
Abstract
Aims:
Inflammasome activation plays a pivotal role in many inflammatory diseases. Given that connexin (Cx) channels regulate numerous cellular events leading to inflammasome activation, we determined whether and how connexin affected inflammasome activation and inflammatory cell injury.
Results:
Exposure of mouse peritoneal macrophages (PMs) to lipopolysaccharide (LPS) plus ATP caused NLRP3 inflammasome activation, together with an increased connexin43 (Cx43). Inhibition of Cx43 blunted inflammasome activation. Consistently, PMs from the Cx43 heterozygous mouse (Cx43+/−) exhibited weak inflammasome activation, in comparison with those from the Cx43+/+ mouse. Further analysis revealed that inflammasome activation was preceded by an increased reactive oxygen species (ROS) production, nicotinamide adenine dinucleotide phosphate hydrogen (NADPH) oxidase 2 (NOX2), protein carbonylation, and mitogen-activated protein kinase (MAPK) activation. Suppression of ROS with antioxidant, downregulation of NOX2 with small interfering RNA (siRNA), or inhibition of NADPH oxidase or MAPKs with inhibitors blocked Cx43 elevation and inflammasome activation. Intriguingly, suppression of Cx43 also blunted NOX2 expression, protein carbonylation, p38 phosphorylation, and inflammasome activation. In a model of acute renal injury induced by LPS, the Cx43+/− mouse exhibited a significantly lower level of blood interleukin-1β (IL-1β), blood urea nitrogen, and urinary protein, together with milder renal pathological changes and renal expression of NLRP3 and NOX4, as compared with the Cx43+/+ mouse. Moreover, inhibition of gap junctions suppressed IL-1β- and tumor necrosis factor-α-induced expression of NOX4 in glomerular podocytes and tubular epithelial cells.
Innovation and Conclusion:
Our study indicates that Cx43 contributes to inflammasome activation and the progression of renal inflammatory cell injury through modulation of intracellular redox status. Cx43 could be a novel target for the treatment of certain inflammatory diseases.
Introduction
Acute kidney injury (AKI), caused by sepsis, is characterized by a whole-body inflammatory reaction, in which inflammatory mediators play a vital role (4). Currently, the molecular mechanisms controlling the production of these mediators are still incompletely understood. It is highly desirable to have a better understanding of the mechanisms of AKI and find novel therapeutic targets.
Inflammasome activation is one of the major cellular events contributing to the production of inflammatory mediators (13). It takes part in bacterial sepsis, including sepsis-initiated AKI. Activation of NLRP3 inflammasome in AKI and its role in renal cell injury have been reported. Targeting NLRP3 inflammasome has been proposed as a novel therapeutic approach to prevent and treat inflammatory renal diseases (5, 6).
Here, we demonstrated, for the first time, that connexin43 (Cx43) modulated inflammasome activation and renal inflammatory injury through mechanisms involving its regulation of nicotinamide adenine dinucleotide phosphate hydrogen oxidases and redox signaling. Intriguingly, Cx43 itself was also under the control of the intracellular redox state. Our study, thus, identified Cx43 as a key intermediate linking oxidative stress to inflammation and cell injury. Targeting Cx43 could be used to treat certain inflammatory diseases.
Inflammasome is a multiprotein complex that activates caspase 1 (Casp1) and promotes the secretion of the pro-inflammatory cytokines interleukin-1β (IL-1β) and interleukin-18 (IL-18) (6, 13, 56). Inflammasome activation occurs in a variety of pathological situations. Among various types of inflammasomes, NLRP3 inflammasome is the most extensively studied because of its role in antipathogenic responses and in the pathology of immune diseases. NLRP3 inflammasome, composed of the Nod-like receptor protein NLRP3, CARDINAL, the adaptor protein ASC, and Casp1, is activated by multiple stimuli. Its activation involves two steps, that is, the priming and the activation steps. In the priming step, NF-κB is activated after signals from TLRs (56), which leads to upregulation of NLRP3 and pro-IL-1β. The second activation step involves signals arising from cellular stress or danger, such as oxidative stress, lysosomal damage, ATP, and cytosolic K+ efflux.
Reactive oxygen species (ROS) is one of the central mechanisms mediating inflammasome activation (22, 36, 38, 50, 56). Many molecules activating NLRP3 inflammasome stimulate ROS generation. Inhibition of ROS or superoxide-generating nicotinamide adenine dinucleotide phosphate hydrogen (NADPH) oxidases attenuates inflammasome activation (1, 39). Macrophages deficient in NADPH oxidases also exhibit a weak activation of inflammasome (38, 50). ROS is a crucial secondary messenger signaling NLRP3 inflammasome activation.
Gap junctions (Gjs), formed by the specific protein term connexins (Cxs), are intercellular channels that allow the direct exchange of small signaling molecules among adjacent cells. Gjs are formed by two hemichannels consisting of six Cx molecules. Docking of two hemichannels in apposed membranes of two neighboring cells forms an intact Gj channel. Up to now, more than 20 different isoforms of Cxs have been reported. Among them, connexin43 (Cx43) is extensively studied because of its predominant and ubiquitous expression in almost all cell types. Cx43 has been shown to regulate a wide range of cellular processes and functions (49, 62).
Several lines of evidence prompted us to speculate that Cx43 might be critically involved in the regulation of inflammasome activation. First, Cx43 regulates intracellular redox status. We have recently reported that Cx43 regulated oxidative cell responses in glomerular podocytes and renal tubular epithelial cells through mechanisms involving its inhibition of NADPH oxidase 4 (Nox4) and thioredoxin-interacting Protein (TXNIP) (17, 61), two pivotal molecules underlying inflammasome activation (38, 50). Second, increased membrane permeability was intimately related to NLRP3 inflammasome activation. Membrane-permeable channels and molecules, such as pannexin, K+, Ca2+, and ATP, are all crucial regulators of inflammasome activation (13, 56). Fascinatingly, Gj hemichannels share many properties with pannexin and permit intra- and extracellular exchange of Ca2+, ATP and K+ (9, 14, 15, 42, 62). In this context, Gj hemichannels themselves may affect inflammasome activation. Third, Cx43 has been reported to regulate activation, migration, infiltration, adhesion, and cytokine production of immune cells (14, 34, 37, 42, 44). Many of these responses are influenced by inflammasome activation. Recently, Cx43 has been implicated in the renal inflammatory processes in several models of renal diseases (2, 29). However, the mechanisms involved are not clearly defined. Based on these backgrounds, we hypothesized that Cx43 might regulate inflammasome activation and affect the renal inflammatory process. This study was conducted to address this hypothesis.
Here, we present our results that activation of inflammasome is associated with an increased expression of Cx43, which, in turn, contributes to inflammasome activation through the regulation of intracellular redox status. Our study, thus, characterizes Cx43 as a determinant of inflammasome activation and inflammatory renal injury. Cx43 might be a novel therapeutic target for the treatment of inflammatory diseases.
