
Abstract
Aims:
It is known that mitochondrial reactive oxygen species generation ([ROS]m) causes the release of Ca2+ via ryanodine receptor-2 (RyR2) on the sarcoplasmic reticulum (SR) in pulmonary artery smooth muscle cells (PASMCs), playing an essential role in hypoxic pulmonary vasoconstriction (HPV). In this study, we sought to determine whether hypoxia-induced RyR2-mediated Ca2+ release may in turn promote [ROS]m in PASMCs and the underlying signaling mechanism.
Results:
Our data reveal that application of caffeine or norepinephrine to induce Ca2+ release increased [ROS]m in PASMCs. Likewise, exogenous Ca2+ augmented ROS generation in isolated mitochondria and at complex III from PASMCs. Inhibition of mitochondrial Ca2+ uniporter (MCU) with Ru360 attenuated agonist-induced [ROS]m. Ru360 produced a similar inhibitory effect on hypoxia-induced [ROS]m. Rieske iron–sulfur protein (RISP) gene knockdown inhibited Ca2+- and caffeine-induced [ROS]m. Inhibition of RyR2 by tetracaine or RyR2 gene knockout suppressed hypoxia-induced [ROS]m as well.
Innovation:
In this article, we present convincing evidence that Ca2+ release following hypoxia or RyR simulation causes a significant increase in MCU, and the increased MCU subsequently RISP-dependent [ROS]m, which provides a positive feedback mechanism to enhance hypoxia-initiated [ROS]m in PASMCs.
Conclusion:
Our findings demonstrate that hypoxia-induced mitochondrial ROS-dependent SR RyR2-mediated Ca2+ release increases MCU and then RISP-dependent [ROS]m in PASMCs, which may make significant contributions to HPV and associated pulmonary hypertension.
Introduction
Hypoxia causes strong vasoconstriction in pulmonary arteries (PAs), termed hypoxic pulmonary vasoconstriction (HPV). This unique response is an important adaptive mechanism for pulmonary ventilation/perfusion matching in the lungs but can also become a crucial pathological factor for pulmonary hypertension. HPV may result from an increase in intracellular Ca2+ concentration ([Ca2+]i), which is mediated by multiple ion channels in pulmonary artery smooth muscle cells (PASMCs). A series of our studies and others have revealed that ryanodine receptors (RyRs), the Ca2+ release channels localized on the sarcoplasmic reticulum (SR), are important for the hypoxia-induced increase in [Ca2+]i and contraction in PASMCs (13, 33, 40, 41, 52). We have further shown that all three subtypes of RyRs (RyR1, RyR2, and RyR3) are involved in hypoxic Ca2+ and contractile responses in PASMCs; however, RyR2 is the most valuable player (17, 18, 51).
Innovation
We and others have disclosed that hypoxia induces Rieske iron–sulfur protein (RISP)-dependent mitochondrial reactive oxygen species generation ([ROS]m) and then Ca2+ release from the sarcoplasmic reticulum (SR) via ryanodine receptor-2 (RyR2) in pulmonary artery smooth muscle cells (PASMCs), thereby leading to hypoxic pulmonary vasoconstriction (HPV) and pulmonary hypertension. However, it is unknown whether and how RyR2-mediated Ca2+ release may in turn promote [ROS]m in PASMCs. In this article, we present novel evidence that Ca2+ release from the SR following the opening of RyR by its agonist caffeine or the opening of inositol-1,4,5-trisphosphate receptor (IP3R) by the vascular neurotransmitter norepinephrine significantly increases mitochondrial Ca2+ uptake and RISP-dependent [ROS]m in PASMCs. Hypoxic stimulation produces a similar response. Agonist- and hypoxia-induced Ca2+ release-mediated RISP-dependent [ROS]m are RyR2 determined and uniquely occur in PASMCs, but not in systemic arterial smooth muscle cells. Collectively, the interaction of RyR2-mediated Ca2+ release with RISP-dependent mitochondrial ROS serves as an important signaling mechanism for HPV and associated pulmonary hypertension.
The hypoxic increase in [Ca2+]i in PASMCs has been thought to be attributed to the enhanced intracellular reactive oxygen species generation ([ROS]i) due to the specific effects of ROS on ion channels (31, 33, 40, 41, 52). The hypoxic increase in [ROS]i mainly occurs in mitochondria and at NADPH oxidase. We have revealed that mitochondria are a primary source of the hypoxic increase in [ROS]i, which enhances the activity of protein kinase C-ɛ and then NADPH oxidase, leading to further [ROS]i, termed ROS-induced ROS generation (RIRG) in PASMCs (28, 29, 37). The hypoxia-induced mitochondrial ROS generation ([ROS]m) is predominantly produced at complex III, in which Rieske iron–sulfur protein (RISP) acts as an indispensable molecule (16, 45).
