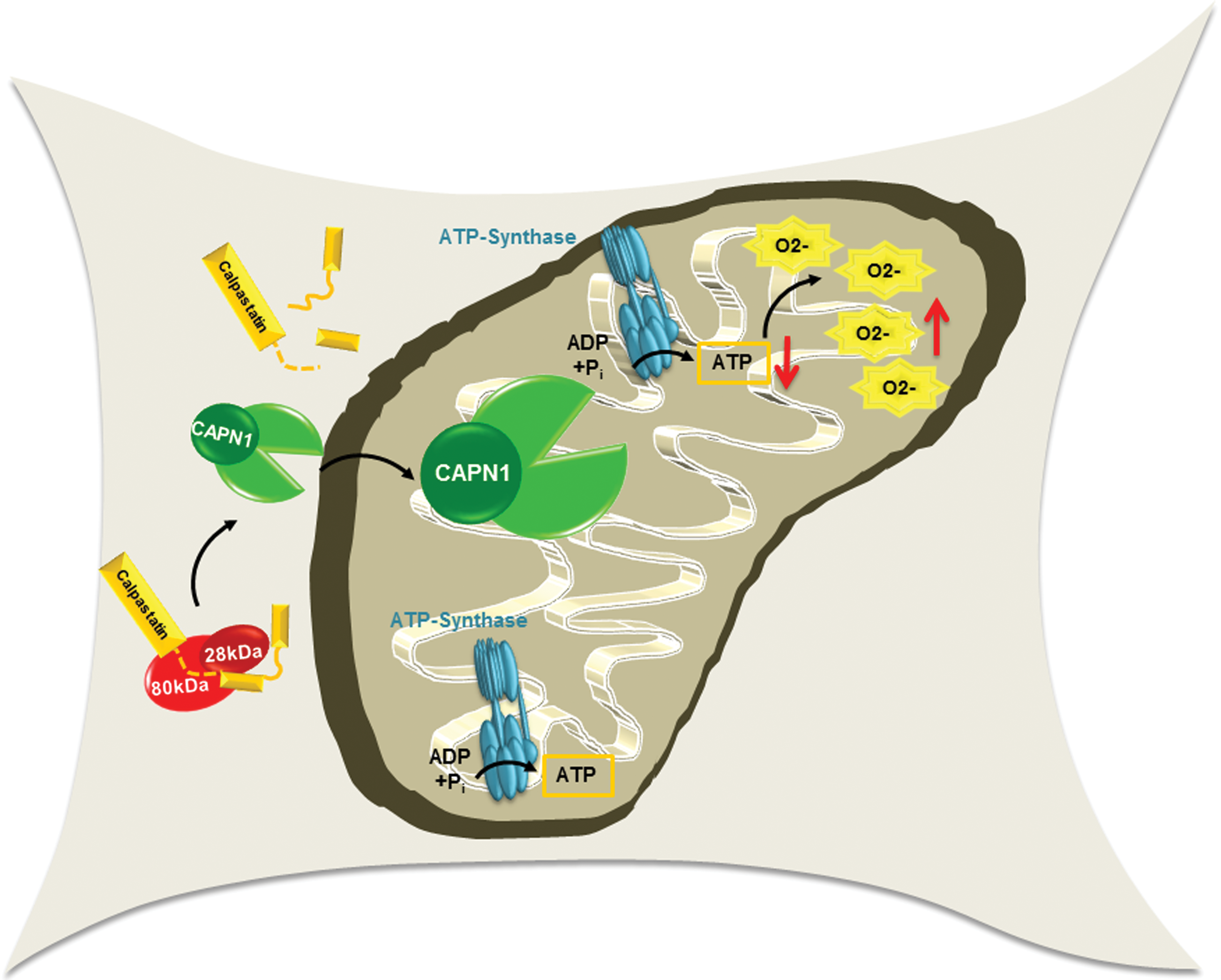
CAPNs have been reported to participate in ROS generation and to increase endothelial cell H2O2 as a consequence of enhanced mitochondrial O2 − generation (29). It seems that mitochondria are the main source of CAPN-induced ROS production, and selectively, scavenging mitochondrial O2 − was shown to increase the NO bioavailability in endothelial cells cultured in the presence of high concentrations of glucose. Moreover, high-glucose-stimulated ROS production in endothelial cells could be prevented by CAPN inhibition and could be reversed by the overexpression of CAST, leading to the improvement of endothelial function (29). Given that CAPN has been shown to translocate from the cytosol to mitochondria (98) and that several mitochondrial proteins are CAPN substrates (20,61,107), it is very likely that CAPN-mediated oxidative stress may be the result of mitochondrial dysfunction. Certainly, CAPN can target the optic atrophy 1, a mitochondrial inner membrane GTPase important for the regulation of mitochondrial dynamics. Indeed, the CAST-sensitive cleavage of the optic atrophy 1 was associated with mitochondrial fragmentation (61). More recently, it was reported that CAPN1 levels and activity were elevated in mitochondria from diabetic mouse hearts and that CAPN specifically targeted the mitochondrial ATP synthase to mediate O2 − production in cardiomyocytes (102) (Fig. 3). A link to oxidative