Results
Cx43 contributes to inflammasome activation
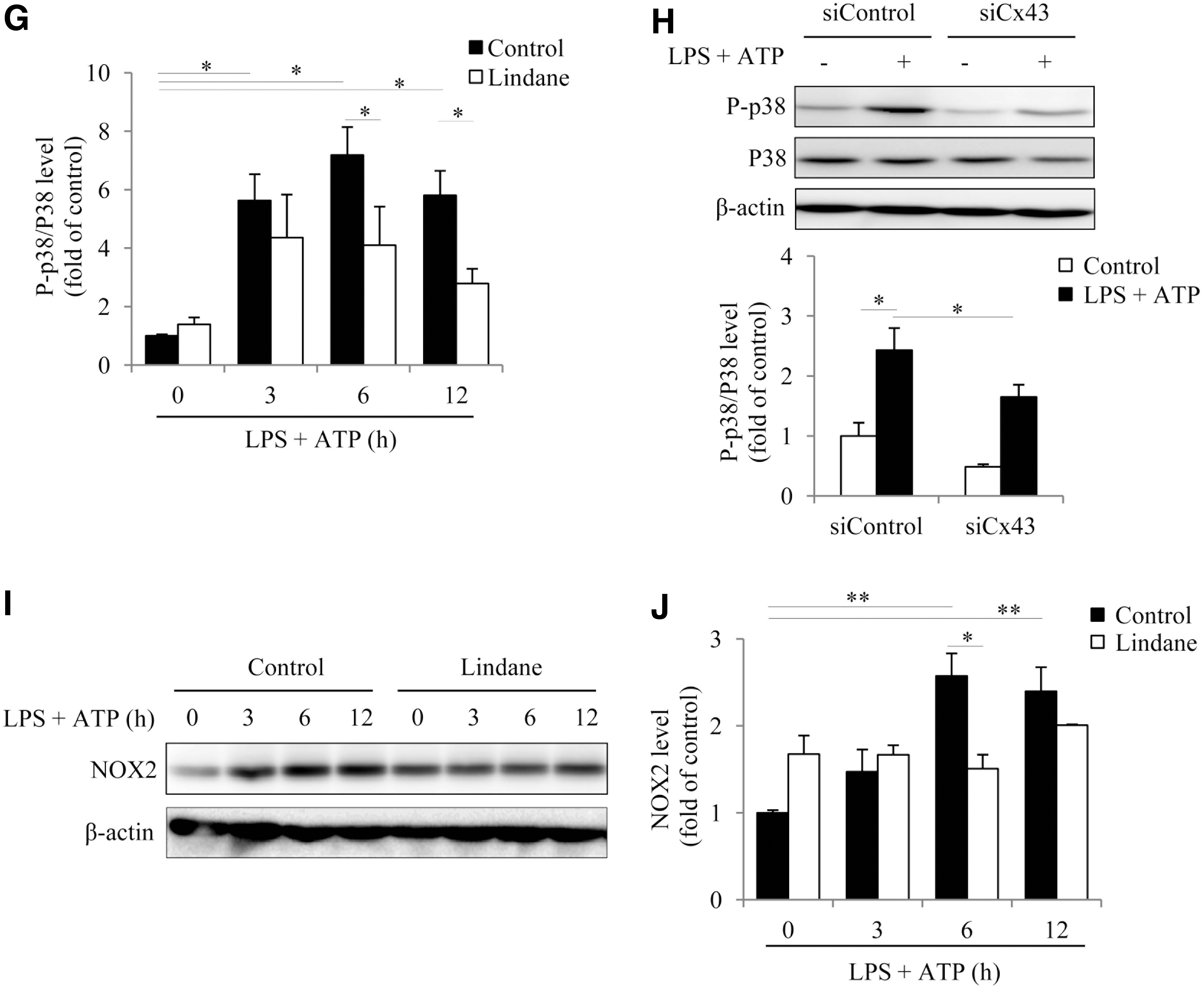
To determine the role of Cx43 in NLRP3 inflammasome activation, we first examined whether inflammasome activation was associated with an altered Cx43. For this purpose, peritoneal macrophages (PMs) were primed with 10 μg/mL lipopolysaccharide (LPS), followed by stimulation with 3 mM ATP to activate inflammasome. This treatment led to an activation of inflammasome, as evidenced by the elevated secretion of IL-1β and Casp1, as well as an increased expression of NLRP3 (Fig. 1A, B). The increment occurred 3–6 h after the stimulation and lasted for at least 12 h. Intriguingly, these changes were associated with a marked elevation in Cx43 level (Fig. 1A, C).
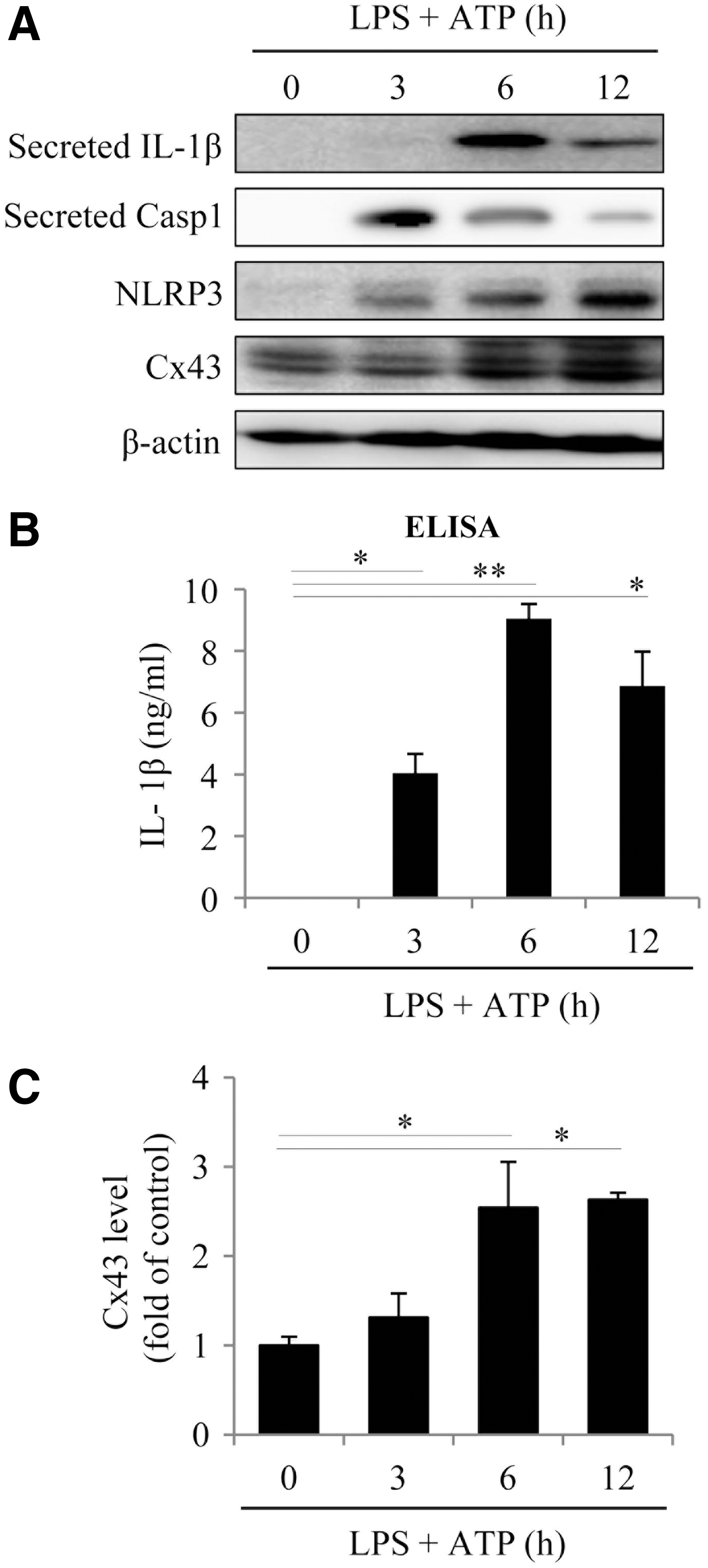
To determine the role of Cx43 in inflammasome activation, we compared the difference between PMs from wild-type (WT, Cx43+/+) and heterozygous (Cx43+/−) mice. Polymerase chain reaction (PCR) analysis revealed that these mice exhibited a typical pattern of Cx43+/+ and Cx43+/− genotype (Fig. 2A, upper inset). Stimulation of PMs with LPS plus ATP elevated IL-1β and NLRP3, which was significantly higher in Cx43+/+ PMs than in Cx43+/− PMs (Fig. 2A, B), indicating the participation of Cx43 in inflammasome activation. To further establish the role of Cx43, we used Cx43 small interfering RNA (siRNA) and Gj inhibitors. Figure 2C–F show that Cx43 siRNA or Gj inhibitors, lindane and flufenamic acid (FFA), markedly suppressed inflammasome activation in Cx43+/+ macrophages.

Of note, in cultured bone marrow-derived macrophages (BMDMs), similar results were also achieved (Supplementary Fig. S1). These results indicate that Cx43 regulated NLRP3 inflammasome activation.
Oxidative stress mediates inflammasome activation
To explore the mechanisms underlying the effect of Cx43, we focused on intracellular oxidative status. Previous studies have shown that ROS is a central factor mediating inflammasome activation (56) and that Cx43 modulates cell responses to oxidative stress (17, 61). To establish the role of oxidative stress in inflammasome activation under our experimental settings, we examined the influence of inflammasome activators on ROS generation and protein carbonylation. Figure 3A shows that stimulation of macrophages with LPS plus ATP increased ROS and superoxide generation, as revealed by the markedly increased fluorescence intensity after incubation of cells with the fluorescent probes. Consistently, the level of protein carbonyl, an index of oxidative protein damage, was also elevated (Fig. 3B, C). Besides, suppression of oxidative stress with a general antioxidant glutathion (GSH) or a mitochondria-targeted antioxidant Mito-TEMPO abolished inflammasome activation (Fig. 3D–G). These observations, thus, indicate a vital role of oxidative stress in inflammasome activation.
