
Abstract
Significance:
Vascular dysfunction plays a key role in the development of arteriosclerosis, heart disease, and hypertension, which causes one-third of deaths worldwide. Vascular oxidative stress and metabolic disorders contribute to vascular dysfunction, leading to impaired vasorelaxation, vascular hypertrophy, fibrosis, and aortic stiffening. Mitochondria are critical in the regulation of metabolic and antioxidant functions; therefore, mitochondria-targeted treatments could be beneficial.
Recent Advances:
Vascular dysfunction is crucial in hypertension pathophysiology and exhibits bidirectional relationship. Metabolic disorders and oxidative stress contribute to the pathogenesis of vascular dysfunction and hypertension, which are associated with mitochondrial impairment and hyperacetylation. Mitochondrial deacetylase Sirtuin 3 (Sirt3) is critical in the regulation of metabolic and antioxidant functions. Clinical studies show that cardiovascular disease risk factors reduce Sirt3 level and Sirt3 declines with age, paralleling the increased incidence of cardiovascular disease and hypertension. An imbalance between mitochondrial acetylation and reduced Sirt3 activity contributes to mitochondrial dysfunction and oxidative stress. We propose that mitochondrial hyperacetylation drives a vicious cycle between metabolic disorders and mitochondrial oxidative stress, promoting vascular dysfunction and hypertension.
Critical Issues:
The mechanisms of mitochondrial dysfunction are still obscure in human hypertension. Mitochondrial hyperacetylation and oxidative stress contribute to mitochondrial dysfunction; however, regulation of mitochondrial acetylation, the role of GCN5L1 (acetyl-CoA-binding protein promoting acetyltransferase protein acetylation) acetyltransferase, Sirt3 deacetylase, and acetylation of specific proteins require further investigations.
Future Directions:
There is an urgent need to define molecular mechanisms and the pathophysiological role of mitochondrial hyperacetylation, identify novel pharmacological targets, and develop therapeutic approaches to reduce this phenomenon.
Introduction
Hypertension is a multifactorial disorder (52); however, in almost all experimental models of hypertension, production of reactive oxygen species (ROS: O2 •− and hydrogen peroxide [H2O2]) is increased in multiple organs. In the brain, ROS promote neuronal firing, increasing sympathetic outflow (78, 132). In the kidney, ROS act in multiple sites to promote sodium resorption and volume retention (114). In the vasculature, ROS promote vasoconstriction and remodeling, increasing systemic vascular resistance (72). There are several sources of ROS contributing to hypertension, including the NADPH oxidase, uncoupled nitric oxide (NO) synthase, and the mitochondria (28) and we defined their interaction (25, 27). ROS overproduction leads to oxidative stress, which promotes target-organ-damage in hypertension (22); however, antioxidant therapy is not currently available and common antioxidants such as ascorbate and vitamin E are ineffective in preventing cardiovascular diseases and hypertension (47). These agents unlikely reach important sites of ROS production such as the mitochondria whereas therapies specifically targeted at mitochondria represent promising strategies to reduce target-organ-damage (95).
Mitochondrial dysfunction contributes to the pathogenesis of hypertension and cardiovascular disease (34, 95); however, despite the central role of mitochondria in human health and disease, there are no approved drugs that directly target mitochondria (118). Mitochondrial dysfunction is characterized by impaired adenosine triphosphate (ATP) production and increased oxidative stress, leading to cell dysfunction and apoptosis (36). Mitochondrial permeability transition pore (mPTP) plays a key role in mitochondrial dysfunction (48) and target-organ-damage in hypertension (36, 95). We have recently reported that depletion or inhibition of cyclophilin D (CypD), a regulatory subunit of mPTP opening (37), improves vascular function and attenuates hypertension (59). Previous studies implicated CypD in cell death (68, 111) and we showed that CypD is critical in the regulation of cytokine-induced vascular oxidative stress and endothelial dysfunction (59). Meanwhile, the precise regulation of CypD is elusive and specific CypD blockers are not available.
Experimental studies have shown an important role of mitochondrial ROS in the development of endothelial dysfunction, hypertension, and atherosclerosis (86, 94), and the overexpression of the key mitochondrial antioxidant, Mn-superoxide dismutase (SOD2), attenuates hypertension (30). SOD2 expression is regulated by peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) (65), and mitochondrial deacetylase Sirtuin 3 (Sirt3) activates SOD2 by deacetylation of specific lysine residues near active center (55, 93, 131). Interestingly, human SOD2 expression is not changed with age but the activity of Sirt3 and SOD2 is diminished (12), suggesting that SOD2 inactivation by acetylation contributes to human hypertension. These data suggest Sirt3 depletion in endothelial dysfunction and hypertension. Indeed, we have found that hypertension is associated with diminished Sirt3 level, a profound increase in SOD2-K68 acetylation in humans with essential hypertension, and reduced SOD2 activity in the vasculature of hypertensive mice and that SOD2 mimetics improve vasorelaxation and reduce blood pressure (30, 31).
Sirt3 is a key node in the regulation of mitochondrial function (53). It activates mitochondrial metabolism by deacetylation of Krebs cycle (103), complex I (1, 90), and fatty acid β-oxidation enzymes (7, 56), and it maintains mitochondrial NADPH-GSH redox status by deacetylation of isocitrate dehydrogenase 2 (IDH2) (128). Sirt3 deacetylation is opposed by spontaneous acetylation of mitochondrial protein lysine residues with acetyl coenzyme A (Acetyl-CoA) (61, 91) and GCN5L1 (acetyl-CoA-binding protein promoting acetyltransferase protein acetylation) mediated acetyltransferase (97). An imbalance between mitochondrial acetylation and Sirt3 activity leads to mitochondrial hyperacetylation and contributes to impaired metabolism and oxidative stress (16, 24). Interestingly, hypertension and endothelial dysfunction are associated with mitochondrial hyperacetylation (31); however, molecular mechanisms and the pathophysiological role of mitochondrial hyperacetylation are not completely understood.
In this work, we reviewed specific pathways involved in the metabolic and redox regulation of mitochondrial functions. These data suggest a potential pathophysiological role of crosstalk between mitochondrial hyperacetylation and oxidative stress. We suggest that targeting of this feed-forward vicious cycle between metabolic disorders and mitochondrial oxidative stress can be beneficial for the treatment of vascular dysfunction and hypertension.
