
Abstract
Aims:
Diabetic nephropathy (DN) is the principal cause of mortality and morbidity in diabetic patients, the progression of which correlates best with tubulointerstitial fibrosis (TIF). Advanced oxidation protein products (AOPPs) have been detected in patients with chronic renal failure, causing injuries to proximal tubular epithelial cells. CD36, a known receptor for AOPP, is an important modulator of lipid homeostasis, predisposing to renal tubular damage. However, whether AOPPs induce lipotoxicity via the CD36 receptor pathway remains unknown. Herein, we tested the hypothesis that AOPPs accumulation in diabetes incurs lipotoxicity, causing renal TIF via the CD36 signaling pathway.
Results:
In DN patients and diabetic mice in vivo, AOPPs overload induces lipogenesis (upregulation of CD36 and sterol regulatory element-binding protein 1), fibrosis (upregulation of Fibronectin), and renal function decline (increased serum creatinine and N-acetyl-β-
Innovation and Conclusion:
Our data reveal a major role of AOPPs in triggering lipotoxicity and fibrosis via CD36-dependent Wnt/β-catenin activation, providing new evidence for understanding the role of lipid accumulation in DN.
Innovation
Our data reveals a major role of advanced oxidation protein products in triggering lipotoxicity and fibrosis via CD36-dependent Wnt/β-catenin activation, providing new evidence for understanding the role of lipid accumulation in diabetic nephropathy.
Introduction
Diabetic nephropathy (DN) is the most common microvascular complication of diabetes mellitus and a major culprit causing end-stage renal disease (ESRD) in patients around the globe (7). Aberrant accumulation of lipid droplets has long been reported in metabolic diseases such as obesity and diabetes (12), which induce disorders of lipid metabolism, thus contributing to the development and progression of DN (16, 27).
In the physiological state, lipid deposition in cells or tissues is indispensable for maintaining the healthy state of the body. Lipid-producing cells, such as adipocytes, have the unique ability to store excess free fatty acids (FFAs) in lipid droplets, a plasticity not seen in other cells. In nonadipose tissues, aberrant accumulation of lipid droplets causes excess cytosolic FFAs, which lead to cell dysfunction and death (22), processes collectively designated as “lipotoxicity” (3).
Chronic kidney disease (CKD), associated with heavy albuminuria, accounts for the vast majority of causes for ESRD. Tubular atrophy, more accurately, predicts CKD progression compared with glomerular pathology (4, 19). In the pathophysiology of DN, the progression from microalbuminuria to macroalbuminuria is often so overlooked that tubular injuries progressively deteriorate without timely intervention. It has been suggested that proximal tubule reabsorption and intracellular accumulation of FFAs are cytotoxic and contribute to tubular atrophy pathogenesis (15). Undoubtedly, correction of lipid metabolism disorder would greatly attenuate the progression of tubular damage.
Our previous study showed that advanced oxidation protein products (AOPPs) were detected in vacuolated renal tubular epithelial cells (TECs) (19). AOPPs overload has also been found to be associated with deteriorated renal fibrosis in remnant kidney and experimental
Accumulating evidence has demonstrated that β-catenin activation participates in the emergence and development of various disease pathologies (28, 31, 36). Previous studies have shown that during macrophage differentiation from monocytes, Wnt1 promotes CD36 expression via activation of peroxisome proliferator-activated receptor gamma and β-catenin signaling (32). However, whether CD36 functions through β-catenin activation in renal TECs in DN is largely unknown and warrants further investigation.
In this study, we set out to investigate whether AOPPs induce lipotoxicity and aggravate renal tubular fibrosis via CD36-dependent β-catenin activation in DN.
Results
AOPPs overload induces lipid accumulation in proximal TECs and predisposes to renal function decline in DN patients
To demonstrate the relationship between AOPP and lipotoxicity in DN, the expression pattern of AOPP in correlation with lipid accumulation was examined in renal biopsy samples from DN patients. First, as shown by immunohistochemical staining (Fig. 1A, B), with increased AOPP expression in renal proximal tubular epithelial cells (PTECs), lipid metabolism-related markers CD36 and sterol regulatory element-binding protein 1 (SREBP-1), β-catenin and target gene matrix metalloproteinase-7 (MMP7), and profibrosis-related marker Fibronectin were significantly increased in DN tissues compared with normal controls. Second, there was increased accumulation of lipid droplets in renal TECs, as shown by toluidine blue staining (TBS) in semi-thin epoxy sections (Fig. 1C, D). Next, with lipid accumulation in renal TECs in DN, tubulointerstitial fibrosis (TIF) was worsened with increased fibrotic score, as detected by Sirius Red Stain (Fig. 1E, F).

In addition, the level of AOPP expression was increasing in accordance with the amount of lipid droplets during the disease progression from mild to severe DN (Fig. 2A). The number of lipid droplets in renal TECs was positively correlated with the level of AOPP (Fig. 2B, r = 0.916, p < 0.02). Further, the number of lipid droplets in DN was positively correlated with serum creatinine (Scr) (Fig. 2C, r = 0.634, p < 0.001) and urinary N-acetyl-β-

Biological Parameters for Patients with Diabetic Nephropathy
p < 0.001.
p < 0.05.