stress has also been reported for CAPN3 in melanoma cells and the resulting oxidative modification of phospholipids, and DNA damage was found to impair cell proliferation. However, the molecular mechanisms underlying the CAPN3-mediated oxidative stress are unknown (96). In skeletal muscle, CAPN3 was suggested to be important for mitochondrial integrity since its deficiency lead to mitochondrial dysfunction, that is, decreased ATP production and increased ROS production. In the latter study, the beta-oxidation enzyme, very long-chain acyl-CoA dehydrogenase, was shown to be protein cleaved by CAPN3 (69).
Role of CAPNs in the Regulation of Vascular Function
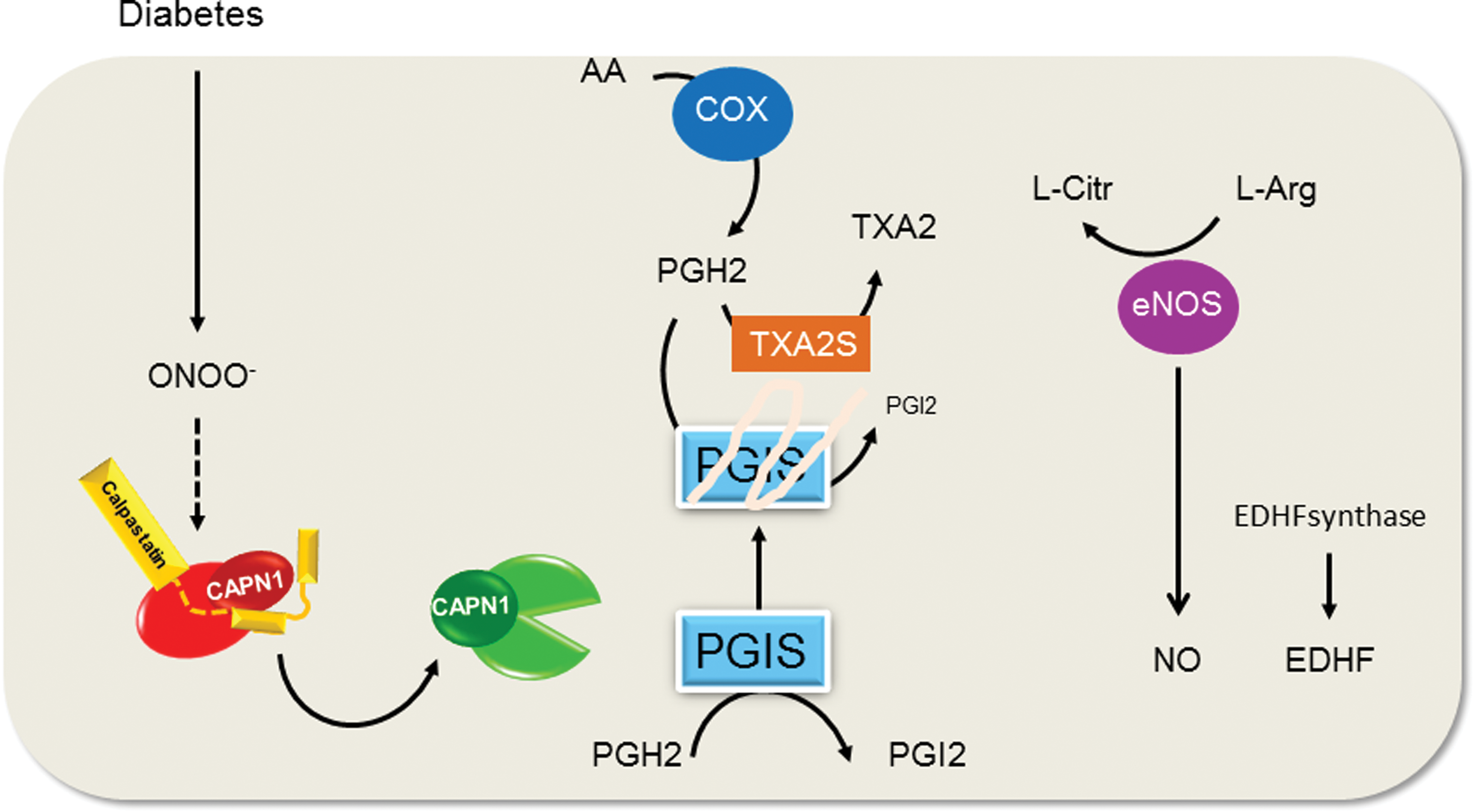
The ubiquitous isoforms CAPN1 and CAPN2 are found in endothelial cells, smooth muscle cells, and circulating blood cells. In endothelial cells, CAPNs are involved in the maintenance of vascular integrity and angiogenesis. Indeed, vascular endothelial growth factor and fluid shear stress are physiological activators of CAPNs and were involved in endothelial cell proliferation, migration, and in the regulation of endothelial barrier (17,19,50,90). However, the enhanced and/or sustained activation of CAPNs have been associated with different pathological conditions, including hypertension, atherothrombosis, and diabetes. In particular, increased activation of CAPNs in endothelial cells has been repeatedly shown to account for diabetes-associated