Mitochondrial Acetylation as a Major Post-Translational Modification
Mitochondrial metabolism generates high levels of Acetyl-CoA (0.1–1.5 mM), which is 3–50 times higher than cytosol and nucleus Acetyl-CoA concentrations (51). High concentration of Acetyl-CoA and high pH in mitochondrial matrix drive non-enzymatic acetylation of lysine residues (116). Acetylation can be more favorable at the active site lysine residues, with reduced pKa values suggesting that regulatory lysine residues can attract acetylation (43). Recently, Michael Murphy group showed that non-enzymatic N-acetylation of lysine residues in mitochondrial proteins frequently occurs via a proximal S-acetylated thiol intermediate (61). It is clear that metabolic disorders are associated with high levels of Acetyl-CoA and promote mitochondrial acetylation; however, these data do not exclude the possibility of enzyme-mediated protein acetylation in mitochondria. Indeed, Michael Sack group has described GCN5L1-mediated acetyltransferase, which plays a critical role in the acetylation of key mitochondrial enzymes such as SOD2 (97, 98). It has been proposed that GCN5L1-mediated acetylation counterbalances the Sirt3 deacetylase activity. Both enzymatic and non-enzymatic pathways account for protein acetylation as a major post-translational modification in the mitochondria, and ∼35% of all mitochondrial proteins are acetylated (4).
Mitochondrial Deacetylase Sirt3 in Regulation of Metabolic and Antioxidant Functions
Sirtuin family of nicotinamide adenine dinucleotide, oxidized form (NAD+)-dependent histone deacetylases catalyzes deacetylation of both histone and non-histone lysine residues and consists of seven isoforms (123). Mitochondria contain one known enzyme with deacetylase activity, Sirt3 (80). It plays a key role in the regulation of mitochondrial metabolism and the activity of mitochondrial antioxidants. Sirt3 activates a key fatty acid β-oxidation enzyme, long-chain acyl coenzyme A dehydrogenase (LCAD) (7, 56), Krebs cycle (103), nicotinamide adenine dinucleotide, reduced form (NADH) oxidase activity by complex I (1, 90), NADPH-producing IDH2 (128), and critical mitochondrial antioxidant SOD2 (110) by deacetylation of specific lysine residues (55, 56, 93, 131). Sirt3 can potentially activate AMP protein kinase via deacetylation/activation of LKB1 (serine/threonine liver kinase B1) (40), and it is conceivable that crosstalk between Sirt3 and adenosine monophosphate-activated protein kinase (AMPK) contributes to metabolic regulations. It has been suggested that both nuclear deacetylase Sirtuin 1 (Sirt1) and Sirt3 induce mitochondrial biogenesis via the PGC-1α pathway (79); however, the role of Sirt3 in mitochondrial biogenesis has not been confirmed. Of note, activation of different Sirtuin isoforms not only regulates distinct pathways but also may have opposing effects. For example, Sirt4 negatively affects fatty acid oxidation and Sirt4 depletion increases expression of Sirt1 and Sirt3 (130).
Sirt3 is associated with human longevity (5, 44). Variable number tandem repeat (VNTR) enhancer in Sirt3 gene is associated with human longevity, and two non-synonymous human SIRT3 single-nucleotide polymorphisms impact SIRT3 activity and stability (33). Activation of the angiotensin II/AT1R pathway reduces Sirt3 (15), and Sirt3 is downregulated in the metabolic syndrome, hyperlipidemia, diabetes, aging, and smoking (Fig. 1) (17, 75, 129). Reduced Sirt3 expression is associated with cell aging, as measured by diminished telomerase reverse transcriptase (hTERT) and increased senescence-associated β-galactosidase (SA-β-gal) (66, 121). Animal studies indicate that Sirt3 deficiency promotes tissue fibrosis (60) and cardiac hypertrophy (108), which are attenuated by Sirt3 activation (2, 107); however, the cell-specific role of Sirt3, its alterations in human pathological conditions, and Sirt3 targeting translational potential are not clear.
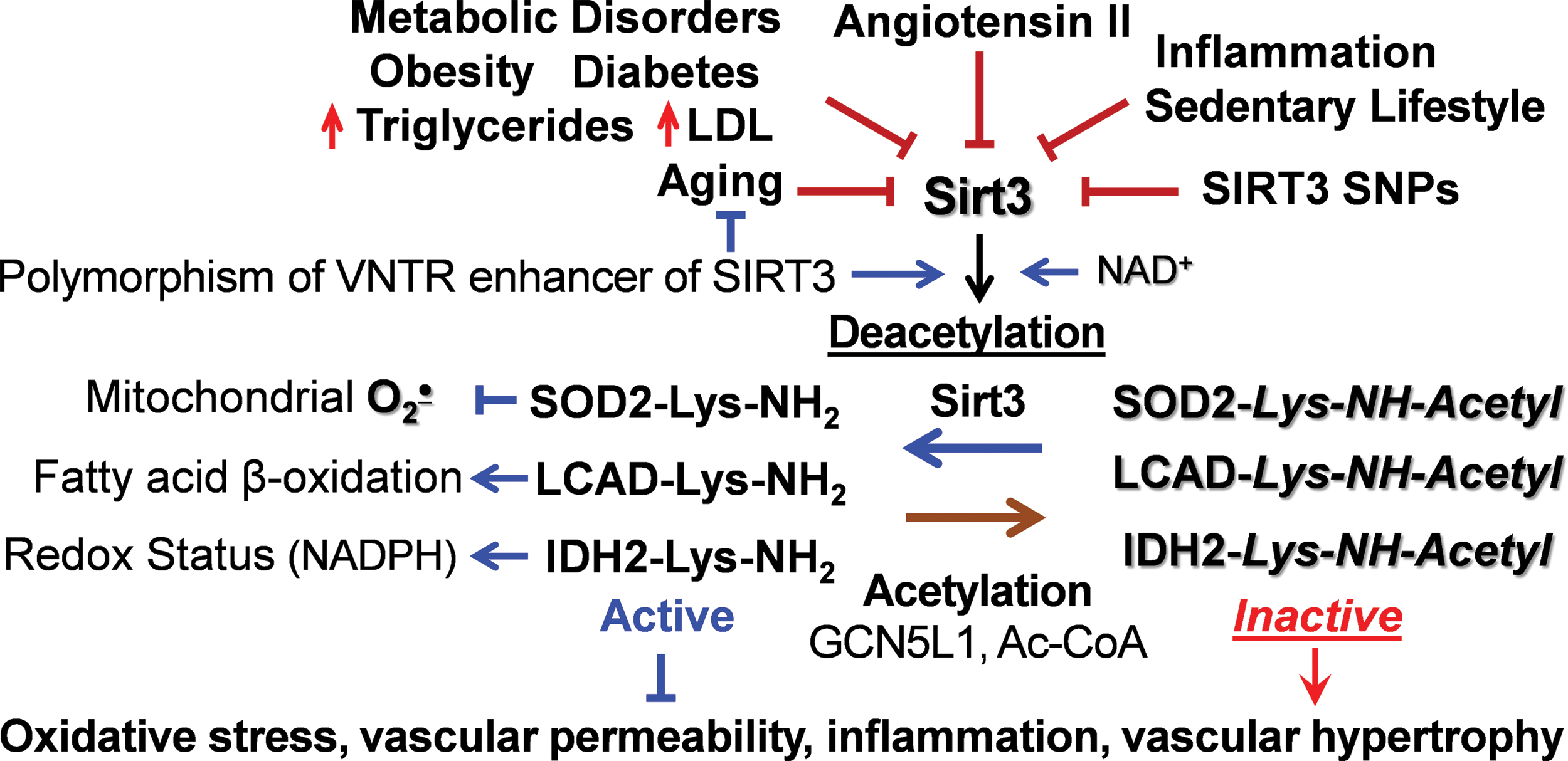
