
Abstract
Aims:
The management of myocardial ischemia has been challenged by reperfusion injury. Reactive oxygen species (ROS) production is the critical cause of reperfusion injury, but antioxidant treatment failed to gain satisfactory effects. We hypothesized that improvement of redox homeostasis by preconditioning regulation should potentiate the ability of antioxidants to protect the heart from reperfusion injury.
Results:
By phenotype-based screening, we identified that dihydrotanshinone I (DT) and protocatechuic aldehyde (PCA) potently protected cardiomyocytes through preconditioning regulation and antioxidant activity, respectively. DT induced transient ROS generation via reversible inhibition of mitochondrial respiratory complex I and thereby stabilizing HIF-1α, while PCA elevated the levels of reduced glutathione (GSH) by providing reducing equivalents to scavenge ROS. HIF-1α, stabilized by DT, transcriptionally upregulated Nrf2 and thereby activated antioxidant enzymes, potentiating PCA to protect cardiomyocytes from reperfusion injury by strengthening intrinsic ROS scavenging capacity. In rat ischemia/reperfusion (I/R) model, sequential administration of DT and PCA, but not in reverse, additively protected the heart from I/R injury, manifested by reduced infarct size and improved cardiac function. These results were further supported by sequential administration of metformin and vitamin E in the rat and porcine I/R models.
Innovation and Conclusion:
Our work demonstrates that preconditioning regulation of redox state is essential for antioxidants to protect the heart from I/R injury, providing a new direction for the treatment of myocardial injury.
Introduction
Ischemia impairs the heart due to inadequate blood supply to the myocardium. Although reperfusion potentially allows the salvage of viable ischemic myocardium, it has been challenged by additional injury (49). During the ischemic stage, disrupted metabolism activates succinate dehydrogenase and thereby increasing succinate accumulation, which drives mitochondrial reactive oxygen species (ROS) generation (9). It is now generally accepted that increased ROS production in response to reperfusion is a critical pathological factor during the ischemia/reperfusion (I/R) process (23). However, previous strategies to alleviate oxidative stress in cardiovascular diseases with antioxidants failed to exert significant beneficial effects in the clinic (42). The exact reasons remain elusive, and one possible reason might be that ROS production on reperfusion is in the context of altered metabolisms, and the impaired redox homeostasis would hinder the efficacy of antioxidants.
Myocardial ischemic preconditioning, made up of one or more brief cycles of ischemia and reperfusion, has been considered a promising therapeutic strategy to protect the heart from acute myocardial infarction and reperfusion injury (35). Ischemic preconditioning provides multifaceted endogenous protection, including improved metabolism, upregulated intrinsic antioxidative enzymes, and maintained calcium homeostasis, consequently establishing resistance against I/R injury (16). Investigation of signaling pathways underlying ischemic preconditioning has identified potential signaling molecules, including HIF-1α, AKT, GSK-3β, and Nrf2 (6, 21), which may be targeted to develop pharmacological preconditioning agents. Although clinical cardioprotection research has been challenged by complicated pathogeny, myocardial preconditioning, including ischemic preconditioning and pharmacological preconditioning, provides novel endogenous therapies for the treatment of I/R injury and might also serve as an adjunct to other cardioprotective strategies (24).
Innovation
Antioxidants lack sufficient effect in combating ischemia/reperfusion (I/R) injury partially due to the impaired metabolism during ischemic stage. Dihydrotanshinone I (DT) reversibly inhibits mitochondrial respiratory chain complex I to induce HIF-1α activation. As a preconditioning regulation, HIF-1α shifts mitochondrial oxidative phosphorylation to glycolysis and transcriptionally upregulates Nrf2 signaling, resultantly potentiating the ability of antioxidant protocatechuic aldehyde (PCA) to protect the cardiomyocytes from oxidative stress on reperfusion injury. This finding raises the possibility that sequential administration of preconditioning and antioxidants might be a prospective therapeutic strategy to protect the heart from I/R injury.
Since multiple pathological factors are involved in I/R injury, only targeting individual mediators of lethal reperfusion injury is considered insufficient to produce satisfactory results, and therefore, a more effective approach may be to target more than one mediator at a time. Indeed, some recent studies show that combinatorial therapeutic approaches targeting different players may provide more effective cardioprotection than a single-target approach (14, 37). As dysregulated metabolism and impaired redox state during ischemic stage contribute to ROS production on reperfusion (9), it is reasonable to speculate that myocardial preconditioning, with the capability of metabolism and redox adaptation, in combination with an exogenous antioxidant, is able to exert enhanced efficiency in attenuation of I/R injury.
Natural medicines have long been a treasury for medication, providing drug candidates for the treatment of cardiovascular diseases (44, 45). Because I/R injury is characterized with distinct pathological events between ischemia and reperfusion stage, we hypothesized that certain cardioprotective herbs, such as Danshen (Salvia miltiorrhiza Bge.) (20), may contain compounds that target different pathological events and ultimately reach an additive/synergistic effect in I/R injury. By phenotype-based screening, we identified that dihydrotanshinone I (DT) and protocatechuic aldehyde (PCA) potently protected cardiomyocytes through different mechanisms. We wondered whether a combination of DT and PCA could potentially protect the heart from I/R insult. We observed that DT activated HIF-1α and Nrf2 signaling to potentiate the action of PCA in alleviating I/R insult, indicating that maintained redox homeostasis is essential for antioxidants to attenuate oxidative stress in I/R injury.
Results
Phenotype-based screening for cardioprotective compounds
Due to the lack of commonly accepted and validated targets for cardioprotection, we used a phenotype-based screening approach to identify potential cardioprotective compounds that might specifically target the ischemic and reperfusion stages, respectively (Fig. 1A). To increase screening efficiency, we first constructed a chemical library containing a total of 304 chemical compounds. The detailed information about individual agents is shown in Supplementary Table S1. Cell viability determined by CCK-8 activity and cell death represented by lactate dehydrogenase (LDH) leakage were recorded, and the ratio of normalized CCK-8 activity to the normalized LDH leakage (SCCK-8/LDH) was used as an index to select specific cardioprotective compounds. For drug screening, oxygen and glucose depletion (OGD) model was developed in H9c2 rat cardiac myoblasts, a well-defined cardiac cell line that shares similar ischemic response with primary cardiomyocytes (30). Because we aimed to discover agents with the efficacy of preconditioning regulation, cells were pretreated with tested compounds for 8 h. As a result, 129 compounds showed positive results, indicated by increased cell viability and reduced LDH leakage compared with vehicle-pretreated cells (Fig. 1B). According to the score of SCCK-8/LDH, these compounds were ranked and listed (Fig. 1C). Among them, we were particularly interested in 15,16-DT with high SCCK-8/LDH score. DT is capable of activating Nrf2 to protect human skin cells (43) and inducing metabolic reprogramming in colon cancer through regulating AKT/HIF-1α pathway (46), indicative of the potential role of DT in preconditioning regulation. DT pretreatment potently protected neonatal rat cardiomyocytes from OGD-induced cell death in a concentration-dependent manner (Fig. 1F). A similar protective effect was found in H9c2 cells (Supplementary Fig. S1A).