Our studies have further shown that hypoxia causes activation of RyRs and Ca2+ release in PASMCs, which are blocked by pharmacological and genetic inhibition of [ROS]m, and mimicked by exogenous ROS (18). Thus, the specific mitochondrial ROS-induced RyR2-mediated Ca2+ release, that is, ROS-induced Ca2+ release (RICR), serves as an essential mechanism for hypoxic cellular responses in PASMCs.
RyR2-mediated Ca2+ release can be taken up by mitochondria, which is likely to be central to mitochondrial functions including ROS generation in cardiac myocytes (7, 11). These important findings in cardiac cells, with our revolutionary discoveries on the essential role mitochondria-SR interaction-mediated RICR in PASMCs as described above, have led us to propose a novel hypothesis that the hypoxia-induced ROS-initiated RyR2-dependnet Ca2+ release may in turn promotes [ROS]m, termed Ca2+-induced ROS generation (CIRG), which provides a positive feedback mechanism to further enhance the hypoxic ROS generation, thereby contributing to attendant Ca2+ and contractile responses in PASMCs. Indeed, Ca2+ uptake is correlated with ROS generation in mitochondria isolated from cardiac ventricular myocytes (35), and Ca2+ addition results in a significant increase in ROS generation at mitochondrial complex III isolated from the heart (3).
To test our hypothesis, we first sought to determine whether direct Ca2+ addition could increase ROS generation in isolated mitochondria and complex III from PASMCs. RyR-mediated Ca2+ release plays an important role in cellular responses in PASMCs (36, 52); thus, we next examined whether Ca2+ release from the SR following application of caffeine, a classic and widely used RyR agonist (12, 21, 30), augmented [ROS]m in PASMCs. In complement to the effect of caffeine-evoked RyR activation, subsequent experiments were performed to test the effect of blockage of RyR2-mediated Ca2+ release with its antagonist tetracaine and gene knockout (KO) on hypoxia-induced [ROS]m in PASMCs.
With the intention of defining a potential mechanism for the enhanced [ROS]m by caffeine-induced RyR-dependent Ca2+ release, we investigated whether Ca2+ release-dependent [ROS]m might be a result of the increased mitochondrial Ca2+ uniporter (MCU) by assessing the effect of its inhibitor Ru360 on caffeine-induced ROS generation. RISP in mitochondrial complex III is an essential molecule for [ROS]m in PASMCs as stated above. To further determine the molecular mechanism for RyR Ca2+ release-mediated [ROS]m, we accordingly conducted a set of experiments to assess whether genetic suppression of RISP using specific lentiviral short hairpin RNA (shRNA)-mediated gene knockdown (KD) could block caffeine-caused ROS generation in PASMCs and mitochondria.
Finally, we sought to determine whether hypoxia-induced mitochondrial ROS-initiated RyR2-dependent Ca2+ release might further enhance the hypoxic [ROS]m in PASMCs and also whether this hypoxic response is attributable to the MCU-associated RISP signaling.
Results
Application of caffeine, norepinephrine, and hypoxia to elevate [Ca2+]i significantly increased [ROS]i and [ROS]m in PASMCs
Here, we first examined whether RyR-mediated Ca2+ release might induce [ROS]i, that is, CIRG in PASMCs. In these experiments, cells were loaded with chloromethyldihydrodichlorofluorescein diacetate (CM-H2DCF/DA) to measure [ROS]i (mainly H2O2) (37, 41) and then treated with the classic RyR agonist caffeine (20 mM) for 5 min to induce Ca2+ release from the SR. As shown in Figure 1A, DCF-derived fluorescence intensity was increased remarkably in caffeine-treated cells compared with untreated cells, indicating that caffeine-induced RyR-mediated Ca2+ release causes a significant increase in [ROS]i in PASMCs.

Norepinephrine, a major vascular neurotransmitter, can increase [Ca2+]i by largely inducing Ca2+ release from the SR via inositol-1,4,5-trisphosphate receptors (IP3Rs) in vascular smooth muscle cells (SMCs) (21, 24, 50, 51). Accordingly, we sought to test whether treatment with norepinephrine also increased ROS generation in PASMCs. Like caffeine, the application of norepinephrine (100 μM) for 5 min resulted in a large increase in [ROS]i in PASMCs (Fig. 1A).
We also found that caffeine at 3 and 10 mM caused qualitative increases in DCF-derived fluorescence in PASMCs (Fig. 1B). Taken together, our findings provide evidence that caffeine can, in a concentration-dependent manner, increase [ROS]i in PASMCs.
