
Abstract
Aims:
Vascular calcification is associated with cardiovascular death in patients with chronic kidney disease (CKD). Peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α) plays an important role in various cardiovascular diseases. However, its role in vascular calcification remains unknown.
Results:
Adenine-induced rat CKD model was used to induce arterial medial calcification. The level of PGC-1α decreased in abdominal aorta of CKD rats. Overexpression of PGC-1α significantly ameliorated calcium deposition in rat abdominal aorta, isolated carotid rings, and cultured vascular smooth muscle cells (VSMCs). Mitochondrial reactive oxygen species (mtROS) increased in calcifying aorta and VSMCs. Upregulation of PGC-1α inhibited, whereas PGC-1α depletion promoted β-glycerophosphate-induced mtROS production and calcium deposition. Moreover, PGC-1α increased superoxide dismutase 1 (SOD1) and SOD2 contents in vivo and in vitro, whereas SOD2 deletion eliminated PGC-1α-mediated mtROS change and promoted calcium deposition. Mechanistically, sirtuin 3 (SIRT3) expression declined in calcifying aorta and VSMCs, while PGC-1α overexpression restored SIRT3 expression. Inhibition of SIRT3 by 3-TYP or siRNA (small interfering RNA) reduced PGC-1α-induced upregulation of SOD1 and SOD2, and abolished the protective effect of PGC-1α on calcification of VSMCs. Importantly, PGC-1α was reduced in calcified femoral arteries in CKD patients. In phosphate-induced human umbilical arterial calcification, upregulation of PGC-1α attenuated calcium nodule formation, while this protective effect was abolished by SIRT3 inhibitor.
Innovation:
We showed for the first time that PGC-1α is an important endogenous regulator against vascular calcification. Induction of PGC-1α could be a potential strategy to treat vascular calcification in CKD patients.
Conclusions:
PGC-1α protected against vascular calcification by SIRT3-mediated mtROS reduction.
Introduction
Vascular calcification, the deposition of calcium phosphate on the medial and intimal layers of vasculature, is an actively regulated process of mineral metabolism, similar to that of bone formation. Emerging evidences suggest that vascular calcification is directly linked to cardiovascular morbidity and mortality in patients with atherosclerosis, chronic kidney disease (CKD), and diabetes mellitus (13, 14, 27, 28, 31). As a key part of the calcification process, vascular smooth muscle cells (VSMCs) undergo phenotypic transition from the contractile phenotype to osteogenic phenotype (8, 31). This transition is accompanied by a loss of contractile markers, including smooth muscle 22α (SM22α) and smooth muscle-α-actin (α-SMA), as well as an increase in chondro/osteogenic genes, including osteopontin (OPN), bone morphogenetic protein 2 (BMP2), runt-related transcription factor 2 (Runx2), sex-determining region Y-box 9 (Sox9), and collagen type II α1 (Col2A1) (39). Numerous pathogenic factors, including oxidative stress, high glucose, and inflammation, contribute to the development of vascular calcification (39).
Innovation
The present study provides the first evidence that peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α) acts as a critical endogenous protective factor against vascular calcification. Overexpression of PGC-1α attenuates vascular calcification in vivo, ex vivo, and in vitro. Mechanistically, PGC-1α-driven antioxidative defense attenuates high phosphate-induced osteogenic transition of vascular smooth muscle cells, and SIRT3 (sirtuin 3) functions as a key mediating factor for both the antioxidation and anticalcification effects of PGC-1α. Our findings reveal a novel mechanism that decreased PGC-1α is involved in chronic kidney disease (CKD)-induced vascular calcification, with consistent data on human vessels together, indicating that the PGC-1α/SIRT3 pathway might represent a promising therapeutic target for the treatment of vascular calcification in CKD patients.
Peroxisome proliferator-activated receptor-γ coactivator-1 (PGC-1) is a family of transcriptional coactivators consisting of PGC-1α-, PGC-1β-, and PGC-1-related coactivators (24). PGC-1α is abundantly expressed in liver, fat, skeletal muscle, heart, and blood vessels. As a metabolic master regulator, PGC-1α plays a key role in mitochondrial biogenesis, fatty acid oxidation, and adipogenesis through recruiting coactivators to regulate downstream target genes in both cell nuclei and mitochondria (24, 33, 36, 52). Recent evidences indicate that PGC-1α may be implicated in the pathogenesis of vascular diseases. PGC-1α induction relieves endothelial inflammation and neointimal formation, while preventing the development of atherosclerotic lesions (17, 19, 21, 37). Therefore, PGC-1α is considered a promising therapeutic target in the treatment of vascular diseases.
Oxidative stress is closely associated with several key pathogenic processes in cardiovascular diseases, including vascular inflammation, atherosclerosis, and calcification. Increasing oxidative stress leads to arterial medial calcification in patients with CKD (30, 32), while elevated reactive oxygen species (ROS) production induces osteogenic transition in VSMCs and promotes hyperphosphatemia-induced vascular calcification in CKD rats (45, 57). Antioxidants, such as resveratrol (RSV) and tempol, suppress the progression of vascular calcification (51, 56). PGC-1α serves as a critical component of the mitochondrial defense system against vascular oxidative stress, by mediating induction of ROS detoxifying enzymes and antioxidative enzymes (19, 42), such as superoxide dismutase 1 (SOD1), SOD2, and catalase (12, 37, 48). Although PGC-1α is reported to induce osteogenesis in osteoblasts (47, 52), little is known on the role of PGC-1α in vascular calcification.
In the present study, we found that PGC-1α protected against vascular calcification in vitro and in vivo in both CKD rats and human specimens. Furthermore, PGC-1α induction suppressed mitochondrial ROS (mtROS) production associated with vascular calcification via promotion of antioxidative enzymes, including SODs. Mechanistically, sirtuin 3 (SIRT3) mediated the protective effect of PGC-1α on mitochondrial oxidative stress and vascular calcification.