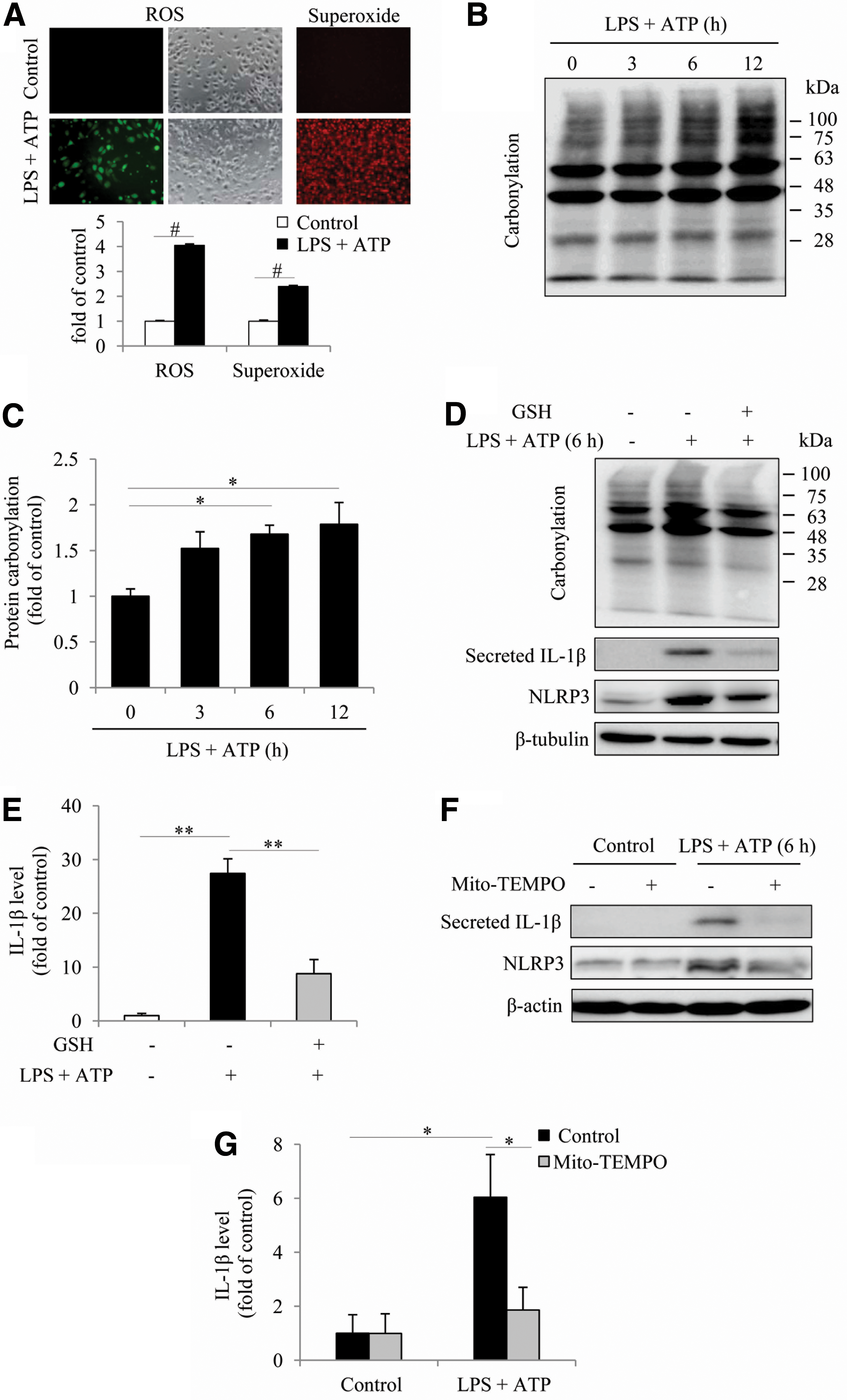
Further analysis revealed that induction of inflammasome activation was associated with a time-dependent elevation in NOX2 (Fig. 4A, B), a phagocytic NADPH oxidase (39, 50). Downregulation of NOX2 with specific siRNA or inhibition of NADPH oxidase with apocynin or diphenyleneiodoniumchloride (DPI) significantly suppressed inflammasome activation (Fig. 4C–F). These results indicate that NADPH oxidase played a crucial role in inflammasome activation.

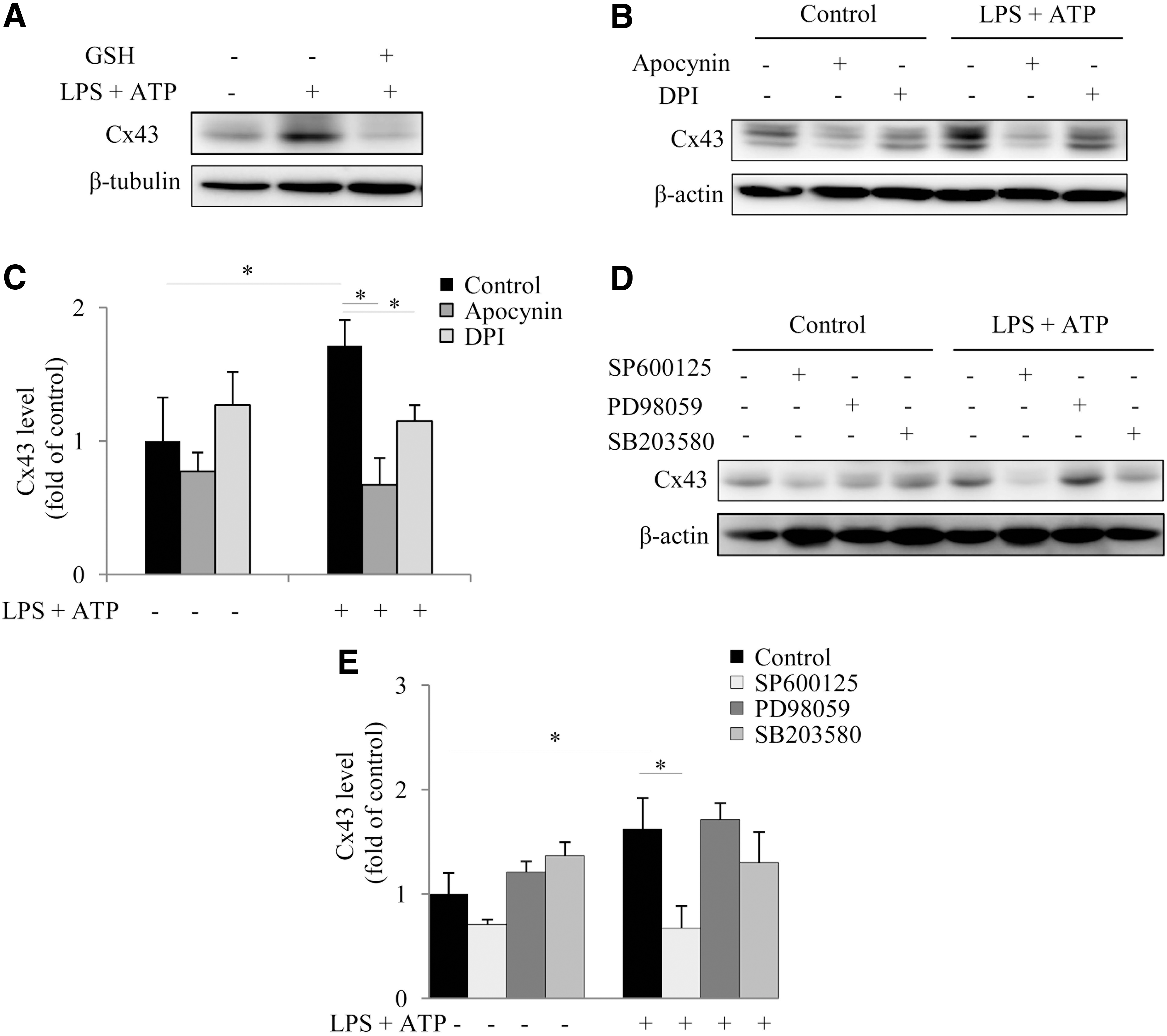
It is reported that mitogen-activated protein kinases (MAPKs) are activated by oxidative stress and mediate oxidative cell responses (56); we, therefore, examined the possible involvement of these kinases. Figure 5A shows that activation of inflammasome was associated with increased activation of p38, ERK, and JNK without great influence on their protein levels. In the presence of antioxidant GSH or NADPH oxidase inhibitor apocynin and DPI, MAPK activation at the 6-h point was significantly blocked, indicative of a causative role of oxidative stress in activation of these kinases (Fig. 5B–D).

