
Abstract
Significance:
Redox homeostasis consists of an intricate network of reactions in which reactive molecular species, redox modifications, and redox proteins act in concert to allow both physiological responses and adaptation to stress conditions.
Recent Advances:
This review highlights established and novel thiol-based regulatory pathways underlying the functional facets and significance of redox biology in photosynthetic organisms. In the last decades, the field of redox regulation has largely expanded and this work is aimed at giving the right credit to the importance of thiol-based regulatory and signaling mechanisms in plants.
Critical Issues:
This cannot be all-encompassing, but is intended to provide a comprehensive overview on the structural/molecular mechanisms governing the most relevant thiol switching modifications with emphasis on the large genetic and functional diversity of redox controllers (i.e., redoxins). We also summarize the different proteomic-based approaches aimed at investigating the dynamics of redox modifications and the recent evidence that extends the possibility to monitor the cellular redox state in vivo. The physiological relevance of redox transitions is discussed based on reverse genetic studies confirming the importance of redox homeostasis in plant growth, development, and stress responses.
Future Directions:
In conclusion, we can firmly assume that redox biology has acquired an established significance that virtually infiltrates all aspects of plant physiology.
I. Introduction
The research field of redox regulation and signaling in aerobic organisms, including humans and microbes, has received a great impetus from early studies conducted on plants. During the 60s and the 70s of the past century, a decade after the discovery of the photosynthetic CO2 fixation cycle, now known as the Calvin–Benson (CB) cycle, it was observed that some CB cycle enzymes were activated in the light and inactivated in the dark, indicating that the CB cycle was temporally coupled to the light reactions of photosynthesis (397) [for a recent review see (339)]. Light activation in vivo was first demonstrated for chloroplast glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (13, 595), and in the next years for phosphoribulokinase (PRK) (279), and the two phosphatases, namely fructose-1,6-bisphosphate phosphatase (FBPase) (23) and sedoheptulose-1,7-biphosphate phosphatase (SBPase) (12). A mechanistic explanation of these results was essentially provided by Bob Buchanan and collaborators (Peter Schürmann and Ricardo Wolosiuk in primis) in a series of articles that marked the birth of the plant redox field (57, 58, 455, 457, 538, 540, 541). Light activation of CB cycle enzymes was proposed to depend on a novel electron chain made by the interaction of three types of stromal proteins: ferredoxin (FDX, an iron–sulfur [Fe-S] protein, where electrons come in from photosystem I [PSI]), FDX:thioredoxin reductase (FTR, a protein containing an Fe-S cluster functionally and physically connected with a disulfide), and thioredoxin (TRX), which also contains two cysteines (Cys) able to reversibly form a disulfide bond (Fig. 1A). By means of this transduction chain, target enzymes are reduced and hence activated in the light (Fig. 1B). In the absence of light, electrons were believed to return to oxygen leaving oxidized enzymes in the inactive form (456). Interestingly, at that time, TRX was only known as a protein involved in ribonucleotide reduction in bacteria and the demonstration of its role in the regulation of chloroplast metabolism opened a wide array of possibilities for the development of redox biology concepts in all aerobic organisms (54).
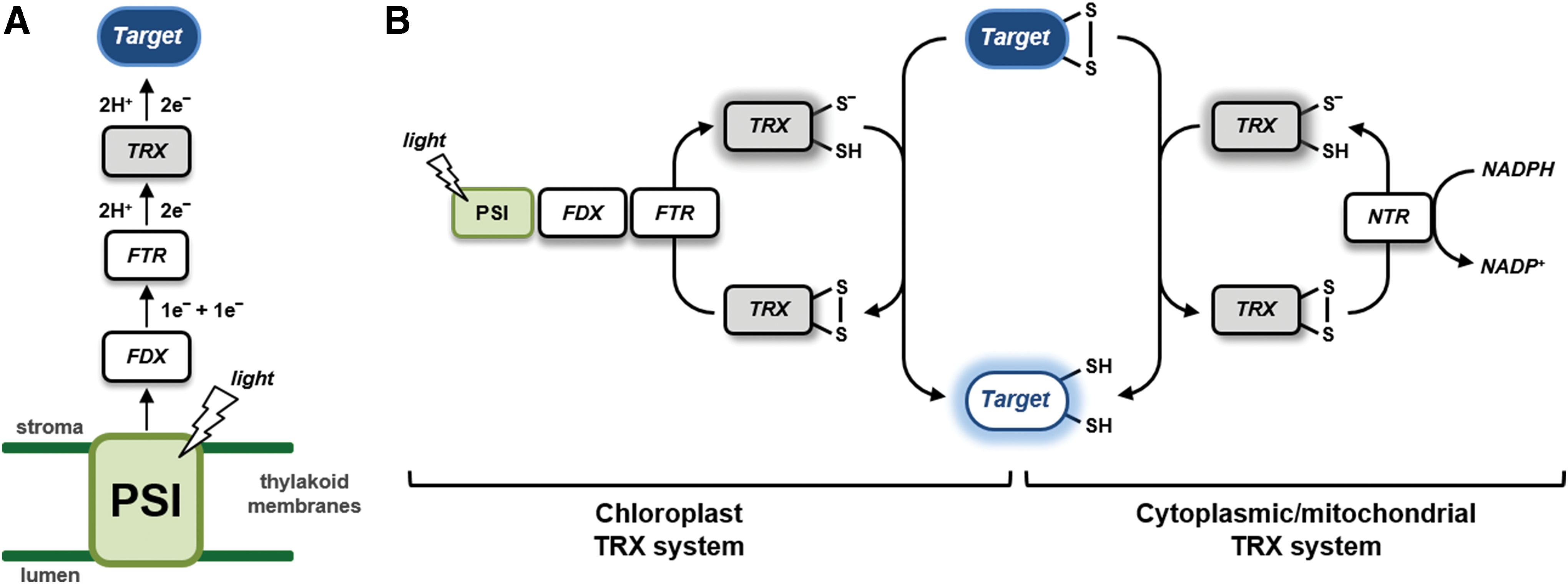
Once established the FDX–FTR–TRX system (hereafter named FDX–TRX system) in plants, new discoveries in the field were obtained in the following decades. By the end of the century, the targets of the system approached the number 25, including 4 enzymes and 2 regulatory proteins of the CB cycle (529, 539, 587), several other metabolic enzymes including NADP-malate dehydrogenase (NADP-MDH) (240, 448) and glucose-6-phosphate dehydrogenase (G6PDH), the latter remaining the prototypical example of enzymes that are inhibited, rather than activated, by disulfide reduction in plants (448). Moreover, the FDX–TRX system was found to be operative also in amyloplasts (nonphotosynthetic plastids) where FDX is reduced by metabolically produced NADPH rather than by light (25). Knowledge on TRX diversity was limited to chloroplastic TRX f and m, with the addition of cytoplasmic TRX h, which can be reduced by NADPH:TRX reductase (NTR) using NADPH as electron donor (Fig. 1B). The first structural studies on TRX-regulated enzymes (FBPase and NADP-MDH) appeared in the late 90s providing nice explanations of how redox regulation could operate at the atomic level, at least in these proteins (55, 286, 339, 456). NADP-MDH, in particular, constituted an interesting case. Its mechanism of regulation, based on C- and N-terminal extensions containing Cys pairs able to form internal disulfides under the control of TRXs, was found to be similar to other proteins such as GAPDH (143) and CP12 (144). Another important achievement of the recent past was the ability to determine, in vitro, the redox potential of the different dithiol–disulfide interchange reactions (223), which allowed the development of hypotheses concerning the reciprocal influence between TRX and target proteins redox states in vivo (92, 93, 222, 223, 266, 267, 322).
Besides the chloroplast pathway for regulatory disulfides reduction, mechanisms of disulfides formation were also investigated. Current knowledge suggests that formation of regulatory disulfides in chloroplasts may involve particular types of TRXs (111, 133, 561) that shuttle electrons from reduced target proteins to 2-Cys peroxiredoxin (2-Cys PRX) and then to hydrogen peroxide (H2O2) (see section VII). These findings imply that H2O2, rather than oxygen, may be the terminal electron acceptor used for downregulating the TRX-activated enzymes. This example nicely fits into the general concept, largely developed in the past decades, that the manifold interactions between reactive molecular species (RMS) and active protein thiols often play essential physiological roles. However, protein disulfides may also play structural rather than regulatory roles, and the formation of structural disulfides is a compulsory step in the correct folding of several proteins. Systems controlling the oxidative protein folding generally rely on two types of proteins, isomerase and oxidase, forming an electron chain that connects the target protein (where the disulfide is formed) to the terminal acceptor (430). In plant cells, systems of this type are present, at least, in the lumen of the endoplasmic reticulum (190), in the lumen of thylakoids (256), and in the intermembrane space of mitochondria (72). Different protein components and final electron acceptors are used in different locations. For detailed analyses of oxidative protein folding in plants, the reader might refer to other reviews that cover the subject (7, 192, 334, 384).
At the end of the past century, redox regulation in plants was perceived as an established physiological mechanism somehow limited in scope, as it appeared to be essentially required for separating photosynthetic carbon fixation occurring in the light, from catabolic reactions occurring in the dark in the same organelle, thereby preventing dangerous futile cycles (54). Twenty years later, the concept is still valid and strongly supported by experimental data, but the field of redox regulation in plants has witnessed an incredible expansion in many new directions. In this context, this comprehensive invited review tries to give the right credit to the recent explosion of thiol-based redox regulation and signaling studies in plants.
The review is organized in sections (sections II–VII) focused on the topics that in our view represent most significantly the scientific developments achieved in the plant redox field in recent times. The section on redox biochemistry of protein thiols (Section II) recognizes the recent transition from a redox biology dominated by TRXs and disulfides to a more articulated subject that takes into consideration how reactive oxygen, nitrogen, and sulfur species (ROS, RNS, and RSS, respectively) may induce up to 10 different post-translational modifications (PTMs) of protein Cys, in a complex interplay that involves also glutaredoxins (GRXs) and glutathione, besides classical TRXs. Section III witnesses the impressive development of redox proteomic techniques that occurs during the past two decades. Emphasis is given to the methodological principles and future technical developments in redox proteomics. To date, these approaches have already allowed the list of putative redox targets to include hundreds or thousands of members with different known redox PTMs on specifically identified Cys in different photosynthetic organisms. The biodiversity of plant TRXs and GRXs and their reducing systems is described in Section IV. Note that before the genomic revolution that in plants started with the sequencing of the genome of Arabidopsis thaliana in 2000, the different known TRXs could be counted on one hand and GRXs were almost unknown. With 20 classes of TRXs and 6 classes of GRXs, photosynthetic organisms are now believed to contain a potential for redox regulation and signaling that seems to largely exceed that of nonphotosynthetic organisms. The state of the art of the structure–function relationships studies in TRXs and GRXs, including their mechanisms of action and interactions with the targets, are included in Section V. Section VI deals with the determination of redox couples in vivo by means of genetically encoded probes and fluorescence microscopy. This section witnesses the adaptation of green fluorescent protein (GFP)-based techniques in the redox field, leading, for the first time, to dynamically determine redox states in vivo. Most of the section is dedicated to glutathione and the popular roGFP probes. Finally yet importantly, Section VII shows that only recently the original model of redox regulation of chloroplast enzymes is receiving experimental confirmation by reverse genetic data. These experiments open the new avenue of redox plant physiology in vivo, including the role of redox regulatory systems in primary productivity, development, and environmental adaptation.
II. Redox Biochemistry of Protein Thiols
A. Production and detoxification of RMS in plants and algae
Redox regulation mainly occurs through different types of PTMs of Cys residues that may occur either through dithiol–disulfide exchange reactions or through reactions in which particular proteins Cys are attacked by RMS. Biologically relevant RMS are based on oxygen (ROS), nitrogen (RNS), or sulfur (RSS), and plant cells may properly synthesize or accidentally release different RMS types by many different mechanisms, both under stress and nonstress conditions.
1. Reactive oxygen species
Light reactions of photosynthesis constitute a fundamental source of ROS in plants. On the one hand, it is believed to be a consequence of the sessile nature of plants since ROS may be produced when the amount of energy obtained from light harvested by photosystems exceeds the combined capacity of downstream metabolic activities and heat dissipation mechanisms (112, 123, 442). On the other hand, ROS are signals that illuminated chloroplasts continuously produce, even in the absence of stress, as the energetic state of the photosynthetic electron transport (PET) chain is affected by varying environmental or metabolic conditions (184). ROS signals produced by altered states of the PET are involved in controlling nuclear gene expression by chloroplast retrograde signaling, leading to long-term acclimation responses (184).
Photosynthesis can produce different types of ROS with different mechanisms (Fig. 2). When light energy absorbed by chlorophylls is not rapidly dissipated, photo-excited chlorophylls in the triplet state accumulate in photosystems II and may generate singlet oxygen (1O2) by interacting with molecular (triplet) oxygen (Fig. 2) (148). This reaction is prevented in light-harvesting antennae where chlorophyll triplet states are quenched by xanthophyll-type carotenoids that dissipate the excitation energy as heat (442). Tocopherols and carotenoids provide a primary protection against the destructive action of 1O2, which primarily results in lipid peroxidation, but also oxidative modification of protein residues including Cys (137, 270, 391).
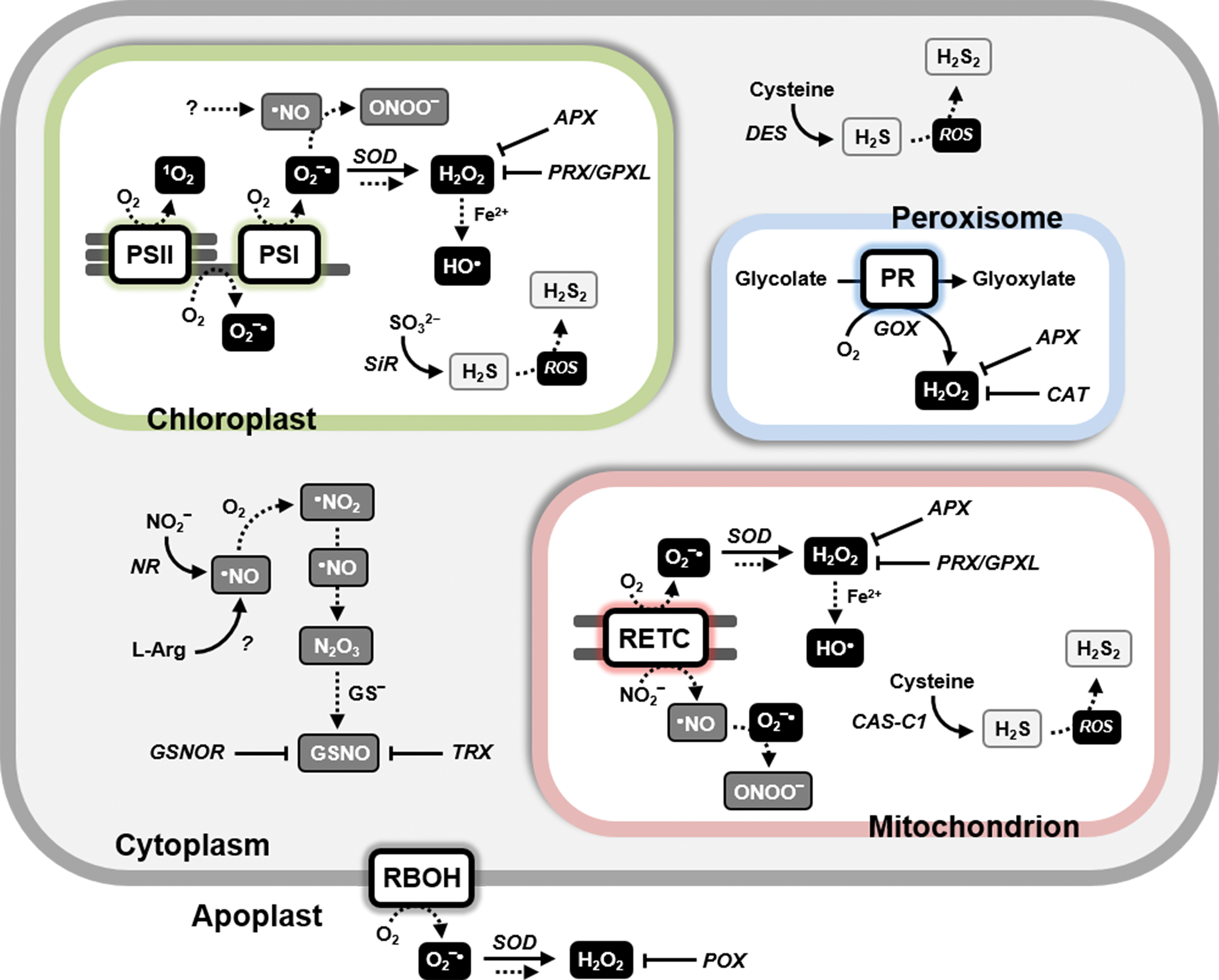
PSI is also a potential source of ROS because it contains low potential Fe-S clusters that easily reduce molecular oxygen to the superoxide ion (O2 −•) (Fig. 2), when downstream acceptors of the PET chain are limiting because they are already reduced. This condition notably arises when carbon fixation by the CB cycle is limited by partial activation of its light-dependent regulated enzymes or low CO2 supply from the atmosphere due to stomata closure. Chloroplast superoxide dismutase (SOD) isoforms guarantee a rapid conversion of O2 −• to H2O2 that ascorbate peroxidases (APXs), glutathione peroxidases-like (GPLXs), and PRXs may then reduce to water (Fig. 2) (377). Ascorbate, glutathione, pyridine nucleotides, TRXs, and their reductases constitute an interlinked powerful system of chloroplasts that tries to keep under control the unavoidable production of H2O2 during photosynthesis (155, 377). Under particular conditions, H2O2 can react with ferrous ion leading to the formation of hydroxyl radical (•OH) (Fig. 2), the most reactive and damaging ROS molecule.
Although iron-containing components of PSI are the major source of O2 −• in chloroplasts in the so-called pseudocyclic electron transfer, photosynthetic oxygen reduction may also occur by other mechanisms. These include a long suspected role of the plastoquinone pool in generating ROS signals (524). However, it is still uncertain whether oxygen reduction might depend on the activity of the plastid terminal oxidase (365) or occur at the site of plastohydroquinone oxidation on cytochrome b6f (31) or even result from the direct reaction between the plastohydroquinone pool and oxygen or O2 −• (516) (Fig. 2).
Another important source of ROS is peroxysomal glycolate oxidase (GOX) that, in the photorespiratory pathway, generates H2O2 in stoichiometric amounts with the oxygenase activity of ribulose-1,5-bisphosphate carboxylase/oxygenase (RubisCO) (Fig. 2). Given the relevant share of photorespiration on photosynthetic metabolism in C3 plants [up to half of carboxylation at 30°C (594)], this is arguably one of the most important sources of ROS in green cells, at least in organisms with no CO2-concentrating mechanisms. Moreover, photorespiration of C3 plants is also another way by which photosynthesis unavoidably produces ROS independently from stress conditions (378). However, huge amounts of catalase (CAT), together with APXs, limit H2O2 from escaping peroxisomes (Fig. 2) (337, 377).
Similar to animal systems, mitochondria are also in plants a potential source of ROS (Fig. 2) (230). Complexes I and III are able to transfer single electrons to oxygen, thereby producing O2 −•, particularly under conditions of low adenosine diphosphate or low oxygen availability (358, 418). Similar to chloroplasts, mitochondria contain SODs and H2O2 detoxifying systems relying on APXs, GPLXs, and PRXs (Fig. 2).
Like H2O2, also O2 −• may be enzymatically produced in plant cells. NADPH oxidases of the respiratory burst oxidase homologue (RBOH) family being probably the major source (Fig. 2). A gene family of about 10 members in higher plants encodes these NADPH-dependent flavocytochromes. Some of them at least reside at the plasma membrane and release O2 −• in the apoplast in response to either abiotic or biotic stress and developmental processes (310). In Arabidopsis, RBOH is responsible for the oxidative burst triggered by incompatible pathogens. Together with nitric oxide (•NO), the resulting superoxide O2 −• orchestrates the hypersensitive response against the pathogens (117). Interestingly, •NO is also involved in a feedback loop that inhibits Arabidopsis RBOH subunit D activity via S-nitrosylation of Cys-890 (570). Except for the presence of SOD and low concentrations of ascorbate, the apoplast is poor in antioxidant systems (19, 157), suggesting that apoplastic H2O2 may accumulate more easily than in other cell compartments.
2. Reactive nitrogen species
Sources of RNS in photosynthetic organisms are diverse and still not fully described. In land plants, reductive pathways converting nitrite (NO2 −) to •NO seem to prevail over oxidative pathways that release •NO from arginine (Fig. 2) (21). Nitrate reductase (NR) can slowly produce •NO by reducing NO2 −, instead of its normal substrate nitrate (NO3 −), using NADH as an electron donor. Since the affinity of NR for NO3 − is higher than for NO2 −, and since NO3 − inhibits the reduction of NO2 −, •NO production by NR is expected to be favored by stress conditions that lead to toxic nitrite accumulation (418). In any case, the role of NR in •NO production in Arabidopsis is supported by reverse genetic studies (21). Alternatively to NR, NO2 − can be also reduced to •NO by components of the mitochondrial electron transport chain (complexes III and IV) (Fig. 2) (196), particularly when oxygen is scarce. Recently, a complex involving NR and NO-forming nitrate reductase (NOFNiR) was shown to constitute a new •NO biosynthetic system in the green microalga Chlamydomonas reinhardtii (74). The role of NR in the complex is to transfer electrons from NAD(P)H to NOFNiR. Whether a similar complex also exists in land plants is currently unknown.
Oxidative pathways for •NO production from arginine seem to be operative in plants (Fig. 2), but the proteins involved remain to be identified. An ortholog of animal NO synthases is found in the alga Ostreococcus tauri (151) but not in other algae and higher plants, where the oxidative release of •NO from arginine may involve distinct mechanisms (21).
Similar to biogenesis, regulation of intracellular •NO levels may also follow different pathways. Nonsymbiotic hemoglobins convert •NO to NO3 − (356), but as part of •NO in the cell is bound to reduced glutathione (GSH) to form nitrosoglutathione (GSNO), the activity of GSNO reductase (GSNOR) that releases ammonia from GSNO (300, 575) is potentially very relevant to modulate •NO availability and also the levels of GSNO, an important transnitrosylating agent (see section II.C.4).