Results
The level of PGC-1α decreased in calcified arteries of CKD rats
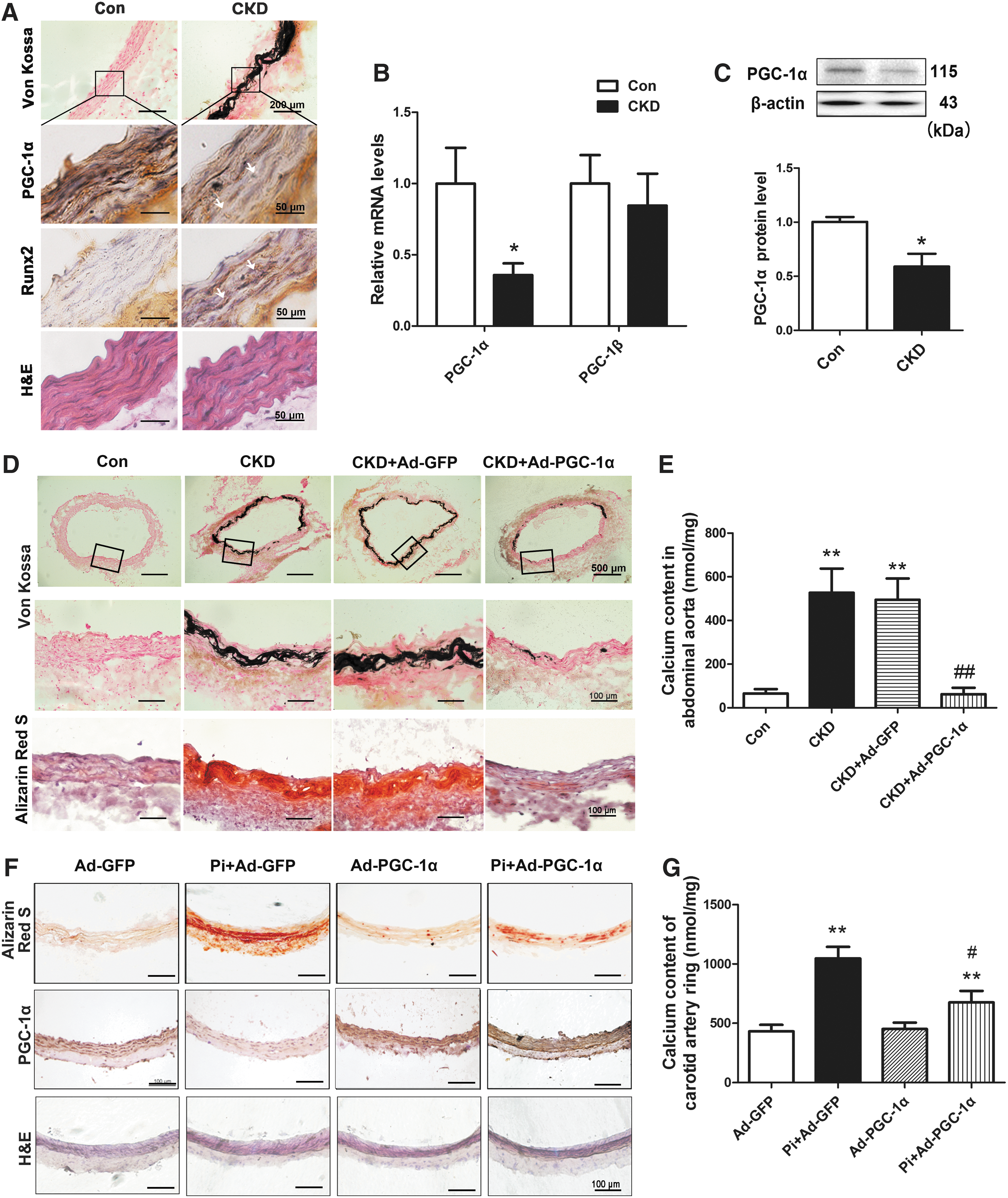
Experimental rat CKD model was induced by adenine diet for 6 weeks to mimic the process of arterial calcification as described previously (57). As shown in Table 1, the adenine-fed rats had severe renal failure with a significant increase in blood urea nitrogen (BUN) and creatinine (Cr) levels compared with age-matched controls (p < 0.01). Serum concentrations of inorganic phosphate (Pi) and magnesium were increased (p < 0.01 and p < 0.05), whereas serum levels of calcium, alanine transaminase, and albumin did not change significantly in CKD rats, consistent with our previous report (59). Histological assessments with von Kossa staining showed extensive linear calcification in the media of abdominal aortas in CKD rats (Fig. 1A), which confirmed the successful induction of vascular calcification.
Weight and Serum Biochemical Parameters in Rats With or Without Adenine Diet
p < 0.05 and ** p < 0.01 versus Control.
Ad-GFP, adenovirus carrying GFP gene; Ad-PGC-1α, adenovirus carrying PGC-1α cDNA; ALB, albumin; ALT, alanine transaminase; BUN, blood urea nitrogen; Ca, calcium; CKD, chronic kidney disease; Cr, creatinine; Mg, magnesium; PGC-1α, peroxisome proliferator-activated receptor-γ coactivator-1α; Pi, inorganic phosphate.
To investigate whether PGC-1α was involved in vascular calcification, immunohistochemical staining was used to assess PGC-1α level in calcified abdominal aortas of CKD rats. In control and CKD rats, calcified areas showed lower expression of PGC-1α and higher levels of Runx2 (as arrows pointed in Fig. 1A). Moreover, the expressions of PGC-1α mRNA and protein in abdominal aortas of CKD rats were 64.2% (p < 0.05) and 41.0% (p < 0.05) lower than those in controls, respectively, whereas the mRNA expression of PGC-1β remained unchanged (Fig. 1B, C, and Supplementary Fig. S1). These results suggest that PGC-1α might be related to aortic medial calcification in the progression of CKD.
Overexpression of PGC-1α suppresses vascular calcification in CKD rats
To explore the role of PGC-1α in vascular calcification, we established adenovirus containing either PGC-1α with fused GFP coding sequence (Ad-PGC-1α) or GFP coding sequence (Ad-GFP). The adenovirus was delivered periadventitially as previously described (59). Expression of PGC-1α mRNA significantly increased at 1 and 4 weeks in rat abdominal aortas (Supplementary Fig. S2A). The protein expression of PGC-1α increased by 1.9-fold 1 week after transfection and increased by 0.28-fold 6 weeks after transfection (Supplementary Fig. S2B, C), suggesting that the PGC-1α adenovirus was efficiently transfected into rat abdominal aortas.
Next, we observed the effect of Ad-PGC-1α transfection on vascular calcification. Calcified nodule formation (Fig. 1D) and calcium deposition (Fig. 1E) significantly decreased with Ad-PGC-1α delivery in abdominal aortas of CKD rats, but not with control Ad-GFP. Moreover, Ad-PGC-1α delivery did not affect calcium content in non-CKD rats (Supplementary Fig. S2D). In addition, blood biochemical parameters were not affected by either Ad-PGC-1α or Ad-GFP transfection compared with age-matched CKD rats (Table 1), suggesting that decreased vascular calcification induced by Ad-PGC-1α delivery was not due to alleviation of renal function injury.
Overexpression of PGC-1α suppresses vascular calcification in high-Pi-cultured carotid arterial rings ex vivo
The studies in vivo and in vitro have shown that high Pi is a crucial inducer of vascular calcification (15, 38, 40). To clarify the direct effect of PGC-1α on vascular calcification, Ad-PGC-1α-transfected carotid arterial rings were cultured with or without high-Pi (3.8 mM, PO4 3−) for 6 days. High-Pi treatment markedly induced calcification and decreased PGC-1α expression in carotid arterial rings (Fig. 1F). By contrast, Ad-PGC-1α transfection attenuated calcium nodule formation (Fig. 1F) as well as calcium deposition in carotid arterial rings (Fig. 1G). Transfection of Ad-PGC-1α alone in low-Pi conditions did not affect calcium content in carotid arteries. These results indicate that overexpression of PGC-1α inhibits vascular calcification induced by high-Pi in carotid arterial rings ex vivo.
Overexpression of PGC-1α inhibits β-glycerophosphate-induced VSMC calcification in vitro
We further explored the effect of PGC-1α on calcium deposition in VSMCs in vitro. ZLN005, a selective PGC-1α transcriptional activator (55), increased the expression of endogenous PGC-1α in VSMCs (Fig. 2A and Supplementary Fig. S3). As shown in Figure 2B and C, β-glycerophosphate (βGP) treatment for 12 and 15 days induced calcium nodule formation and increased calcium content in VSMCs. ZLN005 at 5, 10, and 20 μM significantly inhibited βGP-induced calcification. Besides, RSV treatment (10 μM), which is reported to increase PGC-1α activation and expression in adipocytes and VSMCs (22, 44, 46), increased PGC-1α expression and alleviated calcified nodule formation induced by βGP (Fig. 2A, B, and Supplementary Fig. S3). While the fluorescence mainly localized in the cytoplasm in control Ad-GFP-transfected cells, PGC-1α was localized in the nuclei in Ad-PGC-1α-treated cells (Supplementary Fig. S4A). The expression of PGC-1α protein was increased by 2.8-fold in Ad-PGC-1α-transfected VSMCs (Supplementary Fig. S4B). Overexpression of PGC-1α by Ad-PGC-1α transfection markedly inhibited calcium nodule formation and decreased calcium content (Fig. 2D, E).