To determine the role of these kinases in inflammasome activation, we treated cells with various kinase inhibitors (JNK inhibitor SP600125, ERK inhibitor PD98059, and p38 inhibitor SB203580). Interestingly, all the inhibitors potently suppressed inflammasome activation, as observed at the 6-h point after exposure to LPS and ATP (Fig. 5E, F), suggesting a mediating role of these kinases in inflammasome activation.
We and others have reported that Cx43 expression and function are regulated by intracellular redox status (7, 61). It is possible that the elevated Cx43 was also caused by oxidative stress. Consistent with this notion, antioxidant GSH abolished the elevation of Cx43 (Fig. 6A). Consistently, NADPH oxidase inhibitors achieved the same effect (Fig. 6B, C). To determine the kinases involved in the induction of Cx43, we used MAPK inhibitors. Only JNK inhibitor SP600125 significantly prevented the elevation of Cx43 (Fig. 6D, E), indicating a possible implication of JNK in mediating the oxidative stress-induced increase of Cx43.
Cx43 regulates intracellular redox status
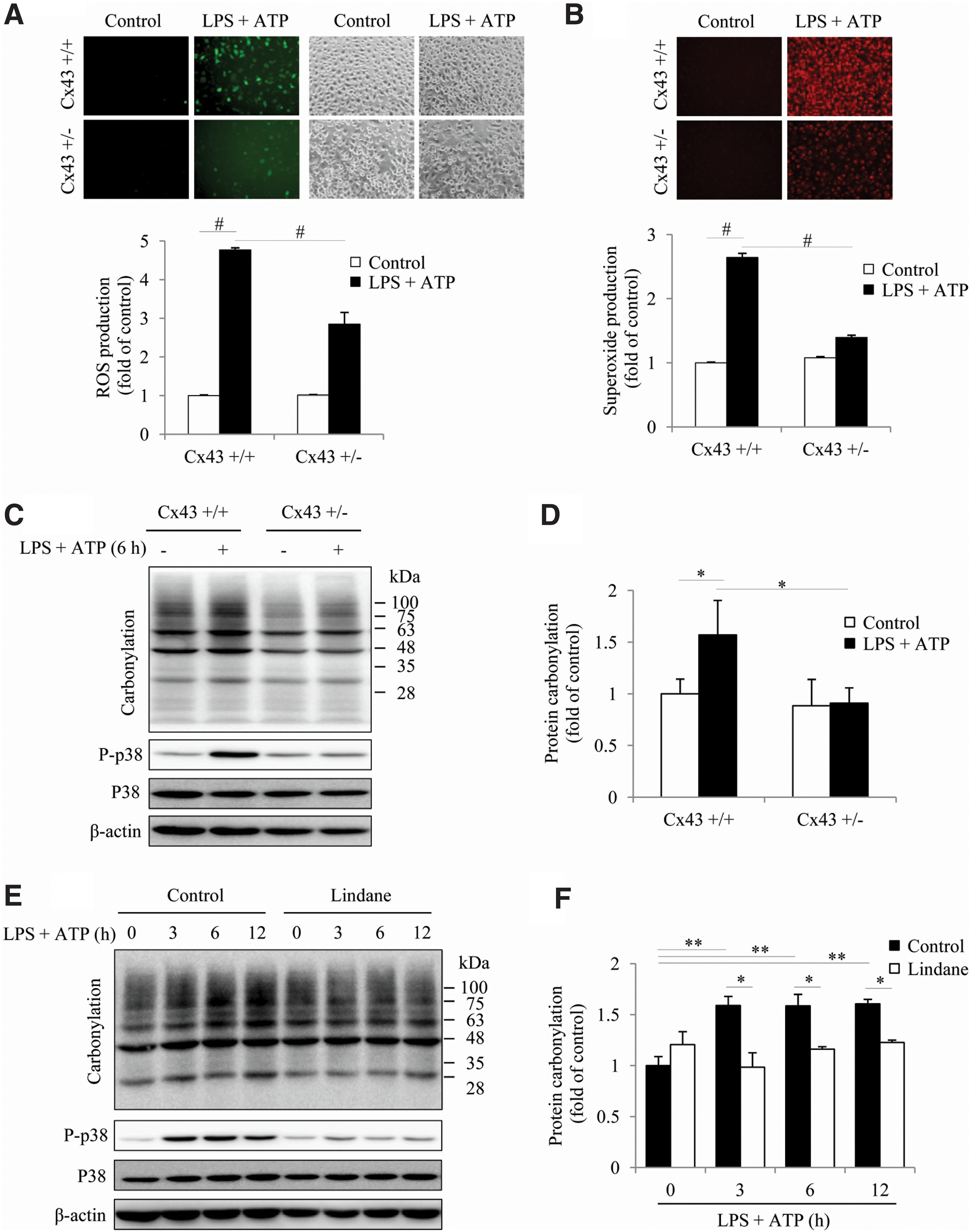
Considering the importance of ROS in inflammasome activation (56), we, therefore, tested whether Cx43 altered oxidative status. For this purpose, we compared the level of ROS generation, protein carbonylation, and activation of oxidative sensitive P38 between Cx43+/+ and Cx43+/− PMs. Figure 7A and B show that LPS+ATP treatment caused an elevation in ROS and superoxide, as indicated by the increased fluorescent intensity. In comparison with Cx43+/+ PMs, the fluorescent intensity in Cx43+/− PMs was obviously lower. Consistently, Cx43+/− PMs also exhibited a lower level of protein carbonylation and a weaker activation of oxidative sensitive p38 in response to LPS and ATP (Fig. 7C, D), indicating an involvement of Cx43 in the regulation of intracellular redox status. In further support of this notion, inhibition of Gjs with lindane or silencing Cx43 with siRNA also achieved similar effects (Fig. 7E–H). Further analysis revealed that inhibition of Gj suppressed NOX2 expression (Fig. 7I, J). These observations indicate that Cx43 regulated NADPH oxidase and affected intracellular redox status in macrophages.
Cx43 contributes to LPS-initiated inflammatory injury and renal dysfunction
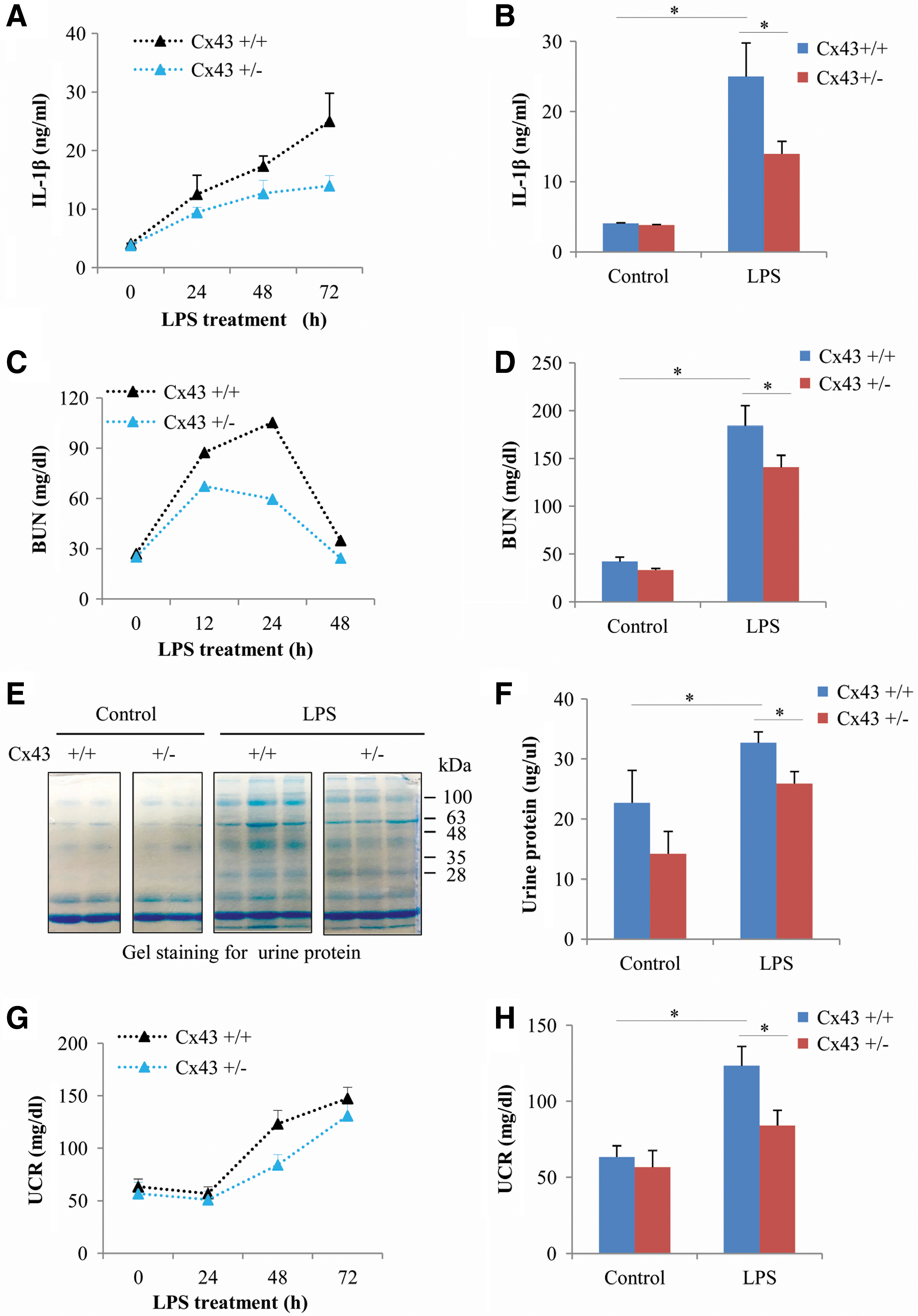
Because Cx43 affected inflammasome activation and many inflammatory responses (9, 24, 34, 37, 40, 44, 57, 60), we speculated that it could contribute to the inflammatory renal injury. To test this possibility, we have used an LPS-induced AKI model, in which the participation of NLRP3 inflammasome has been reported (11, 35). Figure 8A and B show that administration of 40 mg/kg LPS to mice caused a time-dependent increase in serum IL-1β, which was significantly lower in Cx43+/− mice than that in Cx43+/+ mice. Consistently, renal dysfunction, as indicated by blood urea nitrogen (BUN), urinary protein, and urinary creatinine (UCR), was also significantly milder in Cx43+/− mice (Fig. 8C–H).