Redox and Metabolic Regulations of Sirt3
Sirt3 requires NAD+ for its deacetylase activity, and “healthy” mitochondria have high NAD+ level and only a small fraction of reduced NADH form (122). Pathological conditions such as hypoxia and metabolic disorders are associated with (i) reduction of NAD+ to NADH diminishing NAD+ level, and (ii) depletion of NAD pool due to PPARγ activation (92). For example, inhibition of mitochondrial complex I NADH oxidase activity reduces NAD+ level, leading to Sirt3 inactivation and mitochondrial hyperacetylation (1). It has been suggested that NAD+ depletion contributes to Sirt3 inactivation in cardiovascular conditions and supplementation with NAD+ donors is beneficial due to Sirt3 activation (58); however, oral supplementation with NAD+ donors has limited pharmacological effect, potentially due to rapid liver metabolism (77).
Sirt3 activity depends on mitochondrial function and matrix pH (127); therefore, reduced membrane potential downregulates Sirt3 activity. Sirt3 is important for metabolic flexibility and Sirt3 depletion induces a switch of skeletal muscle substrate utilization from carbohydrate oxidation toward lactate production (62). Meanwhile, increased substrate utilization leads to high NADH/NAD+ ratio and elevated Acetyl-CoA, which inhibits Sirt3 activity and provides “on-demand” On/Off metabolic switch.
In recent years, it has become clear that Sirtuins can be inactivated by oxidative stress. For example, the activity of Sirt1 is regulated by reversible S-glutathionylation at Cys204 in the catalytic region (11). Sirt1 cysteine reaction with H2O2 drives S-glutathionylation, and glutaredoxin 2 activates Sirt1 by deglutathionylation of cysteine residue in the conserved catalytic region (11). Because the NAD+-dependent deacetylases are highly homologous, we hypothesized that Sirt3 might undergo redox inactivation in a manner similar to Sirt1. Indeed, incubation of human recombinant Sirt3 in the presence of H2O2 and reduced glutathione caused a dose-dependent inactivation of Sirt3, which was associated with Sirt3 S-glutathionylation (31). We reasoned that Sirt3 S-glutathionylation would be reduced in transgenic mCAT (mice expressing mitochondria-targeted catalase) mice with mitochondria-targeted expression of catalase. Indeed, angiotensin II-induced hypertension was associated with robust Sirt3 S-glutathionylation in the mitochondria of wild-type mice but scavenging of mitochondrial H2O2 in mCAT mice abrogates Sirt3 S-glutathionylation, prevents SOD2 inactivation by acetylation, diminishes mitochondrial superoxide, and attenuates hypertension (31). These data support the pathophysiological significance of Sirt3 S-glutathionylation in vascular dysfunction and hypertension.
It is important that glutaredoxins 1/2 in the mitochondrial intermembrane and thioredoxin-thioredoxin reductase in the matrix are critical for reversing mitochondrial protein S-glutathionylation in the vasculature and heart (42, 54). Acetylation of these proteins affects their activity and alters the redox maintenance (124). Further, hyperoxidation of cysteine residues into sulfenic, sulfinic, and sulfonic acids may cause irreversible loss of enzymatic activities and impair the recovery from oxidative stress (45).
Redox and Metabolic Regulations of CypD
CypD plays a dual function in mitochondria: peptidyl prolyl cis-trans isomerase F and a regulatory subunit of the mPTP acting as a Ca2+ sensitizer for mPTP opening (83). Previous studies were focused on the important role of CypD in the regulation of cell death (68, 111), but recent data implicate CypD in the regulation of mitochondrial metabolism (71). CypD is exquisitely H2O2 sensitive via its cysteine 203 residue, which acts as a redox switch when it is S-glutathionylated (76). Further, CypD acetylation at lysine 166 promotes mPTP opening and mitochondrial Sirt3 deacetylates CypD-K166 (49). These data implicate both redox and metabolic regulations of CypD activity (Fig. 2).