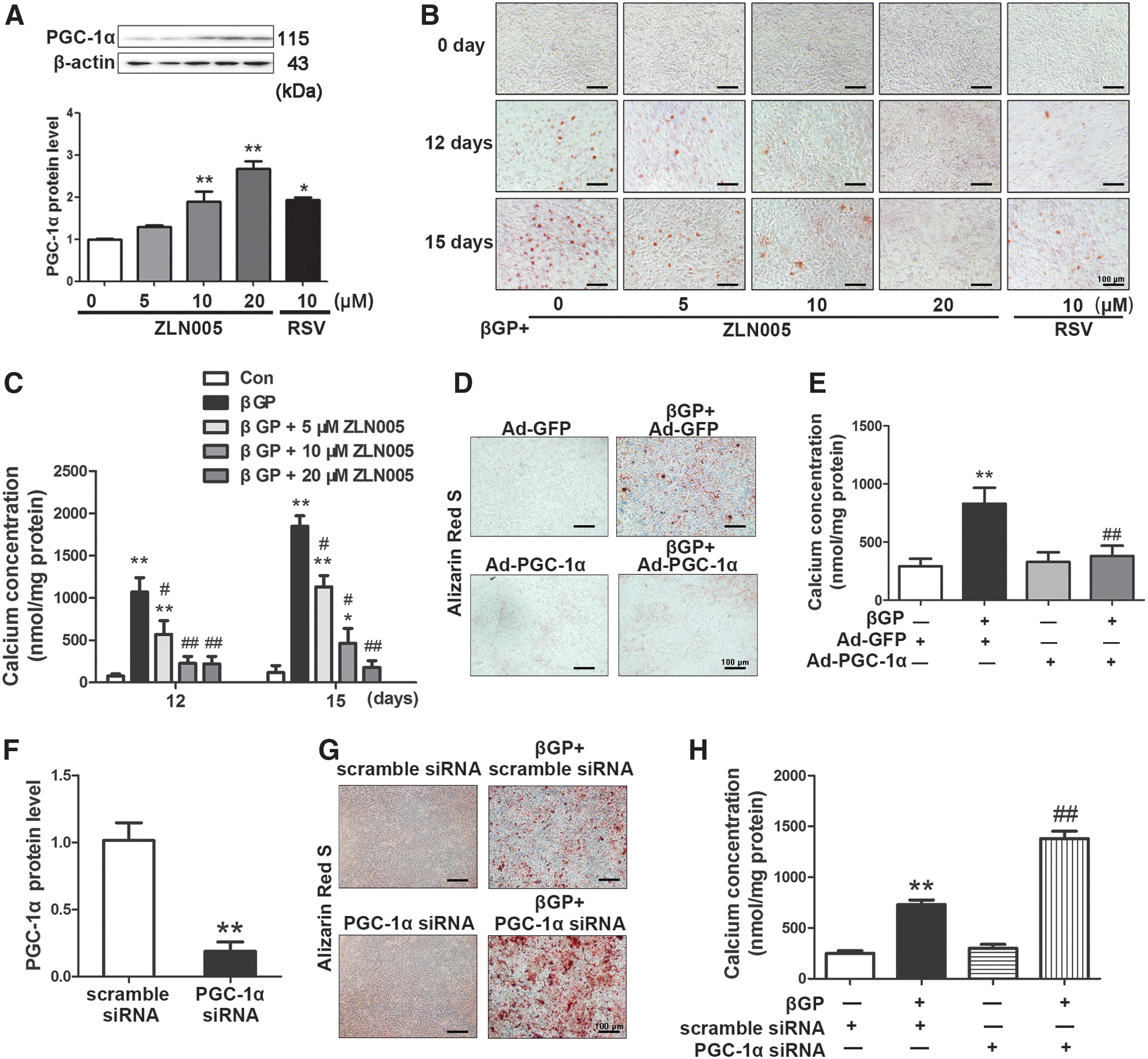
Knockdown of PGC-1α promotes βGP-induced VSMC calcification in vitro
To confirm the direct effect of PGC-1α on vascular calcification, we knocked down PGC-1α expression with smartpool small interfering RNA (siRNA) (2). Efficient knockdown of PGC-1α by 81.3% was confirmed by Western blot analysis (Fig. 2F). PGC-1α depletion enhanced βGP-triggered calcium nodule formation and increased calcium content in VSMCs, whereas PGC-1α knockdown alone did not induce calcium deposition (Fig. 2G, H). These results indicate that endogenous PGC-1α exerts a protective effect against calcium deposition.
Overexpression of PGC-1α inhibits CKD/βGP-induced VSMC osteogenic phenotypic transition in vivo and in vitro
The transition from contractile to osteogenic phenotype of VSMCs plays a key role in vascular calcification (41). Hence, we evaluated the role of PGC-1α in the phenotypic transition of VSMCs both in vivo and in vitro. As shown in Figure 3A–C (Supplementary Fig. S5A), the expressions of contractile markers (α-SMA and SM22α) significantly decreased, whereas the levels of osteogenic markers (Runx2 and OPN), as well as the levels of chondrogenic markers (Col2A1 and Sox9), markedly increased in the abdominal aortas of CKD rats. Overexpression of PGC-1α in abdominal aortas of CKD rats significantly reversed the changes of contractile and chondro/osteogenic markers. Ad-GFP alone did not affect the expression of contractile and chondro/osteogenic markers in CKD rats. These results indicate that overexpression of PGC-1α inhibits chondro/osteogenic transition of VSMCs in CKD rats.
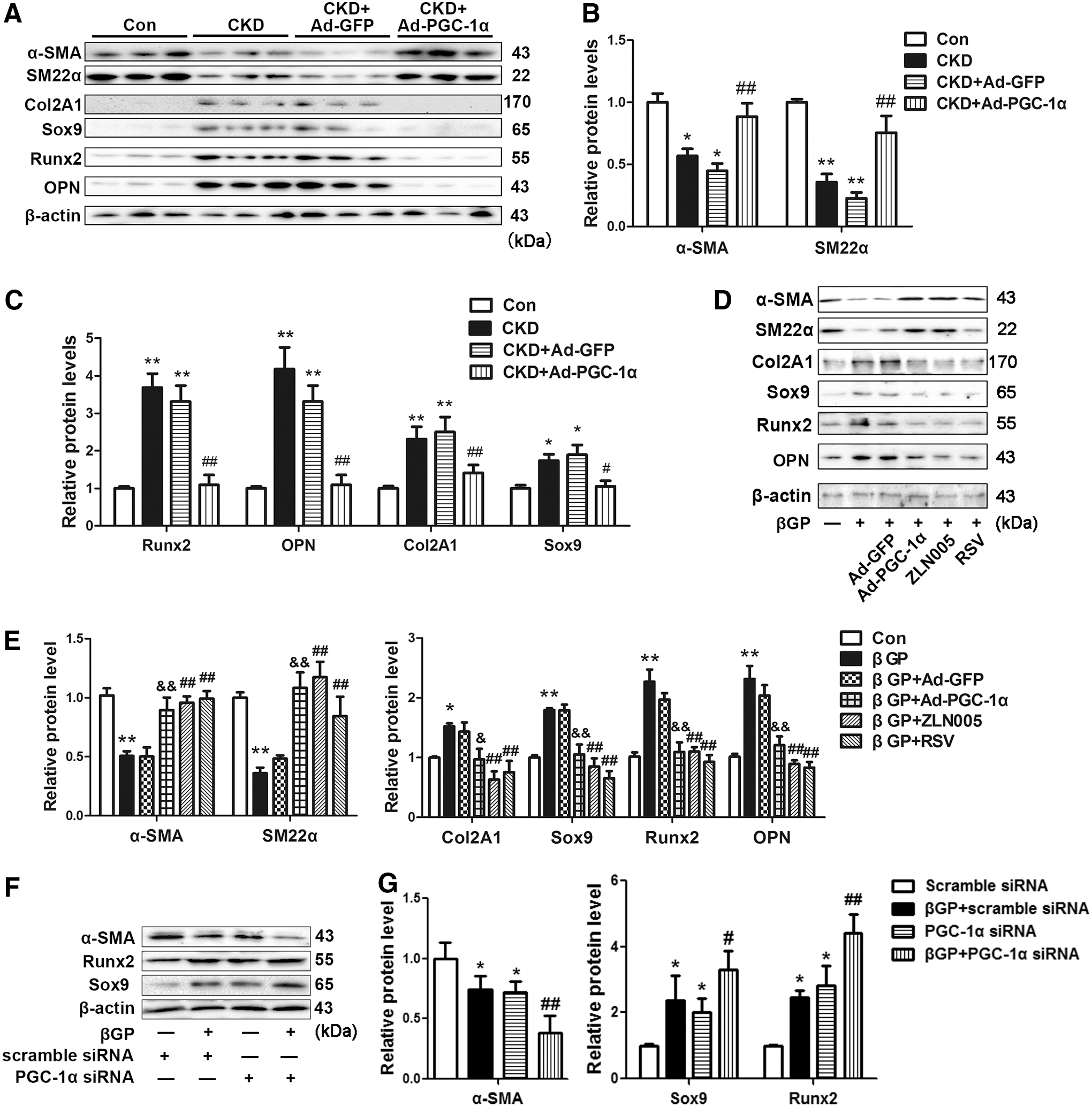
We further evaluated the direct effect of PGC-1α overexpression on phenotypic transition in vitro. As shown in Figure 3D and E, the expressions of α-SMA and SM22α significantly were decreased, whereas the levels of Col2A1, Sox9, Runx2, and OPN were markedly upregulated after βGP treatment for 48 h. Ad-PGC-1α transfection restored the βGP-induced changes in contractile and chondro/osteogenic marker expression. Ad-GFP transfection did not alter the expression levels of these markers. In parallel, both ZLN005 (10 μM) and RSV (10 μM) decreased the expression of chondro/osteogenic marker genes, while enhancing the levels of contractile markers that were reduced by βGP (Fig. 3D, E, and Supplementary Fig. S5B). Moreover, knockdown of PGC-1α enhanced βGP-induced phenotypic transition by further reducing α-SMA expression and increasing Runx2 and Sox9 levels (Fig. 3F, G, and Supplementary Fig. S5C). These results indicate that PGC-1α alleviates vascular calcification through inhibiting chondro/osteogenic transition in VSMCs.