3. Reactive sulfur species
In plants, hydrogen sulfide (H2S) generation occurs through three pathways that differ in the underlying mechanisms and the subcellular compartments in which they take place. The primary source of H2S is the chloroplast where it is produced in the reductive sulfate-assimilation pathway through the action of sulfite reductase (SiR, Fig. 2) (488). Alternative pathways occur in both mitochondria and cytoplasm. β-Cyanoalanine synthase (CAS-C1), catalyzing the conversion of cyanide and Cys to β-cyanoalanine and H2S, is found in mitochondria (Fig. 2) (11). In the cytoplasm, the enzyme L-Cys desulfhydrase (DES1) catalyzes the desulfuration of Cys yielding sulfide, ammonia, and pyruvate (Fig. 2) (9, 10, 185). In any case, the production of H2S in subcellular compartments where ROS or RNS may also be produced can result in nonenzymatic reactions, including the one-electron oxidation of H2S to hydrogen disulfide (H2S2) (Fig. 2), which may lead to persulfidation of protein Cys (see section II.C.5).
B. Reactivity of Cys is strictly controlled by the protein microenvironment
In plants, RMS (including ROS, RNS, and RSS) actively participate in redox homeostasis. In this context, proteins play an essential role as central mediators of RMS-dependent signaling events. Many of these proteins rely on modifications of Cys residues for modulating their redox activity, whereas a few of them use other residues (e.g., methionines or tyrosines) for the same purpose, but knowledge on methionine- and tyrosine-dependent signaling pathways is still limited to a few studies (35, 237, 238, 265, 327).
Cys-based redox modifications have been extensively investigated and they are widely accepted to play a prominent role in regulatory and signaling networks that support plant development, metabolic functions, and responses to varying environmental conditions. The functionality of Cys residues in redox biology depends on the chemical reactivity and structural flexibility of their sulfur atom. Sulfur can form covalent bonds with different types of atoms present in living organisms (C, H, O, P, and N) and establish stable complexes with transition metals (Zn, Fe, and Cu). In addition, being weak acids, Cys thiols (−SH) are found in equilibrium with the deprotonated thiolate form (−S−) over a physiological range of pH to flexibly optimize the function of specific protein Cys (Fig. 3A). Compared with the protonated forms, Cys thiolates are more sensitive to the intracellular redox environment and susceptible to RMS-dependent oxidative modifications. Altogether, these features allow Cys residues to play fundamental structural and catalytic roles, and to function in RMS-mediated redox signaling as reversible molecular switches (321, 508, 537).
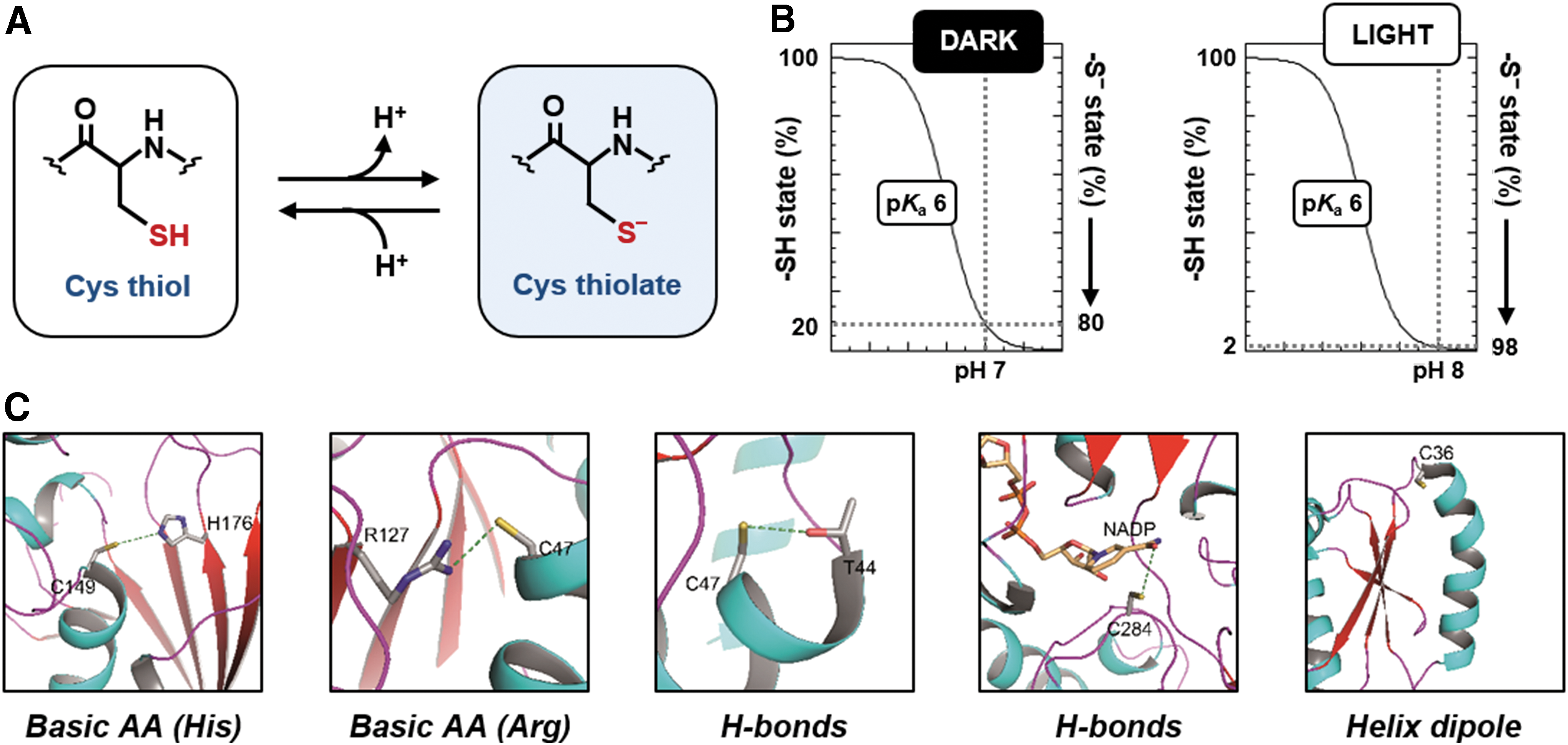
The acid dissociation constant (pK a) of a Cys designates its tendency to dissociate. The pK a of the sulfhydryl groups of free Cys is ∼8.3 (395, 434, 502). A slightly higher pK a value [8.8, (440)] is attributed to the Cys thiol of GSH. These pK a values imply that these Cys thiols are largely found in the protonated form at neutral pH, whereas thiolate forms might progressively accumulate only at alkaline pH values. For example, the percentage of GSH thiolate (GS−) at pH 7 is only 2%, but this value increases to 14% when the pH raises to 8. This variability is particularly important in subcellular compartments that experience a shift from neutral to slightly alkaline pH as observed in the chloroplast stroma during dark to light transitions (215, 221, 503).
Although the vast majority of protein Cys harbors a pK a >8, some of them are acidic due to the microenvironment in which they are located (395, 508). Selected protein Cys involved in thiol switching reactions have pK a values ranging between 3 and 6.5 (508), allowing these residues to be predominantly or fully deprotonated at physiological pH (Fig. 3B). The structural features that contribute to modulate the acidity of Cys thiols mainly include the proximity of amino acids such as lysine, histidine, or arginine, which by attracting the proton of the thiol become positively charged and form an ion pair with the negatively charged thiolate (Fig. 3C) (96, 508). These types of interactions are found in enzymes such as GAPDH (36, 576), isocitrate lyase (37),and PRXs (368). In other proteins, hydrogen-bonding networks may also be relevant (Fig. 3C); in TRXs and GRXs, for instance, the hydrogen-bonding network is believed to be the major structural determinant of the acidity of the catalytic Cys (434). Finally, the location of the Cys residue at the N-terminus of an α-helix generating an electric macrodipole may also contribute to its acidity (Fig. 3C) and, in general, desolvation can also have an impact on thiol pK a by decreasing the dielectric constant of water and thus enhancing electrostatic interactions that occur in catalytic sites (146). In many other cases, the relative influence of each structural factor to the thiol pK a is still undefined and difficult to derive from the protein tridimensional structure, such that it needs to be determined experimentally (508).
Although thiolates are stronger nucleophiles than thiols, it should be remembered that the nucleophilicity of a thiolate actually decreases with decreasing pK a of the Cys. In other words, the most reactive Cys are often Cys that are acidic enough to be largely deprotonated at neutral pH, but not too acidic to lose completely their nucleophilicity (146, 508). Moreover, the protein microenvironment affects the reaction between Cys and RMS also in other ways, not directly dependent on Cys pK a.
The H2O2-dependent oxidation of Cys thiolate nicely exemplifies the latter point. By comparing the reactivity toward H2O2 of two thiolate-containing proteins, namely PRX and GAPDH (pK a values of ∼5 and ∼6, respectively), it was observed that PRX reacts with H2O2 104–105 times faster than GAPDH (508, 536). Since the catalytic Cys of both PRX and GAPDH are fully or almost fully deprotonated at neutral pH, other factors than thiolate availability and exposure should be taken into account to explain the vastly different reactivity. Indeed, the stabilization of the transition state (−S···O···O···H) by active-site residues was recently proposed to sustain the catalytic power of PRX (207, 362). A counter example is given by GRX S12, which contains a highly acidic catalytic Cys [pK a value <4.0; (102, 573)] but exhibits a reactivity toward H2O2 that is comparable with GAPDH [pK a ∼6; (508, 573, 576)]. Based on these observations, we can conclude that although oxidation mainly affects acidic Cys, the Cys microenvironment can control the reaction kinetics with H2O2 and possibly other RMS, as detailed in the following subsections.
C. Cys residues may be modified in many different ways by RMS or enzymes
The cellular capacity for RMS-mediated regulatory pathways depends on different types of Cys modifications that allow oxidant signals to be transduced into biological responses. In the following subsections, the chemistry and mechanisms of oxidative modifications induced by each class of RMS molecules, namely ROS, RNS, and RSS, are discussed. Alternative mechanisms of protein Cys oxidation catalyzed by enzymatic systems or mediated by intermediate Cys oxoforms (i.e., sulfenic acids and nitrosothiols) or oxidant molecules (e.g., oxidized glutathione, GSSG) are also described.
1. ROS-dependent redox modifications of protein thiols
Protein Cys thiol can be oxidized by both radical (O2 −•, •OH) and nonradical ROS molecules (1O2, H2O2). Singlet oxygen is a nonradical molecule that can react with sulfur-containing amino acids (i.e., Cys and methionine) but also with histidine, tryptophan, and tyrosine residues (391). The oxidation of Cys thiols by 1O2 occurs via formation of a short-lived zwitterionic intermediate (RS+(H)–OO−), which decomposes yielding oxidized sulfur species such as sulfonic acids (−SO3H) or alternatively, disulfides if another Cys residue is able to react with the initial intermediate (Fig. 4) (360, 391). Although 1O2 is believed to play a signaling role in chloroplasts (276), the molecular bases of its action are not fully understood.
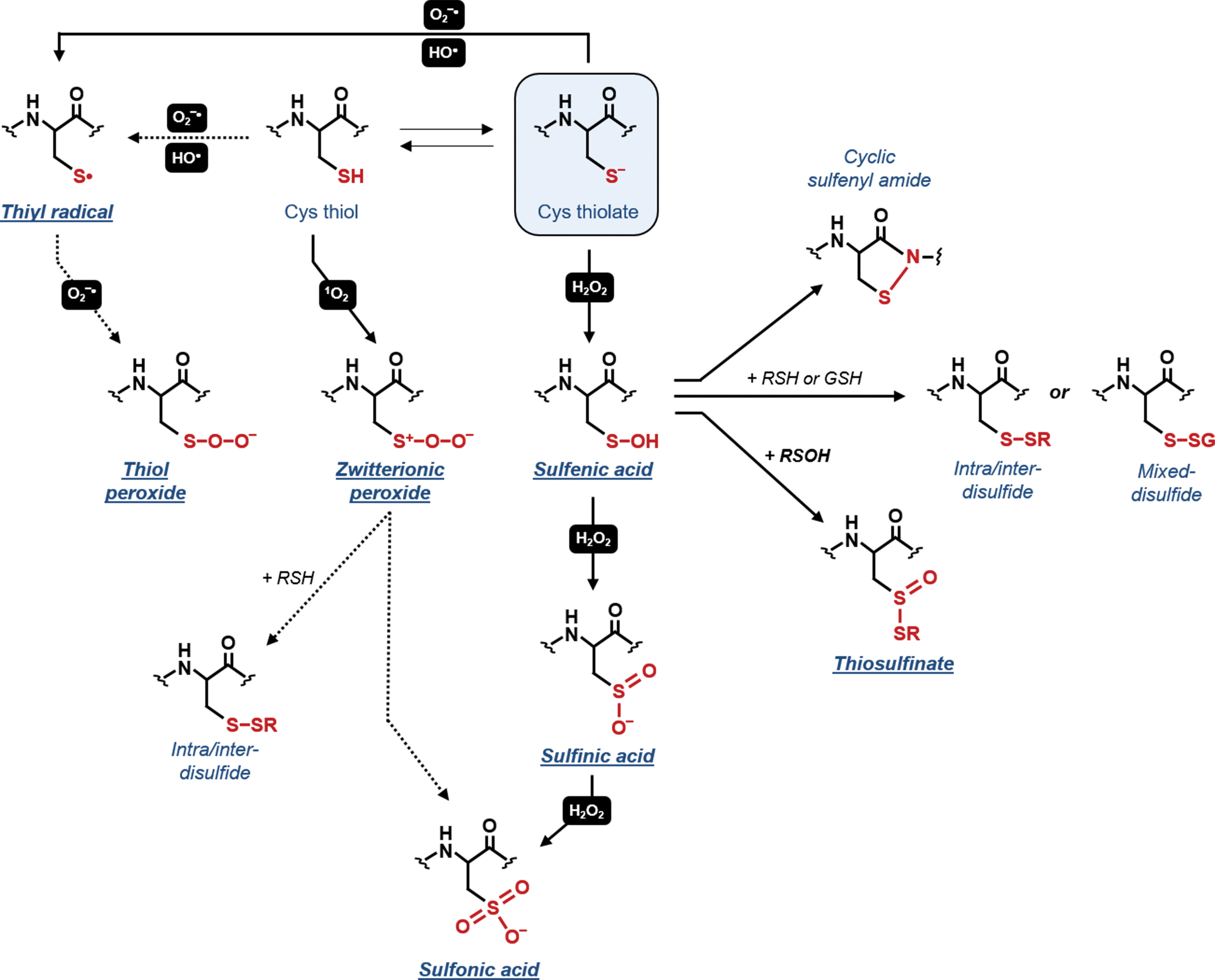
The radical superoxide (O2 −•) is a relatively unreactive radical and its preferential targets appear to be other radical species such as •NO (395). ln proteins, O2 −• can react with Fe-S clusters and some transition metals (113, 537), and shows low reactivity toward protein side chains, Cys being one of the less sensitive amino acids (113). However, if this reaction occurs, Cys may undergo cysteinyl (thiyl) radical (−S•) formation and possibly peroxidation (i.e., thiol peroxide formation) (Fig. 4) (169, 454). In contrast to O2 −•, •OH is highly reactive and is capable to oxidize nearly all protein residues with second order rate constants near the diffusion limit (i.e., 109–1010 M −1s−1) (113). Protein Cys oxidation mediated by •OH is postulated to occur through hydrogen atom abstraction from S–H bonds yielding thiyl radicals (−S•, Fig. 4) (15, 113, 477, 519).
The aforementioned reactions are likely to occur under physiological conditions but their relevance in thiol-based redox signaling networks might be limited. These ROS molecules (1O2, O2 −•, and •OH) have high reactivity with biological macromolecules other than proteins. The abundance of these targets in vivo results in very short lifetimes and limited diffusion from the sites of generation. Therefore, oxidation by these ROS is restricted to proteins located at the proximity of production sites. In addition, they react with diverse protein side chains and display no specificity for reactive Cys.
Among ROS, H2O2 has the longest lifetime and is highly selective toward sulfur-containing residues, Cys thiolates being the most sensitive (226, 395, 453). The H2O2-dependent two-electron oxidation of reactive Cys leads to the formation of a sulfenic acid (−SOH) (Fig. 4). Sulfenic acids are emerging as redox signaling hubs implicated in different types of secondary modifications. Owing to their reactive nature, sulfenic acids are often considered as an unstable intermediate subjected to several alternative fates (Fig. 4). In the presence of excess H2O2, sulfenic acids can act as a nucleophile and be further oxidized to sulfinic (−SO2H) and sulfonic acid (−SO3H) Fig. 4), with reaction rates that are generally slower (0.1–102 M −1s−1) than the primary oxidation event (10–107 M −1s−1) (395, 508). Sulfinic and sulfonic acids are usually considered irreversible forms except for sulfinated 2-Cys PRX (PRX-SO2H), which can be reversibly reduced to the thiol form by sulfiredoxin (243). Sulfenic acids can alternatively serve as electrophiles reacting with the backbone amide group of a neighboring residue forming a reversible cyclic sulfenamide or condensate with an interfacing additional sulfenic acid to generate a thiosulfinate (Fig. 4). In most cases, however, sulfenic acids react with a proximal thiol from a protein Cys or a GSH (Fig. 4) leading to the formation of intra-/intermolecular disulfide bonds (−S−S–) or a mixed disulfide (−S−SG, S-glutathionylation). Besides protein Cys, H2O2 can also react with GSH yielding glutathione sulfenate intermediates (GSOH) but, owing to its pK a, this reaction proceeds very slowly (∼1 M −1s−1) (395).
2. Plant cysteine oxidases catalyze the enzymatic oxidation of protein Cys to sulfinic acids
Besides protein disulfides, other oxidative modifications are found to be catalyzed by specific enzymes. Indeed, Cys oxidation to sulfinic acids can occur in the presence of plant Cys oxidases (PCOs). These enzymes are nonheme Fe2+-dependent dioxygenases catalyzing an essential step of the N-end rule pathway in plants that controls, for example, the stability of group VII ethylene response factors (ERF-VIIs). Whereas ERF-VIIs are rapidly degraded in normoxia, flooding-induced hypoxic conditions reduce the activity of PCOs allowing ERF-VIIs stabilization and consequently transcriptional adaptative responses (509, 531, 533). The molecular mechanisms underlying PCO activity have been recently established and Cys sulfinic acids are generated via an oxygen-dependent reaction (532, 533). Besides oxygen, ROS and likely •NO are postulated to be involved in such reactions but the mechanisms are still not clarified (418).
3. RNS-dependent redox modifications of protein thiols
In biological systems, •NO and derived compounds [i.e., nitric dioxide (•NO2), dinitrogen trioxide (N2O3), and ONOO−] can also induce oxidative modifications of protein residues including Cys thiols (Fig. 5). Similar to O2 −•, •NO is a relatively unreactive radical and preferentially reacts with other radical species and with metals. By reacting with O2 −•, •NO generates ONOO−. Besides binding to heme-containing proteins (395), •NO is involved in a covalent modification of protein Cys termed S-nitrosylation (575). This reversible modification does not directly involve •NO and three major mechanisms have been proposed to account for S-nitrosothiol (−SNO) formation (575). The reaction of •NO with transition metals of metalloproteins yields unstable metal–nitroxyl complexes that can then transfer the NO moiety to a Cys residue that generally belongs to the same protein (Fig. 5). Alternatively, •NO2, which is spontaneously generated by the reaction of •NO with molecular oxygen, can induce the one-electron oxidation of Cys thiolates (Fig. 5). This reaction leads to the formation of thiyl radicals that can undergo radical–radical combination with •NO to yield S-nitrosothiols. S-nitrosothiols formation can also be generated by the nitrosating compound N2O3 that is spontaneously formed by the radical reaction between •NO and •NO2 (107, 395). N2O3 can subsequently transfer its nitrosonium group (+NO) to proteins or low-molecular weight thiolates generating S-nitrosothiols and releasing NO2 −.
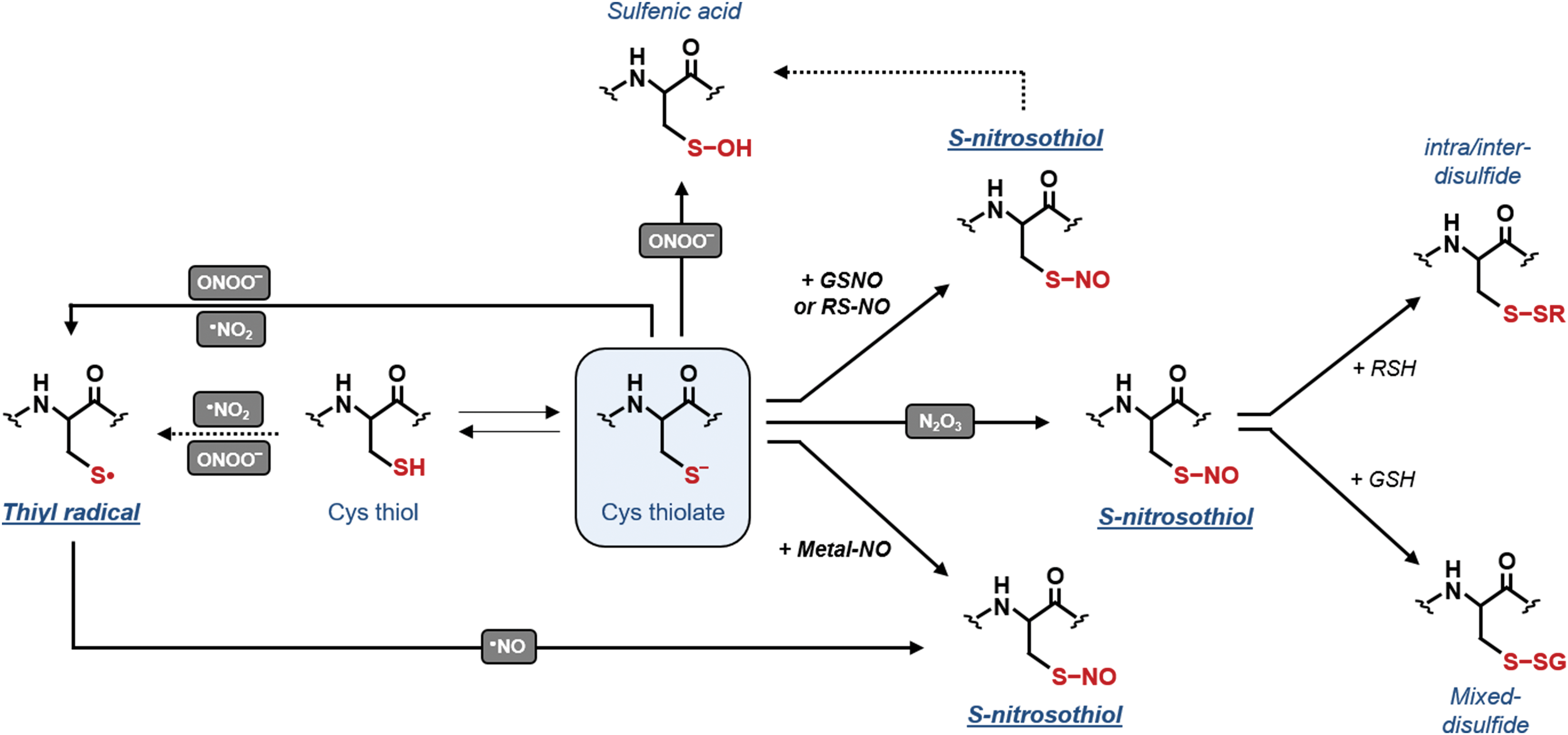
Owing to its high intracellular concentration [1–5 mM, (156, 373, 440)], GSH might be a primary target of N2O3-dependent nitrosylation yielding GSNO (Fig. 6). This molecule along with S-nitrosylated proteins can transfer the NO moiety to another Cys in a process termed trans-nitrosylation (Fig. 5). Within cells, the equilibrium between GSH and GSNO controls the level of S-nitrosylation in some proteins at least (Fig. 6) (43, 580). TRXs efficiently reduce GSNO in vitro [(369); Zaffagnini et al., personal communication] and catalyze protein denitrosylation of specific targets in vivo (262). However, TRX-dependent reduction of GSNO or protein-SNO releases a nitroxyl (HNO) that is highly reactive and still able to interact with Cys residues (49). To date, the foremost enzyme known to control the intracellular concentration of GSNO is GSNOR (300, 575) (see Section II.C.4).