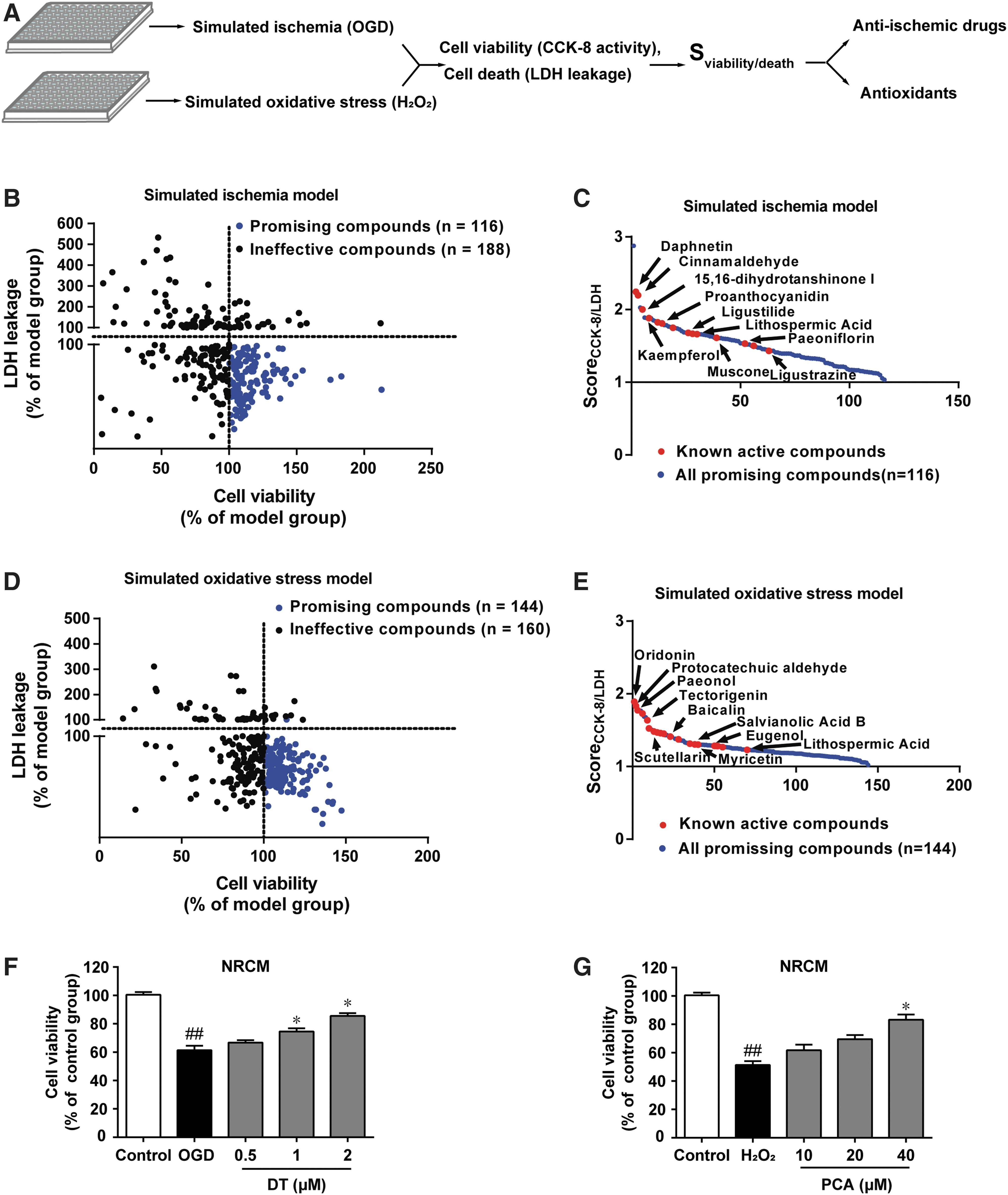
Increased ROS production during reperfusion is an important contributor to heart reperfusion injury (23), and therefore, we used H2O2-induced oxidative injury model in H9c2 cell line to screen for compounds that might combat reperfusion injury. As a result, 154 of the chemicals showed positive effects, indicated by increased cell viability and reduced LDH leakage compared with cells from the model group (Fig. 1D). According to the calculated SCCK-8/LDH, ranks of the compounds were listed (Fig. 1E). PCA particularly attracted our attention because PCA is a natural phenolic compound with the ability to scavenge ROS by providing reducing equivalents. Interestingly, both DT and PCA are major active components in Danshen. In support, PCA improved cell survival against H2O2 challenge concentration dependently in neonatal rat cardiomyocytes and H9c2 cells (Fig. 1G and Supplementary Fig. S1B) without influencing gene expressions of Nrf2, superoxide dismutase (SOD) 2, and catalase (Supplementary Fig. S1C–E).
In addition, we examined the potential cytotoxicity of DT and PCA in nonmyocytes (human umbilical vein cells hy926, mouse neuroblastoma cells N2a, liver cells L02, human embryonic kidney 293T cells) at a given working concentration, and no toxicity was observed (Supplementary Fig. S1F, G), excluding the possible cytotoxicity in DT and PCA treatment.
DT preconditions cardiomyocytes via reversible inhibition of mitochondrial complex I
DT treatment induced transient mitochondrial ROS production in H9c2 cells and the significant effect was observed at 2 h after DT treatment (Fig. 2A). Consistently, DT induced mitochondrial ROS production in the rat heart in a similar way (Supplementary Fig. S2). Bioenergetic analysis of oxygen consumption rate (OCR) showed that DT reduced the maximal respiration in H9c2 cells, despite no observable influence on the basal respiration (Fig. 2B). Because inhibition of complex I is proposed to induce mild superoxide production (41), we tested whether DT induced transient ROS production via inhibiting mitochondrial respiratory complex I. In permeabilized cardiomyocytes, DT-mediated respiratory inhibition was observed in the presence of the complex I substrates pyruvate and malate instead of the complex II substrate succinate (Fig. 2C). In contrast to the irreversible inhibitor rotenone, inhibition of complex I activity by DT was restored by washing, indicating that inhibition of complex I by DT was reversible (Fig. 2D). Tiron, a cell-permeable antioxidant, largely abolished the cell-protective effects of DT (Fig. 2E, F), indicative of the involvement of ROS. These results raised the possibility that transient mitochondrial ROS production might act as one signaling molecule in the preconditioning regulation.