Overexpression of PGC-1α inhibits vascular oxidative stress in CKD rats
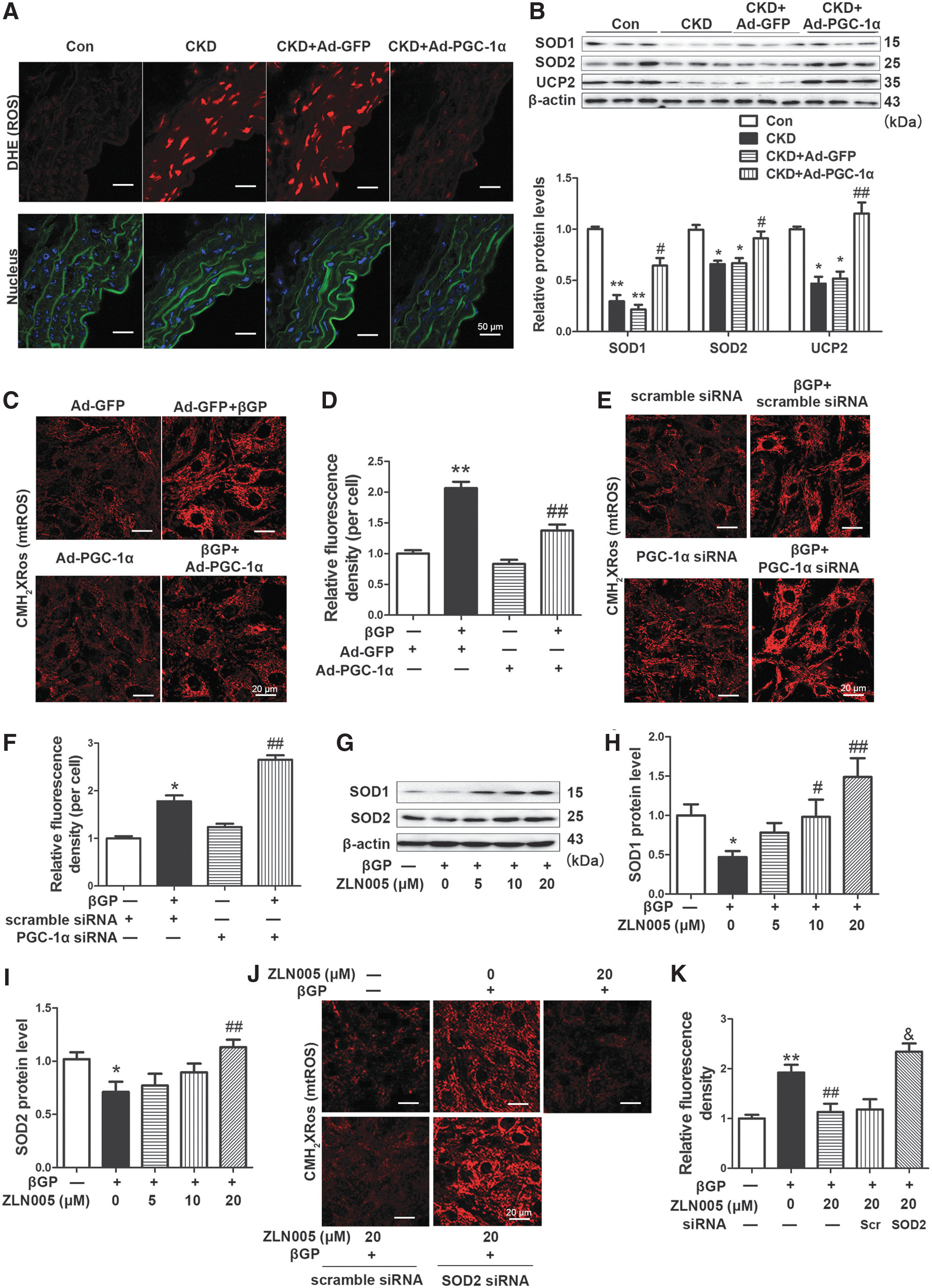
Oxidative stress plays a key role in vascular calcification in CKD rats (57). As shown in Figure 4A and B (Supplementary Fig. S5D), the level of ROS (red) was markedly elevated as assessed by dihydroethidium (DHE, 1 μM) staining, and the protein expressions of antioxidative enzymes, including SOD1, SOD2, and uncoupled protein 2 (UCP2), significantly decreased in calcified abdominal aortas of CKD rats. Ad-PGC-1α transfection markedly reduced ROS level and recovered the expressions of SOD1, SOD2, and UCP2 in the aortas of CKD rats.
PGC-1α decreases the level of mtROS through upregulating SOD expression in VSMCs
MtROS is the main source of ROS production induced by Pi and is essential for high-Pi-triggered VSMC phenotype transition and calcium deposition (57). Here, we used the MitoTracker Red CMH2XRos probe (200 nM) to explore the role of PGC-1α in mtROS production. βGP treatment for 30 min significantly elevated mtROS in VSMCs, but this increase was inhibited by Ad-PGC-1α transfection (Fig. 4C, D), which was also detected by MitoSOX (1 μM) (Supplementary Fig. S6A). On the contrary, knockdown of PGC-1α by siRNA enhanced βGP–induced mtROS production, whereas it did not change mtROS level without βGP treatment in VSMCs (Fig. 4E, F).
To explore the mechanism by which PGC-1α reduced mtROS level, we measured the expressions of antioxidative enzymes. The levels of SOD1 and SOD2 significantly decreased by 54.0% (p < 0.05) and 28.9% (p < 0.05) after βGP treatment for 48 h, whereas this suppression was abrogated by preincubation with ZLN005 (Fig. 4G–I and Supplementary Fig. S6B). Furthermore, siRNA knockdown of SOD2 by 81.2% further promoted βGP-triggered calcium deposition (Supplementary Fig. S7). ZLN005 (20 μM) incubation for 24 h significantly inhibited βGP-induced mtROS increase, and this inhibitory effect of ZLN005 on mtROS was diminished by SOD2 siRNA (Fig. 4J, K). Besides, DHE staining evaluated by high-performance liquid chromatography (HPLC) is a more reliable method for ROS measurement. We detected alteration of DHE, 2-E+OH, and E+ on VSMCs by HPLC under the same conditions as reported by Fernandes et al. (11). As shown in Supplementary Figure S7, βGP stimuli notably increased 2-E+OH and E+ level and decreased DHE level, whereas overexpression of PGC-1α recovered the alterations of DHE and its oxidative products. These data suggest that PGC-1α reduces mtROS level through upregulating SOD2 expression in VSMCs.