The sensitivity of a particular Cys thiolate to trans-nitrosylation seems to depend on different factors including Cys reactivity, the accessibility to NO donors and the local Cys microenvironment (e.g., acid–base motif and hydrophobic residues) (129, 153, 304, 320, 469, 579). In general, trans-nitrosylation is considered not only as a prominent mechanism of protein S-nitrosylation but also as a mechanism that allows propagating the NO signal far away from the site of •NO production (395). Compared with sulfenic acids, nitrosothiols cannot further react with oxidants but can generate sulfenic acids by spontaneous hydrolysis (Fig. 5) or, alternatively, form disulfides in the presence of protein or GSH thiolates (Fig. 5).
Peroxynitrite (ONOO−) and its protonated form (ONOOH) are highly reactive nonradical species that can cause oxidation of several protein residues including Cys, methionine, tryptophan, and tyrosine. The most relevant peroxynitrite-mediated reaction is tyrosine nitration but its physiological relevance in signaling pathways still requires further confirmation. Similar to •OH, the reaction of ONOO− with protein Cys yields thiyl radicals (Fig. 5) (113, 486) but other oxidation products such as sulfenic acids are also generated (Fig. 5) (584).
4. GSNO reductase controls the level of nitrosothiols in plants
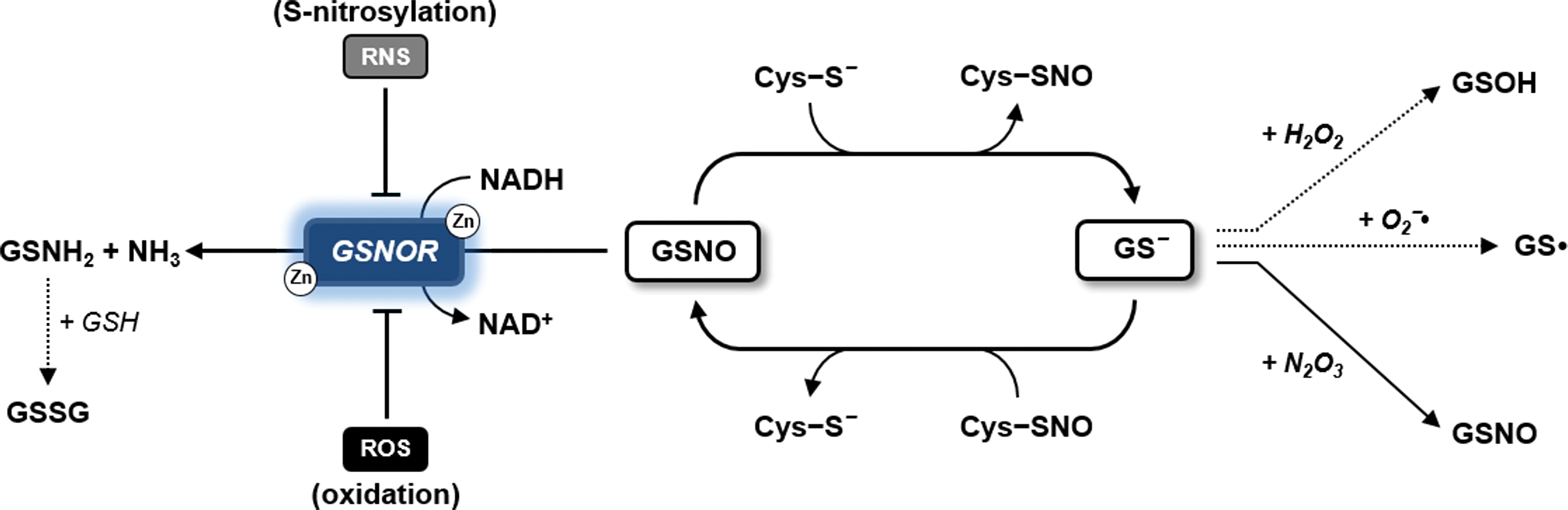
GSH can efficiently reduce protein S-nitrosothiols (181, 433, 575). However, although this nonenzymatic reaction restores reduced proteins, it also generates GSNO (Fig. 6), which can further react with reactive Cys thiols yielding de novo S-nitrosothiols (97, 575). Consequently, GSH by acting as an efficient reducing system can also promote further S-nitrosylation via GSNO. To date, the foremost enzyme known to control the intracellular concentration of GSNO is GSNOR (300, 551, 575). This enzyme is highly conserved in most bacteria and all eukaryotes including plants (303). GSNOR belongs to the class III alcohol dehydrogenase family and catalyzes the reduction of GSNO using NADH as an electron donor (268, 271, 303). The effective contribution of GSNOR in degrading GSNO relies on its catalytic ability to reduce GSNO into glutathione sulfenamide (GSNH2), which spontaneously forms GSSG and NH3 in the presence of GSH (Fig. 6). Consequently, GSNOR acts as a specific scavenging system for GSNO and indirectly controls the extent of GSNO-dependent protein S-nitrosylation.
In plants, the role of GSNOR in S-nitrosothiols metabolism was demonstrated by Loake and colleagues (139). Arabidopsis mutants that do not express GSNOR (gsnor) have more low-molecular weight nitrosothiols (e.g., GSNO) and high-molecular weight nitrosothiols (e.g., S-nitrosylated proteins). The function of GSNOR was also associated with various physiological processes including pathogen response, thermotolerance, plant growth, flowering, hypocotyl elongation and germination, and resistance to cell death. Whether these effects are also mediated by S-nitrosylation, however, still need to be clearly established (139, 272, 281, 300, 443).
The activity of plant GSNOR itself has been recently reported to be altered by redox modifications (Fig. 6). Arabidopsis and poplar GSNOR were found to undergo S-nitrosylation in vivo under conditions of increased endogenous NO availability (83, 162). Intriguingly, this modification causes partial inhibition of GSNOR activity (162, 193). More recently, AtGSNOR was also found to be negatively affected by in vitro treatment with H2O2 or exposure of Arabidopsis plants to paraquat (268). Altogether, these pieces of evidence suggest that the transient inhibition of plant GSNOR by oxidative modifications might reinforce NO signaling by favoring GSNO accumulation (193, 268, 300).
5. RSS-dependent redox modifications of protein thiols
The prototypical inorganic RSS is H2S, which is the most stable RMS with a half-life in the minute time scale (485). Based on its chemical properties [pK a1 = 7 and pK a2 = 12–15; (80, 343)], H2S can easily dissociate under physiological conditions and it is, therefore, assumed that H2S pools mainly include H2S and HS−. In plants, the involvement of H2S as a signaling molecule is receiving growing attention because of its ability to interact with proteins and possibly with other RMS (16, 17, 79). Given its nucleophilic properties, H2S can scavenge reactive intermediates including •NO, O2 −•, ONOO−, or H2O2, suggesting that it can play protective effects against oxidative stress (249, 534). However, a biological relevance for this activity is largely speculative because of its limited reactivity compared with GSH and its intracellular concentration, which is considered low (174, 249, 485). With proteins, H2S can interact with some heme groups but also with Cys residues in a process called persulfidation. This oxidative modification consists in the conversion of a protein Cys into a persulfide (−S−SH) and it is suggested to modulate protein functions (259, 361, 392, 393) by increasing the nucleophilicity of the Cys (106, 392). Noteworthy, this reaction can involve both Cys thiolates and oxidatively modified Cys intermediates such as sulfenic acids (395). Although persulfidation has been proposed as a new key player in redox signaling, the underlying mechanisms are poorly understood and the physiological relevance of H2S-related mechanisms in plants is still largely unknown.
Three major mechanisms for protein persulfidation have been postulated (Fig. 7), none of which involves a direct reaction between H2S and Cys residues (249, 505). The first two mechanisms involve a nucleophilic attack of H2S on oxidized protein Cys, either present as sulfenic acid or engaged in disulfide bonds (i.e., intra/inter or mixed disulfide) (Fig. 7). However, disulfide-mediated persulfide formation is uncertain mainly because H2S is a poor reductant compared with GSH and this reaction may proceed very slowly in vivo (70, 395). Another possibility is that alternative intermediate Cys oxoforms (e.g., S-nitrosothiols or sulfenylamides) can react with H2S yielding persulfides. The third mechanism involves the ROS-mediated oxidation of H2S to H2Sn (n = 2 or higher), which can subsequently undergo a nucleophilic attack by a protein thiolate to give rise to a persulfide (Fig. 7).
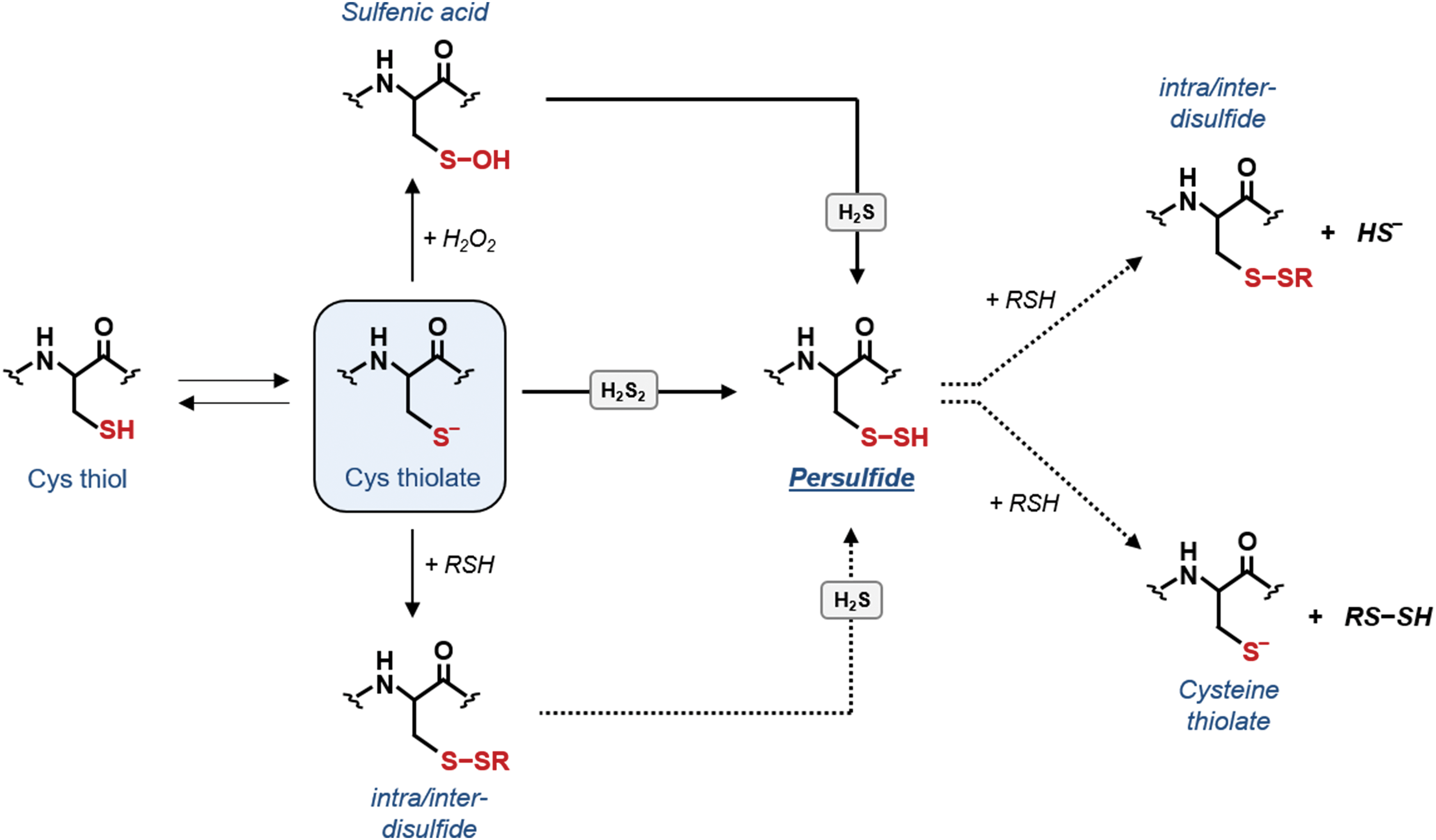
Similar to nitrosothiols and sulfenic acids, persulfides contain two electrophilic centers and can react with another protein thiol yielding a disulfide or facilitating trans-persulfidation (Fig. 7). The latter route is reminiscent to trans-nitrosylation and is likely to be highly protein specific (395).
6. S-glutathionylation as a special type of disulfide formation
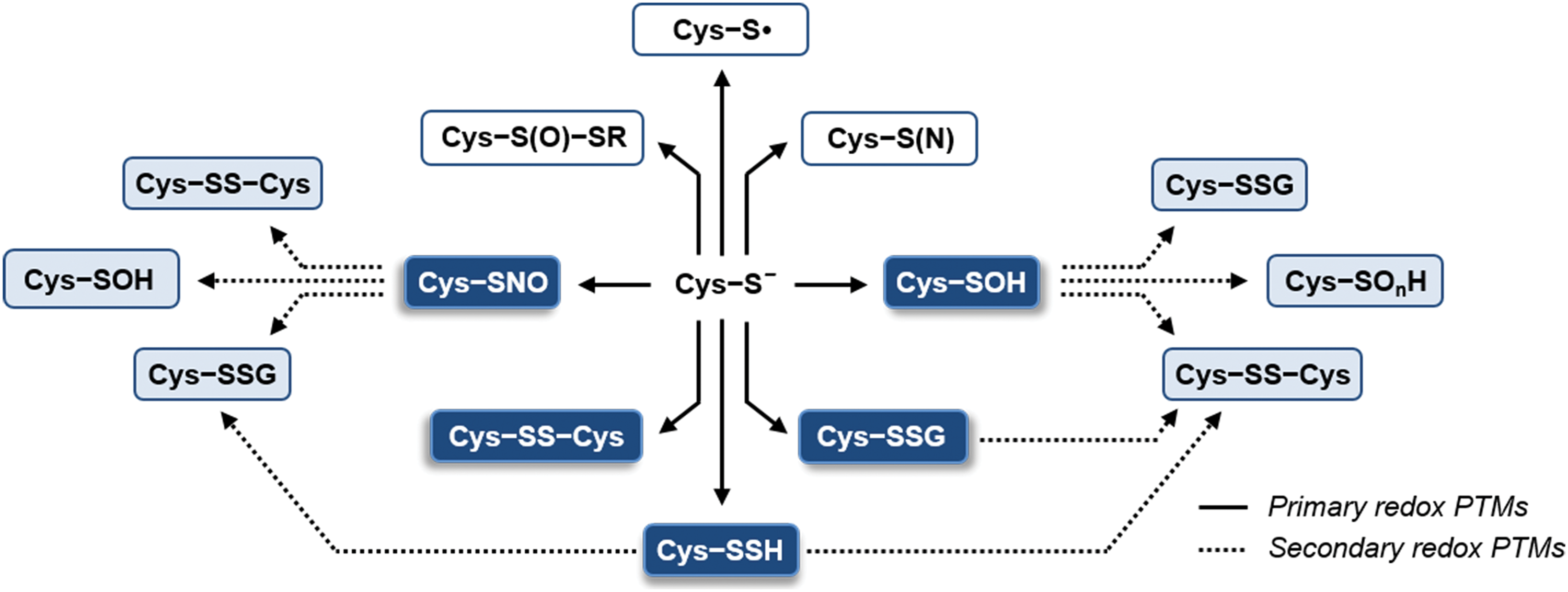
Disulfide bond formation is the best characterized Cys-based redox modification. It consists in the covalent bonding between two Cys residues belonging to the same or different polypeptides. Besides the well-known role of TRXs in dithiol–disulfide interchange reactions (see section I) (Fig. 8A), disulfide formation may also involve RMS. One possible route relies on the primary oxidation of a Cys to sulfenic acid or S-nitrosothiol, followed by thiol condensation with an additional Cys (Fig. 8A; see sections II.C.1 and II.C.3).
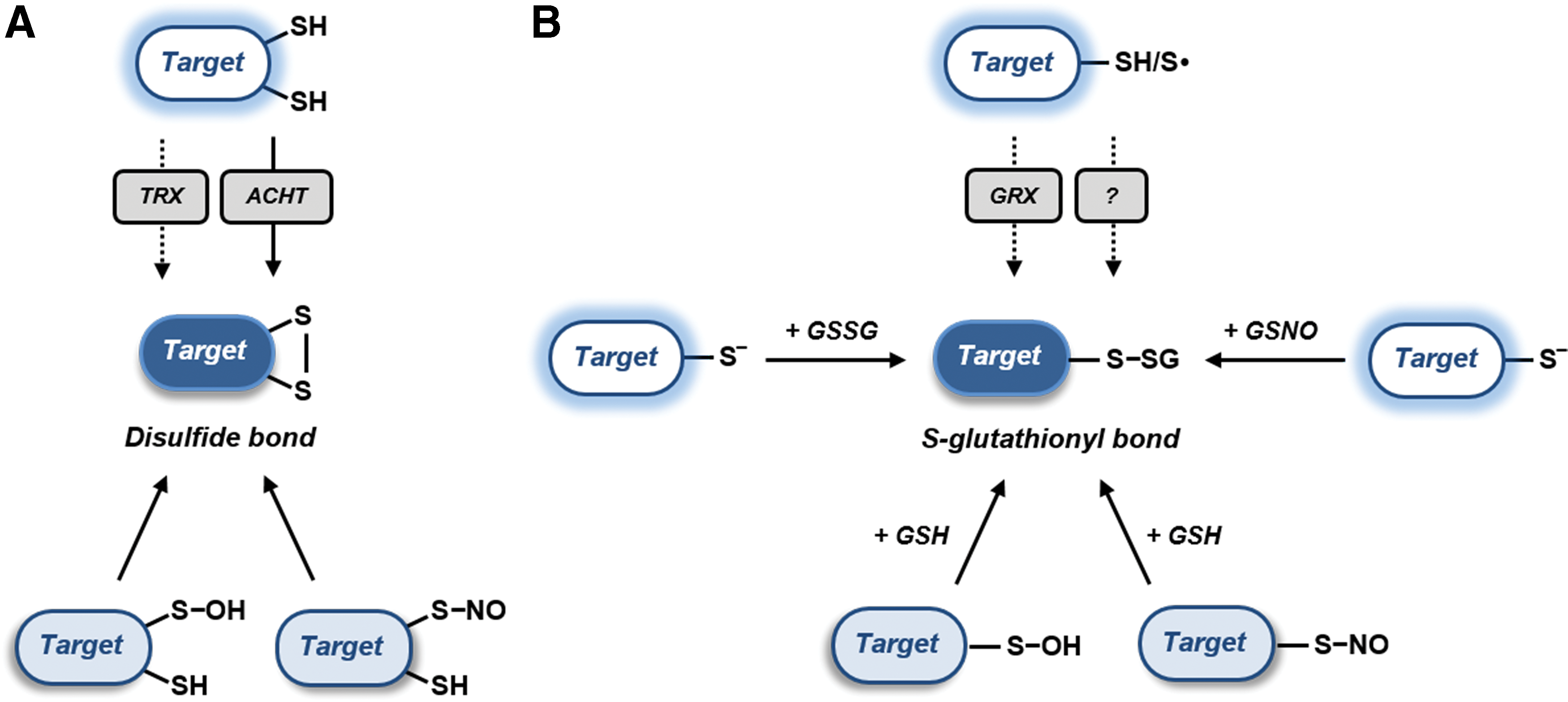
Protein S-glutathionylation has emerged as a widespread oxidative modification involved in the modulation of protein function but also in the protection of protein Cys from irreversible oxidation (i.e., sulfinic and sulfonic acid formation) (572, 574). As already mentioned, one potential mechanism of protein S-glutathionylation is the condensation of GSH with an intermediately oxidized Cys (i.e., sulfenic acid or S-nitrosothiol; see sections II.C.1 and II.C.3, respectively). The electrophilic nature of these oxidative intermediates favors the nucleophilic attack of GSH thiolates, leading to the formation of protein mixed disulfides (Fig. 8B).
Another mechanism of protein S-glutathionylation involves a thiol–disulfide exchange between GSSG and a protein Cys thiolate (Fig. 8B). Typically, this reaction proceeds very slowly and is supposed to be thermodynamically prevented by the high GSH/GSSG ratios of most plant subcellular compartments (see section VI) (155, 157, 458). Nevertheless, we cannot exclude a priori the possibility that specific proteins might undergo GSSG-dependent glutathionylation as a consequence of limited fluctuations (i.e. oxidation) of the glutathione redox pool. Plastidial GRXS12 for instance is glutathionylated in vitro at GSH/GSSG ratios of 102−103 that fully prevent the glutathionylation of other targets such as cytoplasmic GAPDH (36, 573).
As an alternative to GSSG, protein glutathionylation can occur in the presence of GSNO (Fig. 8B). This molecule can allow the formation of S-nitrosothiols but can also transfer its GS moiety to a target Cys. The structural features controlling one reaction over another are still uncertain and are likely related to the local environment surrounding the target Cys residue. GRXS12 is an example of a protein that is glutathionylated by GSNO, rather than nitrosylated (573).