DT increases HIF-1α induction dependent on ROS generation
To determine how DT activated the preconditioning signaling pathway, we performed a digital gene expression (DGE) profiling analysis of DT-treated cardiomyocytes. Neonatal rat ventricular myocytes were exposed to DT for 8 h and genes with a p-value <0.05 found by DGE-Seq were assigned as differentially regulated. The DT-regulated genes were enriched to representative pathways by KEGG analysis (Fig. 3A). Among this list signs, HIF-1 signaling pathway is extensively related with the regulation of glycolysis. HIF-1α signaling pathway is important in preconditioning-conferred cardioprotection (13, 22). In line with this, HIF-1α-targeted genes for pyruvate dehydrogenase (PDK) and hexokinase-II (HK-II) were upregulated by DT pretreatment (Supplementary Fig. S3A). ROS are able to stabilize HIF-1α by impairing HIF-1α hydroxylation (31). We observed that DT pretreatment caused increased HIF-1α accumulation along with reduced hydroxy-HIF-1α in H9c2 cells (Fig. 3B, C). Moreover, the confocal microscopy analysis showed that DT promoted HIF-1α translocation into the nucleus (Fig. 3D), a step necessary for transcriptional regulation. Although DT increased HIF-1α levels in both the nucleus and cytosol, more retention was observed in the nucleus (Fig. 3E), supporting that DT promoted HIF-1α translocation into the nucleus for transcriptional regulation. In neonatal rat cardiomyocytes, DT demonstrated similar effects on the regulation of HIF-1α (Supplementary Fig. S3B, C).

Different from tiron, mito-tempol is considered a mitochondrial ROS scavenger. DT impaired the hydroxylation and thereby increased HIF-1α accumulation, but these effects were largely reversed by both tiron and mito-tempol treatment (Fig. 3F, G), indicating the pivotal role of ROS in the HIF-1α stabilizing effect of DT.
DT activates Nrf2 signaling dependent on HIF-1α
Apart from HIF-1α downstream genes, we also found that DT pretreatment time dependently increased Nrf2 protein level in neonatal rat cardiomyocytes (Fig. 4A). Concordantly, the nuclear translocation was also induced by DT in H9c2 cells (Supplementary Fig. S4A). Nrf2 and the downstream genes, including HO-1, NQO1, SOD2, and catalase, were also upregulated by DT in cardiomyocytes (Supplementary Fig. S4B–F). We further hypothesized that as a transcriptional factor, HIF-1α transcriptionally activates Nrf2 by binding to its promoter region. We used promoter analysis using the JASPAR database to predict the binding of HIF-1α to Nrf2 promoter, and a positive result was obtained (Fig. 4C). Luciferase promoter reporter assay further validated that HIF-1α was required for DT to induce Nrf2 induction (Fig. 4B, C). Knockdown of HIF-1α significantly alleviated DT-induced Nrf2 gene expression (Fig. 4D). Consistently, the upregulation of gene expressions for HO-1 and NQO1 was also blocked by HIF-1α knockdown (Fig. 4E, F). These results indicated that DT-induced Nrf2 antioxidant signaling was mediated through HIF-1α induction. DT improved cardiomyocyte survival against OGD insult, but the protective effect was diminished by knockdown of HIF-1α (Fig. 4G). Interestingly, the induction of Nrf2 by DT was largely abolished by antioxidants tiron and mito-tempol (Fig. 4H). Together, these results indicate that ROS-triggered HIF1α-Nrf2 signaling underlies the myocyte protective effects of DT.

In addition, KEGG enrichment analysis also showed an involvement of PI3K-AKT signaling pathway in the biological action of DT (Fig. 3A), known as reperfusion injury salvage kinase (RISK) pathway implicated in promoting cell survival against ischemic insult (22). AKT is commonly regarded as the upstream kinase of GSK-3β by phosphorylation at Ser 9 (10, 11). Western blotting analysis confirmed that DT activated AKT and thereafter inactivating GSK3β by phosphorylation (Supplementary Fig. S4G, H). Bradykinin (BK) is commonly formed in the interstitium and there is also a local kinin–kallikrein system existing in the heart to produce BK (17). BK released from cardiac myocytes is proposed to protect the heart from I/R injury via downstream RISK pathways (26, 38), and we found that DT significantly elevated BK concentration in OGD-treated cells (Supplementary Fig. S4I). All these results support that DT functions as an ischemic preconditioning agent.