Overexpression of PGC-1α recovers reduced SIRT3 expression in vivo and in vitro
SIRT3 functions as a downstream target gene of PGC-1α and mediates the inhibitory effect of PGC-1α on ROS production in C2C12 myotubes (20). Here, we found that the levels of SIRT3 mRNA and protein reduced by 57.5% (p < 0.05) and 64.4% (p < 0.01) in abdominal aortas of CKD rats compared with those in control rats (Fig. 5A, B, and Supplementary Fig. S8A). The levels of SIRT3 protein decreased by 38.5% (p < 0.05), 53.1% (p < 0.05), and 76.5% (p < 0.01) after treatment with βGP for 9, 12, and 15 days in VSMCs, respectively (Fig. 5C, D, and Supplementary Fig. S8B). Ad-PGC-1α transfection significantly reversed SIRT3 levels in calcified abdominal aortas of CKD rats, but did not change sirtuin 1 (SIRT1) level (Fig. 5E and Supplementary Fig. S8C). In cultured VSMCs, βGP-induced inhibition of SIRT3 expression was restored by ZLN005 in a dose-dependent manner (Fig. 5F, G, and Supplementary Fig. S8D). ZLN005 alone increased SIRT3 expression in VSMCs (Supplementary Fig. S9A). These data suggest that SIRT3 may be involved in the protective effect of PGC-1α against vascular calcification.
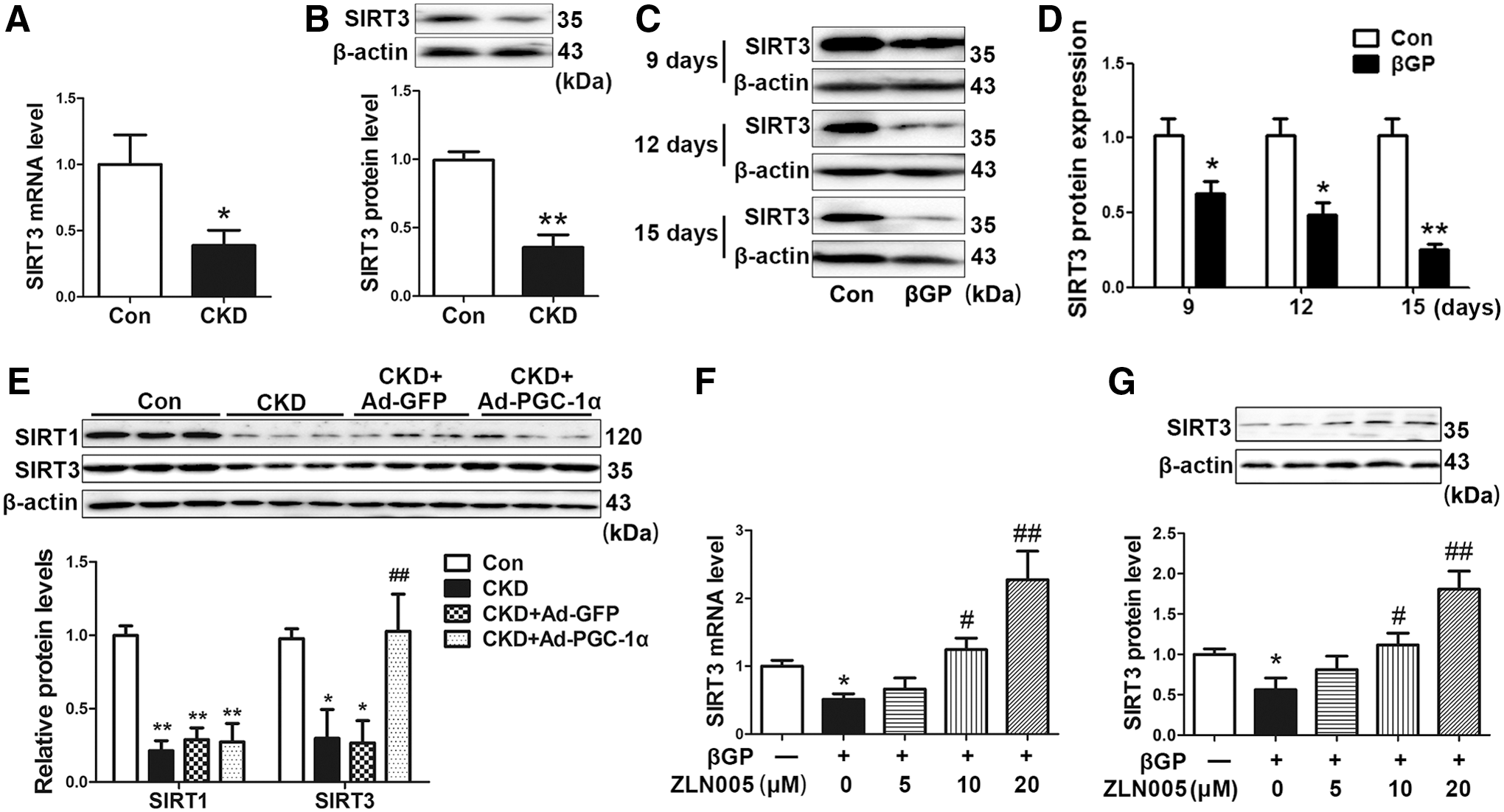
SIRT3 is required for the inhibitory effect of PGC-1α on βGP-induced calcium deposition
To clarify the role of SIRT3 in PGC-1α-inhibited vascular calcification, we knocked down the expression of SIRT3 protein via siRNA in VSMCs (Fig. 6A and Supplementary Fig. S10A). 7C, 3-(1H-1, 2, 3-triazol-4-yl) pyridine (3-TYP, 50 μM), a selective inhibitor of SIRT3, and SIRT3 siRNA abolished the inhibition effect of Ad-PGC-1α transfection on calcium nodule formation and calcium content in VSMCs (Fig. 6B, C). Knockdown of SIRT3 diminished the responses of α-SMA and Runx2 expression induced by Ad-PGC-1α transfection in βGP-treated cells (Fig. 6D–F and Supplementary Fig. S10B). Moreover, SIRT3 inhibition by either 3-TYP or SIRT3 siRNA reversed the reduction of mtROS level induced by Ad-PGC-1α (Fig. 6G, H), while increased expressions of SOD1 and SOD2 by Ad-PGC-1α were abolished by SIRT3 siRNA in βGP-treated cells (Fig. 6I and Supplementary Fig. S10C). These results suggest that SIRT3 is required for the inhibitory effect of PGC-1α on βGP-induced calcium deposition, VSMC phenotypic transition, and mtROS level.

Expression of PGC-1α is decreased in calcified femoral arteries of CKD patients
Femoral arteries were obtained from eight patients (ages 66–88 years, five males) who died of CKD and six individuals (ages 55–90 years, two males) who died of cancer without renal dysfunction (as control). Among eight CKD individuals, vascular calcification was observed from six patients by Alizarin red staining (Fig. 7A). Calcium nodules were mainly in the medial artery, and immunohistochemical staining showed that the levels of PGC-1α were markedly lower compared with control individuals (Fig. 7A). Meanwhile, the expression levels of SIRT3 in femoral arteries of CKD patients were significantly lower than control individuals (Supplementary Fig. S9B, C).