endothelial dysfunction (29,31,117,159). In mice, the induction of diabetes leads to the activation of CAPN1 in mesenteric arteries and a calpain-inhibitor-sensitive development of endothelial dysfunction (attenuated acetylcholine-induced vasorelaxation). However, rather than targeting NO production, one important target of CAPN in this situation was identified as the prostacyclin synthase (117). Indeed, CAPN1 cleaved the C-terminal domain of the prostacyclin synthase close to the catalytic site of the enzyme, resulting in its inactivation and the loss of prostacyclin-induced relaxation. Also in this setting, an increased generation of ROS was upstream of CAPN activation, and it was possible to phenocopy the effects of diabetes by treating mesenteric arteries with the OONO− donor, SIN-1, which activated CAPN1 and induced endothelial dysfunction in a calpain-inhibitor-sensitive manner (117). However, whether or not ONOO− activated calpain by increasing intracellular Ca2+ or via another more direct mechanism remains to be determined (Fig. 4). A further condition associated with calpain activation in vascular tissue and endothelial dysfunction is hyperhomocysteinemia (31). Mechanistically, hyperhomocysteinemia was shown to increase superoxide anion production leading to CAPN1 activation. The latter cleaves and activates PKC resulting to enhanced endothelial nitric oxide synthase (eNOS) phosphorylation on Thr497/495 and inactivation.