Sequential administration of DT and PCA additively combats I/R injury
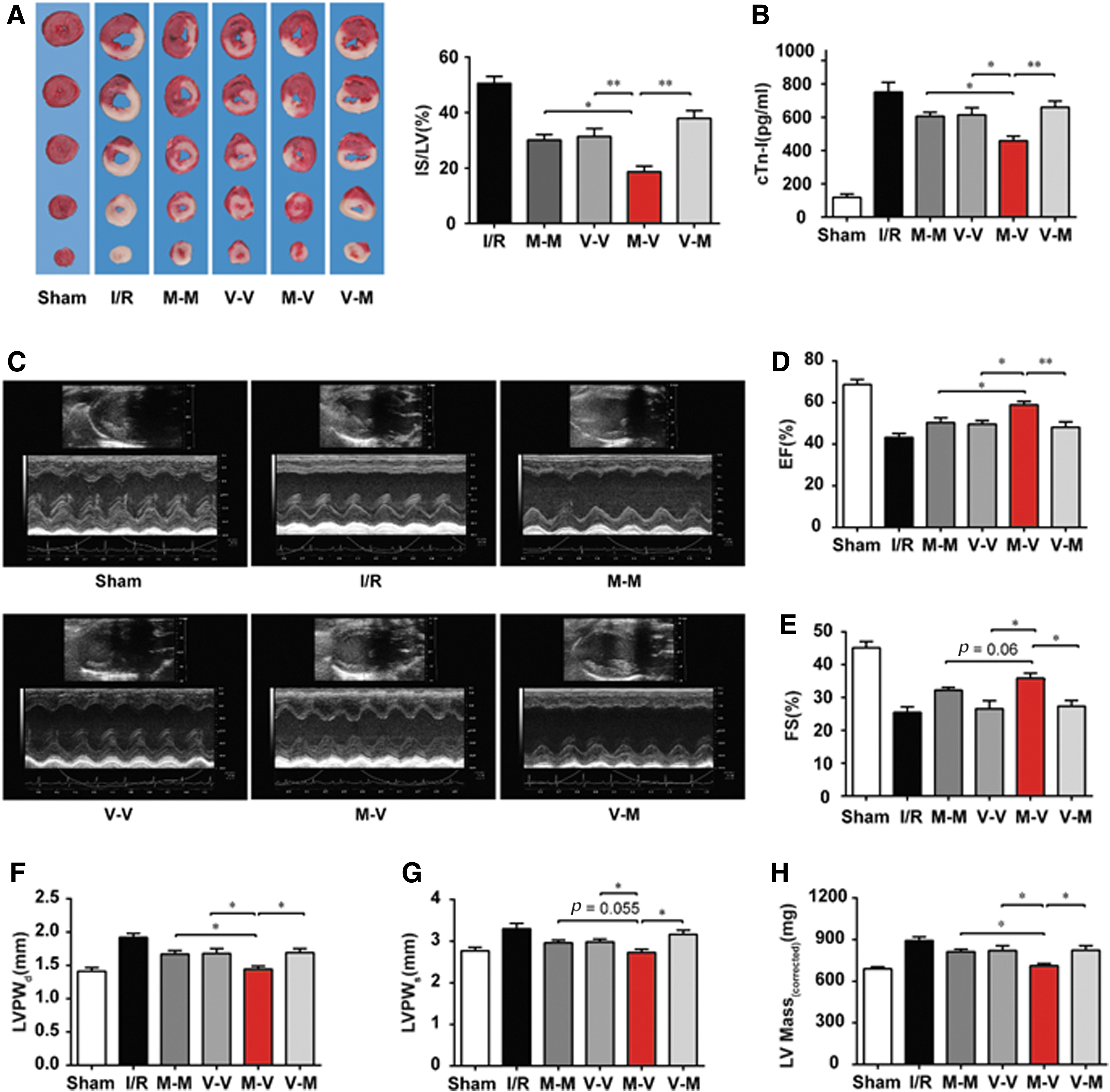
Next, we tested whether sequential administration of DT and PCA could additively protect the heart against I/R injury. DT, PCA, or vehicle was delivered to rats before the surgery or on reperfusion in different sequences (Fig. 5A). The infarct size was detected at 24 h after reperfusion and the cardiac function was determined 2 weeks later. Although DT or PCA treatment alone reduced the infarct size to certain extents, sequential DT-PCA treatment, but not the reverse, resulted in significant improvements (Fig. 5B). Cardiac troponin I (cTnI) release is an indicator of ischemic injury (1), and DT-PCA treatment also more effectively reduced cTnI release when compared with other different sequential treatments (Fig. 5C). Moreover, the analysis of cardiac functions 2 weeks later showed that DT-PCA treatment significantly elevated ejection fractions (EF) and fractional shortening (FS) (Fig. 5D–F), key parameters characterizing the systolic function of left ventricle (LV). Besides, DT-PCA attenuated ventricular hypertrophy, indicated by reduction in the left ventricular posterior wall thickness at the end of diastole or systole (LVPWd, LVPWs) and left ventricular (LV Mass) (Fig. 5G–I). Hematoxylin and eosin (HE) staining results also showed that DT-PCA treatment effectively attenuated myocardial interstitial edema, fiber rupture, and leukocyte infiltration (Supplementary Fig. S5A). These results provided evidence that sequential administration of DT and PCA additively protects cardiac function from I/R insult, much better than that when DT and PCA were applied either alone or in combination with the sequence in reverse. Although exposure to myocardial ischemia increased both HIF-1α and Nrf2 expression in the heart of rats, a regulation likely due to adaptive responses, more significant results were observed in DT treatment, supporting that DT may act as a preconditioning agent for cardioprotection (Supplementary Fig. S5B).

The glutathione redox cycle plays a major role in scavenging H2O2 (7, 8). We speculated that PCA might support the redox cycle by providing reducing equivalents to regenerate glutathione (GSH). Indeed, PCA increased the ratio of GSH/GSSG under basal and H2O2-stimulated conditions (Supplementary Fig. S5C), partially explaining why PCA promoted cell survival against H2O2 challenge in a manner independent of SOD2 (Supplementary Fig. S5D, E). In contrast, in hypoxia and reoxygenation (H/R) model, the effects of DT-PCA treatment on cell survival and ROS suppression were reduced by SOD2 knockdown (Supplementary Fig. S5F, G). Given that the conversion of O2 •− to H2O2 involves SOD, we reasoned that PCA alone had little effects in H/R model but capable of synergizing with DT that activates the endogenous antioxidative system, including SOD. As a support, in cardiomyocytes subjected to H/R, knockdown of Nrf2 reduced the effects of DT-PCA treatment on cell protection (Supplementary Fig. S5H). DT-PCA activated Mn-SOD and catalase; such effects were also largely abolished by Nrf2 silencing (Supplementary Fig. S5J, K). These results indicated that Nrf2 induction by DT may strengthen the endogenous antioxidative activities and cooperate with PCA in combating reperfusion injury.
Sequential application of metformin and vitamin E alleviates I/R injury
Above data indicated that the combination of preconditioning agents and antioxidants could be a promising strategy to prevent I/R injury. To further validate this hypothesis, we investigated whether metformin (MET), an antidiabetic agent with the ability to mildly inhibit mitochondrial complex I, could work together with an antioxidant agent vitamin E (VE) in a sequential administration to combat cardiac I/R injury effectively. In a rat cardiac I/R model, sequential MET-VE treatment effectively limited infarct size (Fig. 6A) and reduced the release of cTnI (Fig. 6B). Concordantly, MET-VE treatment significantly rescued cardiac function, much better than that when MET and VE were administered alone or in reversed sequence (Fig. 6C–H). To characterize gene expression profiles, we performed RNA-Seq assay using samples from the infarct area of the heart. Differences in transcriptomes among MET-VE-treated groups with the other groups were globally examined using hierarchical clustering. As expected, MET-VE treatment recovered the dysregulated genes, including the pathways involving redox status, metabolism, and inflammation, to the greatest extent among all medication groups (Supplementary Fig. S6A). The quantitative real-time PCR (qPCR) results further validated these findings (Supplementary Fig. S6B–P). All these results indicated that sequential MET and VE treatment efficiently protected the heart from ischemia and reperfusion injury.