To further confirm the role of Cx43, we examined the renal histological changes by hematoxylin and eosin (H&E) staining. LPS administration induced renal tubular cell injury, as indicated by the shortened or lost brush border, vacuolization of tubular epithelial cells, tubular cell swelling, and occasional tubular cell detachment in Cx43+/+ mice. Consistent with the less severe deterioration in renal function, Cx43+/− mice also exhibited relatively milder changes in renal histology, as compared with Cx43+/+ mice (Fig. 9A).

To further confirm the difference, we detected renal cell apoptosis by using TUNEL staining and Western blot analysis of caspase 3 (Casp3) activation. We found several clusters of TUNEL-positive apoptotic cells in Cx43+/+ renal tissue. In contrast, in Cx43+/− renal tissue, there was a less number of apoptotic cells that were sporadically distributed (Fig. 9B). Consistently, the Casp3 activation, as indicated by the level of cleaved Casp3, was also significantly lower in Cx43+/− renal tissue than Cx43+/+ tissue (Fig. 9C, D).
Previous studies have shown that LPS-induced acute cell injury in vivo was related to NADPH oxidase and oxidative stress (31). These reports prompted us to ask whether Cx43 affected LPS-induced oxidative stress in vivo. To this end, we examined the renal expression of NOX4, a predominant isoform of NADPH oxidase in the kidney (8, 61). Figure 9E and F show that administration of 40 mg/kg LPS to mice caused an elevation in NOX4 protein in renal tissue, which was significantly more pronounced in Cx43+/+ mice than that in Cx43+/− mice. Moreover, in agreement with our in vitro finding in macrophages, the elevation of NLRP3 induced by LPS in renal tissues was also significantly lower in Cx43+/− mice than Cx43+/+ mice. These observations indicate that Cx43 regulated oxidative status and inflammasome activation in vivo.
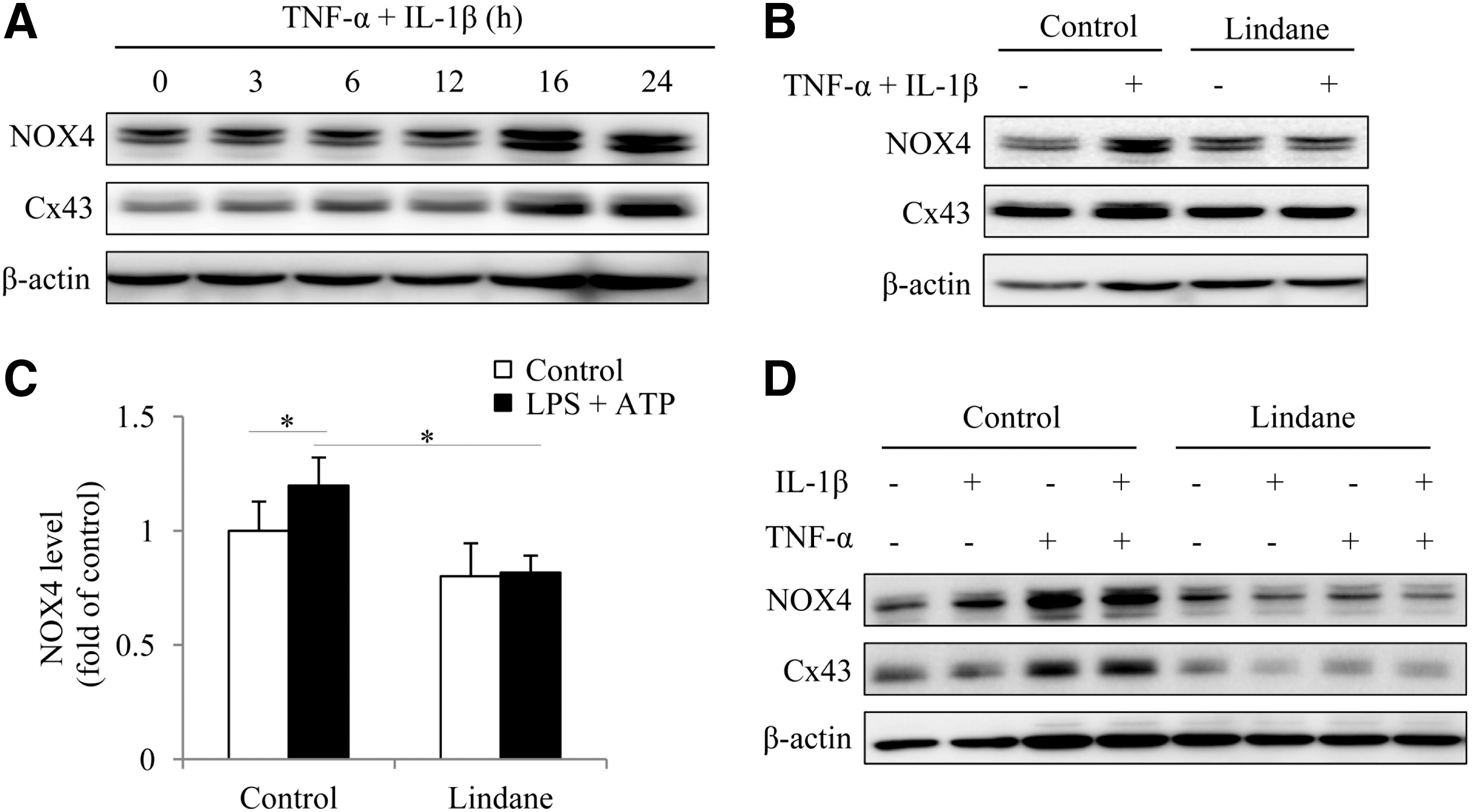
The different renal expression of NOX4 between Cx43+/+ and Cx43+/− mice prompted us to explore the potential mechanisms involved. Theoretically, the difference could be a result of the different blood level of inflammatory mediators (45), or the altered cell response to the mediators caused by Cx43, or both at the same time. To test these possibilities, we examined the expression of NOX4 under the stimulation of inflammatory mediators and its influence by Gj inhibitor in cultured glomerular podocytes and renal tubular epithelial cells. Figure 10A shows that combined stimulation of NRK cells with IL-1β and tumor necrosis factor-α (TNF)-α induced a time-dependent elevation in NOX4, which was associated with increased Cx43. Inhibition of Cx43 with lindane potently suppressed the expression of NOX4 (Fig. 10B, C). Similarly, lindane also suppressed NOX4 in cultured glomerular podocytes (Fig. 10D). These results indicate that Cx43 also contributed to the inflammatory mediator-elicited expression of NOX4 in renal cells.