Caffeine- and norepinephrine-induced [ROS]i were also detected by using pHyPer-cyto, a mammalian expression vector encoding a fluorescent ROS sensor HyPer (specifically assessing H2O2) (2, 16). As shown in Figure 1C, both caffeine and norepinephrine increased [ROS]i in PASMCs.
As depicted in Figure 1D and E, the similar effect of caffeine and norepinephrine on [ROS]i in PASMCs was confirmed by the chemiluminescent reagent lucigenin (mainly for measuring O2 −) and the fluorescent dye RedoxSensor Red CC-1 (for measuring both O2 − and H2O2) (37, 41). Taken together, caffeine and norepinephrine elicited ROS generation by releasing Ca2+ from the SR in PASMCs.
It is known that mitochondria are the primary sources for [ROS]i in PASMCs (18, 28, 29, 37, 42, 43, 46 –48). As such, we examined ROS generation in isolated mitochondria from PASMCs following caffeine or norepinephrine exposure. The results indicated that [ROS]m was increased as well (Fig. 1F).
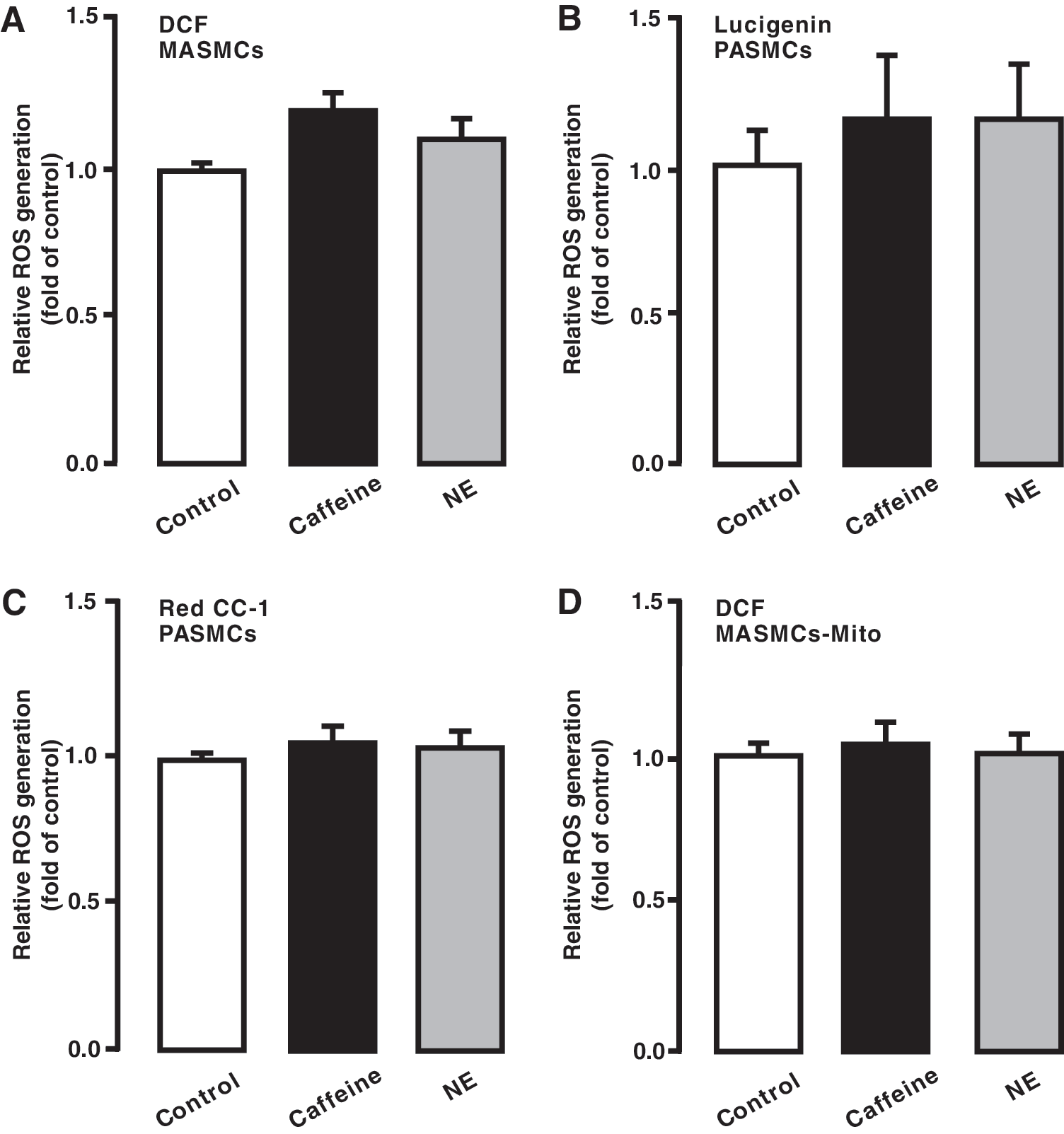
Hypoxia causes differential ROS, Ca2+, and contractile responses in PASMCs and systemic artery SMCs, thereby leading to significant vasoconstriction in pulmonary arteries, but not in systemic arteries (40). As such, we thought that CIRG might only occur in the former cells, rather than in the latter. In support, as shown in Figure 2A
Caffeine, norepinephrine, or hypoxia increased intramitochondrial Ca2+ concentration in PASMCs
To further illustrate the role of Ca2+ in [ROS]m, we investigated the effect of caffeine, norepinephrine, and hypoxia on intramitochondrial Ca2+ concentration [Ca2+]m. The mitochondria-targeted double-mutated aequorin (mit-2mutAEQ) Ca2+ sensor was used to assess [Ca2+]m, as described in a previous publication (6). The correlation of the mit-2mutAEQ-derived relative luminescence (luminescence/total residual luminescence, L/Lmax) with different free Ca2+ concentrations was constructed, as shown in Figure 3A. This correlation curve was then used to calibrate the data.