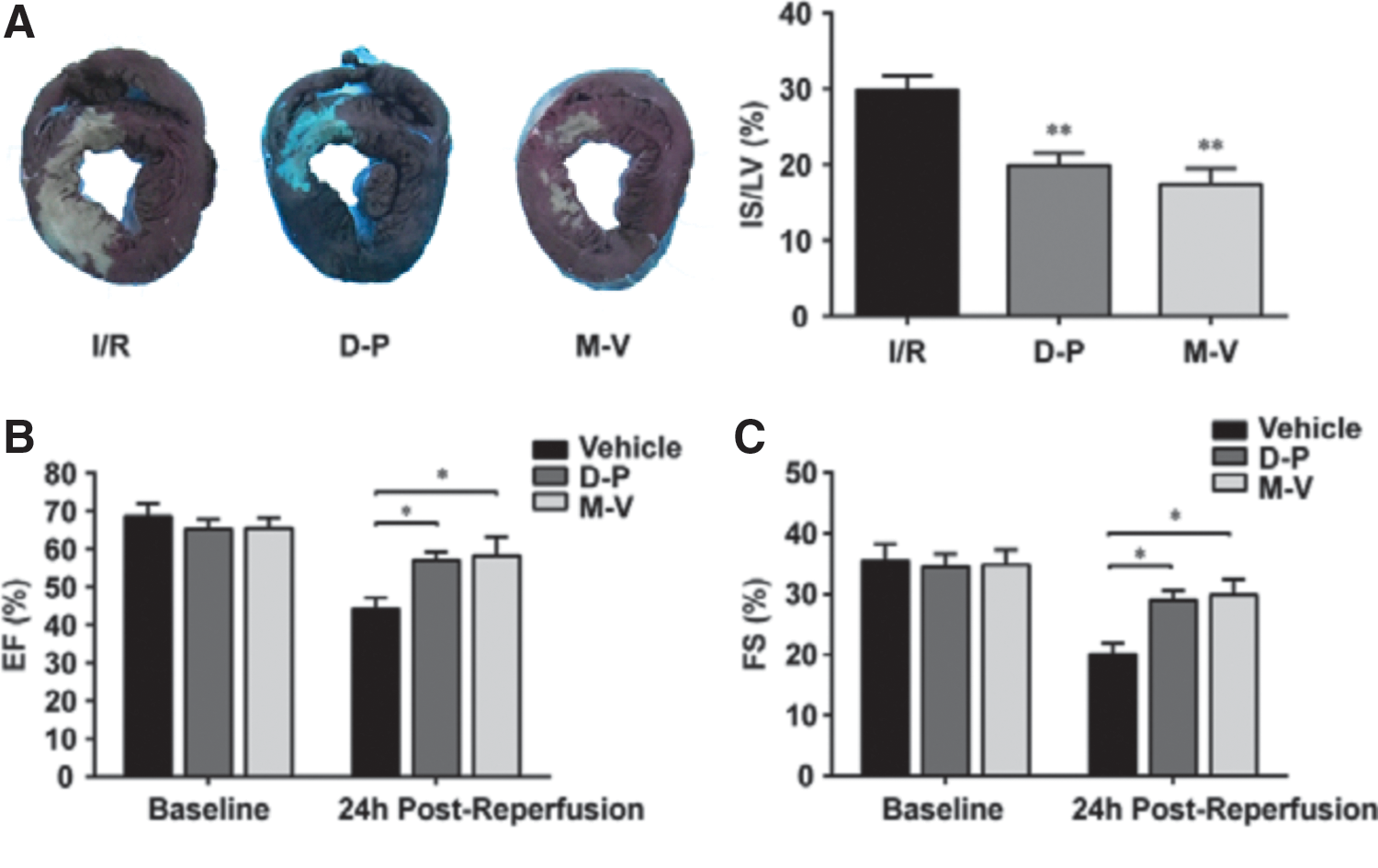
DT-PCA and MET-VE treatment in the porcine model
For further confirmation, a preclinical porcine I/R model was used. The results showed that infarct size of I/R hearts was significantly reduced by DT-PCA and MET-VE administration (Fig. 7A). Consistently, I/R-induced loss of cardiac function, indicated by decreased EF and FS, was restored in both DT-PCA- and MET-VE-treated pigs (Fig. 7B, C). Taken together, these results support the potent efficacy of sequential DT-PCA or/and MET-VE treatment in cardioprotection.
Discussion
In ischemic heart disease, reperfusion is mandatory to salvage the ischemic myocardium from infarction, but this therapeutic strategy has been challenged by reperfusion injury due to ROS production. Most of the antioxidant therapies failed in translation to the clinic, probably due to disrupted metabolism and intrinsic redox homeostasis during ischemic state. The preconditioning phenomena provide cardioprotection via endogenous mechanism, whereas the molecular basis and signal involved remain to be further elucidated (25). In the present study, we demonstrated that, by adaptive regulation of intrinsic antioxidant defense system, the HIF-1α-dependent preconditioning program during ischemia sets cardiomyocytes at a state sensitive to antioxidant activity for the attenuation of reperfusion injury. Cardioprotection by sequential administration of DT and PCA provides evidence to support the rationale.
Most of the monotherapy approaches against myocardial injury have been found unsatisfactory in translation to the clinic (27, 28). One of the major reasons might be that multifaceted factors are involved in the process of cardiac I/R injury that can hardly be addressed by monotherapy (49). To achieve better cardioprotective effects, an emerging concept has been to adopt combined therapy targeting differential stages and/or pathological events in myocardial infarction. For example, glucose-insulin-potassium or exenatide has additive effects with remote ischemic conditioning in reducing infarct size (2, 32). N-acetylcysteine is an antioxidant, while nitroglycerin promotes vasodilation by NO production. A recent study demonstrates that high dose of N-acetylcysteine administered with low dose of nitroglycerin leads to additive effects in reducing infarct size in a clinical trial for patients undergoing percutaneous coronary intervention (36). Nutrient depletion is the cause for cardiomyocyte death during ischemia, while excessive ROS production on reperfusion further impairs cell survival. Therefore, two different cellular models were designed for screening of cardioprotective agents from natural products. We had shown that DT potently protected cardiomyocytes in the OGD model, suggesting that cardioprotection by DT is likely due to the regulation of cellular metabolism. When cardiomyocytes were exposed to H2O2, PCA effectively promoted cell survival, indicating that PCA protected cardiomyocytes from oxidative stress. More importantly, both DT and PCA are the main components in Danshen, allowing us to investigate whether DT and PCA could work together to protect the heart from I/R injury via targeting different pathological events.