Discussion
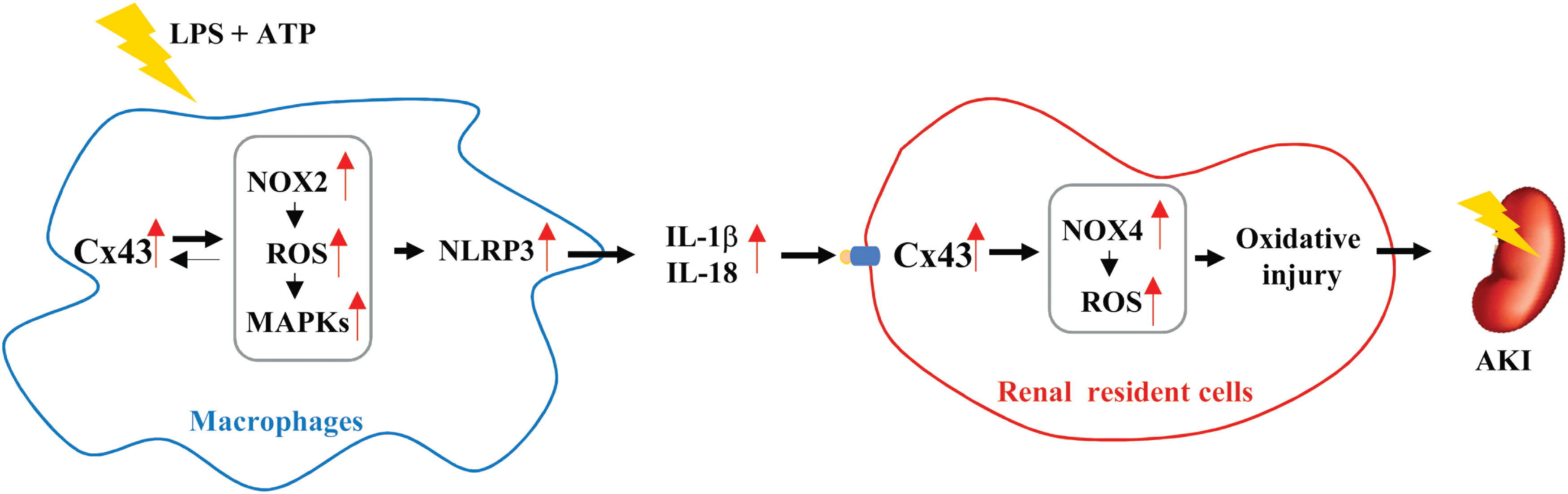
In this study, we demonstrated a pivotal role of Cx43 in the control of NLRP3 inflammasome activation and the development of AKI through mechanisms involving its regulation on intracellular redox status. The graphic depiction of our findings is shown in Figure 11. Because that inflammasome activation is implicated in many inflammatory processes, our finding of the regulation of inflammasome by Cx43 could have important clinical and basic implications.
Cx43 is reported to regulate many immune responses, such as neutrophil recruitment, macrophage function, dendritic cell maturation, as well as B cell and T cell development (34, 37, 44). It was upregulated by several inflammatory mediators (24, 34) and contributed to the inflammatory process (34, 37). It facilitated inflammatory cell activation, stimulated the production of inflammatory mediators, and enhanced the expression of adhesion molecules (2, 29, 32). Also, it promoted the interaction between inflammatory cells and resident cells, and it modulated cell responses to inflammatory mediators via transmission and synchronization of multicellular signals (57). Inhibition or downregulation of Cx43 alleviated the local inflammatory reactions and accelerated the recovery process (40). Currently, the mechanisms behind the effects of Cx43 are still poorly understood. Here, we demonstrated that Cx43 regulated inflammasome activation, a cellular event that is essential for immunity and inflammation. Regulation of inflammasome could be an important mechanism behind the effects of Cx43 on the inflammatory process.
The question naturally occurs as to how Cx43 regulated inflammasome activation. Previous studies have implicated ROS as the central signaling mechanism for NLRP3 inflammasome activation (56). Macrophages deficient in NADPH oxidase displayed weak inflammasome activation (39, 56). In an in vivo sepsis model, inhibition or knockout of NADPH oxidase also reduced inflammatory cytokine production (31). In line with these previous reports, we also demonstrated a requisite of NADPH oxidase and ROS in inflammasome activation. Treatment of PMs with NADPH oxidase inhibitors, NOX2 siRNA, and antioxidant GSH largely abolished inflammasome activation. Moreover, our study also confirmed the previously reported participation of mitochondria ROS in inflammasome activation with the use of a mitochondria-targeted superoxide dismutase mimetic, Mito-Tempo (23, 47). Because the effect of Cx43 on inflammasome was associated with altered ROS generation, NOX2 expression, oxidative kinase activation, and protein carbonylation, it is concluded that the effect of Cx43 was through its modulation on intracellular redox status.
How did Cx43 regulate intracellular redox status? Cx43 has been reported to regulate redox status via communication-dependent and -independent mechanisms. As for the communication-dependent mechanism, Cx43 channels transmitted and amplified Ca2+ signaling, a crucial molecule involved in the induction of ROS (20). Gjs also exaggerated oxidative stress via intercellular transmission and propagation of ROS among adjacent cells. In terms of Gj hemichannels, it was activated under oxidative and inflammatory situations (9, 14, 15, 42). The activated hemichannels further worsened the intracellular oxidative status because of the loss of ATP, and the major antioxidants GSH and NADPH (10, 15). Regarding communication-independent mechanisms, we have reported that Cx43 regulated TXNIP and NOX4 in renal cells via a communication-independent way (17, 18, 61). Collectively, Cx43 may regulate intracellular redox status through multiple mechanisms.
To establish the pathway involved in the effect of Cx43, we have conducted several preliminary experiments and found that the activation of inflammasome did not require direct cell-to-cell contact. There was no substantial difference in inflammasome activation between cells with or without direct cell-to-cell contact (Supplementary Fig. S2). This observation argues against the role of the intercellular communication. Regarding the role of hemichannels, previous studies have implicated pannexin hemichannels in stimulation of ROS generation, activation of inflammasome, and modulation of inflammatory responses through purinergic signaling (9, 19a, 26, 46, 48, 52). Further, the efflux of membrane-permeable Ca2+ and K+ also facilitated NLRP3 inflammasome activation (43, 56). Recently, Dosch et al. reported a pivotal role of Cx43 hemichannels-derived ATP in LPS-induced cytokine production in macrophages. They demonstrated that macrophage-specific deletion or pharmacologic blockade of Cx43 inhibited local and systemic levels of cytokine secretion, and improved sepsis survival in a murine sepsis model induced by cecal ligation and puncture (14). There is also an article describing an important role of Cx43 hemichannel-centered ATP autocrine feedback loop in the amplification and perpetuation of inflammation in the inflammasome/inflammation cycle (42). In this background, it appears that activation of Cx43 hemichannels could be a potentially important mechanism involved in this study.
We have reported that Cx43 modulated NADPH oxidase via a communication-independent way in glomerular podocytes and renal tubular cells (61). In this study, Cx43 also regulated NOX2 in macrophages. Other than NOX2, our preliminary experiments showed that NOX4 was also critically involved in inflammasome activation (Supplementary Fig. S3). It was elevated by inflammasome activators and downregulation of NOX4 with siRNA attenuated inflammasome activation. Moreover, Cx43 also regulated NOX4 in a way similar to NOX2. Macrophages with a high level of Cx43 exhibited a significantly higher level of NOX4 in response to inflammasome activators. These observations indicated that the effects of Cx43 on inflammasome could be ascribed to its effect on NADPH oxidase.
One may wonder why two different isoforms of NADPH oxidase similarly contributed to inflammasome activation in a similar way. One plausible explanation is the existence of a complex interaction between NOX2 and NOX4 in the production of ROS. Several recent publications appear to support this notion. A study by Kim et al. showed that VEGF-induced changes in endothelial phenotype required both NOX2 and NOX4. Downregulation of NOX2 with siRNA abolished the overproduction of H2O2 induced by overexpression of NOX4. They concluded that Nox4-derived H2O2 activates Nox2 to promote mitochondrial ROS production (30). Consistent with this conclusion, another report showed that inhibition of NOX4 with GKT137831 decreased the levels of Nox2 protein (28). Further, the implication of NOX4 in mediating NLRP3 inflammasome activation in macrophages has also been documented (38). In the current investigation, we also explored the possible interactions between NOX2 and NOX4 in macrophages (Supplementary Fig. S4). We found that downregulation of NOX4 with siRNA or inhibition of NOX4 with specific inhibitors obviously blunted the inflammasome activator-initiated elevation of NOX2. Intriguingly, treatment of cells with NOX2 siRNA also attenuated the elevation of NOX4. Collectively, our results, together with the previous reports, indicate that inflammasome activation requires the concerted interactions of multiple informs of NADPH oxidases.