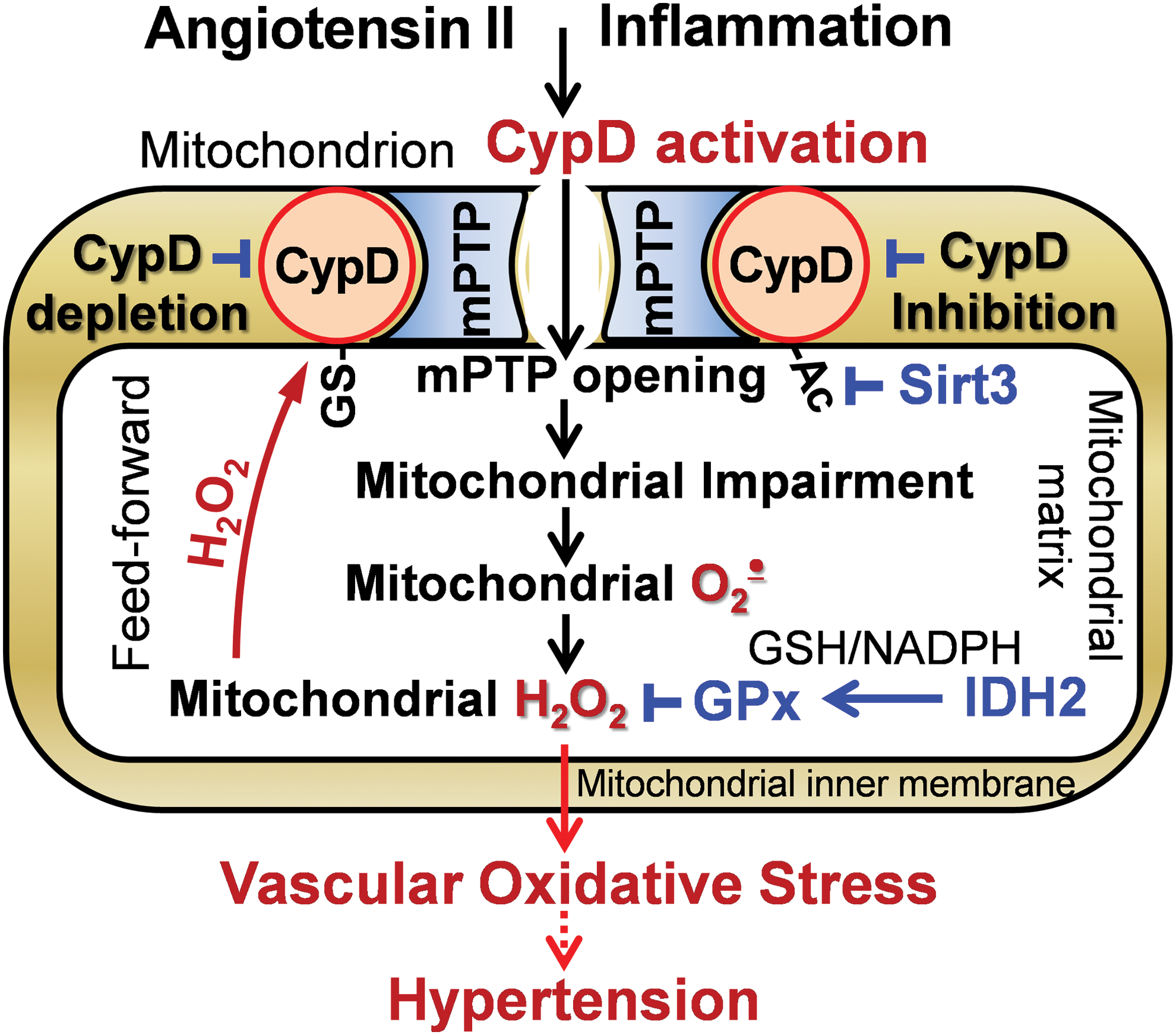
Recent data suggest a feed-forward regulation of CypD by oxidative stress. It has been shown that inhibition of CypD in isolated endothelial mitochondria reduces superoxide production and CypD deficiency attenuates superoxide production in leukocytes (32, 69). We tested whether mitochondrial H2O2 activates CypD by S-glutathionylation and this induces overproduction of mitochondrial ROS in the electron transport chain. Indeed, treatment of isolated aortic segments with H2O2 significantly increased mitochondrial superoxide and induced CypD S-glutathionylation whereas supplementation with the specific CypD inhibitor Sanglifehrin A or treatment with the complex I inhibitor rotenone blocked H2O2-induced mitochondrial superoxide production (59). Further, scavenging mitochondrial H2O2 by mitoEbselen (mitochondria-targeted glutathione peroxidase mimetic) or mitochondrial-targeted catalase in aorta isolated from mCAT mice completely prevented CypD S-glutathionylation and reduced mitochondrial superoxide. These data support the pathophysiological role of CypD redox modulation; however, specific molecular mechanisms of CypD S-glutathionylation and deglutathionylation are not clear.
David Sinclair group reported that global Sirt3 depletion induces acetylation of CypD at lysine 166 (K166), which promotes age-dependent mPTP opening, cardiac hypertrophy, and fibrosis (49). Authors suggested that age-associated Sirt3 depletion causes CypD hyperacetylation, which increases induction of the mPTP opening and the decline in cardiac function with age. CypD-K166-directed mutagenesis and Sirt3 overexpression in cardiomyoblasts prevented CypD acetylation, limited PTP opening, and reduced cell death in response to hypoxia-reoxygenation (9). Meanwhile, the effects of global Sirt3 depletion or Sirt3 overexpression are not limited by CypD acetylation, and further studies are necessary to define the specific regulation of CypD acetylation/deacetylation and its tissue-specific pathophysiological role.
Deacetylation of IDH2 in Regulation of Redox Status
Several mitochondrial enzymes maintain thiol redox status by NADPH-dependent reduction of glutathione, S-glutathionylated proteins, and protein disulfides such as glutathione reductase and thioredoxin reductase (63). Glutathione and thioredoxin systems serve parallel and non-redundant functions to maintain the dynamic mitochondrial redox balance and disruption of this redox organization is a common basis for disease (63, 64). Meanwhile, the maintenance of mitochondrial redox balance requires NADPH produced mainly by nicotinamide nucleotide transhydrogenase and IDH2 (74, 128). Depletion of either nicotinamide nucleotide transhydrogenase or IDH2 induces endothelial dysfunction and promotes hypertension (74, 88). Meanwhile, Sirt3-mediated deacetylation of IDH2 at lysine 413 is critical for IDH2 activity (128). Site-specific, genetic incorporation of N(ɛ)-acetyllysine into position 413 of IDH2 causes a dramatic 44-fold loss of activity.
Disruption of IDH2-mediated cellular redox balance enhances H2O2-induced apoptosis and hypertrophy (70). IDH2 deficiency increases mitochondrial superoxide, leading to mitochondrial dysfunction and diminished endothelial NO, which can be rescued by SOD2 mimetic mitoTEMPO (88). IDH2 functions as the principal source of NADPH for the mitochondrial GPx (Fig. 2) and thioredoxin antioxidant defense (50), and IDH2 depletion accelerates age-dependent hearing loss and renal dysfunction (73, 119). Meanwhile, IDH2 overexpression leads to “reductive stress” and contributes to genome instability and cancer (21). Therefore, Sirt3-mediated regulation of IDH2 plays an important role in cellular homeostasis.
Regulation of Mitochondrial Oxidative Stress
Mitochondrial oxidative stress is commonly defined as an imbalance between mitochondrial ROS production and antioxidant activity, which is associated with oxidative damage and cell dysfunction (29). It is frequently confused with redox signaling by thiol redox reactions serving as redox sensors in response to oxygen, metabolic, and oxidant fluxes (46, 101). Initial ROS production can induce specific cell signaling pathways mediated by protein phosphorylation and transcriptional factors such as NO synthase and NRF2 (transcription factor nuclear factor erythroid 2-related factor 2), which later provide a feed-back to downregulate the ROS production (13, 102). The dysregulation of redox signaling (wrong time-wrong place) leads to a feed-forward ROS-induced-ROS production and development of oxidative stress (25). Indeed, mitochondrial oxidative stress is associated with altered thiol redox status and scavenging of mitochondrial H2O2 reduces production of mitochondrial superoxide (28, 32). It has been suggested that both redox and metabolic alterations contribute to the development of mitochondrial oxidative stress (101).