Finally, in addition to nonenzymatic mechanisms, protein glutathionylation might also be catalyzed by specific oxidoreductases (Fig. 8B). This was shown for human GRX2 that appears to promote protein S-glutathionylation after a reaction mediated by either GSSG or GS• radical (38, 163). Both mechanisms rely on the formation of glutathionyl GRX intermediates and the ability of GRX to transfer the glutathionyl adduct to an acceptor protein thiolate in a trans-glutathionylation reaction. To date, no evidence suggests the ability of plant GRXs to catalyze such reactions in vivo. However, a remarkable example of enzyme-assisted glutathionylation occurring in plants involves the genetically encoded probe roGFP2 fused to human GRX1 [GRX1–roGFP2; (333, 458)]. This chimeric protein has been developed to monitor the glutathione redox state and its functioning is specifically related to reversible trans-glutathionylation reactions between the probe and GRX1.
III. Redox Proteomics: Methodological Principles and Future Developments in the Plant Field
Despite the latest improvements of mass spectrometry (MS) in terms of sensitivity and resolution over the past decade, direct analysis of redox-modified proteins remains highly challenging. As shown in Figure 9 (see also section II), >10 thiol-based redox PTMs are currently known (101, 182, 395). Owing to their lability, their low stoichiometry, and their possible interchange during sample processing as exemplified in Figure 9 (black and gray boxes corresponding to primary and secondary modifications), the redox proteomics field has to face different biochemical, methodological, and instrumental challenges to get insights about the in vivo dynamics of redox PTMs. In complex systems, redox proteomic strategies currently rely on the differential labeling of Cys according to their modification state followed by MS analyses at the peptide level after an affinity enrichment step.
Nontargeted quantitative strategies, such as OxICAT (283, 470) and OxiTMT (474), were developed to determine oxidation levels of hundreds of Cys upon oxidative treatments. To date, these approaches have been applied to quantitatively identify oxidative-prone Cys in the marine diatom Phaeodactylum tricornutum (436) and the cyanobacteria Synechocystis sp. PCC 6803 (194). In the latter organism, 20% to 40% of proteins were found to contain oxidized Cys in the dark. Nevertheless, these strategies are unable to distinguish which reversible redox PTM is at the origin of the modification of the Cys. In this section, we focus on approaches trapping selectively the different reversible redox PTMs with a special emphasis on their advantages, drawbacks, and limitations, and their use in photosynthetic organisms.
A. Thioredoxome
Two main proteomic strategies have been employed to identify hundreds of proteins containing disulfide bonds reduced by TRX (56, 299). The first and most common approach takes advantage of the ability of a monocysteinic TRX variant (Fig. 10), where the C-terminal active site Cys is replaced by serine or alanine, to covalently bind oxidized target proteins (for the mechanism, see section V). The monocysteinic TRX is most often grafted on a chromatographic resin and TRX-bound targets are eluted with a chemical reductant such as dithiothreitol (DTT). This type of column has been applied to numerous protein extracts from the cyanobacterium Synechocystis sp. PCC 6803 (298, 402, 404) and also different photosynthetic eukaryotes (6, 24, 27, 28, 32, 187, 208, 227, 285, 317, 319, 353, 543, 552, 565). This approach has several drawbacks. First, it lacks specificity as several TRX classes (f, m, y, h) immobilized to the resin retain the same targets while they have distinct specificities in solution at more diluted conditions (see section IV). This may be due to the high concentration of TRX or to peculiar properties of the monocysteinic variants (339). Moreover, depending on the washing conditions, proteins interacting with TRX targets may be eluted together with genuine TRX targets, thereby increasing false-positive rates. Nevertheless, the major drawback of the column approach is that it only identifies the target protein, whereas the exact Cys targeted by TRX remains unknown.

The second main strategy, named “reductome” approach, is based on the in vitro reconstitution of the enzymatic TRX system (NADPH, NTR, and TRX) within a cell-free protein extract followed by labeling of newly exposed Cys with fluorescent (311, 559), radioactive (318), or biotinylated probes (317) (Fig. 10). This strategy was applied to total or subcellular soluble protein extracts from different land plants (6, 26, 27, 208, 311, 312, 325, 542, 543, 558). Biotinylated tags allow enrichment of Cys-containing peptides by affinity purification and allow identification of TRX-targeted Cys, a major advantage of the reductome approach. Unfortunately, to increase the number and diversity of targets, the in vitro reduction has to be performed using relatively high TRX concentration for which isoform specificity is mostly lost. Therefore, the lack of specificity is common to both the affinity column and reductome approaches. The two approaches are complementary as the targets identified only partially overlap (317, 405, 543).
Recently, quantitative adaptations of the reductome approach were developed for MS analyses based on chemical labeling with cleavable isotope-coded affinity tag reagents (cICAT) (205, 206) or with Cys-reactive tandem mass tag (Cys-TMT) (588). The most recent study combined the column with the quantitative reductome approach to investigate the thioredoxome of the unicellular green alga C. reinhardtii and identified 1188 proteins and 1052 Cys regulated by TRX. The quantitative approach based on differential cICAT labeling allowed to decrease false positives by filtering out the noise due to incomplete thiol blocking of the protein extract and thereby retain only proteins that are effectively reduced by TRX (405). Nevertheless, the targets identified remain putative and the presence of a TRX-reduced disulfide bond needs to be confirmed experimentally. Some TRXs were also shown to function, on specific targets, as denitrosylase (41, 42, 46, 487) and deglutathionylase (36, 189, 482). However, such activities should not impact the identification of TRX targets in both approaches as the vast majority of nitrosylated proteins are denitrosylated by GSH rather than TRX (44, 388, 433, 580), and TRX targets were analyzed in conditions wherein S-nitrosylation and S-glutathionylation are limited or absent (350, 571). Moreover, the reduction of S-nitrosylated or S-glutathionylated proteins by monocysteinic TRX is considered to yield nitrosylated or glutathionylated TRX rather than mixed disulfide with the target (36, 262, 405). Finally, both the proteomic identification of already established TRX targets and the biochemical confirmation of targets previously identified by proteomics strongly support the reliability of proteomic approaches to identify TRX targets. Biochemically confirmed TRX targets previously identified by proteomic studies include at least 2-Cys PRX (187, 353), phosphoglycerate kinase (349) magnesium chelatase CHLI subunit (232), β-amylase 1 (478), methionine sulfoxide reductases (494, 517), glucan water dikinase (342), uricase (130), and cytosolic NAD-MDH (212).
B. Nitrosylome
The identification and the quantification of S-nitrosothiols and S-nitrosylated proteins in biological samples remain highly challenging due to the lability of the −SNO bond (242) whose stability is strongly influenced by multiple factors, including light, metals, and reducing compounds such as GSH or TRXs. Such an instability of S-nitrosothiols precludes their direct detection by matrix-assisted laser desorption-ionization MS (250) and even by electrospray ionization MS (211) unless ionization parameters are carefully optimized (525). Therefore, high-throughput analysis of nitrosylated proteins is based on indirect methods for which the NO moiety is replaced by a more stable tag that allows an enrichment step.
Most studies rely on the biotin switch technique (BST) developed in 2001 (241) that was the first approach allowing detection and identification of S-nitrosylated proteins at the proteome scale (Fig. 10). This method consists in the replacement of the NO moiety of S-nitrosylated Cys residues by a disulfide-bonded biotin tag in a three step process: (i) initial blocking of unmodified Cys thiols under denaturing conditions, (ii) “specific” reduction of −SNOs by ascorbate, and (iii) labeling of the nascent thiols with the biotinylating reagent N-[6-(biotinamido)hexyl]-3′-(2′-pyridyldithio)-propionamide (biotin-HPDP). The replacement of the −SNO moiety by a disulfide-bonded biotin tag allows detection of previously S-nitrosylated proteins by immunoblotting or purification by avidin-based affinity chromatography and DTT elution for MS-based identification (301). Many variants of the original BST approach have been proposed such as the −SNO site identification (SNOSID) approach that includes a trypsin digestion step before enrichment (210) or the −SNO resin-assisted capture (SNO-RAC) method that takes advantage of a thiol-reactive resin for capturing nascent thiols after ascorbate reduction (Fig. 10) (152). The two methods allow identification of both the modified proteins and the modified Cys. The BST was applied to a wide range of photosynthetic organisms [reviewed in (269, 420, 463, 575)] and allowed identifying nitrosylated proteins in different organs and subcellular compartments (73, 385, 389, 463), in mutant lines (229, 296), and in plants exposed to exogenous NO donors (301, 350, 389) or affected by biotic (20, 432) or abiotic stresses (1, 64, 136, 217, 296, 421, 462, 463, 491, 492, 512). The most extensive studies identified 492 proteins and 392 sites in C. reinhardtii cells subjected to 15 minutes GSNO treatment (350) and 926 proteins and 1195 sites in Arabidopsis Col-0 and KO mutants for GSNOR [gsnor1-3 lines; (229)].
Despite its popularity, BST is a very difficult technique with inherent limitations and biases that are not sufficiently taken into account. A major drawback relies on the identification of false positives due to incomplete blocking and loss of targets due to spontaneous denitrosylation during sample handling. Moreover, the specificity of the ascorbate-dependent reduction step is difficult to establish unambiguously toward either disulfide bonds (105) or by-products of reactions of classical thiol blocking agents with other species such as sulfenic acids (426). Overall, the signal-to-noise ratio is low and variable due to differences in biological material, growth conditions, experimental design, sample handling, instrument setup, and bioinformatic data analysis. This strongly decreases the reproducibility and sensitivity of the method.
Several quantitative BST approaches allowing quantification of nitrosylation levels have been proposed. They are based on the combination of BST with chemical labeling strategies such as ICAT or related molecules (136, 167, 388, 421), Cys-TMT (359), iodo-TMT (422) or isobaric tag for relative and absolute quantification (iTRAQ) using the SNO-RAC method (152), stable isotope labeling with amino acids in cell culture (593), or label-free spectral counting (589). Such quantitative approaches will certainly improve the confidence into data generated by BST-based studies and allow uncoupling protein levels from nitrosylation levels. We believe that a method more reliable than BST is probably required for analysis of nitrosylation at a dynamic level. More direct and promising approaches based on direct capture of S-nitrosocysteine residues have been proposed but need further confirmation of their potential for quantitative proteomic studies (129, 135, 520).
C. Glutathionylome
Proteomic analysis of S-glutathionylated proteins has been initially performed using radiolabeling of the glutathione pool in cell cultures in the presence of 35S-cysteine and protein synthesis inhibitors (Fig. 10). Radiolabeled proteins are visualized by fluorography after separation on 2D gels. The spots disappearing in the presence of reducing agent, which correspond to S-glutathionylated proteins, are then identified by MS. Originally developed for human cells (160), this method allowed identification of 25 proteins in C. reinhardtii (340) but proved unsuccessful in Arabidopsis due to low levels of radiolabeling (126). This method has numerous drawbacks: (i) the protein synthesis inhibitors perturb cell physiology; (ii) this method cannot distinguish S-glutathionylated proteins (protein-SSG) from other forms of S-thiolation such as S-cysteinylation; (iii) it is limited by the necessity to perform 2D gels; (iv) it can only be used with cell cultures, thereby precluding studies on whole plants; (v) it can only detect proteins undergoing glutathionylation during treatment excluding proteins already glutathionylated under basal conditions; and (vi) finally it precludes high-throughput identification of glutathionylated sites.
An alternative method is based on biotinylated glutathione (BioGSH/BioGSSG) or the membrane permeant biotinylated glutathione ethyl ester (Fig. 10). The presence of the biotin tag allows detection of S-glutathionylated proteins by immunoblotting or enrichment by affinity chromatography. The latter can be coupled to MS for identification of not only S-glutathionylated proteins but also S-glutathionylated Cys if proteins are trypsin-digested before enrichment, as in the SNOSID approach (see section III.B). The major drawback of such methods is that proteins are not S-glutathionylated by the cellular GSH itself but by an exogenous sterically different molecule. The presence of the biotin tag on the glutathione molecule might perturb the function of glutathione-dependent enzymes and especially GRXs (Zaffagnini et al., personal communication). Another drawback, shared with the 35S labeling method, is that proteins glutathionylated under basal conditions are not detected. Originally used in mammals (483), this approach allowed identification of >70 S-glutathionylated proteins in Arabidopsis (126, 236), 225 proteins and 56 S-glutathionylation sites in Chlamydomonas (571), and 349 proteins and 145 sites in Synechocystis sp. PCC 6803 (76).
Several additional methods have been employed but not yet used in photosynthetic organisms. Commercial antiglutathione antibodies that can be useful for analysis of isolated proteins lack specificity and sensitivity, precluding application for high-throughput proteomics. S-glutathionylation can also be studied using an adaptation of the BST where the reduction step is performed with GRXs instead of ascorbate (Fig. 10) (175, 209, 253, 297). This approach has roughly the same drawbacks as the BST. In addition, the blocking of free thiols under denaturing conditions is difficult to combine with the enzymatic reduction of S-glutathionylated proteins by the GRX system (NADPH, glutathione reductase, GRX; see section V) that has to be performed in the absence of detergents.
Overall, despite the fact that S-glutathionylation is more stable than S-nitrosylation, the methods currently employed have numerous caveats and drawbacks, and the development of new approaches is most probably required for proteome-wide quantitative analysis of glutathionylation. A “chemobiology” approach based on click chemistry (417) may be possible since biosynthesis of a click analogue of glutathione seems experimentally feasible (141, 254, 445, 446). Such approaches have proven very efficient for proteomic analysis of S-palmitoylation (323, 592), N-myristoylation (545), or glycosylation (292, 496).
D. Sulfenylome
Proteomic analysis of sulfenic acids follows two major strategies that are based on either chemical or genetically encoded probes (4, 413, 554). Current chemical probes are mostly based on 1,3-carbonyl scaffold such as the cyclic dimedone (5,5-dimethyl-1,3-cyclohexanedione) (198, 424). At physiological pH, dimedone is in equilibrium with its enolic form that itself performs a nucleophilic attack on sulfenic acid. Dimedone tagged peptides can be detected by MS, and due to the generated mass increase, the involved Cys can be easily characterized. Nevertheless, dimedone has limited application for complex samples as it lacks a functional group for enrichment. Therefore, molecules harboring a dimedone conjugated with a fluorescent tag (DCP-Rho and DCP-FL series) or a biotin tag (DCP-Bio series) have been developed (Fig. 10) (77, 415). These probes have proven efficient but the presence of a bulky tag may alter cell permeability or prevent interaction with sulfenic acids that are not fully solvent accessible (413, 466).
Recently, small biorthogonal probes derived from dimedone have been developed such as DAz-1/DAz-2 (289) and DYn-1/DYn-2 (396). These probes can be biotinylated through click chemistry allowing enrichment of sulfenylated peptides. Used at lower concentrations than the classical dimedone, they are nontoxic and do not influence the intracellular redox balance (396, 555). Analysis of sulfenylated Cys with dimedone-based probes is compatible with classical quantitative MS-based strategies such as iTRAQ or TMT, which introduce reporter tags on tryptic peptides. Another way consists in synthesizing light and heavy isotope-coded forms of DYn-2 (556). Such a strategy allowed identification, in human cells, of 1000 sulfenylated Cys in 700 proteins (555). Despite their selectivity, these probes suffer from poor reaction kinetics under physiological conditions compared with biological reactions of sulfenic acids (197). New probes with faster reaction rates are, therefore, being considered to further expand our ability to monitor the sulfenome (198, 199, 302, 416). Biotinylated strained bicyclo[6.1.0]nonyne derivatives appear promising tools as they show reaction rates two orders of magnitude higher than dimedone even at low concentrations (μM range) (416).
The second approach is based on the yeast transcription factor Yap-1 that naturally interacts with the sulfenic acid formed on the Orp1 protein through formation of a transient mixed disulfide (Fig. 10) (116, 544). An engineered monocysteinic His-tagged version of Yap1 has been developed and shown to covalently trap sulfenylated proteins in Escherichia coli (489) and Saccharomyces cerevisiae (490). The major advantage of this type of probe is that their reaction kinetics is, at least theoretically, faster than dimedone-based chemical probes (4). Moreover, since they are genetically encoded, they can be controlled through genetic circuits and can be targeted to explore the sulfenylome of diverse subcellular compartments. Yap1-based methods also have several drawbacks, including a low efficiency that may be linked to in vivo reduction of Yap1 target mixed disulfides and a selectivity bias due to the Yap1 protein backbone and its steric effects.
In photosynthetic organisms, few studies addressed the question of the sulfenylated proteome in vivo. A combination of DCP-Bio and Yap1 probe allowed identification of 91 proteins in Medicago truncatula and 20 in its symbiont Sinorhizobium meliloti (381). More recently, the YAP1 probe was combined with a tandem affinity purification tag to detect 97 sulfenylated proteins in Arabidopsis cell suspensions under H2O2 stress (527). The DYn-2 probe was also recently employed in Arabidopsis and allowed identification of 226 sulfenylated proteins (3). Interestingly, a low overlap (17%) was observed between the two Arabidopsis sulfenylomes obtained by the same groups, suggesting that both approaches are highly complementary.
E. Persulfidome
Persulfides exhibit a reactivity similar to thiols, rendering their analysis at a proteome scale challenging. Some BST-based proteomic strategies aiming at unravelling persulfidation in complex samples have been recently developed. In the pioneering method, free thiols are blocked by methyl methanethiosulfonate (MMTS), whereas unblocked persulfides are subsequently biotinylated by HPDP-Biotin before enrichment by avidin-based affinity chromatography (361). Nevertheless, the assumed selectivity of the strong thiol-alkylating agent MMTS is questionable as it was shown to react indifferently with thiols and persulfides (390). Another approach combines initial blocking of all free thiols and persulfides with N-ethyl maleimide (NEM), DTT reduction, and labeling of nascent thiols with NEM-biotin (511). This strategy should be used with care as many proteins can undergo multiple DTT-reducible redox PTMs (186, 405).
A more innovative proteomic approach, called Tag-switch, allows persulfide biotinylation without the use of any reductant (585). In this strategy, both thiols and persulfides are first blocked with the alkylating reagent methylsulfonyl benzothiazole (MSBT), but only the activated disulfide bond of MSBT-derivatized persulfides is able to react in a second step with the biotinylated electrophile methyl cyanoacetate (Fig. 10). After enrichment using avidin-based affinity purification, persulfidated proteins are eluted under nonselective denaturing conditions that may lead to contaminations with proteins tightly bound to avidin such as endogenously biotinylated proteins. Another issue is linked to the selectivity of the Tag-switch approach as methyl cyanoacetate can cross-react with other forms of protein oxidations (sulfenic acid, sulfenylamide, and carbonyls) (585).
The last strategy recently developed consists in the direct alkylation of persulfidated proteins with biotinylated cysteine alkylating reagents (127, 165). In this case, both persulfides and thiol groups are indiscriminately biotinylated and persulfidated proteins retained on avidin affinity columns are specifically eluted in the presence of DTT. Nevertheless, to avoid that true persulfidated proteins remain linked to the column due to the presence of other biotinylated surface-exposed Cys in their sequence, low concentration (50 μM range) of biotinylated alkylating reagents should be employed as these conditions are known to promote alkylation of hyper-reactive thiols such as persulfides and thiolates rather than thiols (165, 530). The persulfide site identification approach, which is equivalent to the SNOSID approach, circumvents these pitfalls by identifying persulfidated peptides and thus Cys instead of proteins (165). Moreover, it remains compatible with classical quantitative MS techniques to compare persulfidomes (307).
In photosynthetic organisms, data about protein persulfidation are limited. Only two studies attempted to characterize the persulfidome in the model plant A. thaliana. By using either the pioneering approach (17) or the Tag-switch assay (16), these studies allowed the identification of 106 and 2015 persulfidated proteins, respectively. These proteins are localized in different subcellular compartments but mainly reside in chloroplasts and cytoplasm (65%) and are involved in a wide variety of pathways and processes, suggesting that persulfidation may be an important thiol switching mechanism as other redox PTMs in photosynthetic organisms.
F. The Cys proteome: a complex dynamic network
Before the advent of omics strategies, research in cell signaling has been conducted using ingenious analytical approaches. It is becoming clear now that proteomes are so intricate that we cannot understand the cellular functional organization using only a reductionist approach studying a limited number of cellular components. This is especially relevant for redox signaling that coordinates large number of redox elements involved in a multitude of pathways and cellular processes to allow resistance and adaptation to environmental challenges (182). This Cys proteome can be considered as an interface between the functional genome and the external environment (183). This highly dynamic network probably involves spatial and temporal regulation of multiple interconnected redox PTMs on hundreds of protein thiols with flexible reactivities (395, 414, 530). Therefore, global approaches are required to fully understand the entire molecular complexity of redox signaling pathways and their links with numerous pathophysiological features. Among global approaches, MS-based strategies have benefited lately from tremendous technological improvements, and are now ready to face the challenge of comprehensive and quantitative proteomic approaches at the level of protein expression, protein interactions, or PTMs (372).
Combinations of multiple redox PTMs act as a cellular network rather than as insulated elements. Understanding the organization of these networks will require to unravel the determinants of the specificity of the diverse redox PTMs for proteins and Cys. Indeed, it remains unclear whether multiple redox PTMs occur on a limited number of proteins containing reactive Cys or whether each modification targets a distinct redox network. Recently, the identity of redox-modified Cys belonging to proteins undergoing at least two different redox PTMs (among targets of TRX, S-glutathionylation, and S-nitrosylation) was compared in Chlamydomonas (405). This analysis revealed that 86% of these Cys were modified by only one type of redox modification. This comparison indicates, on one hand, that the Cys proteome does not represent a subset of highly reactive Cys that are modified indiscriminately, and highlights, on the other hand, a strikingly high specificity of each modification for distinct Cys residues (405). A similar high specificity with a limited overlap between Cys targeted by multiple PTMs was also reported in human and mouse (186, 289). These results indicate that the Cys proteome does not represent a small subset of highly reactive Cys that are modified through indiscriminate interaction with the molecules they encounter but represent a complex system of redox PTMs that are specific toward distinct interconnected protein networks (405).
The complexity of the network likely provides the robustness and specificity required to allow simple molecules such as ROS, RNS, and RSS to play a signaling role. This redox network is presumably a major component of signal integration and constitutes the molecular signature of the ROS/RNS/RSS cross talk whose importance in cell signaling has been recognized (158, 161, 191, 347, 471).