CAPN activation was also linked with increased permeability in the microcirculation in Zucker diabetic fatty rats (126), through an unknown mechanism. CAPN activation also accounts for the upregulation of intercellular adhesion molecule-1 and leukocyte adhesion on the endothelium of streptozocin-induced diabetic rats (137). CAPN2 was induced in human and mouse vascular endothelial cells during atherosclerosis development where it leads to the proteolytic disorganization of vascular endothelial (VE)-cadherin resulting in the dissociation of β-catenin from the VE-cadherin complex, disorganization of adherence junctions, and endothelial cell hyperpermeability (62,92). Interestingly, in nonvascular cells such as colon cancer cells, hepatoblasts, as well as in human pluripotent stem cells, β-catenin has been shown to be a direct substrate of CAPN (13,67,72,75), implying a role in the regulation of Wnt signaling. The CAPN-mediated degradation of β-catenin has been also shown to account for the thromboxane A2-mediated repression of the differentiation of human adipose-derived stromal cells into endothelial cells (130), linking CAPN with endothelial cell homeostasis. Additionally, CAPN mediates thromboxane-induced cytotoxicity of neural microvascular endothelial cells by activating the translocation of Bax into the mitochondria, mitochondrial depolarization, cytochrome C release into the cytosol, and depletion of cellular ATP (113). Finally, in lipopolysaccharide-induced sepsis, ROS produced from NADPH oxidase have been shown to stimulate CAPN1 activation and to induce apoptosis of pulmonary microvascular endothelial cells, a process that contributes to sepsis-associated endothelial injury (56).
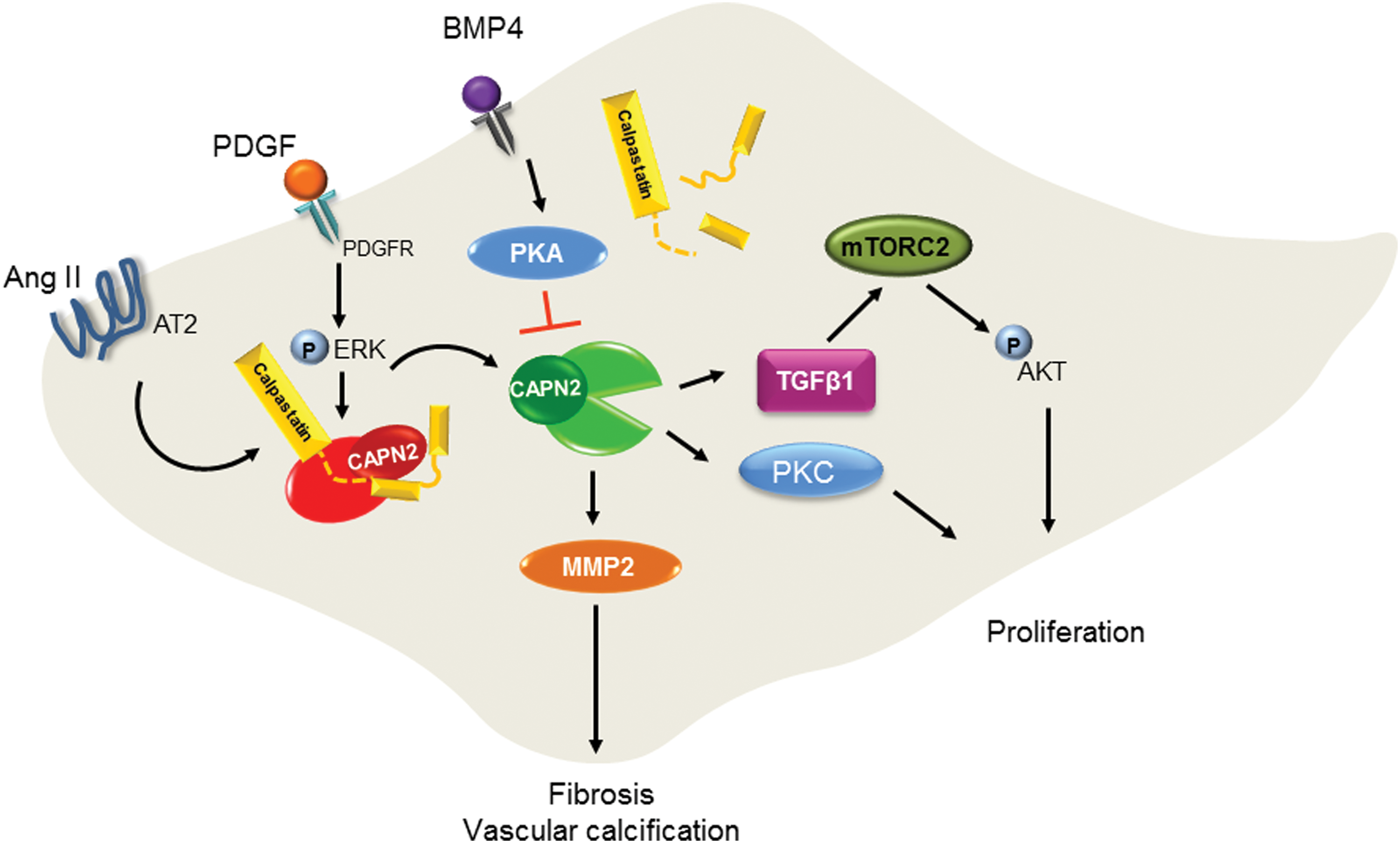
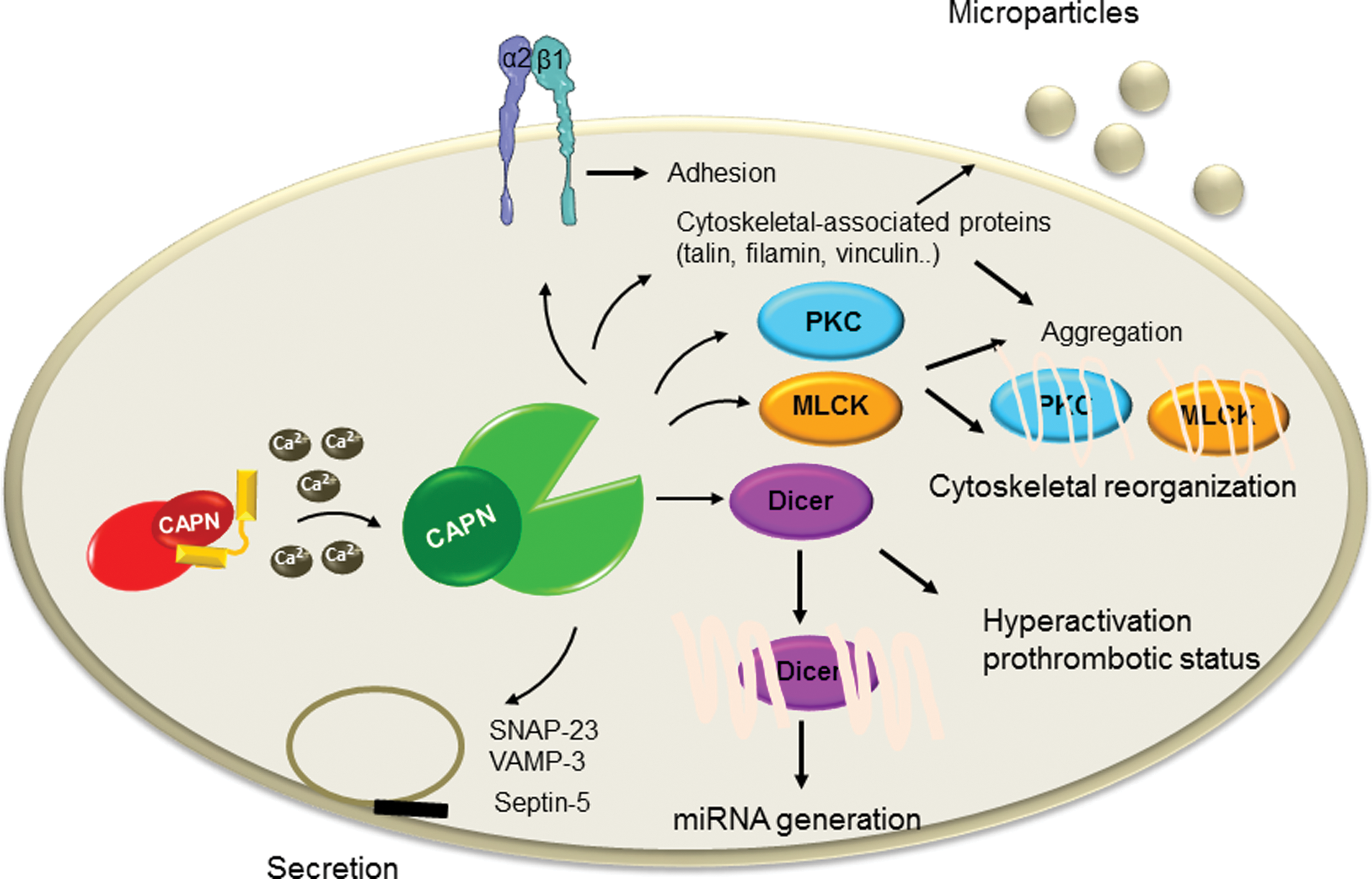
More recently, CAPN activation has been shown to mediate myeloperoxidase-induced endothelial dysfunction (43). Myeloperoxidase was shown to reduce eNOS phosphorylation on Ser1147 and NO production resulting to the decrease of CAPN nitrosylation, which consequently increases the protease activity. The active CAPN was responsible for the increased vascular cell adhesion molecule 1 expression and the adhesion of leukocytes. Numerous studies have shown that CAPNs are also involved in several cellular processes of VSMCs, including cell growth, proliferation, and vascular remodeling—course of events that play an important role in the pathogenesis of hypertension (74) and atherosclerosis (91). One of the early studies investigating CAPN in VSMC demonstrated