Production of mitochondrial superoxide via reverse electron transport (RET) is of particular importance in vascular and cardiac oxidative stress (20, 84). Inhibition of mitochondrial complex II with malate reduces RET to complex I, inhibits superoxide production at complex I, and reduces endothelial oxidative stress (84, 87). Mitochondrial ROS promote T cell activation and prohypertensive immune response (29). RET is induced by high mitochondrial membrane potential and mitochondrial matrix alkalization, which can be associated with opening of the ATP-sensitive potassium channel (84) and CypD-mediated mPTP (59). Indeed, blocking the ATP-sensitive potassium channel and treatment with complex I and complex II inhibitors prevent endothelial oxidative stress (84). Further, CypD depletion or CypD inhibition prevents the rotenone-sensitive superoxide production (59). Interestingly, scavenging of mitochondrial H2O2 prevents CypD redox activation by S-glutathionylation, reduces mitochondrial superoxide production, and prevents cytokine-induced endothelial dysfunction (59) (Fig. 2). Meanwhile, the specific roles of redox and metabolic alterations of complex I and CypD in RET and mitochondrial dysfunction are still obscure.
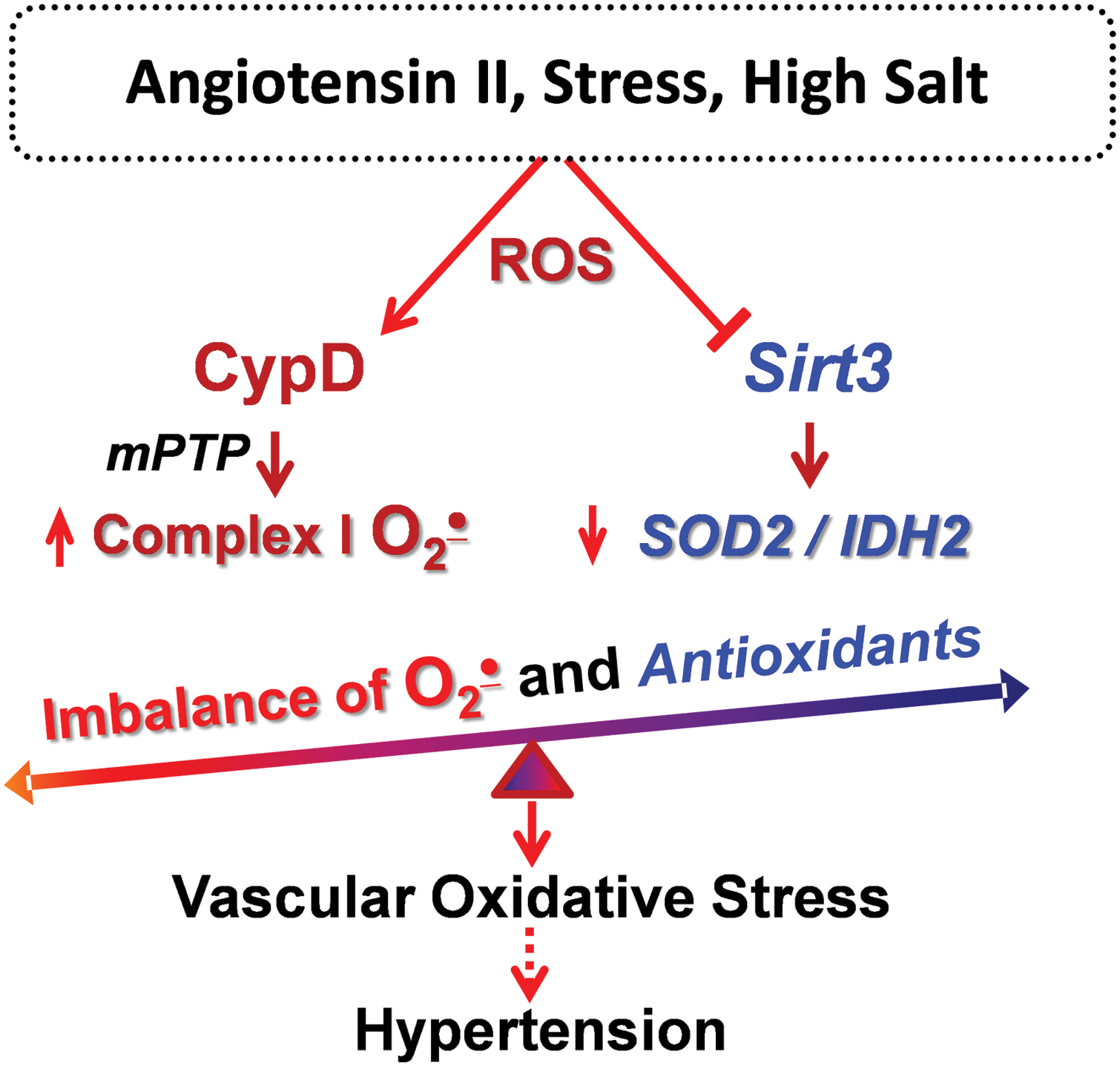
In recent years, it has become clear that metabolic conditions drive mitochondrial hyperacetylation, which induces mitochondrial oxidative stress and vascular dysfunction (41, 126). Hypertension is associated with metabolic impairment, and we showed that vascular oxidative stress and hypertension are associated with inactivation of key mitochondrial antioxidant enzyme, SOD2, due to SOD2 hyperacetylation. Treatment of mitochondrial lysate with recombinant Sirt3 deacetylates SOD2 and restores SOD2 activity (31). The impairment of Sirt3 in hypertension is likely mediated by Sirt3 depletion and Sirt3 S-glutathionylation (31). Sirt3 depletion in endothelial cells increases superoxide level in these cells and promotes endothelial dysfunction, whereas treatment of Sirt3-depleted mice after onset of hypertension with SOD2 mimetic mitoTEMPO rescues endothelial function and reduces hypertension (31). We suggest that Sirt3 inactivation results in imbalance between CypD-dependent superoxide production and SOD2/IDH2 activities, leading to mitochondrial oxidative stress, which contributes to vascular dysfunction and hypertension (Fig. 3).