Understanding this complex network will require to determine the stoichiometry and dynamics of multiple redox PTMs under diverse physiological conditions or in different genetic backgrounds, and at different time scales. This should be favored in the future by the development of sensitive and accurate redox quantitative MS approaches combined with the development of new chemospecific probe molecules (554). These chemical probes will have to (i) be specific for a given modification with no interference with other biological molecules, (ii) be compatible with quantitative MS, (iii) be nontoxic and membrane permeable to allow in situ or in vivo labeling, (iv) be highly sensitive to allow detection of low abundant proteins or low levels of modifications, (v) allow efficient enrichment methods using, for example, click chemistry, and (vi) exhibit fast reaction rates compatible with the half-life and reactivity of the species studied. New types of modifications may also become amenable to proteomic analysis with the development of new probes such as NO-Bio, a recent biotin-tagged probe for proteomic analysis of sulfinic acids (306). Future redox proteomic studies will have to take advantage of isotope-coded multiplex reagents such as TMTs to monitor multiple modifications or multiple samples simultaneously. Progress in the sensitivity of MS instruments and proteomic methods will allow analyses on limited amounts of biological samples and thus foster the development of single cell redox proteomic approaches to decipher the redox signaling network rather than unravel averaged redox signals from multiple cells. In other words, temporal quantitative redox proteomics on limited number of cells is certainly the grail that will allow us to discriminate redox modification events from noise and thus shed light on the functioning of the redox network.
In addition, computational structural genomic approaches will be required to integrate the Cys proteome at the structural level. Finally, besides redox PTMs, the integration of the signal implicates a myriad of other molecules and processes acting at multiple levels (326). In photosynthetic organisms, several redox PTMs are linked to signaling pathways controlled by hormones (140, 262, 497, 522, 528, 567) or calcium (506), and in mammals, nitrosylation was shown to interfere with signaling processes mediated by phosphorylation, ubiquitylation, sumoylation, acetylation, or palmitoylation (214). Therefore, a strong effort is required to integrate redox networks with other signaling pathways and to analyze their impacts on the cellular responses at multiple levels. This will certainly be crucial to unravel how environmental challenges are encoded into a biochemical signal than can be exploited to trigger the appropriate responses in terms of localization, duration, and intensity, at the genome, transcriptome, proteome, and metabolome levels to allow adaptation and survival.
IV. The Remarkable Diversity of Redoxins in Photosynthetic Organisms
A. A general introduction on plant TRX superfamily (redoxins)
The TRX superfamily encompasses several protein families (notably TRXs, GRXs, protein disulfide isomerases [PDI], and glutathione-S-transferases [GSTs]), the members of which have in common a specific structural arrangement named the TRX fold (see Section V) and often a typical XCXXC/S signature containing the redox active Cys pair.
The number of PDI genes found in plant genomes is comparable with that in mammals and higher than that in fungi (465). For the TRX, GRX, and GST gene families, algae and terrestrial plants have an expanded number of representatives, which is explained, in part, by the existence of additional classes (87, 100, 274, 286, 287, 336). Hence, in the next subsections, we focus our attention on the remarkable diversity found in TRX and GRX families, describing their subcellular distribution and how comparative genomics led to a rather exhaustive and refined classification of these genes/proteins and to a better understanding of their evolution.
B. Classification and evolution of redoxins and their reductases
TRXs and GRXs were initially defined by quite strict signatures, for example, WC[G/P]PC and YCP[F/Y]C, respectively, but the sequencing of numerous genomes pointed to the existence of a large variety of other combinations. These variations are usually still compatible with an oxidoreductase activity, although some are associated with the capacity to bind Fe-S clusters as observed initially for GRXC1, which possesses a slightly divergent YCGYC active site signature and then with several other GRXs (441). In the PDI family, the majority of plant isoforms possess a WCGHC signature, but variations also appeared in some representatives (465). There is no such universal signature for GSTs and actually only a very few of them have conserved both Cys. An important number has even lost the first catalytic Cys that has been replaced by a serine. This has led to a change in the type of activity catalyzed by GSTs. Those that kept the catalytic Cys have glutathione-removing activities, whereas those possessing a serine have glutathione-conjugating activities, this residue serving for the activation of the thiol group of the glutathione molecule. Besides, GSTs have a particular structural arrangement with the existence of an all-helical domain fused at the C-terminus of the TRX domain. We invite the reader to refer to the following reviews for detailed information about phylogenomic analyses of PDIs (287, 465) and GSTs possessing the catalytic Cys (274). From now, this section uniquely focuses on the TRX and GRX systems that primarily control the RMS-dependent PTMs of protein Cys.
1. Phylogenetic and sequence diversity within the TRX and TRX reductase families
The TRX family is split into 21 well-defined classes including the NADPH–TRX reductase C (NTRC) fusion proteins that contain a TRX domain and a TRX reductase (TR) domain (Table 1 and Fig. 11). Some TRX family members can unequivocally be distinguished by the active site signature and domain organization. Typical TRX isoforms (TRX f, m, x, y, z, o, and h classes) are formed by a single domain with regular tryptophan-cysteine-glycine-proline-cysteine (WCGPC) or tryptophan-cysteine-proline-proline-cysteine (WCPPC) active site signatures corresponding to that found in ancestral TRXs (Fig. 11). In addition, there are larger proteins that contain either two or more TRX domains (chloroplast drought-induced stress protein of 32 kDa, CDSP32, or nucleoredoxins, NRX) or a TRX domain fused to a domain with other functions (TR domain in NTRC, tetratricopeptide repeat domain in tetratricopeptide domain-containing TRXs (TDXs) (Fig. 11). The active site signatures of the TRX domain(s) are also usually regular or with little variations. In CDSP32, the first domain has lost the Cys, whereas the signature of the C-terminal domain is of the HCGPC type (Fig. 11). Among NRXs, three groups can be distinguished. In NRX1 and NRX3 members, both TRX domains have generally WCGPC or WCPPC active site signatures, whereas in NRX2 members, only the C-terminal domain conserved the Cys and the consensus signature has significantly diverged being of the [W/R]C[L/A]P[C/G] form (Fig. 11). The C-terminal TRX domains in NTRC and TDX have a TCGPC and WCGPC signature, respectively (Fig. 11). Finally, there are atypical TRXs formed by a single domain and divergent active site motifs: CLOT (WCPDC), HCF164 (WCEVC), TRX-like1 (most often WCRVC), TRX-like2 (WCRKC), TRX-lilium1 (GCGGC), TRX-lilium2 (WC[G/A]SC), TRX-lilium3 (SCGSC), TRX s (no conserved signature), and TRX CxxS (often WC[M/I]PS), which are included in the TRX h class (Fig. 11). Lilium-type TRXs are also known as atypical Cys histidine-rich TRXs [ACHT, (110, 111, 133)] because they contain several conserved Cys and histidine residues outside the active site. These chloroplast atypical TRXs are proposed to play a role in the inactivation of light-activated redox targets (see section VII). It is worth mentioning that HCF164 possesses an N-terminal anchoring domain to the thylakoid membrane (Fig. 11). The TRX s class is not presented in Table 1 because it is only found in some leguminosae. There are four members in M. truncatula (428) and they likely possess specific functions for the establishment of symbiotic interactions between plants and bacteria of the rhizobia genus. Interestingly, the TRX s1 is secreted into the microsymbiont although it seems that it derived from plastidial TRX m (428). Therefore, it may be that the plastid targeting sequence evolved into a secretory signal.

Gene Content in the Glutaredoxin and Thioredoxin Families in Representative Organisms of the Green Lineage
Sequences from Oryza sativa (Os), Selaginella moellendorffii (Sm), Physcomitrella patens (Pp), C. reinhardtii (Cr), and Synechocystis sp. PCC 6803 (Syn) have been retrieved from genomic data available through Phytozome V12 portal or cyanobase by BLAST-p analysis using Arabidopsis thaliana (At) sequences as references. The classes in the GRX and TRX families have been previously defined (87, 100).
The five NRXs found in C. reinhardtii have been arbitrarily classified as NRX1 but they group independently from land plant NRXs.
This indicates the existence among NTRA/B from C. reinhardtii of a mammalian-type selenocysteine-containing NTR.
Synechocystis sp. PCC 6803 does not possess an authentic NTR, but another type of diflavin protein of unknown function (59).
CDSP32, chloroplastic drought-induced stress protein; GRX, glutaredoxin; FTR, ferredoxin:thioredoxin reductase; NRX, nucleoredoxins; NTR, NADPH:thioredoxin reductase; NTRC, NADPH:thioredoxin reductase C; TDX, tetratricopeptide domain-containing thioredoxin; TR, thioredoxin reductase; TRX, thioredoxin.
When considering two angiosperms, the dicot A. thaliana and the monocot Oryza sativa; a lycophyte, the fern Selaginella moellendorffii; a bryophyte, the moss Physcomitrella patens; a green alga, C. reinhardtii and a cyanobacterium, Synechocystis sp. PCC 6803, the minimal TRX equipment in photosynthetic organisms, as found in this cyanobacterium and conserved in all other organisms, appear to be formed by four members belonging to the HCF164, TRX m, TRX x, and TRX y types (Table 1) (87). This number increases to 20 in C. reinhardtii with the appearance of TRX f, h, o, z, CDSP32, CLOT, TRX-like, TRX-lilium, NRX, and NTRC. Another increase occurred in terrestrial plants, both nonvascular (mosses) and vascular (ferns and angiosperms) plants. So far, in land plants, the lowest and highest number of reported TRXs have been found in S. moellendorffii (22 isoforms, Table 1) and in Eucalyptus grandis (45 isoforms) (412). This rise is mostly linked to duplications within existing classes, as the sole innovation specific to angiosperms is the TDX class that contains one or two members. These substantial differences in the TRX content among terrestrial plants are appealing although some cautions may be needed for recent automatically annotated genomes.
Why the evolution positively selected complexity in the plant TRX system (on average in the human genome 1 TRX-coding gene is found for every 10,000 protein-coding genes versus 1350 protein-coding genes in Arabidopsis) is unknown. Reasonably more than a single evolutionary cause has contributed to positive selection. In fact, it is generally accepted that in plants several physiological processes are under the control of the TRX-mediated redox mechanisms. Whether as a result of the sessile lifestyle of photosynthetic organisms or due to the greater permissiveness to genome doubling events as well as arising from the existence of three evolutionary distinct genomes (nuclear, mitochondrial, and plastid genome) inside a cell, plant TRX system is indeed more complex and versatile than that of prokaryotes (e.g., bacteria) and heterotrophic organisms (e.g., animal and fungi).
On the contrary, there are only minimal variations concerning the TRs along the green lineage (Table 1). The FTR is composed of a catalytic and a variable subunit (Fig. 12), which is, by definition, difficult to identify based on sequence homology. Hence, concentrating on the FTR catalytic subunit, all analyzed genomes contain a single gene except P. patens that has two genes (Table 1). Concerning NTRs, there is usually a single NTRC isoform and one to two NTRA/B members (Table 1, Figs. 11 and 12). A large number of NTRA/B genes (six) is found in the genome of Quercus robur but the same caution as before applies for this first assembled genome version (412). One particularity is the presence of a mammalian-type NTR in some green algae such as C. reinhardtii (239). These are remnant selenoproteins common to many eukaryotes but not terrestrial plants, since they lost the system for selenocysteine insertion.

The subcellular localization of most Arabidopsis TRXs and TRs has been determined experimentally. The mitochondrion is likely the less rich compartment containing only TRXs o and a TRX h2 in some organisms as poplar and Arabidopsis (172, 275, 330). They should be maintained reduced via NTRA or NTRB, both isoforms having been detected, although NTRB may be more abundant (425). Both TRXs o1 and o2 show double localization, the former in mitochondria and nucleus, the latter in mitochondria and cytoplasm (118, 171). Both NTRA and NTRB are also found in the cytoplasm, whereas NTRA may also be in the nucleus (316, 425). In these compartments, they might reduce a certain number of cytoplasmic TRXs (Clot, TRX-like 1, TRX h1, h3 to h8, TRX CxxS1, and TDX) and nuclear proteins (NRX1, NRX2, and NRX3) (85, 316) but also some membrane-bound TRXs (TRX h9, TRX CxxS2) owing to the existence of N-terminal glycine and Cys residues promoting membrane anchoring through N-myristoylation and palmitoylation, respectively (330, 507). The employed reduction system has not been validated for all of them but TRX CxxS and TRX h9 (or TRX h4 in poplar) use a GSH/GRX system (173, 264, 330). Besides, a myristoylated glycine in A. thaliana TRX h7 and TRX h8 promotes their attachment to the ER/Golgi endomembrane system (507). An orthologous tobacco TRX h is secreted, which raises the question of its reduction (247).
The chloroplast possesses by far the largest TRX equipment. In A. thaliana, there are 20 TRXs taking into account NTRC (52, 61, 92, 110, 288, 330, 468). All regular/typical TRXs (i.e., TRX m, f, x, y, z) are reduced by FTR (87, 564) and some of them, such as TRX z, may be reduced by NTRC as well (563). It is not yet clear how CDSP32 is recycled upon oxidation, whereas poplar TRX-like 2.1 and TRX-lilium2 were shown to be reduced by a GSH system (85). ACHT1 and ACHT4 were proposed to be reduced by TRX-regulated ADP-glucose pyrophosphorylase, thereby contributing to its downregulation (133) (see section VII). HCF164 is attached to thylakoids, likely facing the luminal side (288) and would relay the reducing equivalents from stromal TRXs into the lumen (352), where proteins regulated by disulfide formation are present (475).
As far as their reducing activity is concerned, TRX m and f reduce disulfides on several metabolic targets, including enzymes of the CB cycle, oxidative pentose phosphate pathway, starch metabolism, ATP synthase, and malate valve (68, 92, 200, 349, 367, 478 –480). TRX x and y, together with CDSP32 and ACHT1/4, are more specific for antioxidant enzymes, for example, PRXs and methionine sulfoxide reductases (93, 111, 124, 147, 164, 225, 494, 495).
2. Phylogenetic and sequence diversity within the GRX family
The GRX family can be split into six classes (see below in this section for further details; Fig. 13). Classes I and II are shared by eukaryotes and cyanobacteria (Table 1). Classes III and IV are specific to eukaryotes (Table 1 and Fig. 13). Classes V and VI are specific to cyanobacteria, although they are not present in Synechocystis sp. PCC 6803 (Table 1 and Fig. 13). Therefore, both photosynthetic eukaryotes and cyanobacteria contain GRXs belonging to up to four classes (eukaryotes: classes I, II, III, and IV; cyanobacteria: classes I, II, V, and VI). As for the TRX family, members of these GRX classes differ notably by their active site signature and domain organization (Fig. 13). The nomenclature established previously using A. thaliana members relies on the presence of a Cys or a serine at the last position of the active site signature (439). Therefore, they were named from GRXC1 to C14 and from GRXS1 to S17, although AtGRXS13 possesses a CPLG motif and at the time, the two class IV members (see this section) were not included. The presence of a residue different from Cys or serine at the last position is also observed in a limited number of GRX members in some other species (e.g., O. sativa or Sorghum bicolor).
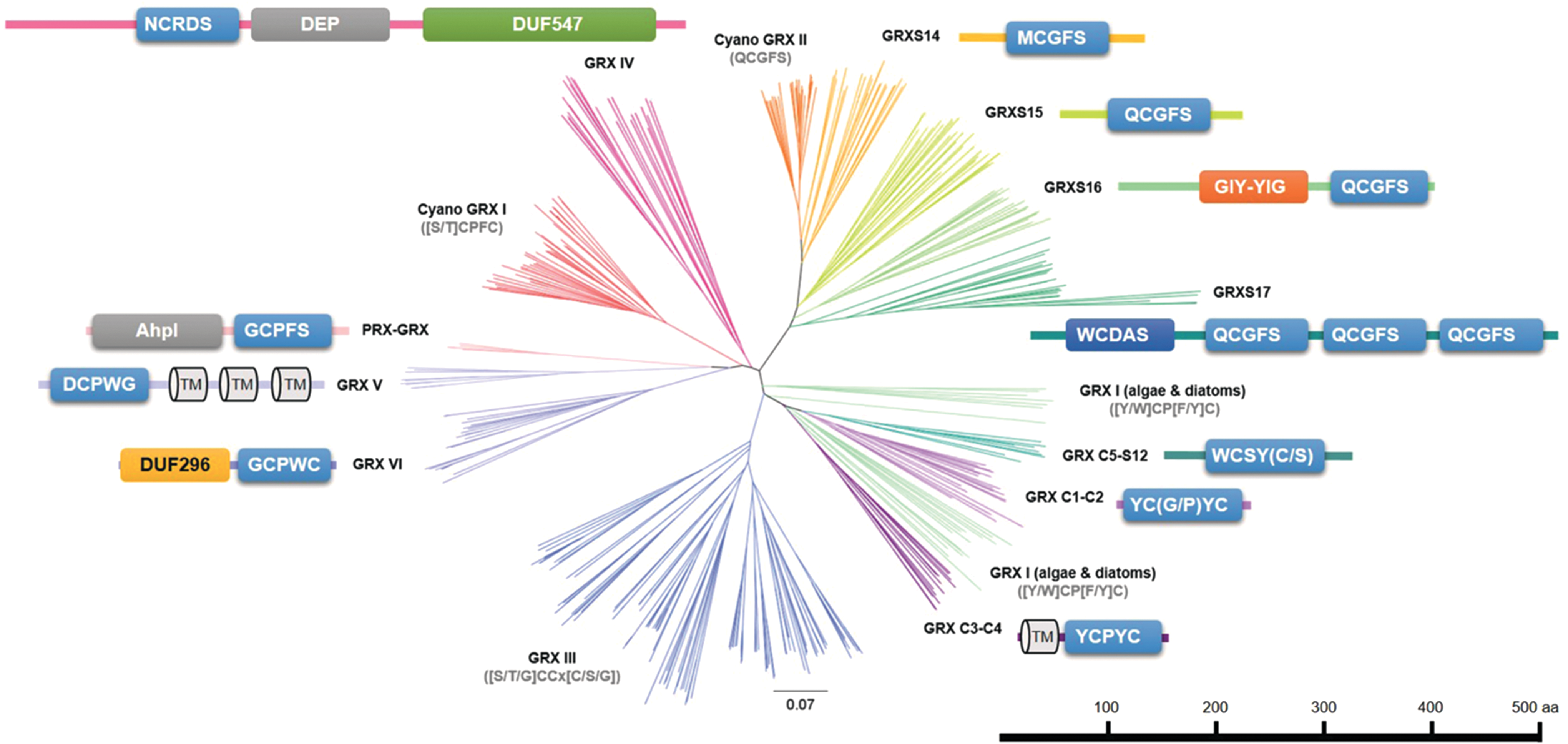
Except for the specific case of the PRX-GRX fusion proteins found in some cyanobacteria, class I GRX isoforms (GRXC1-C5, GRXS12, and cyanobacterial GRX I) are formed by a single domain with a quite regular YC[P/S/G][Y/F]C active site signature with some exceptions as GRXC5 (WCSYC) and GRXS12 (WCSYS) (Fig. 13). The phylogenetic analysis reveals that cyanobacterial and algal GRXs form independent clades, whereas terrestrial plants can be further divided into GRXC1/C2, GRXC3/C4, and GRXC5/S12 subgroups (Fig. 13), and these subgroups also differ in their biochemical and redox properties (100 –102, 104, 573). Only two class I isoforms are found in model nonphotosynthetic organisms such as E. coli and Homo sapiens, but four in S. cerevisiae.
The class II GRXs are typified by their extremely conserved CGFS signature. They can also be further divided into four subclasses (GRXS14, S15, S16, and S17) in eukaryotic photosynthetic organisms according, in particular, to the existence of multidomain proteins, whereas cyanobacterial GRX II isoforms systematically grouped independently (Fig. 13). The GRXS14 and S15 members are only formed by a single GRX domain, as are cyanobacterial orthologs (Fig. 13). The GRXS16 and S17 have a modular organization (Fig. 13). The former possesses an N-terminal domain with some similarity with a certain type of endonuclease and the latter is formed by an N-terminal TRX-like domain with a distorted active site signature fused to one to three GRX domains. It is extremely interesting to point out how the GRXS17 fusion appeared and evolved during evolution. Indeed, haptophytes such as Emiliania huxleyi have isoforms with only one GRX domain, heterokonts and green algae with two GRX domains, sequenced mosses (P. patens and Sphagnum fallax), liverwort (Marchantia polymorpha), and fern (S. moellendorffii) possess isoforms with two and/or three GRX domains, and gymnosperms (Picea abies) and angiosperms have isoforms with three GRX domains (100). Since GRXS16 prototypes are specific to the green lineage, only one to three class II isoforms are found in model nonphotosynthetic organisms, one in E. coli, three in S. cerevisiae, and two in H. sapiens.
The class III GRXs are characterized by the presence of two adjacent Cys forming CCxC, CCxS, or CCxG signatures (Fig. 13). They are uniquely found in terrestrial plants, ranging from 2 isoforms in P. patens to 24 isoforms in Populus trichocarpa (100, 412). The expansion of class III GRXs in angiosperms occurred mainly through paleopolyploidy duplications shortly after the monocot–eudicot split (201) and then proceeded by species-specific duplication, leading to the existence of multiple tandem duplication.
The class IV GRXs (also referred to as GRX-like) are only present in terrestrial plants and they are rarely represented by more than two isoforms in a given species. These proteins have a particular domain organization with the presence of a long N-terminal extension followed by a GRX domain with a quite divergent active site signature (although green algal ancestors have CPYC/CPHC motifs), and two additional domains with unknown function, named DEP (domain found in Dishweller, Egl10, and Pleckstrin) and DUF547 (domain of unknown function 547) (Fig. 13). There are in fact two clades, the first containing sequences with an NCRD[C/S] signature, the second comprising sequences with a GCE[E/D]C signature.
As mentioned previously, the classes V and VI GRXs are found uniquely in cyanobacteria but not in all of them. They are for instance absent in Synechocystis sp. PCC 6803 and thus not included in Table 1. On the contrary, a few species have both of them (100). Members of class V are formed by a GRX domain with a highly conserved CPWG followed by a C-terminal extension predicted to form three to five transmembrane domains (Fig. 13). Members of class VI are formed by an N-terminal DUF296 domain, followed by a C-terminal GRX domain with a CPW[C/S] signature (Fig. 13). From the presence of this DUF296 domain in proteins that contain AT-hook motifs and the conservation of metal-binding histidines, it is predicted that these proteins have DNA-binding properties.
When considering the same set of representative organisms as before, the basal common GRX equipment in photosynthetic organisms as found in Synechocystis sp. PCC 6803 is formed by three members, two belonging to class I and one to class II (Table 1). This number increases to seven in C. reinhardtii because of duplications occurring for class II GRXs and of the appearance of class IV GRXs (Table 1). Another increase occurred in nonvascular plants (mosses) and in ferns with further duplications occurring for class II GRXs and with the appearance of class III GRXs (Table 1). Finally, class III GRX has strongly expanded in seed plants, from 9 in Carica papaya to 24 in P. trichocarpa (Fig. 13) (201, 412). To date, in terrestrial plants, the lowest and highest number of reported GRXs are found in P. patens (15 isoforms) and in P. trichocarpa (38 isoforms), respectively (Table 1) (412).