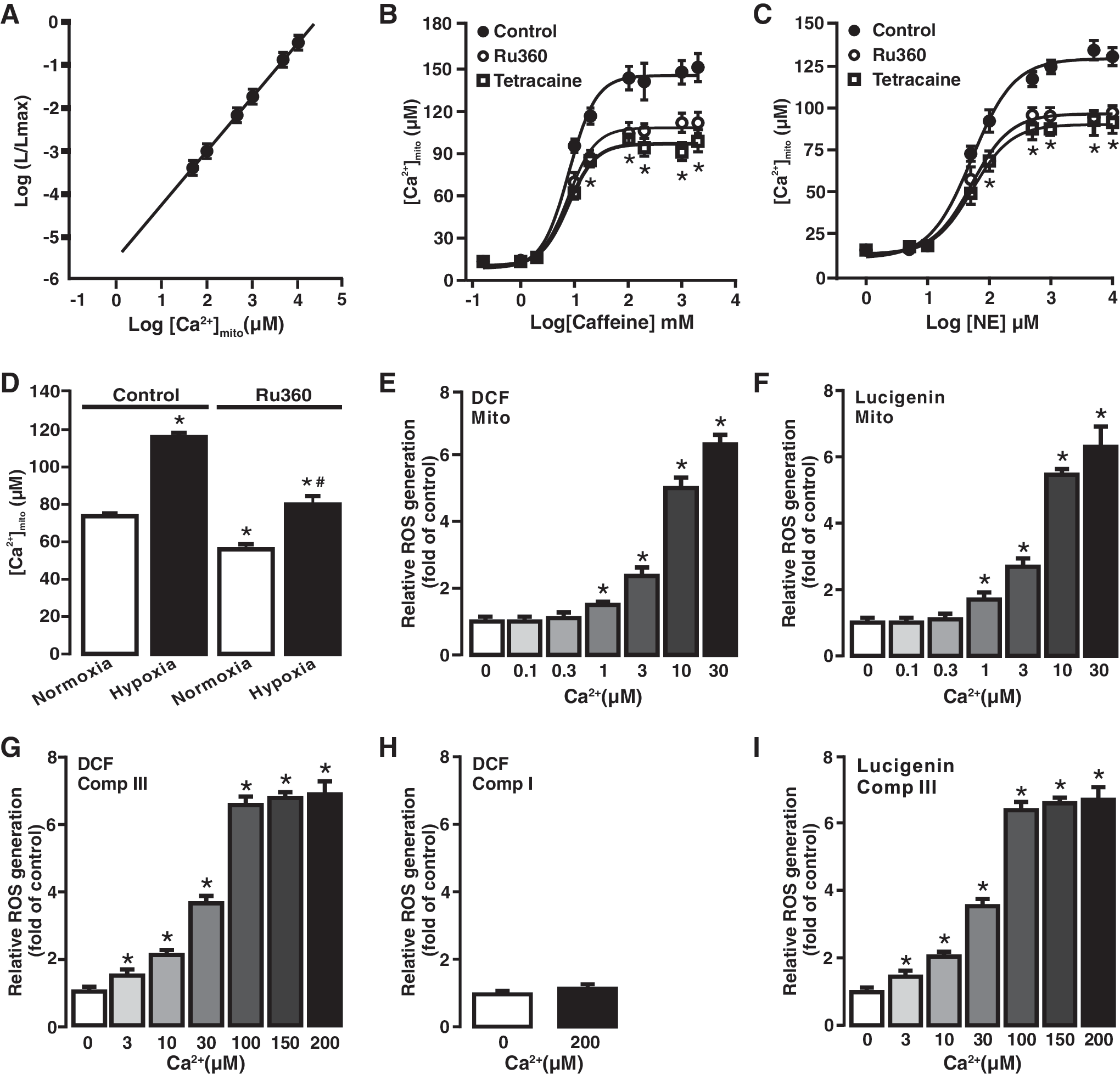
To test the effect of norepinephrine and caffeine, cells were first transfected with pcDNA3.1+/mit-2mutAEQ and then exposed to different concentrations of norepinephrine (1 μM–10 mM) and caffeine (0.2 mM–2 M). As presented in Figure 3B and C, both caffeine and norepinephrine increased [Ca2+]m in concentration-dependent manner. Interestingly, caffeine- or norepinephrine-induced increase in [Ca2+]m was significantly inhibited by pretreatment with the MCU inhibitor Ru360 (10 μM) for 5 min. Similarly, hypoxia also induced a large increase in [Ca2+]m compared with normoxia, and the hypoxia-induced effect was suppressed by treatment with Ru360 (10 μM) for 5 min (Fig. 3D).