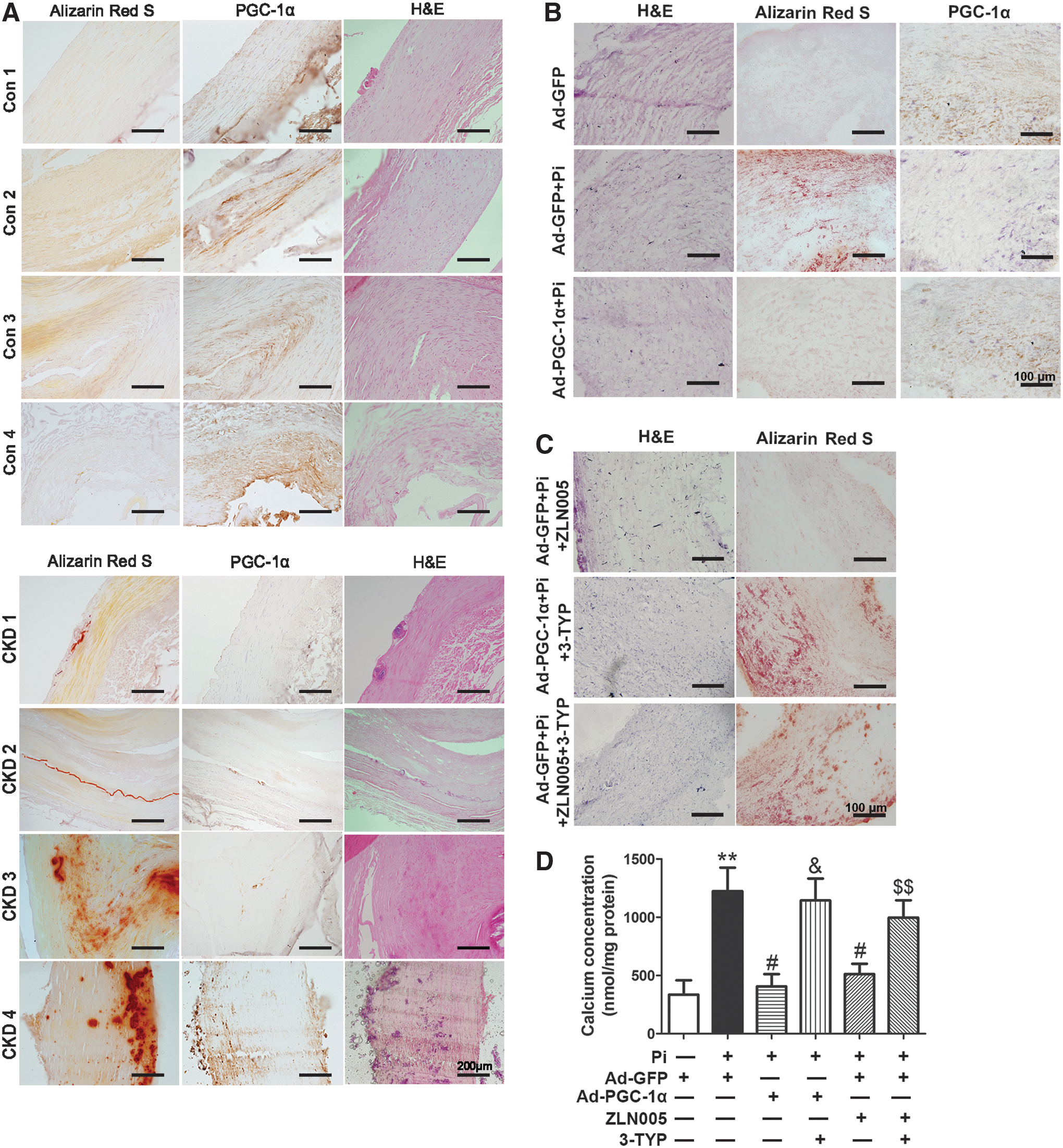
Activation of PGC-1α/SIRT3 pathway suppresses high-Pi-induced vascular calcification in cultured human umbilical arterial rings ex vivo
To confirm the role of PGC-1α/SIRT3 pathway in regulating vascular calcification in human blood vessels, we incubated the human umbilical arterial rings with high-Pi for 6 days. High-Pi treatment significantly induced calcium nodule formation and decreased the expression of PGC-1α. These responses were reversed by Ad-PGC-1α transfection (Fig. 7B). Moreover, ZLN005 treatment eliminated Pi-induced calcium nodule formation, and the inhibitory effect of PGC-1α induction on calcium nodule formation was abolished by 3-TYP pretreatment (Fig. 7C). Consistent with Alizarin red staining, Ad-PGC-1α or ZLN005 inhibited high-Pi-induced calcium deposition in human umbilical arterial rings, whereas these responses were abolished by 3-TYP pretreatment (Fig. 7D).
Discussion
In the present study, we found that PGC-1α level was lower in calcified arteries of both CKD patients and rats, while overexpression of PGC-1α suppressed vascular calcification in CKD rats, high-Pi-cultured carotid rings, and βGP-treated VSMCs. We further identified that reduction of mtROS was involved in PGC-1α-mediated inhibition of vascular calcification in vivo and in vitro. Moreover, SIRT3 was required for the protective effects of PGC-1α on calcification, chondro/osteogenic phenotypic transition, and mtROS production in VSMCs. In high-Pi-treated human umbilical arterial rings, activation of PGC-1α/SIRT3 pathway was shown to suppress vascular calcification. Taken together, these results reveal a novel role of PGC-1α as a negative regulator of CKD-induced vascular calcification by activation of SIRT3 to mediate antioxidative mitochondrial defense.
PGC-1α activates numerous mitochondria-associated genes to regulate energy metabolism and exerts pivotal biological functions in various tissues (17, 33). Overexpression of PGC-1α relieves endothelial apoptosis, VSMC proliferation, and vascular senescence (21, 37, 48, 50). PGC-1α depletion promotes angiotensin II-induced vascular inflammation and dysfunction, and aggravates atherosclerotic lesion in ApoE−/− mice fed with high-fat diet (21, 49). These studies suggest that PGC-1α functions as a protective agent against vascular damages. Furthermore, PGC-1α gene level declined in the peripheral blood mononuclear cells, and it is identified as one of the most differentially expressed genes associated with fatty acid oxidation in CKD patients (6, 54), thereby suggesting that PGC-1α may be involved in the progression of CKD. Our findings provided direct evidence that PGC-1α was downregulated in calcified arteries of CKD patients and rats, and overexpression of PGC-1α by periadventitial adenoviral delivery significantly ameliorated CKD/Pi-induced vascular calcification in vivo and ex vivo.
Moreover, upregulation of PGC-1α by either pharmacological agents or adenoviral transfection inhibited βGP-induced calcium deposition in VSMCs. Consistently, knockdown of PGC-1α aggravated βGP-induced calcium deposition in VSMCs. Our study demonstrates that PGC-1α acts as an endogenous protective factor against vascular deposition. Decreased PGC-1α plays an important role in the pathogenesis of vascular calcification in CKD. However, knockdown of PGC-1α alone did not affect calcification in healthy conditions, indicating that PGC-1α deficiency is not the only mechanism of calcification process. Other mechanisms are involved in the progression of vascular calcification, such as apoptosis and matrix degradation (38). Similarly, without βGP, PGC-1α deficiency alone did not induce cell apoptosis or matrix degradation or other calcification-dependent processes on VSMCs. This might be the reason why the depletion of PGC-1α alone plays a procalcification factor instead of an inducer of vascular calcification.
VSMC transition from a contractile to a chondro/osteogenic phenotype is closely associated with vascular deposition, and plays a crucial role in promoting Pi-induced vascular calcification (38, 41). Here, we found that PGC-1α overexpression reversed the decrease in contractile markers and the increase in chondro/osteogenic markers in abdominal aortas of CKD rats and βGP-treated VSMCs. Furthermore, knockdown of PGC-1α promoted the expressions of Runx2 and Sox9, and reduced α-SMA protein under Pi stimulation. These results indicate that PGC-1α ameliorates vascular calcification by inhibiting chondro/osteogenic transition in VSMCs.
Oxidative stress is one of the major causes of vascular calcification in CKD patients and animals (32). Particularly, mtROS are thought to be the major source of Pi-induced ROS production and act as a signaling molecule to promote the transcription of Runx2 (57). Previous studies showed that PGC-1α overexpression reduces both basal and H2O2-stimulated mtROS production (37), whereas PGC-1α deletion increases mtROS generation in VSMCs (21). Furthermore, PGC-1α upregulates the expressions of SOD1, SOD2, catalase, glutathione peroxidase, and UCP2 protein in human aortic endothelial cells, and upregulates SOD1, SOD2, and hemeoxyenase-1 expressions in VSMCs (25, 37, 48). However, the effect of PGC-1α on Pi-induced oxidative stress and mtROS production remains unknown. Here, we found that overexpression of PGC-1α decreased ROS level in abdominal aortas of CKD rats. Both Ad-PGC-1α delivery and ZLN005 treatment decreased, whereas knockdown of PGC-1α promoted mtROS level in βGP-treated VSMCs. Furthermore, upregulation of PGC-1α restored the protein levels of SOD1, SOD2, and UCP2 in abdominal aortas of CKD rats and in VSMCs, whereas knockdown of SOD2 eliminated the effect of ZLN005. These data suggest that PGC-1α reduces the mtROS level through promoting the expression of antioxidative enzymes.