The subcellular localization of many poplar and Arabidopsis GRXs has been determined experimentally. First, it is important to point out that there might be a single GRX in mitochondria, which is the class II GRXS15 (30, 351). This is extremely surprising because it has no or extremely poor oxidoreductase activity (39, 351), whereas there is an intense GSH-dependent metabolism in this compartment suggesting that a catalyst of protein deglutathionylation is required. In chloroplasts, there are three GRXs in most photosynthetic organisms, GRXS12 (class I) and GRXS14 and S16 (class II) (30, 104). A variation is observed in brassicaceae including A. thaliana due to existence of the close GRXS12 paralog, GRXC5, and in algae, because they have only the two class II GRXs (102, 104, 284). It is again surprising that there is no class I GRX with regular GSH-dependent activity in algal plastids. The fourth class II GRX, GRXS17, is found both in the cytoplasm and in the nucleus as do GRXC1 and C2 and most class III GRXs (30, 228, 263, 366, 431, 441, 549, 550, 553). In fact, there are still some uncertainties about several class III GRXs, for which the targeting has not been experimentally verified or which have N-terminal extensions (104, 439). Interestingly, GRXC3 and C4 have also short N-terminal extensions, which may represent either a signal peptide for secretion or a membrane-anchoring sequence.
In conclusion, the genomic and phylogenetic analyses indicate that the TRX and GRX families constitute a largely diversified group of proteins in plants with numerous plant-specific isoforms or classes, which appeared during evolution, whereas this expansion/diversification did not occur in bacterial, fungal, and animal kingdoms. Although this classification is quite robust, relying on the use of specific motifs for protein identification (for instance, the presence of glutathione-binding residues in the case of the GRX family) (100), one could wonder whether all these proteins adopt a TRX fold and have oxidoreductase activity. Hence, having systematic activity and structural information for isoforms belonging to each class would be mandatory in assessing to which extent the electrostatic surfaces are crucial in determining the specificity of TRXs and GRXs toward their targets as proposed in the case of E. coli 3'-phosphoadenosine-5'-phosphosulfate reductase (48). This may provide clues to refine the classification on an activity/structure basis. Besides this is not detailed at all in previous paragraphs, the presence of extra-Cys residues in some specific GRXs or TRXs is known to interfere with their activity and recycling. To cite only two examples, some TRX h having an additional Cys at position 4 become dependent on GSH and GRX instead of NTR (264), and the glutathionylation of Cys67 of A. thaliana TRX f1 inactivates the protein, preventing its regeneration by FTR (338). Finally yet importantly, besides the punctual changes in key amino acids, many proteins have additional domains, the function of which is often not yet determined although it could considerably affect their localizations, protein–protein interactions, or activities. It would be expected that these protein innovations modify for instance the set of partner proteins. In this regard, it is interesting to see the intricate relationship between class III GRXs and TGACG motif-binding (TGA) transcription factors, sustaining the role of these GRXs in plant stress response and development (notably floral development) (201).
V. Structures and Catalytic Mechanisms of Redoxins
A. The TRX fold and the structural determinants of redoxin reactivity
The TRX fold, common to all TRXs and GRXs, is composed of a central core made of a four to five stranded mixed beta-sheet, flanked by three to four alpha-helices (Fig. 14A, B). The residues forming the typical CXXC/S signature containing the catalytic Cys are positioned at the N-terminus of one of the alpha helices. Another important structural feature of the TRX superfamily members is the presence of an invariant cis-Pro residue that is found about 40 amino acids on the C-terminal side of the CXXC/S motif and, in the tridimensional structures, faces the catalytic Cys in the active site. This fold was first identified in the crystal structure of oxidized E. coli TRX1 (224). Since then, several structures of TRXs and GRXs from different photosynthetic organisms have been solved (Table 2).
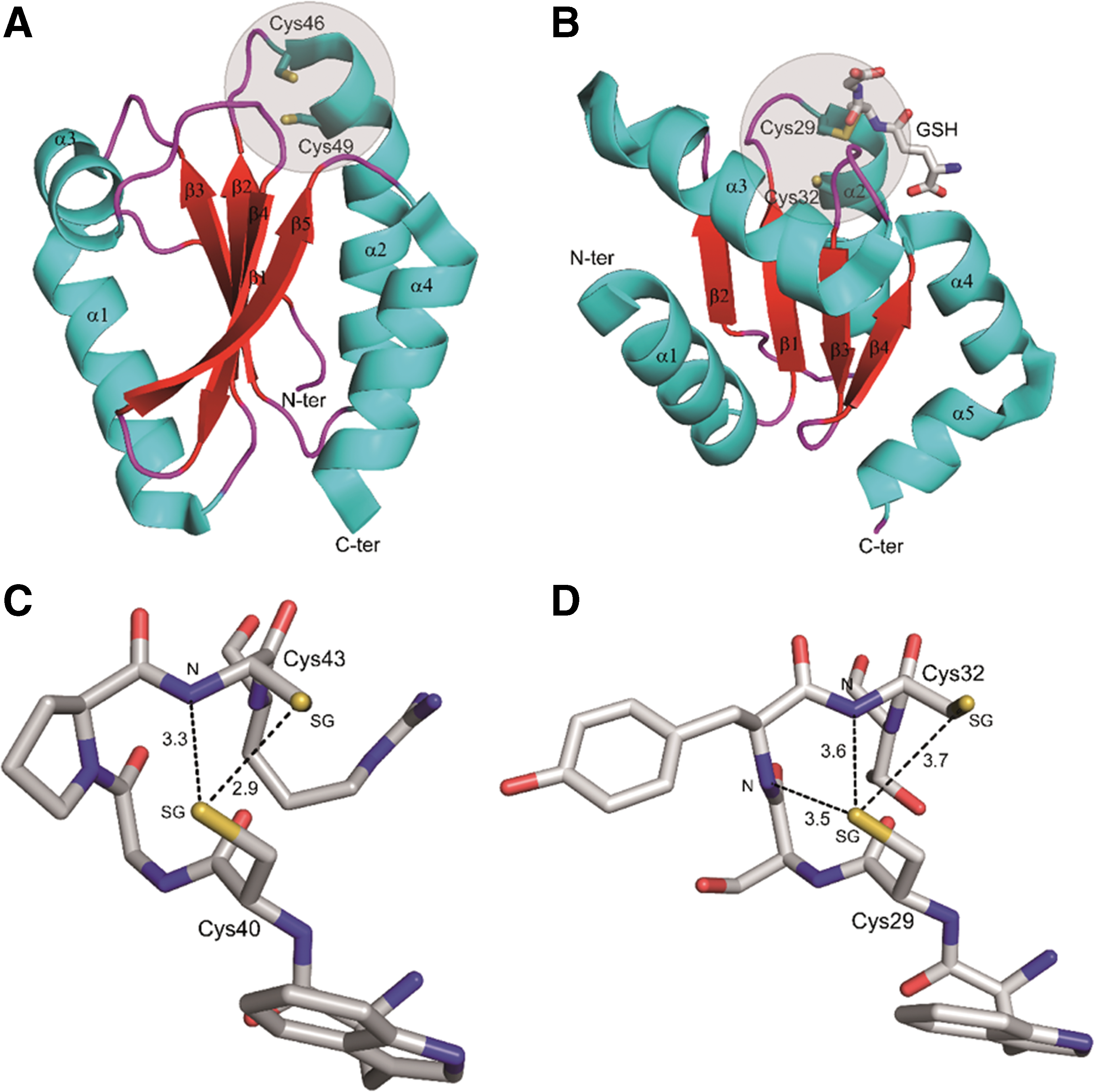
Three Dimensional Structures of Thioredoxins and Glutaredoxins from Photosynthetic Organisms
BME, beta-mercaptoethanol; GSH, reduced glutathione.
The catalytic site containing the redox active Cys is located in a hydrophobic region quite exposed to the solvent. The thiol group of the first Cys of the generic CNX1X2CC/S signature (where CN and CC are the N-terminal and C-terminal Cys, respectively) is accessible and can easily react with disulfides or possibly other forms of oxidized thiols on the target proteins (Fig. 14A, B). On the contrary, the thiol of the second Cys, substituted by serine in some GRX classes (see section IV and Fig. 13), is buried and surrounded by hydrophobic residues. The reactivity of the N-terminal Cys is mainly determined by the pK a of its thiol group (see section II) ranging from 4.0 to 5.0 in GRX (102, 104, 573) and from 6.3 to 7.1 in TRX [(434), Zaffagnini et al., personal communication], indicating partial or complete deprotonation of the N-terminal Cys at physiological pHs. Despite their acidic Cys, neither TRX nor GRX is particularly prone to oxidation by H2O2, confirming that other factors come into play in the thiolate to sulfenic acid conversion [(508, 573), Zaffagnini et al., personal communication]. In contrast, the C-terminal active site Cys, which may be absent or not essential for activity in GRX, shows a pK a that may even be higher than that of free Cys and, therefore, should be relatively unreactive [(88), Zaffagnini et al., personal communication]. Nevertheless, the C-terminal Cys of TRX is involved in the thiol–disulfide exchange reaction. The mechanism proposed for E. coli TRX (89) predicts that a buried and highly conserved aspartic residue (Asp26 in E. coli TRX) works as an acceptor for the proton released by the C-terminal thiol when it attacks the N-terminal Cys bonded with the target protein (see section V.B) (65, 329).
The low pK a of the N-terminal Cys of both TRXs and GRXs is chiefly determined by a hydrogen bond network, whereas the contribution of the helix macrodipole is negligible (150, 435). Crystallographic investigations showed that the N-terminal Cys thiolate is often stabilized by hydrogen bonds with residues belonging to the catalytic sequence CNX1X2CC/S. For example, in barley TRX h1 [PDB ID 2VM1; (314)], the sulfur atom of the N-terminal Cys40 is involved in a double hydrogen bond with the sulfhydryl group and the backbone amide group of the C-terminal Cys (Cys43; Fig. 14C). The lower pK a value of the N-terminal Cys in GRX-like Arabidopsis GRXC5 was explained by an additional third H-bond with the backbone amide group of the X2 residue, which further stabilizes the N-terminal thiolate (Fig. 14D) (104). A similar hydrogen bond network could not be established in most TRX active sites due to the presence of a proline in the X2 position (150).
B. TRX and GRX: mechanisms of disulfide reduction
Although the large superfamily of TRXs include members that do not appear to be redox active, TRXs and GRXs can be considered anyway typical reducing agents for disulfide bonds. The TRX system is older than the GRX system in evolutionary terms (171, 335) and it is more efficient in reducing protein disulfides even under severe oxidative stress, ensuring a reduced environment in the cell. In contrast, GRXs are more versatile being able to reduce protein disulfides compensating if necessary TRXs, but also glutathione-mixed disulfides. Reduced GRXs are regenerated mainly by GSH and in a few cases by TRs (i.e., FTR and NTR), depending on the redox potential and catalytic mechanism of the specific GRX (145, 246, 578). Instead, TRXs are mainly reduced by TRs (FTR and NTR), but a small subgroup of plant TRXs h is uniquely reduced by the GSH/GRX system [see section IV.B.1; (173, 264, 330)].
In the catalytic mechanism of TRXs, the exposed N-terminal Cys of the active site CNX1X2CC signature performs a nucleophilic attack on the disulfide of the target protein forming a mixed disulfide bond with the target protein itself (Fig. 15A). Then, the free C-terminal Cys becomes reactive (deprotonated), thanks to the proton accepting role of a conserved nearby Asp, and attacks the N-terminal sulfur atom involved in the mixed disulfide, generating oxidized TRXs and reduced target proteins (Fig. 15A). The reaction is reversible and its equilibrium is determined by the redox potentials of both TRXs and target proteins (92, 93, 222, 223, 322, 478 –480). The midpoint redox potential of TRX f and m is about −290 mV at pH 7 (223, 339) and several target proteins show midpoint redox potentials that differ from that of these TRXs by < ± 30 mV (92, 93, 222, 223, 322, 478 –480), suggesting that fluctuations in TRX redox state may effectively translate in fluctuations of target protein redox states and vice versa. An exception to this general view is constituted by lilium/ACHT atypical TRXs that possess redox potential about 50 mV less negative than typical TRX f or m (85, 110). Indeed these TRXs have been proposed to shuttle electrons from reduced AGPase to 2-Cys PRX in the presence of H2O2, possibly constituting a pathway of downregulation of chloroplast starch biosynthesis under low-light intensity. A similar role of TRX in the oxidation of reduced chloroplast targets might be expected for TRX y that on one side is less reducing than TRX f and m (93), and on the other hand is a good reductant for 2-Cys PRX (93, 124).

Although typical of TRXs, the dithiol oxidoreductase mechanism just described is also shared by some GRXs (166, 578). Most GRXs, however, are specialized in protein deglutathionylation. In this reaction, the N-terminal catalytic Cys of GRXs attacks the disulfide of the glutathionylated protein, releases the reduced peptide, and becomes itself glutathionylated (Fig. 15B). Afterward, a second GSH molecule reduces back the glutathionylated thiol of GRX (60, 379) generating GSSG (Fig. 15B), in turn reduced to GSH by NADPH and glutathione reductase (GR), all together forming the GSH/GRX reducing system (100, 574). The C-terminal Cys of GRXs when present is not involved in this mechanism, which is, therefore, called monothiol mechanism. The monothiol mechanism requires a single Cys on GRX (the N-terminal of the active site signature) but two glutathione molecules, one bound to the target protein and the other free (39). This mechanism may be used by class I GRXs bearing one or two Cys in the active site (see section IV.B.2). An example of GRX utilizing a monothiol mechanism for deglutathionylation is poplar GRXS12 found in chloroplasts with a WCSYS active site sequence, unique to plants (102, 573).
Some GRXs such as GRX3 from Chlamydomonas (578) use instead a dithiol mechanism for deglutathionylation (Fig. 15C). In this mechanism, the deglutathionylation of the target protein occurs like in the monothiol mechanism. However, the glutathionylated GRX is then deglutathionylated by a second protein Cys that generates an internal disulfide and releases the GSH (Fig. 15C). Depending on the GRX isoform, the second Cys may or may not belong to the active site. The latter is the case of GRX3 from Chlamydomonas, a chloroplast class II GRX whose internal disulfide is very efficiently reduced by FTR, thus constituting a potential link between deglutathionylation and photosynthesis (335, 578).
Unlike TRXs, some plant GRXs (GRXC1, GRXC5, GRXS14-S17) have been identified as Fe-S cluster binding proteins ligating [2Fe-2S] clusters (30, 142, 233, 351, 441). Although the Fe-S clusters bound to class I GRXs (GRXC1 and GRXC5) may modulate GRX activity (the holoforms are inactive) under oxidative stress conditions for instance, class II GRXs (GRXS14-S17) are involved in Fe-S cluster biosynthesis and assembly in specific cell compartments (103).
C. Structural basis of TRX–target interaction and specificity
A detailed comparison of the crystal structures of two plastidial TRXs from the same organism (spinach TRX f and TRX m) showed that despite a quite similar overall structure [rmsd 1.2 Å for 102 superimposed Ca atoms; (65)], they show a different distribution of charges around the active site, with TRX f being characterized by a positive region that is less prominent in TRX m. In addition, TRX f active site is more flexible and the Trp45, the residue preceding the N-terminal Cys, can adopt different conformations. It is plausible that these features contribute to the different specificities shown by these two TRXs toward their targets. Indeed, although many tested targets may be reduced in vitro by either TRX f or TRX m, in some cases a strong specificity for TRX f was documented [(339) and references therein].
The crystal structure of TRX–target complexes provide further information on the interaction between plant TRXs and their targets. One study investigated the complex between barley TRX h2 and barley alpha-amylase/subtilisin inhibitor (BASI) (313). The interface area between the two proteins is quite small (762 Å2). TRX h2 recognizes the target by interacting with the exposed Cys (Cys148) with which it forms the mixed disulfide bond, and two preceding residues (Asp146 and Trp147). This short peptide of BASI that is solvent exposed and free of intermolecular contacts forms van der Walls interactions and three backbone–backbone hydrogen bonds with two TRX residues (Met88 and Ala106) both belonging to loop regions. Therefore, the TRX h2 active site portion (Trp45-Cys46-Gly47-Pro48) plus two additional segments (Ala87-Met88-Pro89 and Val104-Gly105-Ala106) form the so called substrate recognition loop motif, which is also conserved in several other TRXs, but also in some GSTs, few PDIs, and different proteins such as cytochrome c (313). A similar motif is also observed in the cocrystal structure of the E. coli 3′-phosphoadenosine-5′-phosphosulfate reductase covalently bound to E. coli TRX1 (78).
VI. Genetically Encoded Sensors for Detection of Redox Couples In Vivo
A. Detection of RMS and antioxidants in plant cells
Biochemical techniques have been largely applied to study RMS and the redox status of the most important antioxidant pools in plant cells or tissues (375, 376). In most cases, these are still the only methods available allowing analysis of the general redox state of antioxidant molecules such as ascorbate and glutathione in whole tissues or subcellular compartments (157). Data on the subcellular concentrations of ascorbate and glutathione in plant tissues were also obtained by immunogold electron microscopy, a technique that cannot distinguish between reduced and oxidized forms (582, 583). TRX isoforms, for which the subcellular distribution is usually known, were quantified by proteomic methods and their redox state under light and dark conditions examined by redox Western blots (564). Unfortunately, in most cases biochemical assays require tissue homogenization that, on one side, may dramatically reduce the sensitivity of the analysis and, on the other, can introduce artifacts due to the sample manipulation. Since both RMS and antioxidants are unevenly distributed in different subcellular compartments, the meaning of biochemical determinations of concentrations and redox states in raw extracts is intrinsically limited, independently from the precision of the measurements.
To overcome these problems, in the past 15 years, biologists have started to use new in vivo technologies that rely on the use of genetically encoded sensors that enable a real-time monitoring of the dynamics of chemical species and redox couples (333, 458). Although this approach has greatly increased the precision and the flexibility of the measurements that can be performed in vivo, the availability of genetically encoded sensors is still restricted to few chemical species and redox couples. Technical developments are urgently needed for expanding the palette of sensors to a larger number of redox compounds.
B. Genetically encoded sensors for glutathione
In the redox field, the most commonly used genetically encoded sensors are RxYFP, roGFP1, and roGFP2 that are based on modified yellow fluorescent protein (YFP) or GFP (128, 333, 386, 458). In these modified versions of the GFP- or YFP-based sensors, two Cys residues have been inserted in adjacent β-strands on the surface of the protein β-barrel making the protein able to make a disulfide in a cellular context (333, 458). The Cys residues, being positioned in proximity to the chromophore, can form a disulfide bond that causes a structural change influencing the light absorption and fluorescence of the sensor (Fig. 16A).

To reveal the formation or reduction of the disulfide bond, it is required to perform a ratiometric imaging by dual and sequential excitation of the sensor, usually with violet (∼405 nm) and blue (∼488 nm) light (Fig. 16A). The emitted fluorescence is then acquired in a 505–540 nm window. Specifically, the disulfide-induced structural change of the fluorescent sensor has the effect of changing the quantum yield (QY) of its two main absorption peaks with an opposite trend: the QY at 405 nm increases, whereas the QY at 488 nm decreases, hence leading to a ratiometric response. The ratio of the light emitted after excitation at 405 and 488 nm (briefly, the 488/405 nm ratio) provides a direct readout of disulfide bond formation in the sensor population. The higher the 488/405 nm ratio is, the higher the oxidation of the sensor (i.e., the percentage of sensor molecules bearing the disulfide). Most importantly, such ratio can be monitored in real time and in vivo with different grades of resolution depending on the system used for the acquisition (e.g., wide-field and confocal microscopy or a fluorescent-based plate reader).
The field of application of the system depends on whether the sensor in vivo equilibrates with one or more redox couples such that it can be used to measure the redox state of these couples. The sensors of the roGFP family show midpoint redox potentials (E0 roGFP) between −260 and −290 mV and are proposed to provide an accurate determination of the redox potential of glutathione (EGSH) in vivo (331, 387, 459). This implies that glutathione is assumed to equilibrate with the sensor in vivo. Since the midpoint redox potential of glutathione is less negative (E0 GSH −240 mV), the roGFP sensors are intrinsically more adapted to measure highly reduced than oxidized glutathione redox states. Moreover, since GSH dimerizes upon oxidation (GSSG +2 e− + 2H+ → 2 GSH), the EGSH depends on the [GSH]2/[GSSG] ratio. In other words, it depends on both the GSH/GSSG ratio and the total concentration of GSH+GSSG. Therefore, glutathione redox potentials estimated by the roGFP cannot be translated into GSH/GSSG ratios unless the total concentration of GSH+GSSG is known (157).
Key advantages of the ratiometric nature of these sensors are manifold. First, the sensor readout is largely independent of their concentration in the cell. Second, the ratiometric feature of the sensors allows for the correction of focus changes or moving artifacts when samples are imaged by microscopy. Third, by using different promoters, they can be specifically expressed in different tissues and engineered for their targeting to different subcellular compartments or to modify their properties. Considering all these features, these redox genetically encoded sensors have been shown to be suitable for deriving information on redox conditions prevailing in the cell in different plant species, different cell types, and different subcellular compartments.
Concerns about the specificity of the roGFP for glutathione have long been debated. Peroxidases such as yeast Orp1 and mammalian GPX4 appear to oxidize roGFP2 directly in response to H2O2 in HeLa cells, suggesting that the roGFP redox state may be influenced by other factors than glutathione (203). In fact, roGFP is not oxidized by H2O2 in vitro but is rapidly oxidized by H2O2 in vivo. Whether this effect is mediated by glutathione or peroxidases is difficult to tell. Anyway, even if different compounds obviously influence the roGFP redox state in vivo, it is still possible that roGFP and glutathione reciprocally equilibrate under any condition. As briefly discussed hereunder, experimental evidence acquired so far supports this hypothesis.