Pretreatment with DT promoted cardiomyocyte survival from ischemic insult, indicative of preconditioning regulation in cardioprotection. Interestingly, DT mildly inhibited mitochondrial respiratory chain complex I with transient mitochondrial ROS production. Although mitochondria are the main source of ROS production, inhibition of complex I by rotenone is shown to induce transient ROS generation (41). Moreover, complex I inhibition is shown to promote cell survival by reducing ROS generation on reperfusion (9, 18). Different from rotenone, the inhibition of complex I by DT is reversal, ensuring its action is protective, but not deleterious. Consistently, Drp1 inhibitor mdivi-1 reversibly inhibits mitochondrial complex I to improve cell survival against I/R injury (18).
HIF-1α is constantly synthesized and rapidly degraded by prolyl hydroxylase domain dioxygenases (PHDs) under normal oxygen environment to prevent its accumulation (29), while in response to oxygen depletion, HIF-1α induction transcriptionally regulates cellular responses to adapt cellular metabolism to low oxygen tension. Apart from low oxygen tension, ROS are also able to increase HIF-1α accumulation by impairing PHDs (31). DT increased transient ROS production via mild complex I inhibition, well explaining its action on HIF-1α induction. When mitochondrial oxidative phosphorylation is suppressed owing to the limited oxygen supply, HIF-1α induction promotes glycolysis to ensure ATP production, despite less efficiency. PDK inactivates pyruvate dehydrogenase to inhibit mitochondrial oxidation (47), while HK-II binds to mitochondria to ensure efficient ATP production from glycolysis (19). Therefore, the upregulation of PDK and HK-II indicated that DT induced metabolic reprogramming and facilitated cell survival under low oxygen tension. The regulation of genes encoding glycolysis by HIF-1α has been identified, and importantly, we found that HIF-1α transcriptionally upregulated Nrf2 expression. As a transcription factor, Nrf2 binds to antioxidant response elements to increase the transcription of the antioxidant proteins (34). In view of the transient ROS production from mitochondrial complex I, the induction of antioxidant enzymes SOD and catalase through preconditioning regulation should serve as a negative feedback to prevent excessive ROS production. Although PCA treatment alone is shown to combat H2O2-induced oxidative injury in cardiomyocytes and limit I/R-induced myocardial infarct size to a reduced level, given the improved metabolism and redox homeostasis brought by DT, we reasoned that preconditioning should potentiate the efficiency of antioxidant PCA in cardioprotection on reperfusion. As expected, sequential administration of DT and PCA, but not in reverse, more effectively improved mitochondrial function along with cell survival. In rats subjected to I/R insult, sequential administration of DT and PCA effectively reduced heart infarct size with protected heart function. Similar cardioprotection by sequential administration of DT and PCA was also reproduced in ischemic porcine heart. These results provided evidence in vivo to support our findings in cultured cardiomyocytes.
We have shown that Nrf2 induction by DT was essential for cardioprotection in cultured cells. However, we cannot rule out the possibility that other mechanisms may be involved in the cardioprotective effects of DT. In fact, HIF-1α has the potential to maintain oxygen homeostasis during hypoxia and could improve outcomes after ischemia by multiple mechanisms, including reprogramming of metabolism and induction of angiogenesis (4). Herein, we demonstrated that upregulation of cellular antioxidant defense is another pathway for HIF-1α-mediated preconditioning.
Because ROS production is the major contributor to reperfusion injury in the heart, antioxidative strategies have been previously tested in an attempt to reduce I/R injury. Unfortunately, very few of them have been translated to improvements in major clinical endpoints in human trials (15). For example, antioxidants, including vitamin C, VE, edaravone, and coenzyme Q10, have shown disappointing or conflicting outcomes in patients (42). Inappropriate drug dosage regimen together with other unclear mechanisms might be involved in the explanation of failure of antioxidant therapy in reperfusion injury (33). Our study also showed that although PCA alone attenuated myocardial injury to some extent, the combined therapy with sequential DT and PCA treatment led to much better effects. Our study suggests that cardioprotection by antioxidant can be potentiated in the context of improved redox homeostasis. As a support of the concept, we also observed the cardioprotective effects of sequential administration of metformin and VE in ischemic rat heart. Metformin has been identified to be a reversible inhibitor of mitochondrial complex I (48), and is shown to activate Nrf2 in ischemic injury (3). VE, a classical antioxidant, reacts with radicals such as lipid peroxyl radical (LOO·) and singlet oxygen (1O2) (5, 12). Importantly, it has been reported that VE eliminates H2O2 via upregulation of GSH levels (39), indicating that VE serves as a free radical scavenger in a way somewhat similar to PCA. From this consideration, we designed an experiment of sequential application of metformin and VE to validate the concept. Similar to DT-PCA treatment, sequential administration of metformin and VE effectively protected the heart from ischemic injury in a porcine model, supporting that sequential preconditioning agents and antioxidant therapy are an effective approach to I/R injury.
Although preconditioning regulation in experimental models is beneficial, it is difficult to be translated to the clinic as the attack of heart ischemia is unpredictable. Therefore, we must admit that the sequential administration of DT and PCA has a limitation in practical application. Our work just emphasizes that a comprehensive strategy from the aspect of improved metabolism and redox homeostasis is needed for the efficiency of antioxidants in cardioprotection.
In summary, we have demonstrated that preconditioning-induced improvement of metabolism and intrinsic redox homeostasis is essential for antioxidants to protect the heart from I/R injury. DT reversibly inhibited mitochondrial complex I and induced HIF-1α induction to maintain metabolic homeostasis, resultantly potentiating the ability of antioxidant PCA to protect the heart from reperfusion injury (Fig. 8). This finding suggests that sequential administration of preconditioning and antioxidants might be a prospective therapeutic strategy to protect the heart from I/R injury.

Materials and Methods
The information of the materials used in this study is summarized in Supplementary Table S3.
Cell culture and cell models
H9c2 cardiac myoblasts were purchased from the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). Cells were cultured in 1 × Dulbecco's modified Eagle's medium (DMEM; Gibco, Thermo Fisher) supplemented with 10% fetal bovine serum (FBS; GIBCO, Thermo Fisher) and antibiotics (25 U/mL penicillin and 25 U/mL streptomycin) at 37°C in a humidified incubator containing 5% CO2 and 95% atmosphere.
Cells were plated into multiwell plates or Petri dishes and cultured till reaching 90% confluency. For OGD model in culture, cardiomyocytes were cultured in glucose-free DMEM and then incubated at 1% O2, 5% CO2, and 94% N2 in a tri-gas incubator with an oxygen controller (MCO-5M; Panasonic) for 8 h. For H2O2 stimulation, cells were incubated in DMEM with 500 μM H2O2 for 2 h.