BMI, body mass index; DBP, diastolic blood pressure; DN, diabetic nephropathy; eGFR, estimated glomerular filtration rate; FT3, serum-free T3; FT4, serum-free T4; HDL, high-density lipoprotein; LDL, low-density lipoprotein; SBP, systolic blood pressure; Scr, serum creatinine; Serum BUN, serum blood urea nitrogen; TSH, thyroid stimulating hormone; urinary NAG, urinary N-acetyl-β-
Taken together, these data suggest that AOPPs overload promotes lipid accumulation, predisposing to renal fibrosis and renal function decline in DN patients.
AOPPs induce lipid overload and fibrosis in renal epithelial cells of diabetic mice
To demonstrate whether AOPPs play a role in inducing lipid accumulation and fibrosis in diabetic mice in vivo, we peritoneally injected AOPPs into diabetic mice and examined the accumulation of lipid and expression of related molecules in kidney tissues. We found that the expression of AOPP was increased in renal TECs in diabetic mice, with concomitant upregulation of CD36, SREBP-1, β-catenin, MMP7, and Fibronectin (Fig. 3A, B). AOPPs stimulate the accumulation of lipid droplets in renal TECs (Fig. 3C, D) and aggravate renal fibrosis (Fig. 3E, F).

These data indicate that AOPP is involved in lipid metabolism and TIF in
CD36 inhibition abrogates AOPPs-induced lipid metabolism and fibrosis in diabetic mice
To decipher the receptor pathway of AOPPs-induced lipotoxicity and renal fibrosis, CD36 expression was depleted by using small interfering RNA (siRNA) and downstream effects were examined. We found that knocking down CD36 attenuated AOPPs-induced lipid metabolism and renal fibrosis, as demonstrated by decreased expression of CD36, SREBP-1, β-catenin, MMP7, and Fibronectin (Fig. 4A–D) with decreased fibrotic score as detected by Sirius red stain (Fig. 4E, F). Further, CD36 inhibition improved renal function, as illustrated by a decreased level of Scr, BUN, urine albumin creatinine ratio (UACR), and urinary NAG and increased creatinine clearance rate (Ccr). In addition, knocking down CD36 significantly decreased the level of total cholesterol, triglyceride, LDL, and VLDL, and it increased HDL (Fig. 4G–K and Table 2).
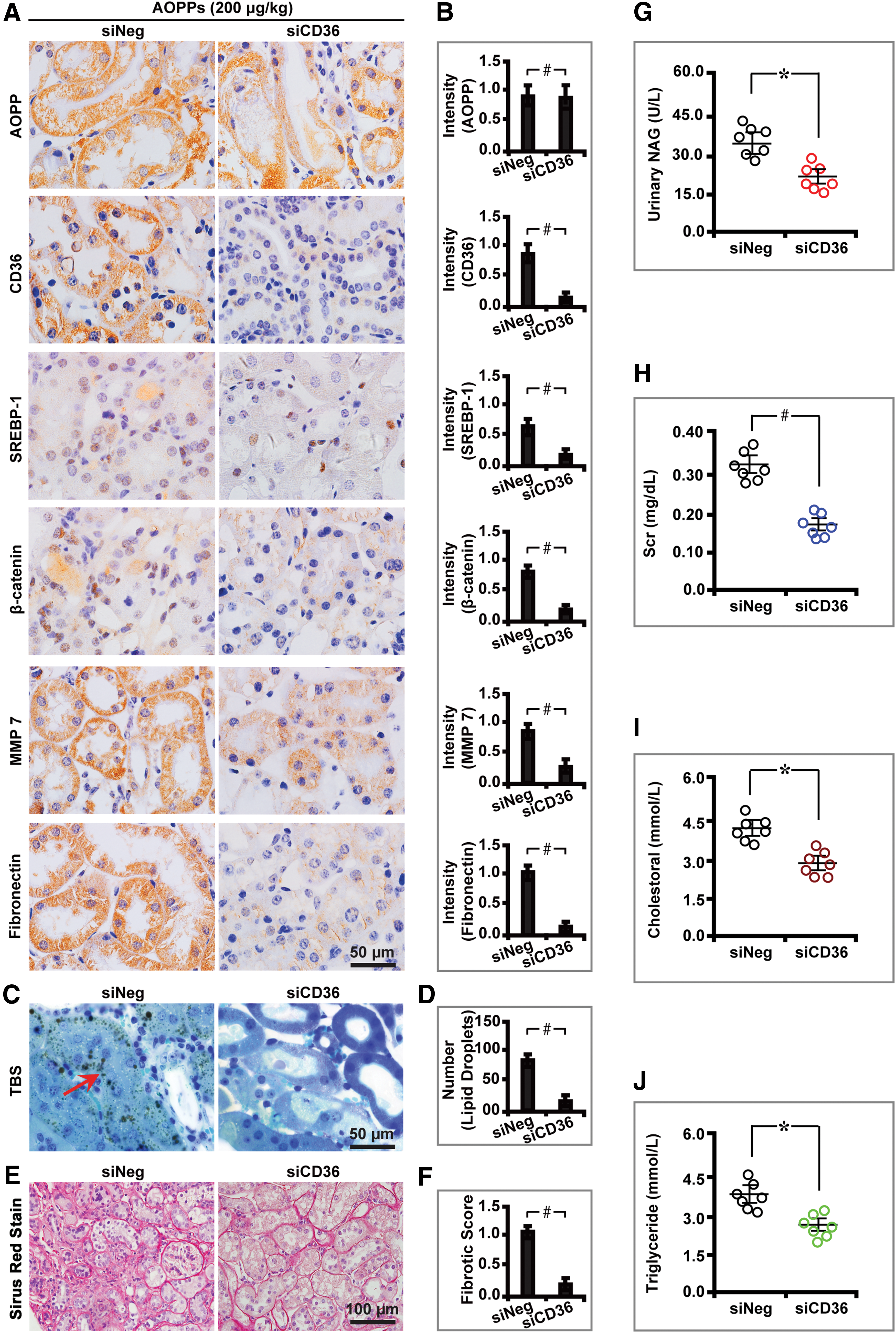
Biological Parameters for Advanced Oxidation Protein Products-Challenged Diabetic C57BL/6 Mice Treated with siCD36 at Week 24
p < 0.05 group STZ+AOPP+siCD36_24w versus group STZ+AOPP+siNeg_24w.
p < 0.05.