The first work reporting the expression of a redox sensor in plants was published in 2006 (244) in a study where the roGFP1 was expressed in the cytoplasm and mitochondria of A. thaliana and performed oxidation and reduction treatments with H2O2 and DTT (244). This pioneering work showed the possibility to monitor dynamically subcellular redox changes by measuring in real time the sensor fluorescent emission ratio. By carrying out a calibration curve, it was possible to convert the sensor ratios into redox potentials, showing that in mitochondria the EroGFP was more reduced than in the cytoplasm (−362 and −318 mV, respectively). Soon after the development of the rxYFP and roGFP sensors and their first applications in animals and plants, it clearly emerged that both redox sensors equilibrated predominantly with glutathione (331, 387, 459) and that equilibration in vivo was accelerated by GRXs that mediated the thiol–disulfide exchange between glutathione and the redox sensor (458). A confirmation of this came in 2007 when Meyer et al. expressed the roGFP2 sensor in Arabidopsis (GFP2 is an enhanced variant of GFP1, see this section). Also in this case, the roGFP2 reversibly responded to redox changes induced by incubation with H2O2 or DTT and, more important, the sensor was severely oxidized in mutants with reduced levels of glutathione and in wild-type plants, in which the glutathione content was depleted by treatments with an inhibitor of glutathione biosynthesis (
As the equilibrium between roGFPs and glutathione was suspected to be mediated by GRXs in vivo, it was reasoned that the availability of endogenous GRXs might limit the fast equilibration between glutathione and the sensor. To overcome this problem, human GRX1 was fused to the roGFP2 (202). This new GRX1–roGFP2 was expressed and tested in plants such as A. thaliana and tobacco and in the phytopathogenic fungus Botrytis cinerea (132, 216, 328). In Botrytis, a side by side comparison of GRX1–roGFP2 and roGFP2 revealed that the oxidation of GRX1–roGFP2 was slightly faster than roGFP2, thereby sustaining the hypothesis that GRX could facilitate the equilibrium between glutathione and the sensor (216). The functional interaction between glutathione and GRX1–roGFP2 is proposed to involve first the glutathionylation of GRX1 by GSSG, then the internal trans-glutathionylation of roGFP2 by GRX1, followed by the formation of the disulfide in the roGFP2 (Fig. 16B). All steps are reversible (Fig. 16B). Note that roGFPs do not contain acidic/reactive Cys; therefore, its glutathionylation can proceed via GRX1-dependent trans-glutathionylation with no requirement of H2O2 and transient sulfenic acid formation (see section II.C.6 and Fig. 8). Neither GRX1 nor roGFP2 is expected to react with H2O2 directly.
Altogether, the results obtained with different redox sensors based on roGFP1/2 expressed in different subcellular compartments of different organisms under physiological conditions show that the sensors are always highly reduced in mitochondria, nuclei, peroxisomes, chloroplasts, and cytoplasm and highly oxidized in the ER lumen (458).
Interestingly, the picture changes in stress conditions. For example, drought stress causes oxidation of roGFP1 in the cytoplasm of Arabidopsis (EGSH shifted from −311 to −302 mV) with reversion to control values after rewatering (248). Strong oxidation of cytoplasmic roGFP2 was also observed in the root tip of Arabidopsis seedlings treated with cadmium (515) and in wounded Arabidopsis leaves (40, 332). In the latter case, an oxidation wave propagating from the wound area preceded a reduction wave in the opposite direction, suggesting a systemic signaling response as previously hypothesized for ROS waves (346). Technically, the in vivo monitoring of the GSH/GSSG status in real time offered an unprecedented spatial and temporal resolution that may be difficult, if not impossible, to reach with other techniques.
The oxidation of cytoplasmic roGFP2 was also reported in Arabidopsis and tobacco leaves infected with the avirulent pathogen Pseudomonas syringae DC3000 (328). The mitochondrial version of roGFP1 and roGFP2 sensors was oxidized in response to heat stress, cadmium, and darkness (437, 458, 460). Dark-induced roGFP2 oxidation occurred also in plastids, peroxisomes, and cytoplasm, but in all cases with a different and slower timing than in mitochondria (437), suggesting that mitochondria may represent the origin of the oxidative stress in the dark occurring during the senescence program. In chloroplasts, treatments with electron transport inhibitors (3-(3,4-dichlorophenyl)-1,1-dimethylurea and 2,5-dibromo-6-isopropyl-3-methyl-1,4-benzoquinone) led to stromal roGFP2 oxidation and, in the same organelle, increased formation of stromules (53). The effect is interesting since stromules were shown to play a role in oxidative signaling (66).
As expected for a sensor sensing the glutathione redox potential, roGFP2 was also found more oxidized in Arabidopsis mutants [rml1, (8); cad2, (332)], in which the total glutathione content is strongly diminished (62, 514), and in mutants of glutathione reductase (gr1, gr2) in which the GSH/GSSG is more oxidized (324, 569).
C. Other redox sensors
The high sensitivity of thiol peroxidases, including PRXs, for H2O2 was exploited to develop a redox sensor with different specificity than roGFP variants. PRXs bear an extremely reactive catalytic Cys that forms a sulfenic acid upon reaction with H2O2. The sulfenic acid is then resolved by a second Cys forming a disulfide. Although in the common catalytic cycle of PRXs, the disulfide is reduced by TRX or GRX, some PRXs harbor an intrinsic and powerful capacity to act as H2O2-dependent protein thiol oxidases when they are recruited into proximity of oxidizable target proteins (203). Hence, the idea of fusing the yeast GPLX protein Orp1 to the roGFP2 to get an H2O2 sensor came (203). However, this probe cannot be considered as a strict H2O2 sensor as the roGFP2–Orp1 is on one side oxidized by H2O2, but on the other is likely to be reduced in vivo by TRXs that have been shown to directly reduce Orp1 (458). Different from the GRX1–roGFP2 probe whose oxidation by GSSG and reduction by GSH are reversible, the oxidation of the catalytic Cys of Orp1 by H2O2 is not reversed by water [(508); sulfenic acids are not easily reduced by water], such that roGFP2–Orp1 cannot equilibrate with the H2O2/H2O redox couple. As a matter of fact, the redox state of roGFP–Orp1 is not only influenced by the level of the oxidant (H2O2), but also by the reductants, GRXs and TRXs. This makes the sensor unsuitable to determine absolute H2O2 levels (458). Nevertheless, the roGFP2–Orp1 expressed in Drosophila showed a different redox state from GRX1–roGFP2 during development and aging (5), suggesting that both sensors provide different information. The roGFP2–Orp1 sensor has been recently used in Arabidopsis, revealing that in guard cells treatment with H2S determines its oxidation via activation of NADPH oxidase (H2O2 production) (461).
Other relevant redox couples important for redox homeostasis in plants are represented by nicotinamide adenine dinucleotides. In vivo monitoring of NADH/NAD+ ratios was attempted by combining a bacterial NADH-binding protein and a fluorescent protein variant, creating a genetically encoded fluorescent biosensor of the cytoplasmic NADH/NAD+ redox state, named Peredox (231). The functionality of Peredox was demonstrated in mammalian cells showing that it efficiently reported the cytoplasmic NADH/NAD+ ratio and that it was sensitive to exogenous administration of lactate and pyruvate (231). Such sensor was also employed in the fungus Ustilago maydis to monitor cytoplasmic NAD redox dynamics (213). More recently, a new ratiometric pH-resistant genetically encoded fluorescent indicator for NADPH (iNap) was also generated (493). The iNap sensors have been used to monitor NADPH fluctuations during the activation of macrophage cells or wound response in vivo (493). Up to now, there are no reports showing the functionality of such NAD(P)(H) sensors in plant cells.
VII. Redox Plant Physiology In Vivo
As outlined previously, plants organize a multiplicity of different low-molecular weight redox couples and redox proteins to regulate cellular redox homeostasis. Whereas research in the past mainly focused on the characterization of these components during in vitro studies, recent progress has been made to resolve the organization and biological significance of this complex redox network in planta. In the following section, we review the emerging roles of this regulatory network in integrating photosynthesis, growth, development, and stress responses of plants to cope with fluctuating environmental conditions.
A. Redox regulation of light acclimation: the FTR–TRX system and light-responsive control of photosynthesis within the chloroplast
Sunlight represents the source of energy for photosynthesis and plant growth. However, photosynthetic cells have to manage strong fluctuations in light intensities that can occur very rapidly in nature. This requires sensitive and rapid light acclimation mechanisms to maintain photosynthetic performance and chloroplast functions in a dynamic manner and to avoid the generation of potentially harmful ROS.
One pathway to transfer light signals to chloroplast target enzymes is provided by the FDX–TRX system (54). It involves sequential transfer of reducing power from photosynthetic light reactions via FDX and FTR to five different TRX classes (f, m, x, y, and z), which activate specific sets of stromal and thylakoid proteins by reducing their regulatory disulfides (Fig. 17) (564).
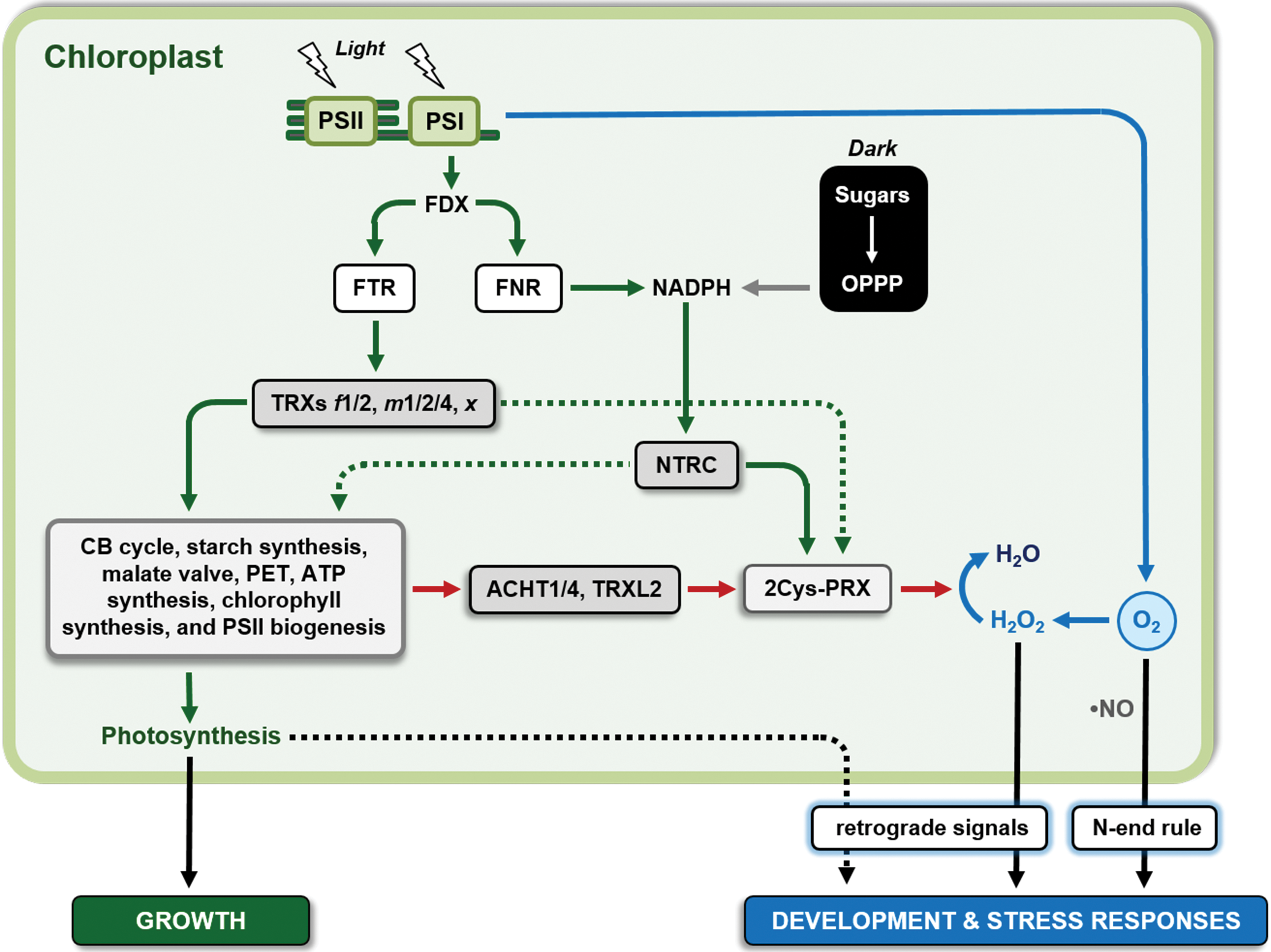
Comparative studies using sets of recombinant purified TRX isoforms and target proteins revealed functional specificities of the different classes of TRXs for their targets in vitro (92, 322, 500, 562). TRXs belonging to f and m classes revealed metabolic functions in activating enzymes of the CB cycle, starch synthesis, redox export via the malate valve, and ATP synthesis, whereas isoforms belonging to the x and y classes revealed antioxidative functions in providing reducing power to PRXs. For a comprehensive overview on the TRX target proteins and their regulatory specificities for different TRX isoforms identified during in vitro studies, see (171).
Although until recently most of our knowledge on the functional diversity of chloroplast TRXs relied on in vitro studies, a boost of genetic studies in the past years specifically in Arabidopsis led to a rapid increase in our knowledge on their roles in vivo. As outlined in section IV, the plant genome contains a complex gene family of TRXs, with up to 21 different TRX classes, including 7 classes containing typical TRXs because of their conserved active site signature and single domain structure. Typical TRXs from five classes reside in Arabidopsis chloroplasts, with different isoforms (TRXs f1-2, m1-4, x, y1-2, and z). The chloroplasts contain also several atypical TRX isoforms that are often little studied with respect to typical isoforms. A quantification of the protein levels of typical TRX isoforms showed that TRXs f and m are the major isoforms, accumulating to 22% and 69% of the total level of typical TRXs in the chloroplast stroma, respectively (383). For the sake of simplicity, when not otherwise specified, the term TRX will be used for typical TRXs in the following text.
Arabidopsis mutants deficient in TRX f1 (lacking 70%–90% of total TRX f proteins) or with combined deficiencies of TRXs f1 and f2 (trxf1f2 mutants) revealed that f class TRXs are important for the rapid activation of carbon metabolism and photosynthesis in response to light. During rapid dark–light transitions, TRX f deficiency led to delayed and incomplete reduction and activation of the CB cycle enzyme FBPase and RubisCO activase, retarded light activation of CB cycle activity, and transient inhibition of PET, whereas thermal dissipation of the absorbed light energy by nonphotochemical quenching (NPQ) was transiently increased (363, 383, 498, 501, 562). This shows a role of TRX f in short-term light adjustment of photosynthetic carbon fixation to optimize photosynthetic efficiency. Deficiency of TRX f also led to an incomplete photoreduction of the small subunit of the key starch synthetic enzyme AGPase resulting in decreased starch accumulation during the day, providing evidence for a role of TRX f in regulating diurnal starch turnover in response to dark–light alterations (363, 498, 500). Interestingly, despite the complete lack of f class TRXs, FBPase and RubisCO activase became partially reduced during illumination (363, 382, 562), indicating functional compensation by other classes of TRXs or thiol-reduction systems (see sections VII.A, VII.B, and VII.C). In line with this, silencing of TRX f did not substantially affect overall rates of photosynthetic carbon fixation and plant growth under long-day conditions, whereas there were only slight growth retardations under short-days or very low light intensities (363, 498).
TRXs of the m-class have more diverse in vivo functions than TRX f. Earlier studies documented a role of the very low abundant TRX m3 in symplastic permeability and meristem development (45), see section VII.F). On the other hand, more recent reports revealed photosynthetic functions for the relatively high abundant TRXs m1, m2 and m4, each representing ∼23% of the total stromal TRX (99, 383, 501, 523). For TRX m4 a role in the regulation of cyclic PET was revealed (99), whereas TRXs m1 and m2 were found to participate in the rapid light activation of NADP-MDH (501) involved in the export of excess reducing equivalents from the chloroplast via the malate valve to prevent photoinhibition (447). This indicates that TRXs m1, m2 and m4 are important to balance the chloroplast ATP/NADPH ratio for optimized photosynthesis. Arabidopsis double mutants with combined deficiencies of TRXs m1 and m2 showed wild-type growth and photosynthesis under constant light conditions, but photosynthetic parameters were strongly modified in fluctuating light environments with rapidly alternating low and high light intensities (501). Combined silencing of TRXs m1 and m2 led to lower photosynthetic efficiency in high light, but surprisingly had the opposite effect in the low light periods. This indicates that TRXs m1 and m2 are involved in dynamic acclimation of photosynthesis, being essential for full activation of photosynthesis in the high-light peaks by rapid induction of the malate valve to prevent photoinhibition, whereas there is a trade-off in photosynthetic efficiency during the low-light phases of fluctuating light (501). The reason for the higher photosynthetic efficiency of the TRXm1 m2 mutants in low light is unclear and requires further investigation.
Interestingly, multiple silencing of TRX m1, TRX m2 and TRX m4 in triple Arabidopsis mutants led to more severe phenotypes, depending on the extent of the decrease in total TRX m protein (383, 523). A decrease in TRX m protein to ∼30% of the level found in wild-type plants led to incomplete photo-reduction of FBPase and SBPase from the CB cycle and NADP-MDH from the malate valve in response to light, resulting in decreased CO2 assimilation rates, inhibition of PET and substantial retardations in plant growth under constant light conditions (383). When compared with trxf1f2 mutants (see earlier in this section), this reveals a high level of redundancy of f- and m-class TRXs in the light activation of the CB cycle and photosynthesis being operational in vivo, which is unexpected given the predominant role of f-class TRXs in regulating enzymes of the CB cycle as proposed by in vitro studies (171). When the total amount of TRX m proteins was decreased to less than 15% of the level found in wild-type plants, mutant plants displayed very severe growth defects, pale green leaves, strongly decreased PSII activity and impaired PSII assembly (383, 523). In line with this, triple silencing of TRXs m1, m2 and m4 in transgenic Arabidopsis led to a decrease in chlorophyll accumulation and in the redox status and activity of Mg-protoporphyrin IX methyltransferase (CHLM), which catalyzes the second step in the chlorophyll synthesizing Mg branch of the tetrapyrrol pathway in the chloroplast (108). Interestingly, studies in pea revealed that simultaneous silencing of TRX f and m genes is required to decrease the in vivo redox status of the Mg chelatase CHLI subunit (CHLI), catalyzing the first step of the Mg branch, as well as chlorophyll content and photosynthetic capacity (309). While interpretation of in vivo results is complicated by the fact that genetic removal of part of the TRX pool is likely to affect the redox state of the remnants, overall, these studies suggest redundant roles of f- and m-class TRXs in, both, rapid light activation of photosynthetic metabolism and more long-term light regulation of the biosynthesis of photosynthetic machineries.
Arabidopsis mutants deficient in the less abundant TRXs x or y showed wild-type phenotypes, despite their proposed roles in reduction of 2-Cys PRX and PRX Q for peroxide detoxification based on in vitro studies (280, 419). This indicates functional compensation by other chloroplast TRX systems in vivo (see sections VII.B and VII.C). In contrast to this, deficiency of the low abundant TRX z led to an albino phenotype with impaired photoautotrophic growth and disturbed chloroplast development, similar to mutants of chloroplast gene expression (18). In confirmation to this, TRX z was found to act as an essential structural component of the plastid-encoded RNA polymerase complex and proposed to be important for the light-dependent expression of photosynthetic genes in the chloroplast. However, the role of TRX z in this context seems to be independent of its redox activity (535) and the in vivo pathways leading to its reduction are currently unclear (563) because the Arabidopsis isoform does not seem to be reduced by FTR in vitro (51) unlike poplar counterpart (86). Further studies will be necessary to elucidate the role of TRX z in the chloroplast dithiol/disulfide network.
B. Redox regulation of light acclimation: chloroplast NTRC, 2-Cys PRXs, and photosynthetic performance under low light
In addition to the light-dependent FDX–TRX system described above, the more recently discovered NTRC forms a separate thiol reduction cascade in the chloroplast stroma, combining both NTR and TRX activities on a single polypeptide (Fig. 17) (468). Unlike FTR, NTRC receives its reducing potential from NADPH and provides electrons to target proteins via its own TRX domain (47). Biochemical and genetic studies established a major role of NTRC in reducing 2-Cys PRXs involved in the scavenging of H2O2 within the chloroplast (409, 563). Comparative studies using Arabidopsis mutants deficient in NTRC and TRX x identified NTRC as the primary electron donor for 2-Cys PRXs in vivo, providing a redox buffer to keep this enzyme in a reduced state for antioxidant functions in the light as well as in the dark (419). Although these studies suggested that NTRC operated as a separate thiol reduction system independently of light, recent work provided in vivo evidence for additional functions of NTRC in the light-dependent regulation of photosynthetic metabolism and thylakoid energy transduction similar to the FDX–TRX system. In Arabidopsis mutants, deficiency of NTRC led to incomplete photo reduction of regulatory disulfides in enzymes involved in chlorophyll biosynthesis [CHLI, CHLM, and glutamyl-transfer RNA reductase1, GluTR1; (407, 429)], starch biosynthesis [AGPase; (498)], CB cycle [FBPase, SBPase, and PRK (371, 382, 498, 563)], ATP synthesis [γ-subunit of CF1-ATP synthase (69, 371)], and NADPH export [NADP-MDH; (501)] in response to dark–light transitions, resulting in impaired chlorophyll accumulation (429, 468), starch turnover (290, 498), CO2 assimilation (498), photosynthetic light energy utilization (69, 364, 371, 498, 501), and plant growth (290, 364, 498). For most of these parameters, silencing and overexpression of NTRC led to opposing effects, indicating that NTRC is limiting for CO 2 fixation, photosynthetic efficiency, and growth in wild-type Arabidopsis plants (371) and may be a promising target for biotechnological strategies to improve crops (370). The role of NTRC to optimize photosynthetic efficiency is specifically relevant under constant (69, 364) and fluctuating low light intensities (69, 501). When light availability is limiting, NADPH-dependent NTRC allows efficient redox activation of proton-coupled ATP synthase, leading to lower acidification of the thylakoid lumen and lower energy dissipation by NPQ, resulting in a more efficient utilization of available light energy for photosynthesis and growth (69, 364). In confirmation to this notion, blocking of NPQ in the ntrc mutant background led to partial recovery of photosynthetic performance and growth, indicating that NTRC promotes photosynthesis by regulating NPQ (364). Under rapidly alternating low and high light intensities, NTRC is indispensable to ensure the full range of dynamic responses of NPQ to optimize photosynthesis and maintain growth in fluctuating light environments occurring frequently in nature (501).
C. Redox regulation of light acclimation: cooperation of FTR–TRX and NADPH–NTRC systems for photoautotrophic growth
The participation of NTRC in the light activation of enzymes known to be regulated by the FDX–TRX system suggests that chloroplast redox regulation depends on the cross talk between both thiol–redox systems in vivo. To dissect the relationship of NTRC with the other TRXs, recent studies investigated Arabidopsis mutants with combined deficiencies of NTRC and TRXs. When the deficiency of NTRC was combined with those of TRX f1/2 (382, 498) or TRX x (382), double/triple mutants showed severe growth retardation phenotypes, almost abolished light activation of FBPase from the CB cycle, severely impaired CO 2 assimilation, starch turnover, photosynthetic efficiency, and chlorophyll accumulation, whereas single mutants were hardly affected. Severe growth retardation and severely impaired chlorophyll synthesis were also revealed when NTRC deficiency was combined with deficiencies of TRXs m1, m2, and m4 in quadruple mutants (108). A double mutant combining the deficiency of NTRC and of the catalytic subunit of FTR was not viable under photoautotrophic conditions (563). This suggests that NADPH-dependent NTRC acts concertedly with diverse other classes of TRXs of the light-dependent FDX–TRX system in photosynthetic redox regulation, with TRXs f1/2, m1/2/4, and x showing a high degree of functional redundancy. A cooperation of both thiol–reduction loops is, therefore, indispensable to sustain light acclimation of photosynthetic metabolism, photosynthetic efficiency, and photoautotrophic growth of plants.