Primary culture of cardiomyocytes
Primary culture of neonatal rat cardiomyocytes was conducted as described previously (40) with minor modifications. Briefly, the ventricles of 1- to 3-day-old Sprague Dawley rat hearts were washed twice with phosphate-buffered saline (PBS), cut into 1 mm pieces, and then digested with collagenase type II (Worthington), repeating five to seven times. Fibroblasts were removed by preincubation for 120 min in a 37°C incubator. After isolation and purification, cardiomyocytes were seeded in Petri dishes or multiwell plates containing DMEM (Gibco, Thermo Fisher), supplemented with 10% FBS (Gibco, Thermo Fisher), 0.05 mM 5-bromo-2-deoxyuridine (BrdU; Sigma-Aldrich), and antibiotics. Cells were preincubated for 24 h before subsequent experiments.
Measurement of cell viability and LDH leakage
Measurement of cell viability and LDH leakage was carried out through colorimetric procedures with the cell counting kit-8 and the Cytotoxicity LDH Assay Kit (Dojindo Laboratories), according to the manufacturer's instructions with minor modifications, respectively. Neonatal rat cardiomyocytes (NRCMs) or H9c2 cells were seeded at a density of 1.0 × 104 cells/well in a 96-well plate and cells were pretreated with indicated stimulations before subsequent measurement. For the measurement of LDH leakage, 30 μL medium/well was collected and 100 μL of LDH assay buffer was added to initiate the colorimetric reaction; the mixture was incubated at 37°C for 30 min and the LDH activity was determined by measuring the absorbance at 490 nm on a microplate reader (Polar Star; Omega). For the measurement of cell viability, CCK-8 reaction solution was prepared by mixing CCK-8 solution and DMEM at the ratio of 1:9, and 100 μL CCK-8 reaction solution was added per well, followed by incubation at 37°C for 2 h; the cell viability was determined by measuring the absorbance at 450 nm on a Polar Star microplate reader.
Rat and porcine model of I/R injury
Adult male Sprague Dawley rats (250–280 g) were used in this study. Rats were anesthetized with pentobarbital (50 mg/kg, i.p.) and monitored with a surface electrocardiogram (ECG; TaiMeng). Ischemia was induced by a left anterior descending (LAD) coronary artery occlusion and was confirmed by an immediate color change of the vessel from light red to dark violet, and of the myocardium supplied by the vessel from bright red to white, as well as the immediate occurrence of ST-elevations in the ECG. After 30 min of ischemia, the occlusion was released and animals were allowed to recover from the surgery.
Pigs were around 25–30 kg. Anesthesia was induced by intramuscular injection of ketamine (20 mg/kg), xylazine (2 mg/kg), and midazolam (0.5 mg/kg), and maintained by continuous intravenous infusion of ketamine (2 mg/kg/h), xylazine (0.2 mg/kg/h), and midazolam (0.2 mg/kg/h). Animals were intubated and mechanically ventilated with oxygen (fraction of inspired O2: 28%). The LAD coronary artery, immediately distal to the origin of the first diagonal branch, was occluded for 60 min with an angioplasty balloon introduced via the percutaneous femoral route using the Seldinger technique. A continuous infusion of amiodarone (300 mg/h) was maintained during the procedure in all pigs to prevent malignant ventricular arrhythmias. In cases of ventricular fibrillation, a biphasic defibrillator was used to deliver nonsynchronized shocks.
For the measurement of infarct size, the heart was excised, frozen and sliced, and then incubated for 10 min in 1% 2,3,5-triphenyltetrazolium chloride to visualize the unstained infarcted region. Sections were digitally photographed, and infarct (IF) area and LV area were quantified with ImageJ. The infarct size was calculated as IF/LV. All animal experiments were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals, following protocols approved by Pharmaceutical Animal Experimental, China Pharmaceutical University.
Echocardiography
Echocardiograms were obtained using a Vevo 2100 Ultrasound System (VisualSonics, Toronto, Canada) equipped with a high-frequency (21 MHz) linear array transducer. Echocardiography was performed before I/R injury, and 2 weeks following surgery. The rats were anesthetized with isoflurane (3% for induction and 1%–1.5% for maintenance) mixed in 1 L/min O2 via a facemask. To determine cardiac structure and function, EF, FS, left ventricular posterior wall end-diastolic thickness (LVPWd), left ventricular posterior wall end-systolic thickness (LVPWs), and left ventricular mass (LV Mass) were analyzed from M-mode.
Measurement of ROS
For the measurement of mitochondrial ROS in the myocardium, hearts were excised, washed, and put into liquid nitrogen. Frozen cross sections (8 μm) through the LV were prepared and stained with a superoxide anion indicator dihydroethidium (DHE; Invitrogen™, Thermo Fisher) or MitoSOX (5 μM, Invitrogen, Thermo Fisher), and the fluorescent images were captured by a confocal microscope (LSM 700; Carl Zeiss) or fluorescent microscope (Ts2R; Nikon); fluorescent intensity was quantified using Image Pro Plus (IPP; Media Cybernetics, Inc.).
For cardiomyocytes, cells were stained with MitoSOX or CellROX. Photomicrographs were taken with a confocal microscope or a live cell station (EVOS® FL; Thermo Fisher Scientific). The fluorescent intensity was calculated using IPP.
Measurement of OCR
Oxygen consumption measurements from intact and permeabilized cells were performed using a Seahorse XF96 bioenergetic analyzer (Seahorse; Agilent Technologies). For intact cell experiments, H9c2 cells were seeded at a density of 1 × 104 cells/well in XF96 microplates. Cellular bioenergetics was measured using a Seahorse XF96 Extracellular Flux analyzer. Cells were resuspended in DMEM for 1 h at 37°C in a CO2-free incubator before OCR measurements. Basal OCR was measured and then oligomycin, FCCP, and rotenone/antimycin A were sequentially injected at 1, 2.5, and 0.5 μM, respectively. For permeabilized cell experiments, cells were resuspended in a mitochondrial assay buffer (70 mM sucrose, 220 mM mannitol, 10 mM KH2PO4, 5 mM MgCl2, 2 mM HEPES, 1.0 mM EGTA, and 0.2% [wt/vol] fatty acid-free bovine serum albumin, pH 7.2) before OCR measurements. Digitonin (30 μg/mL) was coinjected with 1 mM ADP and the indicated mitochondrial substrate(s) to initiate permeabilization and ADP-stimulated respiration in mitochondrial assay buffer. Substrate combinations for complex I-linked respiration consisted of 5 mM pyruvate plus 5 mM malate. A combination of succinate (5 mM) plus rotenone (1 μM) was used to assay complex II-dependent respiration.