Exogenous Ca2+ application significantly increased ROS generation in mitochondria and complex III isolated from PASMCs
A previous publication (22) has reported that cell stimulation largely increases local [Ca2+]i (20–40 μM) in chromaffin cells, whereas the rest of the cell has a lower [Ca2+]i (1–2 μM). To further understand the Ca2+-dependent [ROS]m in PASMCs, we sought to test the effect of different free Ca2+ concentrations on ROS generation in isolated mitochondria. Various free Ca2+ concentrations were obtained using different EGTA and Ca2+ concentrations with a widely used online program (10). Application of exogenous Ca2+ caused a significant increase in [ROS]m, determined by DCF-derived florescence (Fig. 3E) and lucigenin-derived chemiluminescence (Fig. 3F), in a concentration-dependent manner (1–30 μM) in isolated mitochondria from PASMCs.
[ROS]m in PASMCs primarily occurs at complex III (4, 14, 16, 37, 41, 45); thus, we next examined Ca2+-dependent ROS generation in isolated complex III using H2DCF/DA. Application of exogenous free Ca2+ induced a concentration-dependent increase in ROS generation (DCF-derived fluorescence) in isolated complex III (Fig. 3G). However, isolated complex I from PASMCs did not show the same Ca2+-induced ROS response as complex III did (Fig. 3H). This suggests that complex I may not play a significant role in Ca2+-induced [ROS]m in PASMCs. Ca2+-dependent ROS generation in isolated complex III was also detected by lucigenin, similar to H2DCF/DA (Fig. 3I).
RISP KD inhibited caffeine- and hypoxia-induced ROS generation in PASMCs and Ca2+-evoked ROS generation in mitochondria from PASMCs
We and other investigators have shown that RISP is indispensable for the hypoxic ROS generation in PASMCs (16, 45); thus, we sought to determine the role of RISP in CIRG. As described in our previous reports (16, 48), infection with lentiviral shRNAs specific for RISP largely knocked down its protein expression in PASMCs (Fig. 4A). Similar results were observed in three different experiments. In contrast, infection of lentiviral nonsilencing (NS) shRNAs did not significantly suppress RISP expression.