AOPP, advanced oxidation protein product; Ccr, creatinine clearance rate; siNeg, small interfering negative control; STZ, streptozotocin; STZ+AOPP, mice treated with STZ+AOPPs; UACR, urine albumin creatinine ratio.
These results collectively suggest that AOPPs mediate lipid metabolism and renal profibrosis via its receptor CD36 in diabetic mice in vivo.
Apocynin treatment attenuates AOPPs-induced lipid accumulation and fibrosis in diabetic mice
To investigate whether lipid accumulation was induced by AOPPs, we treated AOPPs-challenged diabetic mice with apocynin, an inhibitor of NADPH oxidase, which inhibits the production of AOPPs. Strikingly, we found that apocynin inhibited AOPPs-induced lipid metabolism and renal fibrosis, as illustrated by decreased expression of CD36, SREBP-1, β-catenin, MMP7, and Fibronectin (Fig. 5A, B). In addition, apocynin abrogated AOPPs-induced lipid accumulation and decreased tubular interstitial fibrosis in AOPPs-challenged diabetic mice, as illustrated by immunohistochemistry (Fig. 5A, B) and Western blot analysis (Fig. 5G). Mechanistically, Western blot analysis (Fig. 5H) further showed that AOPPs-induced lipotoxicity and fibrosis were blocked by CD36 depletion, as depicted by a decreased level of lipid-related molecules (CD36 and SREBP-1), inactivation of β-catenin signaling, and fibrosis (MMP7 and Fibronectin).
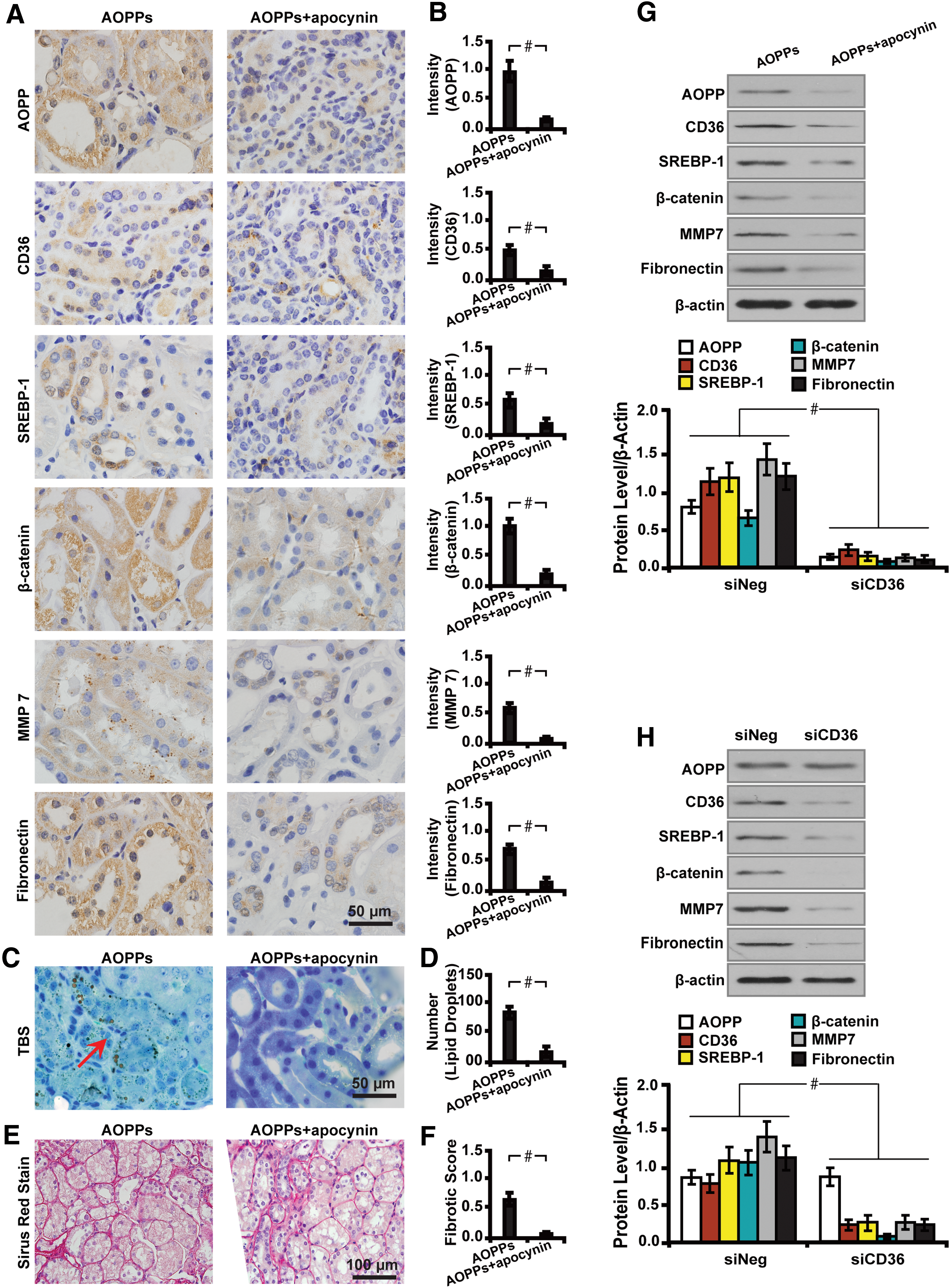
Collectively, these data indicate that AOPPs induce lipotoxicity via a CD36-dependent β-catenin activation mechanism.