Fatty Acid β-Oxidation and Sirt3
Fatty acids derived from triacylglycerols (fat) are important sources of energy and in tissues with high-energy requirement, such as the heart, more than 50% of ATP comes from fatty acid β-oxidation (81). Fatty acids are oxidized in peroxisomes and mitochondria. Mitochondria can oxidize fatty acids all the way to CO2 and H2O; however, peroxisomes are only able to chain-shorten fatty acids and the end products of peroxisomal β-oxidation must be transported to mitochondria for full oxidation (117). Fatty acids are transformed into fatty acyl-CoA and transported via Carnitine shuttle into mitochondrial matrix for β-oxidation. Sirt3 deacetylates and activates a key component of mitochondrial fatty acid β-oxidation, LCAD (7, 55, 56) (Fig. 4). K318 and K322 were identified as an Sirt3-targeted lysines (7). Medium-chain acyl-CoA dehydrogenase and acyl-CoA dehydrogenase 9 have lysines at positions equivalent to Lys-318/Lys-322, which were also efficiently deacetylated by Sirt3 (7).
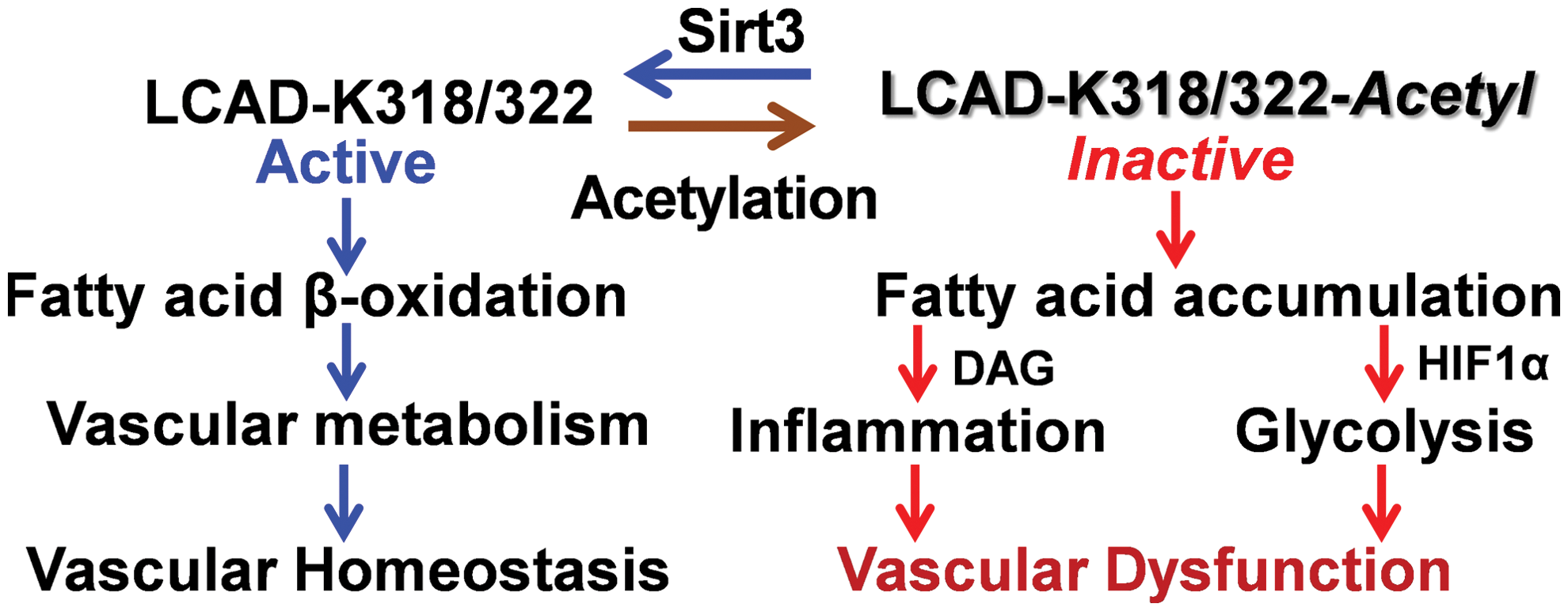
Fatty acid β-oxidation is critical for endothelial and smooth muscle cells function (18, 125). Impaired fatty acid β-oxidation alters mitochondrial function and leads to accumulation of non-oxidized fatty acids, which promotes cell dedifferentiation and inflammation (14, 112). Fatty acid oxidation is critical for endothelial cell function, and disruption of fatty acid oxidation leads to phenotypic switch to endothelial-to-mesenchymal transition associated with vascular permeability and inflammation (19, 125) (Fig. 4). Fatty acids are important components of vascular smooth muscle cell function (100); dysregulation of long-chain fatty acid metabolism causes a shift toward glycolysis (109), downregulates expression of smooth muscle cell marker α-smooth muscle actin, and induces a phenotypic switch of smooth muscle cells (106). Multiple risk factors for cardiovascular disease and hypertension are associated with reduced Sirt3 expression and activity (39), and we suggest Sirt3 inactivation as a convergent mechanism that underlies the interplay of major risk factors leading to impaired fatty acid β-oxidation and mitochondrial dysfunction in these pathological conditions (Fig. 4).
Crosstalk Between Mitochondrial Dysfunction and Oxidative Stress
The sections described earlier provide an important insight into the molecular mechanisms of mitochondrial dysfunction and oxidative stress. It is interesting that oxidative stress is commonly associated with mitochondrial dysfunction and, vice versa, mitochondrial dysfunction causes ROS overproduction and development of oxidative stress. Indeed, enzymes that typically produce ROS are associated with metabolic regulation, and diseases associated with metabolic dysfunction involve changes in redox balance (38). Therefore, we suggest a crosstalk between mitochondrial dysfunction and oxidative stress (Fig. 5).