Western blot analysis
Cells were lysed in RIPA lysis buffer containing 1% protease inhibitor cocktail and phosphatase inhibitors (Pierce Protein Technology, Thermo Fisher). Lysates were incubated with loading buffer for 10 min at 95°C. Samples were resolved on 9% polyacrylamide gels and transferred to PVDF membranes. Membranes were blocked with nonfat milk (5% in Tris-buffered saline, TBS) for 2 h at room temperature and then with a primary antibody in TBS overnight at 4°C. Membranes were washed with 0.1% TBST (3 × 10 min) and then incubated with the appropriate secondary antibody in TBS at room temperature for 2 h and washed again. The protein quantification was performed on an Odyssey Sa Imaging System. The following primary antibodies were used: GAPDH (No. 5174, 1:1000; CST), HIF-1α (ab1, 1:1000; Abcam), HO-1 (ab68477, 1:1000; Abcam), Nrf2 (sc-722, 1:200; Santa Cruz), Lamin B1 (No. 12586, 1:1000; CST), β-actin (sc-47778, 1:200; Santa Cruz), Akt (No. 4691, 1:1000; CST), Phospho-Akt (Thr308; No. 4060, 1:1000; CST), GSK-3β (No. 12456, 1:1000; CST), Phospho-GSK-3β (Ser9; No. 9323, 1:1000; CST), and Hydroxy-HIF-1α (No. 3434, 1:1000; CST). The secondary antibodies were IRDye® 680RD donkey anti-mouse IgG (H+L) (1:10,000; LI-COR Biotech) and IRDye 800CW goat anti-rabbit IgG (H+L; 1:10,000; LI-COR Biotech).
Small interfering RNA transfection
H9c2 cells were seeded at 50% confluency in DMEM containing 10% FBS 24 h before transfection. One hundred picomoles of small interfering RNA (siRNA) per six-well dish were transfected using the Xfect™ RNA transfection reagent (Takara) as recommended by the manufacturer. After 24 h of incubation, cells were used for further experiments. The 21-nucleotide sequence for each siRNA is as follows: HIF-1α sense: 5′-GAGCUCCCAUCUUGAUAAATT-3′, HIF-1α antisense: 5′-UUUAUCAAGAUGGGAGCUCTT-3′; Nrf2 sense: 5′-CCGGAGAAUUCCUCCCAAUTT-3′, Nrf2 antisense: 5′-AUUGGGAGGAAUUCUCCGGTT-3′; and SOD2 sense: 5′-GCCAAGGGAGAUGUUACAATT-3′, SOD2 antisense: 5′-UUGUAACAUCUCCCUUGGCTT-3′.
Luciferase reporter assay
Nrf2 promoter or pGL3-control vector (E1741; Promega) was cotransfected into H9c2 cells using the Xfect transfection reagent (631317; Takara) with HIF-1α or Nrf2 siRNA or control siRNA using Xfect RNA transfection reagent (631450; Takara). The medium was changed to fresh 10% FBS-DMEM 4 h after transfection. The cell lysate and supernatant were harvested 36 h after transfection. Luciferase activity was measured with the luciferase assay system (E1501; Promega) using a luminometer (Polar Star; Omega) according to the manufacturer's instructions. The protein amount of the supernatant was determined with a BCA Protein Assay Kit (23227; Pierce). Relative luciferase activity (firefly luciferase activity/BCA value) was quantified as the fold change relative to the basal values obtained from control transfected cells not exposed to DT treatment.
Quantitative polymerase chain reaction
Total RNA was extracted from frozen tissues or cardiomyocytes using a specific RNA isolation kit (TIANGEN) following the manufacturer's instructions. Complementary DNA (cDNA) synthesis from 1 μg RNA was carried out using a high-capacity cDNA reverse transcription kit (Vazyme Biotech Co., Ltd.). Real-time quantitative polymerase chain reaction (qPCR) was done using a SYBR® Green Master Mix (Vazyme Biotech Co., Ltd.) on a Light Cycler® 96 system (Roche, Germany). 18s ribosomal RNA or β-actin was used as internal reference and the 2ΔΔCT method was used for data analysis. Primers used for qPCR are listed in Supplementary Table S2. The results were from three independent experiments, and each sample was performed in triplicate in each experiment.
Measurement of Mn-SOD activity and catalase activity
The Mn-SOD activity was carried out through a colorimetric procedure with the Cu/Zn-SOD and Mn-SOD Assay Kit (Beyotime) according to the manufacturer's instructions. Cells were collected, washed twice with ice-cold PBS, and homogenized with the Dounce homogenizer. The homogenate was centrifugated (10,000 g, 5 min, 4°C), and the supernatant was collected for the assay of enzyme activities. For the measurement of Mn-SOD activity, sample homogenate was mixed with Cu/Zn-SOD inhibitor for 1 h at 37°C. The WST-8 and reaction solution was then added to initiate the colorimetric reaction. After incubation at 37°C for 30 min, Mn-SOD activity was determined by measuring the absorbance of the solution at 450 nm. For the measurement of catalase activity, sample homogenate was mixed with excess hydrogen peroxide for decomposition, and the remaining hydrogen peroxide coupled with a substrate was treated with peroxidase to generate a red product, N-4-antipyryl-3-chloro-5-sulfonate-p-benzoquinone monoimine, which absorbs maximally at 520 nm. Catalase activity was thus determined by measuring the absorbance of the reaction mixture at 520 nm.
Immunofluorescence
Cells cultured on glass Petri dishes were washed twice with PBS, fixed with 3.7% paraformaldehyde for 15 min at room temperature. After fixation, cells were rinsed and permeabilized with 0.1% Triton X-100-PBS solution on an orbital shaker for 2 × 5 min. Cells were then blocked in PBS containing 5% normal serum and 0.3% Triton X-100 for 60 min. After removal of the blocking buffer, primary antibodies of HIF-1α (1:200; Abcam) were applied and samples were incubated overnight at 4°C. Samples were then washed free of primary antibodies with PBS and incubated with secondary antibodies at room temperature for 1 h, followed by counterstaining with Hoechst (Invitrogen, Thermo Fisher). After washing with PBS, the samples were mounted with antifade mounting medium (KeyGEN) and fluorescent images were taken with a confocal microscope (LSM 700; Carl Zeiss).
Statistical analysis
Significance between two groups was performed by Student's two-tailed t test with GraphPad Prism 6.01. In other cases, significance across more than two groups was done in Prism with one-way analysis of variance. A p < 0.05 was considered significant for all tests. All data are expressed as mean ± standard error of the mean, unless otherwise noted.
Footnotes
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 81421005 and 8181101169), National Key R&D Program of China (No. 2018YFC1704500), and “Double First-Class” University project (CPU2018GF04).
Author Disclosure Statement
No competing financial interests exist.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.