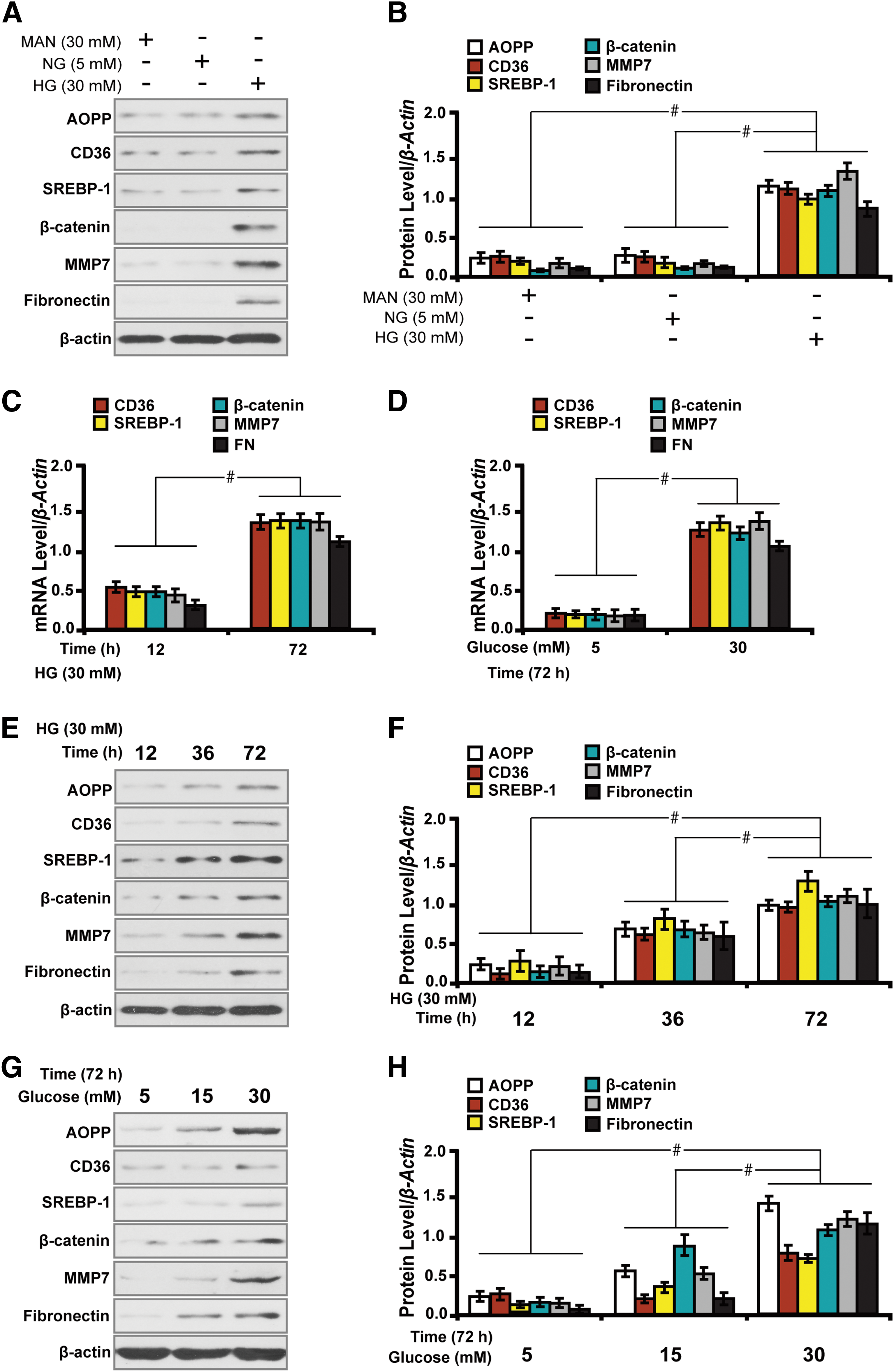
AOPPs overload time and dose dependently activates lipid metabolism and fibrosis in high glucose cultured HK-2 cells
To illustrate whether AOPPs could induce lipid accumulation and the relationship with fibrosis, we then examined the effect of AOPPs on the expression of related molecules in high glucose (HG, 30 mM) cultured HK-2 cells in different conditions for 72 h. Western blot analysis demonstrated that HG induced the accumulation of AOPP and the expression of CD36 and SREBP-1, β-catenin, MMP7, and Fibronectin; whereas mannitol (MAN) had no such effects (Fig. 6A, B). Further, HG time and dose dependently stimulated the expression of AOPP and related markers, as shown by quantitative real-time reverse transcription–polymerase chain reaction (qRT-PCR) (Fig. 6C, D) and Western blot analyses (Fig. 6E–H). These results suggest that HG incurs dysregulation of lipid metabolism and promotes profibrosis in HK-2 cells in vitro.