the role of CAPN2 in cell proliferation. It has been shown that CAPN inhibition altered cell proliferation and was associated with increased expression of the focal adhesion kinase pp125FAK (5). More recent studies investigated the signaling pathways involved in CAPN-mediated cell proliferation. For example, in pulmonary artery smooth muscle cells (PASMCs), platelet-derived growth factor (PDGF) causes the phosphorylation of AKT, which then induces collagen synthesis and cell proliferation. In this study, CAPN2 was reported to be involved in the activation of AKT via transforming growth factor (TGF)-β1–mammalian target of rapamycin complex 2 (mTORC2) pathway (1). However, the exact molecular mechanism linking CAPN2 and TGF-β1 was not shown. A different group has reported similar finding on the role of CAPN in mediating PDGF-induced cell proliferation. In the latter study, bone morphogenetic protein-4, which belongs to the superfamily of transforming growth and differentiation factor, was shown to inhibit PDGF-induced proliferation and collagen synthesis via protein kinase A-mediated inhibition of CAPN2 in PASMCs (24) (Fig. 5). CAPN is also involved in pulmonary vascular remodeling in rodent models of pulmonary hypertension. Indeed, CAPN mediated epidermal growth factor- and PDGF-induced collagen synthesis and proliferation of PASMCs via an intracrine TGF-β1 pathway (81). Additionally, CAPN2-mediated cleavage of PKC—an important player in different signal transduction cascades, was shown to account for the proliferation of PASMCs (127). CAPN1 was also involved in the age-associated angiotensin II-mediated increase in VSMC migration. Indeed, age-associated increase in matrix metalloproteinase 2 activity and migration of VSMC are blocked by calpain inhibitor 1 or CAST, and CAPN was shown to target vimentin to mediate its effect (62). Moreover, transgenic mice constitutively expressing high levels of CAST displayed a marked decrease of angiotensin II-induced hypertrophy of the left ventricle and reduced angiotensin II-dependent media hypertrophy, implying the role of CAPN in vascular remodeling in angiotensin II-induced hypertension (74). CAPN1 has been also linked to vascular calcification. Indeed, it can regulate matrix metalloproteinase 2 activity in VSMCs and facilitate age-associated aortic wall calcification and fibrosis (63) (Fig. 5). CAPN1 is also involved in oxidized low-density lipoprotein-induced vascular calcification by impairing pyrophosphate metabolism (146). The pathophysiological relevance of these