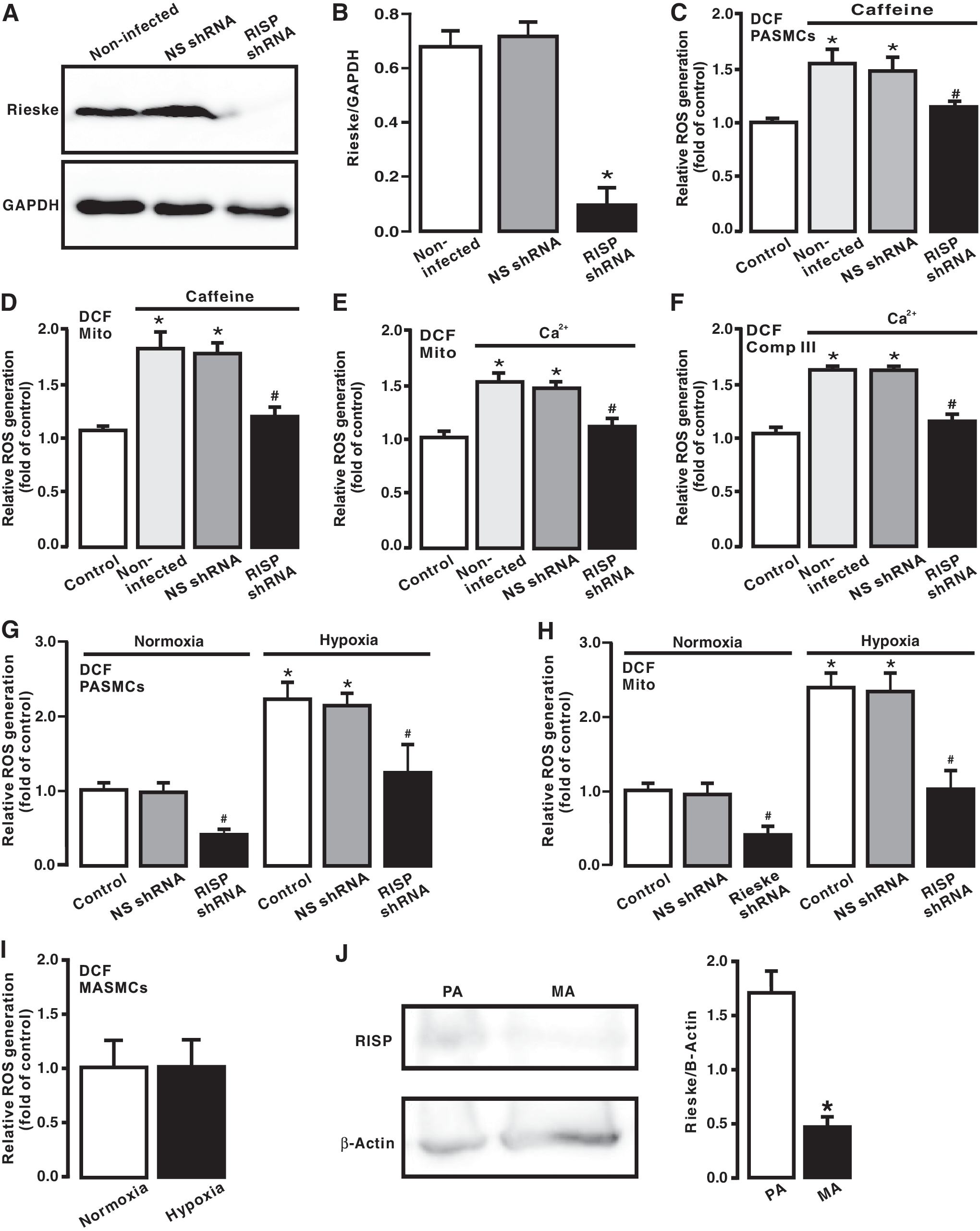
Corresponding to the suppressed RISP expression, caffeine-evoked ROS generation was abolished in PASMCs infected with lentiviral RISP shRNAs (Fig. 4B). Similarly, ROS generation was completely blocked in isolated mitochondria from cells infected with lentiviral RISP shRNAs followed by treatment with caffeine (Fig. 4C).
Moreover, we observed that exogenous CIRG was eliminated in isolated mitochondria and complex III from PASMCs infected with lentiviral RISP shRNAs, whereas ROS generation was not affected in isolated mitochondria and complex III from cells infected with lentiviral NS shRNAs (Fig. 4D, E).
We also demonstrated that hypoxia-induced ROS generation was significantly inhibited in RISP KD PASMCs, isolated mitochondria, and complex III. RISP KD also slightly suppressed ROS generation under normoxic conditions (Fig. 4F, G). Additionally, we showed that hypoxia-induced ROS generation only occurred in PASMCs, but not in MASMCs (Fig. 4H), which are consistent with our previous reports (28, 29, 34, 37, 39, 48, 49). Interestingly, RISP expression level was significantly higher in PASMCs than in MASMCs (Fig. 4I). All these findings support our new concept that CIRG only functions in PASMCs, but not in systemic arterial smooth muscle cells; thus, RISP plays a primary role in mediating hypoxia-induced CIRG in PASMCs.
Inhibition of mitochondrial Ca2+ uptake blocked Ca2+-evoked [ROS]m in PASMCs
We assumed that caffeine-induced Ca2+ release from the SR via RyRs might cause mitochondrial Ca2+ uptake and accordingly mediate mitochondrial CIRG in PASMCs. To test this assumption, we assessed the effect of the MCU inhibitor Ru360. Following treatment with Ru360 (10 μM) for 5 min, cells were exposed to caffeine (20 mM) for 5 min in the continued presence of Ru360. As shown in Figure 5A, treatment with Ru360 attenuated caffeine-elicited ROS generation in PASMCs. ROS generation was also prevented in isolated mitochondria from cells exposed to caffeine after pretreatment with Ru360 (Fig. 5B).