CD36 abolishment attenuates AOPPs-induced lipotoxicity and fibrosis via Wnt/β-catenin activation in HG-cultured HK-2 cells
To gain molecular insights into how CD36 mediates AOPPs-induced lipid metabolism, HG-cultured HK-2 cells were treated with CD36 siRNA and the interaction between CD36 and β-catenin was investigated. First, AOPPs-induced lipid uptake by HK-2 cells was significantly abrogated by CD36 knockdown (Fig. 7A, B). Second, CD36 siRNA remarkably decreased CD36 expression and the level of SREBP-1, β-catenin and target gene MMP7, and profibrosis-related marker Fibronecin by reverse transcription-PCR (RT-PCR) (Fig. 7C) and Western blot analysis (Fig. 7D–E). Notably, the SREBP-1/β-catenin co-localization (Fig. 7F) and positive correlation (Fig. 7G, r = 0.647, p < 0.001) were confirmed by co-immunofluorescence analysis, suggesting the role of lipid metabolism in the activation of β-catenin signaling. Moreover, AOPP co-operated with β-catenin to activate CD36 transcription, as demonstrated by the enrichment of β-catenin binding the CD36 promoter region detected by chromatin immunoprecipitation (ChIP) assay (Fig. 7H).

Taken together, these results suggest that AOPPs-induced lipotoxicity and fibrosis are mediated via CD36-dependent β-catenin activation in HG-cultured HK-2 cells.
CD36 attenuates AOPPs-induced mitochondrial injuries and function in HG-cultured HK-2 cells
To further elucidate the mechanisms of AOPPs in inducing lipotoxicity, we next sought to explore the effects of AOPPs accumulation on mitochondria and its function in the setting of HG exposure. First, CD36 depletion improved the morphology of AOPPs-induced mitochondrial injuries (Fig. 8A) and increased the number of cells with tubular mitochondria (Fig. 8B), as shown by Mitotracker analysis. Second, CD36 abolishment ameliorated AOPPs-induced mitochondrial injuries, resulting in decreased generation of adenosine triphosphate (ATP) (Fig. 8C), decreased oxygen consumption rate (OCR) (Fig. 8D), and decreased ROS production as examined by MitoSOX (Fig. 8E) and dichlorofluorescein-diacetate (DCFH-DA) assays (Fig. 8F).

These data illustrate that CD36 inhibition could reverse AOPPs-induced mitochondrial injuries and, hence, improve mitochondrial function in HG-cultured HK-2 cells in vitro.
Discussion
TEC injuries and TIF are characteristics of advanced
Based on our in vitro and in vivo findings, we propose a unique mechanism that AOPPs overload in HG causes lipid accumulation in renal tubules predisposing to TIF and renal function decline in DN via the CD36 receptor pathway. HG stimulated AOPP expression and augmented CD36-mediated lipid accumulation and profibrosis in HK-2 cells. Further, we provide mechanistic evidence that CD36 upregulation renders renal TECs susceptible to AOPPs-induced lipotoxicity via Wnt/β-catenin activation in DN.
Our conclusions are supported by several key observations. First, we identify a novel functional role for CD36 as an essential receptor for AOPPs-induced lipotoxicity in renal TECs. Previous reports have demonstrated that CD36 mediates macrophage apoptosis, contributing to atherosclerosis (9). CD36 has been shown to co-operate with αM integrin and Toll-like receptor 2 in cell-surface lipid domains on macrophages to promote lipid accumulation and modulate the inflammatory response (2). CD36 palmitoylation has also been shown to disrupt FFA metabolism and promote tissue inflammation in nonalcoholic steatohepatitis (34). In this study, we show for the first time that AOPPs play a critical role in inducing lipid accumulation in renal TECs via the CD36 receptor pathway. Second, we found that AOPP/CD36-induced lipotoxicity and injuries were mediated via Wnt/β-catenin signaling in DN. Previous research has shown a direct role for Wnt5a signaling in the pathogenesis of atherosclerosis, specifically the accumulation of lipid in macrophages and formation of foam cells (1). Third, our findings that apocynin, an inhibitor of NADPH oxidase blocking AOPPs production, abrogates AOPPs-induced lipid accumulation in renal TECs through CD36-mediated and Wnt/β-catenin-dependent mechanism, similar to the interaction between CD36 and TLR2 in macrophages, suggests that multiple, context-dependent extracellular stimuli of lipid metabolism may converge on the CD36 scavenger receptor to activate the Wnt/β-catenin pathway, triggering cascades of events causing renal fibrosis. Last, CD36 inhibition could reverse AOPPs-induced mitochondrial injuries and, hence, improve mitochondrial function in HG-cultured HK-2 cells, suggesting the critical role of CD36 in regulating mitochondrial function. In the context of diabetic milieu and diabetic complications, our findings provide new molecular insights into AOPPs-induced lipotoxicity and injuries of renal epithelial cells.