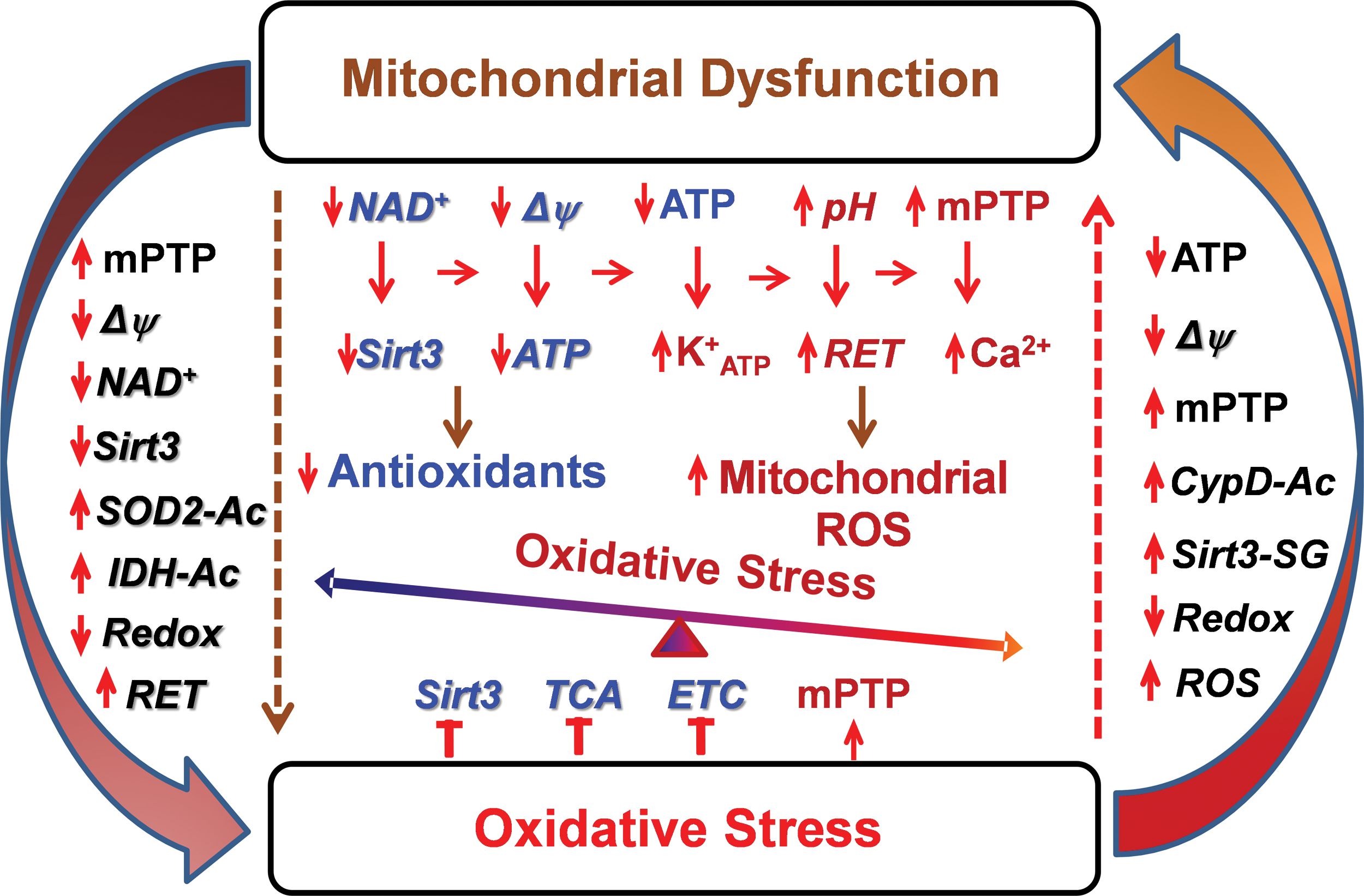
Metabolic disorders and aging cause mitochondrial dysfunction associated with diminished respiration, increased mitochondrial uncoupling, altered membrane potential, and depletion of ATP and NAD+ (10, 23), which promote mPTP and ATP-sensitive potassium channel opening (84, 113), increase ROS production via RET, and reduce Sirt3 activity, decreasing redox state and antioxidant activity. Mitochondrial dysfunction may result from direct impairment by hyperacetylation and accumulation of toxic metabolites in certain metabolic diseases (96, 105). The activation of these pathways leads to development of oxidative stress (Fig. 5). On the other hand, oxidative stress inhibits Sirt3 and tricarboxylic acid (TCA) activity, reduces electron transfer by complex I, and promotes mPTP opening by S-glutathionylation of critical cysteine residues (31, 76, 82), leading to impaired mitochondrial metabolism and mitochondrial dysfunction. These data support a novel concept of crosstalk between mitochondrial dysfunction and oxidative stress. Meanwhile, the precise molecular mechanisms of this feed-forward vicious cycle and its pathophysiological significance in human diseases are still elusive. Further studies are required to define the role of individual pathways to specific human disease and develop mitochondria-targeted therapies.
Targeting Vicious Cycle Between Metabolic Disorders and Oxidative Stress
Both metabolic disorders and oxidative stress contribute to mitochondrial dysfunction, which plays an important role in multiple pathological conditions such as cardiovascular disease, hypertension, and neurodegeneration (38, 85). In the past decade, the focus of many studies was the role of oxidative stress in mitochondria dysfunction (26, 105); however, antioxidant therapy has not been developed. Mitochondria are an important source of superoxide and H2O2, which contribute to mitochondrial dysfunction and hypertension (28, 30, 120). This is likely mediated by S-glutathionylation/inactivation of key metabolic nodes, such as Sirt3 and complex I, redox activation of CypD-mediated mPTP opening (31, 76, 82), and impaired redox recovery by thioredoxin 2 (120). Oxidative stress triggers a vicious cycle where mitochondria are both the site and the target of oxidative damage (25). Oxidative modifications of mitochondrial metabolic targets such as Sirt3 lead to hyperacetylation of key mitochondrial metabolic and antioxidant enzymes such as LCAD, IDH2, CypD, and SOD2 (31), which promote metabolic disorders and disease progression (35, 99). On the other hand, metabolic disorders decrease NAD+/NADH ratio and increase acetyl-CoA/CoA ratio, leading to imbalance in mitochondrial protein acetylation/deacetylation and development of mitochondrial hyperacetylation (6), which contributes to mitochondrial dysfunction (91). This creates a vicious cycle between metabolic disorders and oxidative stress (Fig. 6).