It is worth mentioning that we also reported that Cx43 regulated the expression of TXNIP (17, 18), a thioredoxin inhibitor that activates inflammasome activation via induction of ROS generation and a direct interaction with NLRP3 (65). Cx43-mediated regulation of TXNIP could also contribute to Cx43-mediated regulation of redox status and inflammasome activation. Moreover, we have recently found that Cx43 also regulated the protein level of Nrf2 in macrophages (Supplementary Fig. S5). As a transcription factor critically involved in the cellular defense against oxidative stress, Nrf2 is indispensable for NLRP3 inflammasome activation. It has been shown to participate in the regulation of many inflammatory responses (3, 64). Cx43 may regulate redox state and inflammasome activation via multiple mechanisms.
Our study indicated that MAPKs, ERK, p38, and JNK contributed to NLRP3 inflammasome activation. This result is not surprising because all of them have been reported to take part in the control of cellular events that are essential for inflammasome activation. For example, phosphorylation of ERK1 was necessary for the priming process. Inhibition or downregulation of ERK1 profoundly impaired the priming step of inflammasome activation (19). p38 was required for ASC oligomerization and assembly of the human pyrin inflammasome (16). In terms of JNK, it controlled inflammasome formation and activity through phosphorylation of NLRP3 and ASC (21, 53).
It is worth mentioning that both Cx43 and MAPKs were under the control of oxidative stress and were reciprocally regulated by each other. On the one hand, altered Cx43 affects MAPK activation (15, 17, 33, 61). In podocytes, we have demonstrated that inhibition of Cx43 attenuated p38 activation through mechanisms involving its suppression on NADPH oxidase and TXNIP (17, 61). In this study, inhibition of Cx43 also resulted in a reduction in both NOX2 level and p38 activation, suggesting that the same mechanism may be operated in macrophages. Besides its effect on p38, Cx43 also regulates ERK and JNK activities (15, 33, 54). In this context, some of the effects of Cx43 on inflammasome activation could be mediated by its action on MAPKs. On the other hand, Cx43 is also under the control of MAPK. In this respect, we have reported that JNK mediated oxidative induction of Cx43 and activation of Cx43 hemichannels (15, 33). This also held true for macrophages. Inhibition of JNK similarly abolished Cx43 elevation in macrophages. Of note, there are also studies showing a critical involvement of p38 and ERK in the regulation of Cx43 expression and function (41). It is conceivable that some of the effects of MAPKs on inflammasome could also be the results of its actions on Cx43. In our previous studies, we have proposed the existence of “a vicious reciprocally regulatory loop” among ROS, JNK, and Cx43 in exaggeration of oxidative cell injury (15, 17, 33, 61). It appeared that the same interactive cycle also existed in macrophages, which contributed to inflammasome activation.
Of note, we have used several different Gj inhibitors (lindane and FFA) to confirm the role of Gjs/Cx43 in the regulation of inflammasome activation. These inhibitors have been previously documented to be able to shut down gap-junctional intercellular communication and/or downregulate Cx43 expression (15, 17). Of course, they also have off-target actions. However, the observed effects in this study were most likely the consequence of the actions on Gjs/Cx43. This is because (i) the structurally different lindane and FFA similarly affected inflammasome activation, and (ii) the results obtained from the chemical inhibitors were consistent with those from other approaches, such as using Cx43 siRNA and cells genetically expressing a different amount of Cx43.
In an in vivo model of AKI induced by LPS, Cx43 also participated in the process of renal injury. This conclusion is supported by the fact that the Cx43 heterozygous mouse (Cx43+/−) displayed a reduced level of serum IL-1β, with less severe changes in renal function and structure. As inflammasome activation is critically involved in ischemic and septic renal injury (5), it is conceivable that the effect of Cx43 on inflammatory renal injury could be, at least in part, through its actions on inflammasome activation. In fact, the in vivo activation of inflammasome appeared to be also regulated by Cx43, as revealed by the reduced level of blood IL-1β and renal NLRP3 in the Cx43+/− mouse after LPS administration. Apart from its action on inflammasome, Cx43 also regulates NF-κB activation and NF-κB-controlled production of several mediators, such as tumor necrosis factor-α (TNF-α) and IFN-γ (37, 60), which are known to be involved in cell activation, migration, infiltration, and adhesion of inflammatory cells. Further, Cx43 also facilitates the interaction between inflammatory cells and resident cells, and it modulates renal cell responses to various insults (2, 29), including inflammatory mediator-induced cell injury (40, 58). In this investigation, we found that Gj inhibition reduced the inflammatory mediator-elicited elevation in NOX4 in cultured glomerular and tubular cells. As a predominant isoform of NADPH oxidase in the kidney, NOX4 underlies oxidative and inflammatory renal cell injury (27, 55). Lastly, Cx43 has been characterized as a determinant of cell fate against oxidative stresses (15, 17, 25, 61). Taken together, Cx43 may regulate AKI through multiple mechanisms.
Our study could have significant clinical and basic implications. First, Cx43 regulates many immune responses. In several inflammatory models, a close link between Cx43 and inflammation has been well established. For example, Cx43+/− mice deficient in low-density lipoprotein receptor exhibited markedly less atherogenesis and a reduced number of inflammatory cells in atherosclerotic plaques (32). In LPS-induced acute lung injury, Cx43+/− mice also displayed reduced neutrophil recruitment (51). In several animal models of renal diseases, Cx43 promoted monocyte adhesion, infiltration, and cytokine production (2, 29). Genetic and pharmacologic suppression of Cx43 attenuated local inflammatory responses, as well as improved renal structure and function. However, the regulatory mechanisms of Cx43 on inflammation are still unclear. As inflammasome activation participates in multiple inflammatory and immune responses, our finding of the regulation of inflammasome activation by Cx43 provides novel mechanistic insights into the regulatory action of Cx43 on immune cells. Second, inflammasome activation has been implicated in many inflammatory diseases. The elevated expression of Cx43 in patients with glomerulonephritis has also been reported (2, 24). Thus, targeting Cx43 could be a novel approach to prevent and treat certain inflammatory renal diseases. In support of this notion, a recent study showed that genetic deletion or pharmacological blockade of Cx43 in macrophages decreased LPS-induced cytokine secretion, systemic and local organ inflammation as well as improved sepsis survival (14). Lastly, our study provides additional evidence supporting a critical role of Cx43 in the control of NADPH oxidase expression and intracellular redox status. Regulation of intracellular redox status could be an important mechanism by which Cx43 exerts its regulatory effects on cellular functions.
Collectively, our results indicate that increased Cx43 contributes to inflammasome activation through a mechanism involving its regulation on intracellular redox status. Downregulation of Cx43 ameliorates renal dysfunction and inflammatory cell injury. Our study suggests that strategies targeting Cx43 could be developed to treat inflammatory diseases.
Materials and Methods
Materials
OxyBlot™ protein oxidation detection kit was purchased from Merck Millipore (EMD Millipore, Billerica, MA). BUN kit was purchased from ThermoFisher Scientific (Frederick, MD). IL-1β enzyme-linked immunosorbent assay (ELISA) kit was bought from Peprotech (Rocky Hill, NJ). UCR assay kit was bought from Cayman Chemical (Ann Arbor, MI). PD98059, SB203580, and SP600125 were obtained from Calbiochem (San Diego, CA). Macrophage colony-stimulating factor (M-CSF) was from Tonbo Biosciences (San Diego, CA; 21-8983-U010). Antibodies against β-tubulin, Casp3, phospho-p38, P38, P-JNK, JNK, P-ERK, and ERK, as well as horseradish peroxidase-conjugated anti-rabbit or anti-mouse IgG were obtained from Cell Signaling Technology (Danvers, MA). Anti-NLRP3 (NBP2-12446) and Nox4 (NB110-58849) antibodies were from Novus Biologicals (Littleton, CO). IL-1β and Casp1 antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA; sc7884). Anti-Nox2 was purchased from BD Transduction Laboratories (San Diego, CA; 611414). LPS, thioglycollate, lindane, α-glycyrrhetinic acid (α-GA), apocynin, DPI, FFA, GSH, Mito-TEMPO, anti-Cx43, anti-β-actin, and all other chemicals were from Sigma (Tokyo, Japan). A detailed description about the major chemicals used for this study is given in Supplementary Table S1.