Cellular lipid homeostasis is controlled by balancing lipid uptake, synthesis, metabolism, and discharge processes (10, 35). Any aberrant incidence in the events just described will lead to lipid accumulation in resident cells, as detected in kidney biopsy samples (21). Moreover, clinical and experimental studies have reported that dyslipidemia has been shown to accelerate the rate of renal damage and leads to progressive loss of renal function among diabetic patients (6). Our immunostaining results indicated that in renal biopsy samples, molecules governing lipid uptake (CD36) and synthesis (SREBP-1) were upregulated, suggesting activation of lipid metabolism in renal TECs of DN. In addition, ChIP assay demonstrated that β-catenin was bound to the CD36 promoter region, suggesting activation of β-catenin signaling on AOPPs overload. Further, immunofluorescence microscopy illustrated the co-localization between SREBP-1 and active β-catenin, suggesting the interaction between lipid metabolism and activation of β-catenin signaling. This study also revealed that the increased number of lipid droplets correlated with declined renal function and worsened tubular damage, as illustrated by increased Scr and NAG and decreased eGFR. Of note, increased CD36 expression in renal TECs of human DN in vivo in this study may be caused by hyperglycemia, as we show that HG concentration stimulates CD36 expression in vitro. It is intriguing that CD36 expression was at a low level in renal TECs in diabetic mice in vivo, whereas AOPPs challenge augments its expression, indicating a role for CD36 in mediating AOPPs-induced downstream signaling.
Mechanistically, this study demonstrates that apocynin, an inhibitor of NADPH oxidase, inhibits AOPPs-induced lipotoxicity, suggesting that AOPPs overload triggers lipotoxicity in a CD36-dependent pathway. A series of recent studies have shown that proteinuria results in considerable toxicity and is closely associated with tubulointerstitial damage (24, 25). Multiple investigators have linked proteinuric toxicity to many macromolecules filtrated through the glomeruli, including fatty acids, albumin, transferrin, complement factors, and oxidized LDL (18, 30). More importantly, in DN, the damaged glomerulus permits FFA to be filtered and gain access to the previously unexposed proximal tubule luminal surface, where aberrant reabsorption could then occur. In addition, CD36 abolishment ameliorated AOPPs-induced mitochondrial injuries, resulting in decreased oxidative stress, adding new insights into mechanisms causing kidney injuries in DN.
In summary, this study demonstrates a significant role of AOPPs in triggering lipotoxicity and fibrosis via CD36-dependent Wnt/β-catenin activation, a potential novel therapeutic strategy for treating DN in a preclinical setting.
Materials and Methods
Antibodies and reagents
Monoclonal antibody against AOPPs (Clone 3F2) was generated according to the protocol previously described (19). Antibodies to CD36 (ab80080), SREBP-1 (ab28481), active β-catenin (ab15180), MMP7 (ab176325), Fibronectin (ab2413), and β-actin (ab8226) were purchased from Abcam (Cambridge, MA). Small interfering RNA (siRNA) for CD36 (siCD36) and nontargeting siRNA (small interfering negative control [siNeg]) were from Santa Cruz Biotechnology (Santa Cruz, CA).
Patients and renal biopsy studies
A total of 34 renal biopsy samples were obtained from type 2 diabetic patients from Huayin Diagnostic Center (Guangzhou, China) from 2010 to 2016. The inclusion criteria were: (i) type 2 diabetic patients with no history of using renal toxic or herbal medicine; (ii) the indications for performing the renal biopsy were proteinuria with or without microscopic hematuria and a fast drop in renal function; (iii) diabetic patients with no other renal diseases; (iv) type 2 diabetic patients with a duration of 6–10 years; and (v) all cases are in the advanced stage with nodular-type morphology. The antihypertensive medications these patients have been receiving mainly comprise angiotensin-converting enzyme inhibitors, angiotensin receptor blockers, and calcium channel blockers.
The morphological diagnosis of DN was confirmed by two individual renal pathologists (J.G. and X.B.). Renal biopsy tissues from subjects with thin basement membrane nephropathy (n = 10) were used as controls. Biological, anthropometric, and endocrine parameters, including weight, BMI, Scr, BUN, blood glucose, eGFR, SBP, diastolic blood pressure, 24-h proteinuria, FT3, FT4, and TSH, were recorded and analyzed. The Ethics Committee from Guangzhou Huayin Diagnostic Center (HY2016-004) specifically approved the use of patient tissue samples in this study, and written informed consent was obtained from each patient.
Animal studies
Male C57BL/6 mice (6–8 weeks of age) were kept in the Animal Center of Nanfang Hospital according to the policy of the Committee for Animal Usage. Streptozotocin (STZ, 50 mg/kg) was intraperitoneally injected for five consecutive days to induce diabetes, according to the protocol previously described. AOPPs-mouse serum albumin (MSA) was prepared as previously described (5). AOPPs (200 μg/kg) were injected intraperitoneally to mice once every 2 days after the induction of diabetes. Dimethyl sulfoxide (DMSO, 0.05%) and MSA (200 μg/kg) were included as controls. To investigate the effect of inhibiting CD36 signaling on lipid metabolism and renal fibrosis, 4 μg/g of siCD36 and siNeg were used to treat diabetic mice intraperitoneally once every 2 days, respectively, until the mice were euthanized till 24 weeks. To investigate the intervention of AOPPs in lipid accumulation, we treated AOPPs-challenged diabetic mice with daily intragastric administration of NADPH oxidase inhibitor, apocynin (50 mg/kg; Sigma), for 16 weeks. The Animal Ethics Committee at Nanfang Hospital, Southern Medical University (Guangzhou, China) specifically approved the study protocol according to the Guide for the Care and Use of Laboratory Animals published by the U.S. National Institutes of Health (NIH Publication No. 85-23, revised 1996).