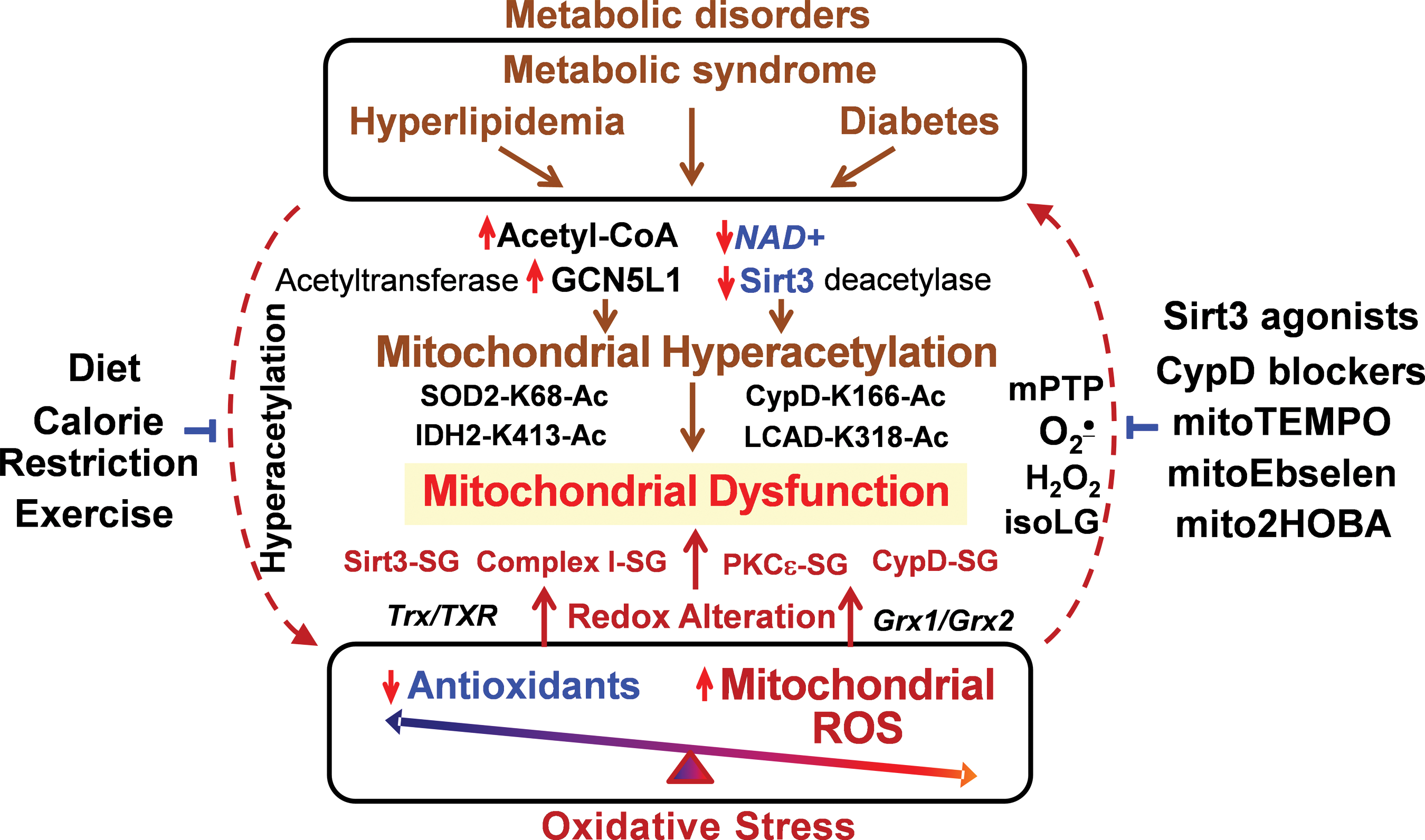
It is important to emphasize that metabolic disorders increase mitochondrial protein acetylation, which directly contributes to mitochondrial dysfunction (Fig. 6) in cardiovascular diseases and heart failure (57). The hyperacetylation of electron transport chain (complex I–V), TCA cycle enzymes, and LCAD inhibits mitochondrial bioenergetics (4). Mitochondrial hyperacetylation directly impairs mitochondrial dynamics (fusion/fission), protein synthesis, mitochondrial protein imports, calcium homeostasis, and cell signaling (3, 49, 89), which may occur without involvement of mitochondrial ROS. Meanwhile, we know that metabolic disorders are commonly associated with increased oxidative stress (8) and it is conceivable that targeting mitochondrial oxidative stress in metabolic disorders can improve mitochondrial function and alleviate these pathological conditions.
Recent studies show that treatment with mitochondria SOD2 mimetic mitoTEMPO, deacetylation of SOD2 by Sirt3 activators, and inhibition of mitochondrial ROS with CypD blockers or mitoEbselen can interrupt this vicious cycle and attenuate the mitochondrial dysfunction and disease progression (31, 59). On the other hand, calorie restriction and exercise increase Sirt3 activity, reduce mitochondrial acetylation, and inhibit mitochondrial oxidative stress (67, 93, 104, 115). Meanwhile, specific molecular targets that can be used for diagnostics and treatments in human disease are not clear, and further studies are warranted for the development of specific mitochondria-targeted therapies to break this vicious feed-forward cycle.
The direct links between the hyperacetylation of specific mitochondrial proteins, cell function, and hypertension have been supported by SOD2 hyperacetylation in human subjects with essential hypertension (31), mitochondrial hyperacetylation in pulmonary hypertension (35), and CypD acetylation in cardiac hypertrophy (49), which are in line with reduced Sirt3 level (39) and activity (12) in cardiovascular conditions, providing new insight into pathogenesis of mitochondrial dysfunction in cardiovascular conditions.
Conclusions
It is known that metabolic disorders increase risk of hypertension and cardiovascular disease. On the other hand, hypertension is frequently associated with metabolic abnormalities such as obesity, glucose intolerance, and dyslipidemia. These pathological conditions are associated with altered mitochondrial function and oxidative stress, suggesting the crosstalk between metabolic disorders and mitochondrial oxidative stress, which can be mediated by mitochondrial hyperacetylation. Metabolic disorders such as hyperglycemia and hyperlipidemia cause mitochondrial hyperacetylation, which leads to mitochondrial dysfunction and overproduction of mitochondrial ROS. On the other hand, mitochondrial oxidative stress in hypoxia and inflammation alter mitochondrial metabolism, causing the development of pathological conditions. We propose a novel crosstalk between mitochondrial hyperacetylation and oxidative stress. This crosstalk identifies potential novel targets for treatment of metabolic disorders and cardiovascular disease. There are many common conditions including aging, atherosclerosis, diabetes, heart failure, and neurodegenerative disorders in which mitochondrial dysfunction seems to play a role. It is conceivable that mitochondria-targeted interventions targeting the crosstalk between mitochondrial hyperacetylation and oxidative stress would be effective in these conditions.
Footnotes
Acknowledgments
This work was supported by funding from National Institutes of Health (R01HL124116) and the American Heart Association (16GRNT31230017).