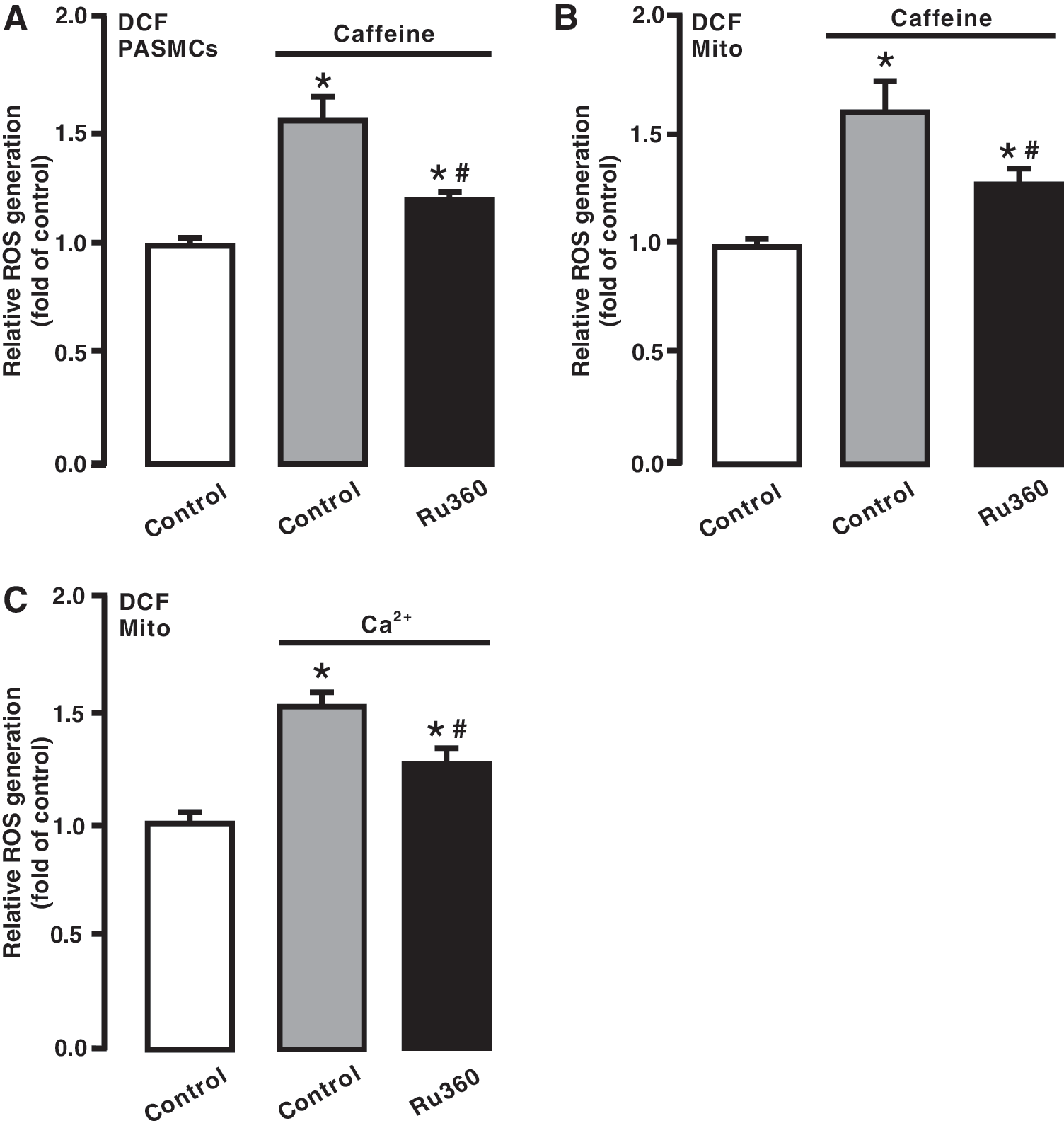
In agreement with its effect on caffeine-induced responses, treatment with Ru360 blocked Ca2+-evoked ROS generation in isolated mitochondria (Fig. 5C).
Inhibition of mitochondrial Ca2+ uptake attenuated hypoxia-induced ROS generation in PASMCs and mitochondria
Consistent with our previous reports (18, 28, 29, 37, 48), exposure to acute hypoxia for 5 min caused a large increase in ROS generation in isolated PASMCs, detected by DCF (Fig. 6A), Red CC-1 (Fig. 6B), and lucigenin (Fig. 6C). The hypoxic ROS generation was inhibited in cells following treatment with Ru360 (10 μM) for 5 min. In addition, Ru360 had an inhibitory effect on the resting ROS generation in PASMCs under normoxic conditions.