Seven groups with six to seven animals each were studied: diabetic rats treated with DMSO, MSA, STZ, STZ+AOPPs, STZ+AOPPs+apocynin, STZ+AOPPs+siNeg, and STZ+AOPPs+siCD36, respectively. The mice were kept and spot urine at 10:00 am was collected for analysis of UACR. Blood glucose level was measured every week. The treatment continued after the STZ injection until the mice were euthanized at 16 or 24 weeks. Blood was drawn from the orbit vein, and plasma samples were prepared for analyzing Scr, BUN, and glucose level by using a Beckman Coulter AU480 Chemistry Analyzer (Beckman, CA). A creatinine assay kit (ab65340; Abcam) was used to determine Scr. An enzyme-linked immunosorbent assay (ELISA) kit specific for mouse albumin (E99-134; Bethyl Laboratories, Montgomery, TX) was used to determine urine albumin. Ccr was calculated as urinary creatinine (μM) × urine volume (mL/min)/Scr (μM), and it was expressed as mL·min−1·kg−1.
At 16 or 24 weeks after the induction of diabetes, mice were anesthetized with pentobarbital sodium (30 mg/kg). The left kidneys were harvested, fixed in 10% formalin in phosphate buffered saline (PBS), and embedded in paraffin for histological and immuhistochemical analyses. The right kidneys were snap-frozen and stored at −80°C for further analysis.
Cell culture studies
Human renal PTECs (HK-2; American Type Culture Collection, Rockville, MD) were cultured in normal glucose for 1 week, supplemented with Dulbecco's modified Eagle's medium/F-12 containing 10% fetal bovine serum, penicillin (200 U/mL), and streptomycin (200 μg/mL) (Gibco BRL, Grand Island, NY). HK-2 cells were grown to 80%–90% confluence and made quiescent by incubation overnight in a serum-free medium before experimentation.
Cells were then maintained in MAN (30 mM), normal glucose (NG, 5 mM), or HG (30 mM) according to each experiment. AOPPs were used to treat HG-cultured HK-2 cells at indicated times and concentrations.
Analysis of mitochondrial morphology and function
Mitochondrial fragmentation studies
HK-2 cells were incubated with rosamine-based MitoTracker probe (Invitrogen, Cambridge, MA) at 100 nM for 30 min to visualize mitochondrial morphology. The cells were then washed with PBS and fixed with 3.7% formaldehyde in growth medium after incubation. Mitochondrial fragmentation was quantified according to a protocol previously described (11, 26). Briefly, mitochondrial morphology was evaluated by an investigator blinded to the experimental procedure. The following criteria were used: tubular (>75% of mitochondria with tubular length >5 mm), intermediate (25%–75% of mitochondria with tubular length >5 mm), or fragmented (<25% of mitochondria with tubular length >5 mm). The categorization of mitochondrial morphology was evaluated.
ATP quantification assay
ATP levels were examined in cells with or without treatment by using a luciferase-based ATP Determination Kit (Invitrogen, Cambridge, MA) according to the manufacturer's protocol. Briefly, HK-2 cells were grown to 70%–80% confluence in a 24-well plate. The cells were then washed with PBS and treated with ATP-releasing agent (Sigma-Aldrich, St. Louis, MO). ATP levels were determined in 10 μL of cells in the ATP-releasing agent, and the remainder was used to quantify protein levels by using a Pierce BCA Protein Assay Kit (Thermo Scientific, Waltham, MA) with bovine serum albumin as a standard.
Oxygen consumption rate
The phosphorescent oxygen-sensing probe MitoXpress (Cayman Chemical, MI) was used to measure extracellular oxygen consumption (20). Briefly, HK-2 cells were first treated with serum-free medium containing either control, AOPPs or CD36. Then, mineral oil was used to prevent the loss of extracellular oxygen. Next, fluorescence was measured with 380 nm excitation every 3 min over a 3-h period. The slope of the curve represented the rate of oxygen consumption (μs/h). The relative change in OCR over control cells was recorded and presented.
Measurement of intracellular ROS
Cells were incubated with 10 μM of DCFH-DA at 37°C for 15 min. Intracellular ROS level was determined by measuring 10,000 events per sample by a flow cytometer (FACS Calibur; BD Biosciences, San Jose, CA) with a 488-nm excitation laser.
Transfection of siRNAs
HK-2 cells were plated in six-well plates at a density of 0.5 × 105 cells/mL and cultured for 24 h before experimentation. SiRNA oligonucleotides were used to knock down the expression of murine genes by using Lipofectamine RNAiMAX (Invitrogen, Carlsbad, CA). According to the manufacturer's protocol, cells were transfected with 1 μg of siRNA in reduced serum medium (OPTI-MEM-I; Invitrogen, Carlsbad, CA) and then harvested for further study after 48 h of transfection. Total RNA and protein were extracted for further analysis.
Semi-qRT-PCR analysis
Total RNA from cells were extracted by using TRIzol reagent (MRC, Cincinnati, OH), and first-strand cDNA was synthesized by using 2 μg of total RNA according to the manufacturer's instructions. Semi-qRT-PCR analysis was performed in triplicate with Power PCR SYBR Green Master Mix (Applied Biosystems, Carlsbad, CA) by using the ABI PRISM 7500 FAST Real-Time PCR System (Applied Biosystems). The ΔΔCT method was used to calculate relative expression, and the results were normalized to β-actin expression. Primer sequences used in RT-PCR are shown in Table 3.