findings is supported by the beneficial effects of CAPN inhibitors in different experimental animal models of cardiovascular diseases, such as atherosclerosis (91) or hypertension (74). In addition to their role in the vasculature, calpains also play an important role in hemostasis by regulating platelet functions. Under normal conditions, the antithrombotic properties of the endothelium as well as the permanent release of antiplatelet factors such as NO and PGI2 prevent the adhesion of circulating platelets (21). Upon vascular injury, platelets roll, adhere, and then firmly attach to exposed subendothelial matrix through membrane receptors. The initial adhesion is triggered by the von Willebrand factor-mediated linkage of the subendothelial-exposed collagen to the platelet glycoprotein Ib/V/IX, which initiates the activation of intracellular signaling in platelets resulting in an increase in intracellular Ca2+ concentration. The latter plays a central role in the regulation of platelet function, including degranulation, shape change, and aggregation. The increase in intracellular Ca2+ also leads to the activation of CAPNs, which are involved in different steps of platelet activation. Indeed, by mediating the limited proteolysis of different cytoskeleton-associated proteins including spectrin, adducin, talin, filamin, vinculin, and cortactin as well as different kinases such as the myosin light chain kinase, PKC, and protein-tyrosine phosphatase 1B, CAPNs are involved in cytoskeletal reorganization, which is important for platelet shape change, aggregation, clot retraction, and microparticle formation (70). Moreover, by targeting SNARE or SNARE-associated proteins such as the synaptosomal-associated protein 23, vesicle-associated membrane protein (VAMP-3), and septin-5, CAPNs amplify platelet secretion. While CAPNs play important role in the physiological platelet activation, sustained activation of CAPN has been reported to mediate platelet hyperactivation and the prothrombotic status associated with hypoxia (149). In the latter study, the authors performed proteomic analysis of platelets from rats exposed to hypoxia and found a significant upregulation of the CAPN small subunit 1 compared with that from normoxia-exposed animals. Moreover, the levels of intracellular Ca2+ and CAPN activity in platelets were significantly enhanced by hypoxia, and platelet hyperreactivity could be reversed by the inhibition of CAPN. Increased CAPN activation also accounts for the diabetes-associated platelet hyperactivation (115,116). Indeed, CAPN induced the change in platelet proteome toward a prothrombotic state. Although the change in platelet proteome was mostly the result of direct CAPN-mediated proteolytic cleavage, it may be also the consequence of a secondary post-translational regulation of protein levels. Certainly, our group has also shown that by cleaving the ribonuclease III Dicer, CAPN activation determines platelet microRNA levels in diabetes (40) (Fig. 6).
Conclusions and Future Directions
It has become clear that CAPNs play a dual role in the vascular system not only as generator of ROS but also as target of oxidative modifications. However, it remains unclear in which condition is vascular oxidative stress the cause or the consequence of calpain activation. It is also unknown whether signals shifting CAPNs from physiological regulators of vascular homeostasis to deleterious proteases exist since under physiological conditions, CAPNs ensure vascular integrity, whereas their sustained activation is associated with vascular dysfunctions. Of special interest is the role of ROS in the regulation of CAPN activity. In particular, the molecular mechanisms underlying the direct regulation of CAST and the different CAPN isoforms by specific reactive intermediates require further investigation. In addition, given that oxidative change of the different members of the CAPN system, that is, the catalytic subunit, the regulatory subunit, as well as CAST, would have different consequences on the CAPN activity, the ROS-mediated changes of the different members should be individually investigated. It would be also interesting to understand whether ROS-mediated modifications of substrate proteins serve as signals for CAPN-mediated protein degradation instead of CAPN-dependent limited proteolysis and whether such mechanisms account for the physiological and pathological roles of CAPNs. Use of isoform-specific knockout cell or mice will help decipher the role of individual CAPN isoforms in different pathophysiological conditions. Finally, given that CAPNs are present in extracellular fluids and are carried by platelet-derived microparticles, research investigating the fate of extracellular CAPNs is expected to result in the identification of new extracellular targets of CAPN and the characterization of their physiological and pathological relevances.
Footnotes
Acknowledgment
The authors' own research was supported by the Deutsche Forschungsgemeinschaft (RA 2435/3-1 to V.R., SFB 815/A16 to I.F., and Exzellenzcluster 147 “Cardio-Pulmonary Systems”).