Cells
For induction of peritoneal-derived macrophages (PDMs), 3% thioglycolate broth (Eiken, Tokyo, Japan) was prepared, autoclaved, and stored for at least 3 months before use. C57BL/6J mice were intraperitoneally injected with 3% thioglycolate broth (2 mL). Three days later, PDMs were harvested by lavaging the peritoneal cavity with RPMI 1640 medium and seeded into a 12-well plate at a density of 1.5–2 × 106 in RPMI 1640 medium containing 10% fetal bovine serum (FBS; Sigma-Aldrich, Carlsbad, CA) and 1% penicillin/streptomycin/antibiotic antimycotic solution (ABAM; Sigma-Aldrich). After 12 h for adherence, the nonadherent cells were washed out. The adherent macrophages were further cultured for an additional 24 h and used for the subsequent experiments.
Primarily, cultured BMDMs were obtained from the femurs of mice, as previously reported (59). Briefly, the femurs isolated from the mice were soaked in Dulbecco's modified Eagle medium (DMEM)/F-12 containing 1% ABAM. The epiphyses were cut off, and the bone marrow cells were flushed out with DMEM/F12 by using a syringe with a 21G needle. The harvested cells were seeded into a 12-well plate at a density of 1.5–2 × 106 and cultured in the presence of 20% FBS and 40 ng/mL M-CSF for induction of BMDMs. Two days later, the culture medium was exchanged. After an additional 3-day culture, the nonadherent cells were removed and the adherent macrophages were used for the experiments.
Renal tubular epithelial NRK cells were purchased from ATCC (American Type Culture Collection, Manassas, VA) and cultured in DMEM/F12 containing 5% FBS and 1% ABAM for maintenance and expansion. Murine podocytes were kindly provided by Dr. Karlhans Endlich (University of Heidelberg, Heidelberg, Germany) and cultured as previously described (61). For experiments, cells were exposed to various stimuli in 0.5%–1% FBS.
Animals
Adult female Cx43 WT (Cx43+/+) and heterozygous Cx43 mice (Cx43+/−) weighing 20–25 g were from the offspring of Cx43+/+ mice that were mated with heterozygous Cx43 mice (Cx43+/−; B6; 129-Gja1<tm1kdr>/J; Jackson Laboratories, Bar Harbor, ME). The mice were housed in the Animal Center of our university and were given free access to food and water in an air-conditioned room with a 12-h light/dark cycle. The genotypes of all mice were determined by PCR, according to the protocol provided by Jackson Laboratories (63). All animal experiments were approved by the animal experiment committee of the University of Yamanashi and performed in accordance with relevant guidelines and regulations.
LPS-induced mouse AKI
The adult female Cx43+/+ and Cx43+/− mice were intraperitoneally injected with saline (control) or with 40 mg/kg LPS for the indicated period. Blood was collected from retro-orbital bleeding or tails by using heparinized microhematocrit tubes at the described time points. Urine was also collected synchronously.
Western blot analysis
The sample preparation and Western blot analysis were performed as previously reported (17, 63). Briefly, kidney tissue was homogenized in urea buffer (8 mol/L urea, 1 mmol/L dithiothreitol, 1 mmol/L ethylenediaminetetraacetic acid, 50 mmol/L Tris-HCl at pH 8.0) on ice. Lysate was sonicated and centrifuged at 12,000 rpm for 30 min at 4°C. Supernatant was collected and assayed for protein concentration with Pierce Micro BCA Protein Assay Kit (Thermo Fisher Scientific, Waltham, MA). For cultured cells, cellular protein was extracted with 1 × sodium dodecyl sulfate (SDS) sample buffer. The same amount of protein was loaded onto SDS-polyacrylamide gels and electrotransferred onto polyvinylidene difluoride membranes. After blocking with 5% nonfat dry milk in phosphate buffered saline (PBS), the membranes were incubated with primary antibody overnight at 4°C. After washing, the membranes were probed with horseradish peroxidase-conjugated anti-rabbit or anti-mouse IgG, and the bands were visualized by using Chemi-Lumi One L (Nacalai Tesque, Kyoto, Japan). The chemiluminescent signal was captured with a Fujifilm luminescent image LAS-1000 analyzer (Fujifilm, Tokyo, Japan). The results were quantified by using Image J software. β-actin or β-tubulin was used as an internal loading control.
Enzyme-linked immunosorbent assay and renal function analysis
The collected culture supernatants and blood samples were assayed for IL-1β by using an ELISA kit from Peprotech following the manufacturer's instructions. To assess renal function, blood and urinary samples were diluted and measured for the concentrations of BUN and UCR by using the commercially available kits.
TUNEL staining
Tissues were fixed in 10% formalin, embedded in paraffin, sliced into 4-μm sections, and finally plated on glass slides. The slides were deparaffinized in xylene and ethanol, which was followed by incubation with proteinase K (20 μg/mL, Tris-HCl 10 mM, pH 7.4) for 30 min. After extensive washing with PBS, the tissues were stained for apoptotic cells by using a DeadEnd Fluorometric TUNEL system (Promega), according to the protocol provided by the manufacturer. The nuclei were counterstained with Hoechst 33342 (Kumamoto, Japan). Afterward, the tissues were mounted with an antifade mounting medium and visualized under an immunofluorescent microscope (BX50; Olympus, Tokyo, Japan). The fluorescent signals were captured with a CCD camera attached to the microscope.
Detection of superoxide anion and ROS production
The generation of superoxide anion (O2 −) and ROS was detected by using a commercially available kit from Enzo (Tokyo, Japan; ENZ-51010) following the manual of the manufacturer, as previously described in our previous reports (15, 17). Briefly, cells in 96-well plates were loaded with O2− detection reagent (orange) and oxidative stress detection reagent (green), followed by stimulation with 10 μg/mL LPS and 3 mM ATP for 3 h. The immunofluorescent images were visualized and captured by using the immunofluorescent microscope.
Renal histology
Renal histology was performed as previously described (63). Briefly, tissues were fixed in 10% formalin, embedded in paraffin, sliced into 4-μm sections, and stained with standard H&E procedure.
Polymerase chain reaction
DNA was extracted from ear punch tissue and was analyzed by PCR following the protocol provided by Jackson Laboratories. PCR mixtures (10 μL) contained distilled water (5.625 μL), Taq buffer (1.25 μL), primer mixture (2 μL), dNTPs (1 μL), and Taq DNA polymerase (0.125 μL) (Invitrogen Life Technology). PCR was carried out for 35 cycles by using a 2720 Thermal Cycler (Applied Biosystems). The reaction products were loaded on an agarose gel (2%, diluted in TAE water). The expected molecular weight of PCR products was 320 and 600 bp for heterozygote mouse, and 600 bp for WT mouse. Once the mice were genotyped, they were separated into the different experimental groups used in this study.
Transient transfection of the cells with siRNA
The cells were transiently transfected with a control (All Stars Negative Control siRNA) or Cx43 siRNAs (Qiagen, Tokyo, Japan) at a final concentration of 20 nM by using the SilentFect transfection reagent (Bio-Rad Laboratories, Hercules, CA) for 48 h, as previously described (17, 63). The cellular protein was extracted and subjected to Western blot analysis for target proteins.
Assessment of protein oxidation
The oxidative modification of the proteins was analyzed by using the OxyBlot Protein Oxidation Detection Kit (EMD Millipore) according to the manufacturer's instructions (17, 63). Briefly, the cellular protein was extracted by suspending the tissue or cells in SDS sample buffer containing 50 mM DTT. The samples were mixed with 12% SDS and 1 × DNPH (2, 4-dinitrophenylhydrazine) solution for denaturalization and derivatization, respectively. After neutralization, the samples were subjected to Western blot analysis.
Statistical analysis
Values are expressed as mean ± standard error. A comparison of two groups was made by Student's t-test. For multiple comparisons, one-way analysis of variance followed by Dunnett's test was employed. Both analyses were performed by using Microsoft Excel (Microsoft, Redmond, WA). p < 0.05 was considered statistically significant.
Footnotes
Acknowledgment
This work was supported by a grant from the Ministry of Education, Culture, Sports, Science and Technology, Japan (17K11176 to J.Y.).
Author Disclosure Statement
The authors have no conflicting financial interests.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Table S1
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.