Primer Sets Used in Real-Time Reverse Transcription–Polymerase Chain Reaction
Immunoblot analyses and immunoprecipitation
Cell lysates were separated in parallel on two 10% denaturing sodium dodecyl sulfate-polyacrylamide gels and probed with anti-AOPP, anti-CD36, anti-SREBP-1, anti-β-catenin, anti-MMP7, anti-FN, and anti-β-actin for 16 h at 4°C overnight. The secondary antibody (horseradish peroxidase-labeled IgG anti-rabbit/mouse antibody, Invitrogen, Cambridge, MA) was used at 1:3000 dilution for 1 h after thorough washing in tris-buffered saline Tween 20 buffer. The supersignal-enhanced chemoluminescent substrate (Pierce Biotechnology, Inc., Rockford, IL) was applied to the probed membrane and exposed before the protein bands were visualized on radiograph films (Super Rx, Fuji Photo Film, Tokyo). Quantification was performed by measuring the intensity of the bands using Image J analysis software (Image J 1.44; National Institute of Health).
Lipid uptake analysis
HG-cultured HK-2 cells were challenged with AOPPs for 48 h and then treated with dil-oxLDL (10 ng/mL; Yiyuan, Guangzhou, China) for 30 min. Next, the cells were washed twice with ice-cold PBS and observed by using the Zeiss Confocal Microscope Imaging System (Carl Zeiss, Germany).
Immunofluorescence and immunohistochemical analysis
HK-2 cells as well as tissue samples from patients and rats were labeled with primary antibodies. For immunofluorescence staining, Alexa Fluor 594-conjugated goat anti-mouse IgG and Alexa Fluor 488-conjugated goat anti-rabbit IgG (1:1000; Invitrogen, Cambridge, MA) were used for secondary antibodies; nuclei were counterstained with 4,6-diamidino-2-phenylindole (DAPI) and coverslipped with aqueous mounting medium (CTS011; BD Bioscience, Minneapolis, MN). For immunohistochemistry, EnVision™ Detection Systems peroxidase/diaminobenzidine, Rabbit/Mouse kit (K4065; Dako, Carpinteria, CA) was used. Nuclei were counterstained with hematoxylin and coverslipped with Permount mounting medium (00-4960-56; eBioscience, San Diego, CA).
Samples were evaluated semiquantitatively by systematically selecting without bias 20 fields for analysis. Images were taken with Zeiss Confocal Microscope Imaging System (Carl Zeiss) with appropriate filters. Staining intensity was measured with Image J analysis software; PBS instead of primary antibodies served as a negative control.
Examination of lipids by toluidine blue stain
For kidney tissues from patients and mice, several 1-mm cubes from the cortex of kidneys were processed as previously described (5, 31). One-micrometer-thick epoxy sections were stained with TBS. Briefly, blocks were cut and 1-μm-thick sections were stained with 2% toluidine blue for 20 s, rinsed in PBS, air-dried, and coverslipped with mounting medium.
Slides were viewed, and representative micrographs were examined and imaged with Olympus Microscope Imaging System fitted with an Olympus digital camera (BX51; Olympus, Japan). The number of lipid droplets was evaluated and analyzed by measuring the number of lipid droplets per total section area examined.
Renal fibrosis evaluation
Formalin-fixed, paraffin-embedded kidney tissues from rats and patients were cut into 5-μm-thick sections and stained with Picric acid-Sirius red staining. The degree of fibrosis was evaluated in eight representative fields (magnification 40 × ) fitted with an Olympus digital camera (BX51; Olympus). The TIF was semi-quantitatively evaluated by calculating the ratio between the Sirius red-positive surfaces and the total section area examined (Image J 1.44; National Institute of Health).
Statistical analysis
Data are presented as mean ± standard deviation (SD) values. Independent-samples t-test and one-way analysis of variance followed by Student-Newman-Keuls post hoc test were used to test statistical significance between groups. Pearson correlation analysis was used to analyze the number of lipid droplets in correlation with patients' biological parameters, as well as the correlation between the expression level of β-catenin and SREBP-1. All statistical tests were performed by using SPSS 12.0 (SPSS, Inc., Chicago, IL). The significance level was set at p < 0.05 to indicate statistical significance.
Footnotes
Acknowledgments
This study was supported by the National Natural Science Foundation of China (no. 81100496, 81873616, 81401180, and 81730019), National Key R&D Program of China (2018YFC1314000), Special Fund from Chinese Society of Nephrology (no. 13030370422), Guangdong Natural Science Foundation (no. 2016A030313581), Guangzhou Science and Technology Planning Project-Key Projects of Scientific Research (no. 201607020019), Guangdong Provincial Science and Technology Planning Project (no. 2014A020209035), “Group-type” Special Support Project for Education Talents in Universities (G619080438, 4SG19002G, 4SG19044G), Outstanding Youths Development Scheme of Nanfang Hospital, Southern Medical University (no. 2015J009) to X.B., President Foundation of Nanfang Hospital, Southern Medical University to X.B. (no. 2017Z001) and X.L. (no. 2017B002), and Guangdong Science and Technology Planning Project to J.G. (no. 2014A020209035) and J.T. (no. 2017ZC0070).
Author Disclosure Statement
No competing financial interests exist.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.