Although Pi-induced ROS production takes place in the mitochondria, several studies focus on the role of nicotinamide adenine dinucleotide phosphate (NADPH) oxidases in vascular calcification. Increased phagocytic NADPH oxidase activity associates with coronary arterial calcification in asymptomatic men (5). NADPH oxidase subunits' p22phox and p47phox expressions are increased in calcified VSMCs of CKD rats, and expressions of NADPH oxidase subunits Nox2, Nox4, p22phox, and protein disulfide isomerase are increased around human calcifying foci (1, 23). While we cannot rule out that NADPH oxidase may be involved in the antioxidative defense mediated by PGC-1α during vascular calcification, our findings strongly supported that PGC-1α activates multiple antioxidative enzymes, including SOD2, to reduce mtROS production, and these enzymes are essential in the prevention of vascular decalcification.
SIRT3, a histone deacetylase rich in mitochondria, is known to protect against cardiovascular diseases. SIRT3-deficient mice develop spontaneous pulmonary hypertension and are more susceptible to transverse aortic constriction-induced left ventricular hypertrophy and myocardial ischemic injury (34, 35). However, the effect of SIRT3 on Pi-induced vascular calcification has not been explored yet. Recent studies show that SIRT3 is required for PGC-1α-regulated mitochondrial biogenesis, adipocyte thermogenesis, and neuroprotection (20). Furthermore, knockdown of SIRT3 suppresses PGC-1α-induced expression of antioxidative genes such as SOD1 and SOD2 in C2C12 skeletal muscle cells (20). Here, we found that SIRT3 expressions were reduced in βGP-treated VSMCs and the arteries of CKD rats and patients. Overexpression of PGC-1α restored the decreased SIRT3 levels in calcified abdominal aortas of CKD rats and calcified VSMCs. Inhibition of SIRT3 by either 3-TYP or siRNA abolished the inhibitory effect of PGC-1α on mtROS production, osteogenic phenotypic transition, and calcium deposition in βGP-treated VSMCs. These results indicate that SIRT3 is required for the antioxidative and anticalcifying effects of PGC-1α.
We further explored the protective effect of PGC-1α on human vascular tissues. Consistent with the results obtained from the rat CKD model and VSMCs, PGC-1α expression was lower while calcium deposition was higher in high-Pi-cultured human umbilical arteries. PGC-1α overexpression diminished calcium deposition, and this effect was reversed by 3-TYP pretreatment. These results again confirm the protective role of PGC-1α in vascular calcification.
Recent studies showed several pathways and reagents could effectively induce PGC-1α. RSV, a polyphenolic compound with antidiabetic properties, increases PGC-1α mRNA expression and induces mitochondrial biogenesis in adipose tissue (4). Adiponectin upregulates PGC-1α expression and protects against glutamate-induced excitotoxicity in hippocampal neurons (53). RSV or adiponectin inhibits vascular calcification by diet supplement or intravenous injection (26, 56). GW4064, a widely used synthetic agonist for the nuclear bile acid receptor, enhances the promoter receptor activity, mRNA, and protein expression of PGC-1α, and this induction of PGC-1α by GW4064 concomitantly increases mitochondrial function (9). C1q/tumor necrosis factor-related protein-3, an adipokine with modulation effects on metabolism and inflammation, upregulates PGC-1α expression and elevates energy production in VSMCs (10). Besides, α-lipoic acid activates PGC-1α transcription and expression in an AMPK-dependent manner, and exerts a protective effect against vascular calcification (3, 18, 29). Accordingly, considering the beneficial effects of PGC-1α activation on the vascular system from our study, future effort will be made to identify regulators of PGC-1α expression and activity in vascular tissues, thus providing a potential therapeutic strategy for vascular calcification.
In summary, we provided the first evidence that PGC-1α serves as an endogenous protective regulator against CKD/Pi-induced vascular calcification. As the schematic hypothesis shows in Figure 8, PGC-1α attenuates vascular calcification through upregulation of antioxidant enzymes, in turn scavenging for mtROS. Mechanistically, SIRT3 mediates the inhibitory effects of PGC-1α on mtROS production and calcium deposition in VSMCs. Therefore, the PGC-1α/SIRT3 pathway may represent a promising therapeutic target for treatment of vascular calcification.

Materials and Methods
Materials
Adenine, βGP, Alizarin red, and ZLN005 were purchased from Sigma-Aldrich (A8626, G5422, A5533, and SML0802, St. Louis, MO). Primary antibody against PGC-1α was obtained from Abcam (ab54481; Cambridge, United Kingdom) and antibodies against SM22α, α-SMA, and OPN were from Proteintech Group (10493-1-AP, 55135-1-AP, and 18933-1-AP; Rosemont, IL). Antibodies against SIRT1, SIRT3, Runx2, BMP2, Sox9, Col2A1, SOD1, SOD2, and UCP2 were from Bioworld Technology (BS6494, BS7772, BS8734, BS1264, BS1597, BS1071, BS6057, BS6734, and BS2917; Minneapolis, MN) and antibody for β-actin was from Easy Bio, Inc. (South Korea). Anti-mouse, anti-goat, and anti-rabbit IgG secondary antibodies were from Jackson ImmunoResearch Laboratories (West Grove, PA). 3-TYP was from Combi-Blocks (San Diego, CA). Von Kossa and Alizarin red staining kits were from Abcam (ab150687 and ab142980).
Ethical approval and patient consent
All experimental procedures were approved by the Ethics Committee of Animal Research, Peking University Health Science Center, and complied with the Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85-23, revised 1996). Human femoral arterial samples were obtained from donated bodies of Beijing Red Cross Volunteer Body Donation Site. Human umbilical artery samples were obtained from healthy donors during parturition in a sterile manner. The research protocol for human tissue was approved by the Medical Ethics Committee of Peking University Health Science Center, which complied with the Declaration of Helsinki principles. All participants had signed an informed consent document before donation and tissue collection.
Adenovirus preparation and in vivo transfection
Adenovirus containing PGC-1α cDNA (PubMed NM_013261) with fused GFP gene was constructed and amplified according to the manufacturer's protocol (BD Biosciences, Clontech, CA). Adenovirus carrying the GFP gene was used as a negative control. For in vivo studies, a single exposure of 1 × 109 pfu of Ad-PGC-1α or Ad-GFP dissolved in 30% pluronic gel solution was periadventitially delivered to rat abdominal or common carotid arteries.
Rat CKD model
All animal research was reported in accordance with the ARRIVE guidelines. Eight-week-old male Sprague-Dawley rats were randomly divided into four groups: control, CKD, CKD+Ad-GFP, and CKD+Ad-PGC-1α (n = 6–8/group). Ad-GFP or Ad-PGC-1α was periadventitially delivered to the rat abdominal arteries in adenovirus-transfected groups under anesthetization (ketamine/xylazine, 80/10 mg/kg, intraperitoneal) (59). Then, the control rats were fed with standard CE-2 chow (containing 1.2% calcium and 0.6% phosphorus), while CKD rats were fed with CE-2 chow containing 0.75% adenine, 1.0% phosphorus, and 2.5% calcium as described previously (43, 58). After 6 weeks, the rats were euthanized and blood was collected to measure BUN, Cr, calcium, and Pi by an autoanalyzer (Hitachi7180; Hitachi, Tokyo, Japan). The sections from each abdominal aorta (6 μm) were processed for von Kossa, Alizarin red staining, and fluorescence dyeing.
Arterial ring calcification model
Arterial ring organ calcification was induced as described previously (7). Briefly, rat common carotid arteries were removed 3 days after adenovirus infection. Medial tissue was separated from segments of carotid arteries, cut into 1-mm rings, and placed in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum (FBS) with or without high-Pi (3.8 mM PO4 3−) at 37°C in 5% CO2 incubator. After 6 days, the arterial rings were prepared for quantitative analysis of calcium content. Besides, human umbilical artery samples were obtained from healthy donors during parturition in a sterile manner. The sections from each rat carotid artery or human umbilical arteries (6 μm) were processed for Alizarin red staining and immunohistochemical analysis.