Different mechanisms have been proposed to explain the functional integration of NTRC and FDX–TRX systems. The phenotypic recovery of the ntrc Arabidopsis mutant by overexpression of redox-inactive forms of NTRC together with bimolecular fluorescence complementation assay-based protein–protein interaction studies provided evidence that NTRC physically interacts with the FDX–TRX system and its targets in vivo (371). However, the functional role of this interaction is still unclear. In vitro studies show that NTRC is very inefficient in reducing TRXs f1, f2, m1, m4, x, and y1 (51, 563) and is not able to reduce the regulatory disulfides of the TRX target enzymes FBPase, SBPase, and NADP-MDH directly (382, 563). This puts forward indirect effects to explain why light activation of the CB cycle and export of excess reducing equivalents require NTRC. Recent studies show that decreased levels of 2-Cys PRX suppress the phenotype of the ntrc single and ntrc-trxf1-trxf2 triple Arabidopsis mutants, indicating that FDX–TRX and NTRC redox systems are integrated via the redox balance of 2-Cys PRX (408). As NTRC is the major system to provide electrons for the reduction of 2-Cys PRXs (419, 563), it will indirectly maintain the reducing capacity of the pool of FDX–TRXs (408) and restrict reoxidation of their targets via an oxidation loop involving H2O2, oxidized 2-Cys PRX, and the atypical TRXs ACHT1/4 (111, 133, 371) and TRX-like2 (TRXL2) (561) to finally increase the reduction state of disulfides in target proteins of the FDX–TRX system (Fig. 17).
D. Redox regulation of light acclimation: integration of redox signals at the cellular level
In addition to intraorganellar cross talk of redox systems within the chloroplast, light acclimation of photosynthesis also requires interorganellar redox communication (380, 449). During acclimation to fluctuating light intensities, chloroplasts communicate information by retrograde signaling to the nucleus, leading to rapid changes in the transcription of nuclear genes coding for proteins involved in light harvesting, electron transport, stromal metabolism, and antioxidant systems to balance input of light energy with photosynthetic capacity (122, 184). There is in vivo evidence that H2O2 acts as an important retrograde signal in this response, sensing excess excitation energy in the chloroplast rather than being a toxic by-product of aerobic metabolism (134, 159, 345). Elevated light leads to increased reduction of oxygen to superoxide radicals at the acceptor side of PSI, leading to increased production of H2O2 via SOD within the chloroplast (134, 355). The elevated level of H2O2 is subsequently transferred from the chloroplast into the nucleus (50, 134, 355), where it leads to induced expression of high light responsive nuclear genes (134, 510) via redox sensitive transcription factors (473). As H2O2 movement from chloroplast to nucleus does not involve the cytoplasm (134), its transfer most likely involves a close physical association of the two organelles (464), allowing efficient aquaporin-mediated transmembrane diffusion (50), or the formation of stromules as direct stroma-filled interorganellar connections (53). In confirmation, stromule formation between chloroplasts and nucleus is specifically increased in response to light and chloroplast ROS production (53). This retrograde signaling pathway will (i) be attenuated by light-dependent reductive signals mediated by chloroplast TRXs and NTRC, which will diminish the production of H2O2 by decreasing acceptor limitation at PSI (501), (ii) increase the scavenging of H2O2 by activation of PRXs in the chloroplast stroma (563), and (iii) restrict the transport of H2O2 to the nucleus by inhibiting stromule formation (53). This is consistent with recent studies on Arabidopsis mutants with combined deficiencies of chloroplast 2-Cys PRXs and APXs revealing increased H2O2 levels and upregulation of H2O2-responsive marker genes in the nucleus (22).
Interorganellar redox signaling also involves the exchange of reducing equivalents via metabolite shuttles, including the triose-P/3PGA shuttle at the chloroplast envelope (518) and the malate/oxaloacetate shuttles at the chloroplast (260, 447), mitochondrial (447), and peroxysomal (219) envelopes/membranes. This allows high light acclimation responses at the cellular level by sensing acceptor limitation at PSI via an increase in the chloroplast NADPH/NADP+ ratio, which is transmitted to cytoplasm, mitochondria, and peroxisomes via a combination of the different redox shuttles. Recent studies suggested that this redox signaling system affects light acclimation responses by (i) translational inhibition of photosynthetic gene expression via TRX-h-dependent regulation of denitrosylation of the repressor protein NAB1 in the cytoplasm (46), (ii) inhibition of protein uptake into chloroplasts via redox regulation of chloroplast envelope translocons (29, 586), (iii) inhibition of CAT in peroxisomes to modulate H2O2 signaling responses (219), and (iv) dissipation of excess reducing equivalents via alternative oxidase in mitochondria (149, 566), probably involving regulation by mitochondrial TRXs (109) to prevent an over-reduction of the photosystems in the chloroplast (malate valve) and to modulate ROS responses. Although direct evidence for the light dependency of mitochondrial TRXs is largely lacking, there is in vivo evidence that the malate valve can act in the reverse direction by transmitting mitochondrial redox signals to the chloroplasts, leading to redox regulation of chloroplast metabolism (71) and import of chloroplast precursor proteins (586) via alterations in the chloroplast NADPH/NADP+ ratio. This implies cross talk of chloroplast, cytoplasmic, and mitochondrial TRX systems to integrate redox signals at the cellular level. Further work is required to resolve the network of interorganellar redox communication and the in vivo roles of its components, ensuring photosynthetic light acclimation and redox balancing at the cellular level.
E. Redox control of abiotic and biotic stress responses: integration of multiple signaling pathways
In addition to its role in light acclimation of photosynthesis, redox regulation is also involved in the control of various abiotic and biotic stress responses. As reviewed recently, ROS and RNS play critical and integrative roles in multiple stress signaling (34, 377, 472), controlling pathogen defense (282, 450), and abiotic stress tolerance of plants (Fig. 17) (138, 423). The complexity in ROS responses to various environmental stimuli is attributable to the intrinsic chemical properties of different ROS, different sites and mechanisms of ROS production, the spatial and temporal coordination of ROS signals, and their integration with other signals related to metabolites, antioxidants, redoxins, hormones, and genetic control elements [reviewed in (34, 218, 251, 341, 374, 377, 423)]. In this context, different subcellular sites of ROS production may define specificity in signaling (341, 377). ROS are produced in the apoplast by activation of plasmalemma RBOHs (484) or cell wall peroxidases (315) and in chloroplasts (123), peroxisomes (115), and mitochondria (230), as a by-product of aerobic metabolism (see section II). It is proposed that specific sets of environmental stress conditions, such as pathogen infection, ozone, UV-B, excess irradiation, salinity, drought, temperature, or low oxygen, will result in specific subcellular ROS, RNS, and redox signatures that will, in turn, lead to the activation of specific defense and acclimation responses (90, 119, 403, 423). However, little is known on the underlying mechanisms allowing subcellular changes in ROS and RNS levels to be sensed and the signal being transduced to specific downstream response elements (158, 377, 423). In yeast and Chlamydomonas, the induction of autophagy in response to numerous stress conditions is associated with ROS production and is regulated by TRX-dependent activation of the ATG4 cysteine protease (398 –401, 406). In plants, recent studies indicate that defense responses to abiotic and biotic stresses involve an interplay between salicylic acid (SA), ROS, RNS, GSH, and TRXs (218, 499). Different environmental stimuli lead to ROS production in different subcellular compartments that precedes SA signaling, causing transcriptional reprogramming of gene expression (218). The ROS–SA interaction is modulated by GSH, which leads to increased SA production, whereas SA causes increased GSH levels and reducing power, which, in turn, is involved in ROS scavenging (131). Upon pathogen infection, ROS is produced in the apoplast (315) leading to elevated SA levels and a subsequent more reduced cellular redox state (or at least glutathione redox state), which is sensed by the NPR1 protein, a master regulator of pathogenesis-related (PR) gene expression (354). Redox regulation involves NTRA-dependent TRX h5, leading to monomerization of the NPR1 oligomer in the cytoplasm to allow its translocation into the nucleus (487), where it activates the expression of PR genes via its interaction with TGA transcription factors, which are also modulated by redox conditions (262). In this context, TRX h5 is facilitating NPR1 monomerization by catalyzing the direct reduction of intermolecular disulfide bonds linking the NPR1 monomers (487) and by denitrosylation of the regulatory Cys156 in antagonistic action to GSNO and NO (262).
A further cytoplasmic mediator of redox signal transduction is the highly conserved glyceraldehyde-3-phosphate dehydrogenase C (GAPC), a key glycolytic enzyme with important noncatalytic functions for various abiotic and biotic stress responses [reviewed in (220, 557, 577)]. Plant GAPCs act as common target proteins of ROS and RNS with their catalytic Cys being subjected to diverse reversible modifications, such as S-nitrosylation, sulfenylation, and S-glutathionylation, which can be reversed/reduced by GSH, TRX, and GRX (36, 46, 102, 166, 575, 577, 580). During cadmium-induced oxidative stress, NO accumulates and GAPC1 is translocated from the cytoplasm to the nucleus, where its role remains to be established (515). Whereas in mammalian cells nuclear relocalization of GAPC depends on nitrosylation of its catalytic Cys, mutation of this residue (Cys155) in Arabidopsis plants led to a stimulation of relocalization, rather than an inhibition (515).
With respect to transcription factors, ERF-VII have emerged as novel regulators of abiotic and biotic stress responses involved in oxygen- and NO-dependent signal transduction in plants (177, 560). Molecular oxygen and NO lead to oxidation of the conserved N-terminal Cys of ERF-VIIs to Cys sulfinic and sulfonic acids facilitated by PCOs targeting ERF-VII proteins to the N-end rule pathway of proteasomal degradation (176, 178, 179, 294, 531, 533). This provides a sensing mechanism for oxygen and NO mediating ERF-VII degradation and reprogramming of gene expression (178, 294, 560), allowing an efficient regulation of central metabolic processes to optimize hypoxic resistance (394) and immune responses of plants (394, 591). Although the N-end rule pathway has emerged as an important regulator of environmental stress responses, further studies are necessary to identify N-end rule substrates beyond ERF-VII and their role in the plant signaling network (125, 176).
Recent studies provide evidence for an emerging role of chloroplasts as integrators of plant stress signals specifically in plant immunity against pathogens [reviewed in (119, 154, 252, 467]. Chloroplasts are important as sensors of available photosynthetic energy to fuel immune responses and serve as major production sites of prodefense molecules such as phytohormones (including SA and jasmonic acid) and ROS providing retrograde signals to modulate nuclear gene expression and plant resistance to pathogens. This involves the chloroplast redox status as a major regulator of defense responses (Fig. 17). Manipulation of ROS buildup in chloroplasts by expression of a plastid-targeted flavodoxin (411), silencing of FTR (295), deficiency of NTRC and PRX (234, 235), and depletion of chloroplast forms of GPLX, GPXL1, and GPXL7 (75), led to changes in the expression of PR genes and pathogen resistance in diverse plant species. Although the interplay between plastidial and extraplastidial ROS sources during plant immunity is still unclear (341), the specificity of chloroplast ROS signaling may be attributable to pathogen-induced formation of stromules, providing physical connections to transport ROS and other prodefense molecules from the chloroplast directly to the nucleus (66). This will allow the transport of chloroplast-derived signaling proteins such as NRIP1 involved in pathogen recognition (67) or WHIRLY1 involved in redox sensing (154) to the nucleus to trigger PR gene expression. As NTRC acts a master regulator of chloroplast redox homeostasis (408), it will affect pathogen-related responses by modulating H2O2 production via 2-Cys PRXs (234) and light-dependent stromule formation (53).
GRXs, represented by members belonging to four different classes in terrestrial plants (see section IV), are also emerging as important redox-active players of plant responses to stress. For example, GRXS12 positively correlates with brassinosteroid accumulation and antioxidant responses under chilling conditions in tomato plants (548). Although poplar GRXS12 is very active in protein deglutathionylation in vitro (573), the relevance of this activity in vivo is still unknown. Class II GRXs are best known for their role in Fe-S cluster biogenesis (30, 351, 481), but they are also implied in stress responses. Class II GRXS14 levels correlate with plant tolerance to abiotic stress conditions in Arabidopsis (81, 427) and tomato (195). Plants with altered class II GRXS17 expression were found to be tolerant to drought and oxidative and heat stresses (82, 546, 547). Recently, GRXS17 was found to be associated with components of the cytoplasmic Fe-S cluster assembly pathway and to be demanded for a proper response to iron deficiency stress (233). Members of class III, namely GRXC7 and GRXC8 (ROXY1 and ROXY2, respectively), participate in pathogen responses, overexpressing lines being hypersusceptible to the infection of the necrotrophic pathogen B. cinerea with a concomitant accumulation of H2O2 (526). Interestingly, plants impaired in class III GRXS13 are less susceptible to B. cinerea infection (273). Transgenic plants with reduced level of GRXS13 and GRXC9 showed increased levels of superoxide radical and reduced tolerance to high light and methyl viologen treatments (278). Overexpression of class III GRXC7 and class I GRXC2 confers increased arsenic tolerance, allowing reduced accumulation of this metal pollutant in both seeds and shoot tissues (513). In general, plant grx mutant analyses point to a positive role of GRXs of any class in different biotic and abiotic stress responses in vivo. Mechanistic models of their function, however, are still largely hypothetical.
F. Redox regulation of plant development: integration of redox signals into molecular networks of developmental control
An increasing number of reports in the literature indicate redox regulation of growth and development as an emerging field in plant biology. This is summarized in many excellent recent reviews documenting emerging roles of oxidation (oxygen, ROS, and RNS) and reduction signals (TRX, GSH, and GRX) in the regulation of the whole plant developmental cycle interfacing with signaling pathways involving phytohormones and transcription factors (95, 377, 438, 451, 452, 504). There is evidence for the role of ROS, GSH, GRX, and TRX in controlling the development of root and shoot apical meristems. ROS production by mitochondria (568) and plasmalemma-located NADPH oxidases (245) together with GRXS17 (263) and ABPH2 (553) are involved in the regulation of transcription factors to determine meristem size and maintenance. Plastid-located TRX m3 (45) and plasma membrane-associated TRX h9 (330) allow cell-to-cell communication and meristem function. Extraplastidic NTRA/NTRB and GSH (33), and chloroplastic TRXs and NTRC (261, 382) are involved in auxin and redox signaling regulating meristem development. These mechanisms may also influence cell cycle progression and cell differentiation, which are associated with oscillations in cellular redox state, involving bursts of H2O2 and subsequent import of GSH into the nucleus, regulating transcription factors through reversible Cys reduction/oxidation via nuclear TRXs (63, 120, 121). Thiol-based regulatory mechanisms are also involved in the molecular networks controlling floral development with GSH and class III GRX proteins regulating petal (549) and anther development and pollen formation (357) by interacting with TGA transcription factors in the nucleus (118).
Although research into hypoxia usually emphasized the response to changes in external oxygen supply during stress responses, there is recent evidence for developmental transitions in the oxygen status of meristems and reproductive plant organs [reviewed in (170, 509)], while conversely local hypoxic conditions may contribute to regulate developmental processes in plants (94, 293, 451). Establishment of internal hypoxic environments will contribute to developmental regulation by maintaining reducing conditions in specific plant tissues. In this context, hypoxia arising naturally within growing anther tissue acts as a positional cue to set germ cell fate (255). Changes in oxygen concentrations may also contribute to plant development by affecting the stability of ERF-VII transcription factors via the N-end rule pathway. As published recently, the N-end rule pathway controls multiple functions during shoot and leaf development (188), probably via its function to sense gaseous signals such as oxygen and NO (179, 294). This may partly involve regulation of ERF-VII by protein degradation, as Arabidopsis plants overexpressing N-end rule insensitive forms of ERF-VII displayed changes in leaf development (180, 394) and photomorphogenesis (2). During leaf development, maturation of chloroplasts regulates transition from cell proliferation to cell expansion (14). Chloroplast development is regulated by NTRC and FDX–TRXs (382), leading to changes in the production of oxidation signals such as oxygen, NO, and ROS that will control the transition in leaf development by acting as retrograde signals (Fig. 17) (14). During cell expansion, the transcription factor KUODA1 inhibits the expression of cell wall peroxidases, lowering the levels of apoplastic ROS to restrict cell wall tightening and promote growth (308).
G. Redox regulation in plant physiology: a brief conclusion
There is a balance of oxidation and reduction signals integrating photosynthesis, development, and stress responses, allowing plants to cope with fluctuating changes in their biotic and abiotic environment. This involves intraorganellar cross talk of redox systems as well as redox communication within and between cells. In this context, NTRC acts as an important hub to control the redox balance between oxidation and reduction pathways within the chloroplast of C3 plants, thereby influencing retrograde signals such as ROS to control light acclimation, abiotic and biotic stress responses, and plant development. There is also an emerging role of gaseous signals such as oxygen and NO, which modulate proteasomal degradation of proteins containing an N-terminal Cys via the N-end rule pathway. Although research into hypoxia usually emphasized stress responses, there are also developmental transitions in the oxygen status, which conversely contribute to the regulation of plant development. Further studies are needed to dissect this complex redox signaling network and its integration with other signals related to metabolites, antioxidants, redoxins, phytohormones, and genetic control elements.
VIII. Concluding Remarks and Future Perspectives
The importance and pervasiveness of redox regulation and signaling in plant biology have currently reached a level that probably Bob Buchanan and collaborators could not even vaguely imagine when they first discovered the principles of TRX-mediated regulation of photosynthetic metabolism in plants >50 years ago. This comprehensive review tries to account for the fact that we now know that redox regulation involves not only one but many different types of PTMs of protein Cys, different RMS, a large number of redoxins (TRXs, GRXs, and NTRC), and an enormous number of protein targets belonging to virtually every metabolic or signaling pathway and located in virtually every subcellular compartment, either of photosynthetic and nonphotosynthetic plant cells. As a result, redox homeostasis infiltrates all aspects of plant physiology. The recent development of plant redox biology has provided the material of this comprehensive review, but also opened many questions that need to be answered in the future.
Combination of traditional biochemical approaches and redox proteomics is showing that redox-regulated proteins are organized in complex networks that we are just beginning to understand. A large part of the redox targets that have been identified by proteomics still have to be analyzed to understand the effect, if any, of the redox modification. Moreover, most of our knowledge is derived from nonquantitative studies performed under conditions that favor the redox modification of the proteome. We have little information of the real status of the redox proteome under different physiological or pathological conditions, and even for the best characterized targets, we rarely know which is the relative abundance of the redox-modified proteins in the cell. To this end, quantitative proteomic methods are being developed and will allow determining the stoichiometry and dynamics of multiple redox PTMs in few or even a single cell, under diverse physiological conditions and time scales. The final goal will be to understand, besides the pervasiveness, the relevance of redox regulation and signaling for plant physiology, thereby digging below the surface that we have just started to scratch.
Beyond quantitative proteomics, a field of redox biology that will hopefully grow more and more in the future regards the genetically encoded redox sensors. At the moment, we have powerful tools to determine the dynamics of the glutathione redox state. Other important redox players (NADPH, ascorbate, TRXs, GRXs, and RMS) are still waiting to be assayed in vivo by similar methods. In the absence of accurate information on their localization, dynamics, and redox state, we will hardly get a comprehensive picture of how redox homeostasis influences plant's life. Basic research and genetic engineering will have a fundamental role in the development of new genetically encoded sensors, and the continuous improvement of fluorescent microscopy imaging techniques will likely provide further support to this field in the future.
Structural biology is also promising to contribute significantly to our global understanding of plant redox homeostasis. Once discovered that plants contain tens of different redoxins coexisting in the same subcellular compartment, which, in the same time, also contains hundreds of proteins potentially targeted by different redox PTMs, we still have only a vague idea of which are the principles that govern specificity in all possible interactions. Such principles will be derived from computational analyses of atomic structures of interacting partners and redox-modified proteins. Our available repertoire of complex structures is still limited in number. Solving the tridimensional structure of large complexes of interacting proteins proved difficult in the past because of the intrinsic limitations in obtaining crystals of sufficient quality for X-ray diffraction analysis, but cryoelectron microscopy techniques allow to bypass the crystallization step and permit to unravel large complex structures at atomic resolution.
At the end, what we really would like to know best is how redox regulation and signaling works in the context of plant physiology. Whereas past research into redox regulation was mainly focused on biochemical studies, a recent boost of genetic studies elucidated the organization and biological significance of the redox network in planta. These recent studies have fully confirmed the original model of light-dependent regulation of the CB cycle as mediated by TRXs, but have also opened new fields of research and new levels of understanding.
Results from reverse genetic studies clearly indicate that redox regulatory and signaling pathways contain multiple branches and interconnections. Although reverse genetic approaches are the most powerful way to currently demonstrate the function of a protein in vivo, complex networks may hinder a clear-cut interpretation of the results. This is particularly true when a hub element of the network, like, for example, a TRX, is knocked out. Indeed, TRX knock out mutants are likely to show pleiotropic effects. Moreover, the cross talk between redox signaling pathways requires the combination of different knock out mutations to obtain a reliable interpretation of the emerging phenotypes.
Besides the master redox regulators, also the targets should be investigated in vivo and mutagenic approaches specifically directed to the redox-active Cys are arguably the best way to tackle this problem. Also, this approach has its own limitation, and in some cases, the substitution of a single Cys was found to affect protein stability in vivo besides redox regulation, thereby significantly complicating the emerging picture (204, 476). Nevertheless, this approach seems promising and will possibly be boosted by genome editing techniques that are becoming available. Overall, we firmly believe that the integration of in vitro biochemical data with in vivo physiological evidence will provide the strongest basis to a general understanding of plant redox homeostasis.
Fifty years after germination, thiol-based redox biology in photosynthetic organisms has developed into a deeply rooted well-established plant that grows and expands its foliage in all directions: it is still in its infancy but the future looks bright and full of opportunities.
Footnotes
Acknowledgments
The author M.Z. gratefully acknowledges support of this work from the University of Bologna (Alma Idea Grant). P.G. thanks the Deutsche Forschungsgemeinschaft (SFB-TR 175 B02) for funding. The authors thank Dr. Stephan Wagner, Prof. Bruce Morgan, and Prof. Markus Schwarzländer for providing the original for ![]() .
.