Cell culture and cell calcification model
Rat VSMCs were obtained by an explant method as previously described (16). After removing the adventitia and endothelium, the vascular media were cut into pieces (1–2 mm2) and placed in a 10 cm culture dish and cultured for 2 weeks in DMEM containing 4.5 g/L of glucose supplemented with 20% FBS, 10 mM sodium pyruvate, 100 U/mL of penicillin, and 100 μg/mL of streptomycin at 37°C in a humidified atmosphere containing 5% CO2. Cells that had migrated from the explants were collected and maintained in 10% DMEM. The cells up to passages 3–8 were used for experiments. Confluent VSMCs were incubated in medium containing 2.5 mM Ca2+ (0.7 mM CaCl2 was added into DMEM containing 1.8 mM CaCl2) and 10 mM βGP calcification medium for 9–15 days. The medium and reagents were replenished every 3 days (16). For drug treatment experiments, 0–20 μM ZLN005 or 50 μM 3-TYP was added into the incubation solution for 30 min before Pi or βGP treatment.
Measurement of calcium content
Arteries were dried, weighed, and extracted with 65% (w/w) HNO3 for 24 h at 180°C to dissolve the minerals, and then redissolved in LaCl3 (27 μM) and KCl (27 nM). VSMCs were treated with 0.6 M HCl overnight at 4°C. After removing the HCl supernatant, the remaining cell layers were then dissolved in 0.1 M NaOH and 0.1% sodium dodecyl sulfate (SDS) for protein concentration analysis. Samples were measured by spectrometry at 600 nm using the QuantiChrom Calcium Assay Kit and were normalized to tissue weight or protein content.
Immunohistochemical analysis
Arteries were fixed in 4% formaldehyde and embedded in Tissue-Tek O.C.T. compound to be frozen in liquid nitrogen. Frozen artery sections (6 μm) were stained with an antibody for PGC-1α overnight at 4°C and then with the horseradish peroxidase-conjugated secondary antibody for 2 h at 37°C followed by 3,3-diaminobenzidine. Nuclei were stained with hematoxylin. Negative controls that omitted the primary antibody were routinely performed. Images were captured by EVOS FL Auto microscope (AMAFD1000; Life Tech, WA).
Western blot analysis
Rat arteries or cultured VSMCs were homogenized with lysis buffer (containing 50 mM Tris-HCl, 0.1% sodium deoxycholate, 1% Triton X-100, 5 mM EDTA, 5 mM EGTA, 150 mM NaCl, 40 mM NaF, 2.175 mM sodium orthovanadate, 0.1% SDS, 0.1% aprotinin, and 1 mM phenylmethylsulfonyl fluoride, pH 7.2). The homogenates were centrifuged at 1000 g for 10 min at 4°C, and the protein concentrations of the supernatants were measured by the Bradford method. Equal amounts of proteins (40 μg) were separated on a 9% SDS-polyacrylamide gel electrophoresis (PAGE) and transferred to polyvinylidene difluoride membrane. The membranes were blocked with 5% nonfat milk for 1 h at room temperature and then incubated with primary antibodies and horseradish peroxidase-conjugated secondary antibodies. The immunoreactive bands were visualized by use of an enhanced chemiluminescence kit (Amersham Biosciences, Inc., Piscataway, NJ). The densities of bands were quantified by use of the LEICA550IW image analysis system (Leica, Mannheim, Germany).
Quantitative real-time reverse transcription–polymerase chain reaction
Total RNA was isolated from VSMCs using the TRIzol reagent (Invitrogen Co., Carlsbad, CA) followed by cDNA synthesis with the First-Strand cDNA Synthesis kit (Fermentas, Burlington, ON, Canada). Quantitative real-time reverse transcription-polymerase chain reaction (qRT-PCR) was performed using the forward and reverse primers of sequences (rat): PGC-1α (F) GGAAAATCAGATACATCCAGCAACC, (R) TAGCTCACCTACAAATCGCCCTTAG; SIRT3 (F) GCCCAATGTCGCTCACTA, (R) CGTCAGCCCGTAGTCTT; and β-actin (F) TATCGGCAATGAGCGGTTC, (R) AGCACTGTGTTGGCATAGAG. Amplifications were performed for 40 cycles using an Opticon continuous fluorescence detection system (MJ Research, Inc., Waltham, MA) with SYBR green fluorescence (Molecular Probes, Eugene, OR). Each cycle consisted of a 45 s at 94°C, a 45 s at 56°C, and a 60 s at 72°C. All data were quantified by use of the comparative CT method and normalized to β-actin.
siRNA sequences
VSMCs were cultured to 80% confluence and transfected with interested siRNA by use of Lipofectamine 2000 (Invitrogen Co.). The siRNA against PGC-1α or SIRT3 mRNA was designed and purchased from Sigma-Aldrich. Rat PGC-1α siRNA smartpool contained four components of siRNA sequences: (i) sense 5′-GAAGAUAGAUGAAGAGAAU-3′, antisense 5′-CUUCUAUCUACUUCUCUUA-3′; (ii) sense 5′-UCAAGUAUCUGACCACAAA-3′, antisense 5′-AGUUCAUAGACUGGUGUUU-3′; (iii) sense 5′-GAACAAUUCUCCAAACUAC-3′, antisense 5′-CUUGUUAAGAGGUUUGAUG-3′; and (iv) sense 5′-CAACUAGACUUCAAAGAUG-3′, antisense 5′-GUUGAUCUGAAGUUUCUAC-3′ with a final siRNA concentration of 50 nM each as described previously (2). Rat SIRT3 siRNA sequence: sense 5′-GAGAGCAUCUGGUAUCCCUdTdT-3′, antisense 5′-AGGGAUACCAGAUGCUCUCdTdT-3′. The scramble siRNA was served as a negative control.
ROS and mtROS level detection
The level of ROS in abdominal aortas was detected using DHE (1 μM) (R001; Vigorous, Beijing, China) that emits red fluorescence on oxidation by ROS. mtROS level was detected by the MitoTracker Red CMH2XRos probe (200 nM, M-7512; Molecular Probes) or MitoSOX (1 μM, M36008; Thermo) (57). All were examined by Leica TCS SP5 confocal system (Leica, Wetzlar, Germany). The level of DHE and its oxidative products 2-E+OH and E+ were detected by HPLC (Agilent HPLC 1260, Santa Clara, CA). Samples were pretreated as reported by Fernandes et al. (11), and detected under 245 nm wave length within 20 min.
Statistical analysis
All data are presented as mean ± standard error of the mean. Differences were analyzed by Student's two-tailed unpaired t-test for two groups or one-way ANOVA for multiple groups followed by Tukey's multiple comparison post hoc tests by use of GraphPad Prism 5.0 software. A value of p < 0.05 was considered statistically significant.
Footnotes
Acknowledgments
We thank Prof. Yong-Fen Qi and Ming Zheng (Peking University Health Science Center, Beijing, China) for providing valuable suggestions. We thank Dr. Ming-Jiang Xu (NIH research fellow, MD, USA) for providing the probe MitoTracker red CMH2XRos. This work was supported by grants from the National Natural Science Foundation of China to L.L.W. (81470398) and Y.Z. (81400356).
Authors' Contributions
H.F. designed, performed, and interpreted the experiments, and wrote the article. J.-Y.W., X.C., and L.L. assisted in the design and interpreted the experimental work. B.Y. assisted in the design of the experiment, performed siRNA sequence, and wrote the article. W.-G.Z., and P.-L.G. performed the experiments of CKD patients. L.-M.L., C.-L.Z., and Y.Z. assisted in animal experiments and data analysis. L.-L.W. designed and interpreted the experimental work and wrote the article.
Author Disclosure Statement
No competing financial interests exist.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Figure S7
Supplementary Figure S8
Supplementary Figure S9
Supplementary Figure S10
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.