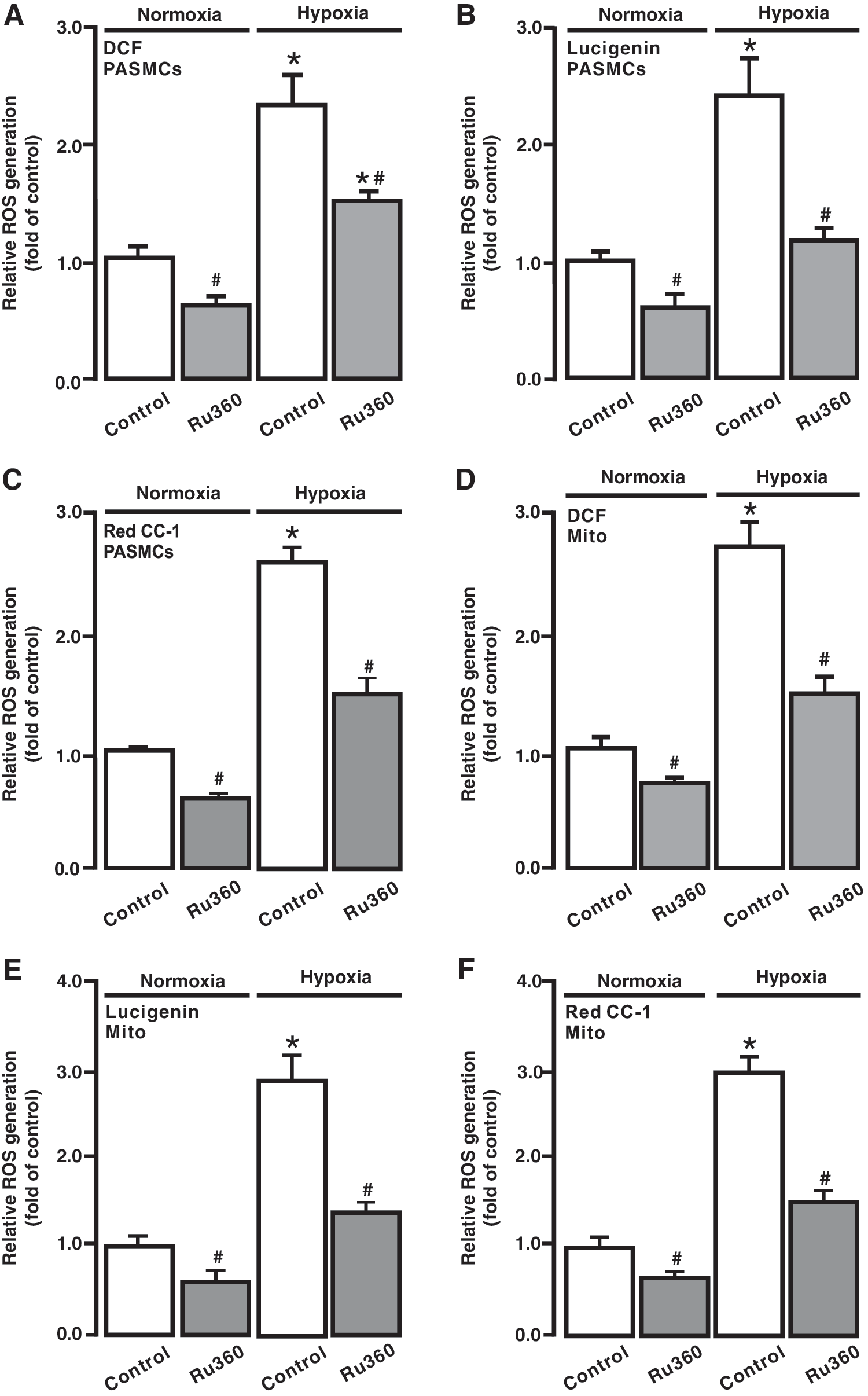
To further evaluate [ROS]m, isolated mitochondria were treated with Ru360 (10 μM) for 5 min followed by hypoxic exposure for 5 min. As shown in Figure 6D
Inhibition of RyRs attenuated the hypoxic ROS generation in PASMCs
ROS-dependent RyR-mediated Ca2+ release is essential for the hypoxic increase in [Ca2+]i in PASMCs (41, 52); thus, we studied whether RyRs may play a significant role in the hypoxic ROS generation in PASMCs. Cells were treated with the RyR antagonist tetracaine (10 μM) for 5 min and then exposed to hypoxia with tetracaine for 5 min. ROS generation was largely suppressed in PASMCs after treatment with tetracaine; the resting ROS generation was also somewhat inhibited in cells under normoxic conditions (Fig. 7A–C).
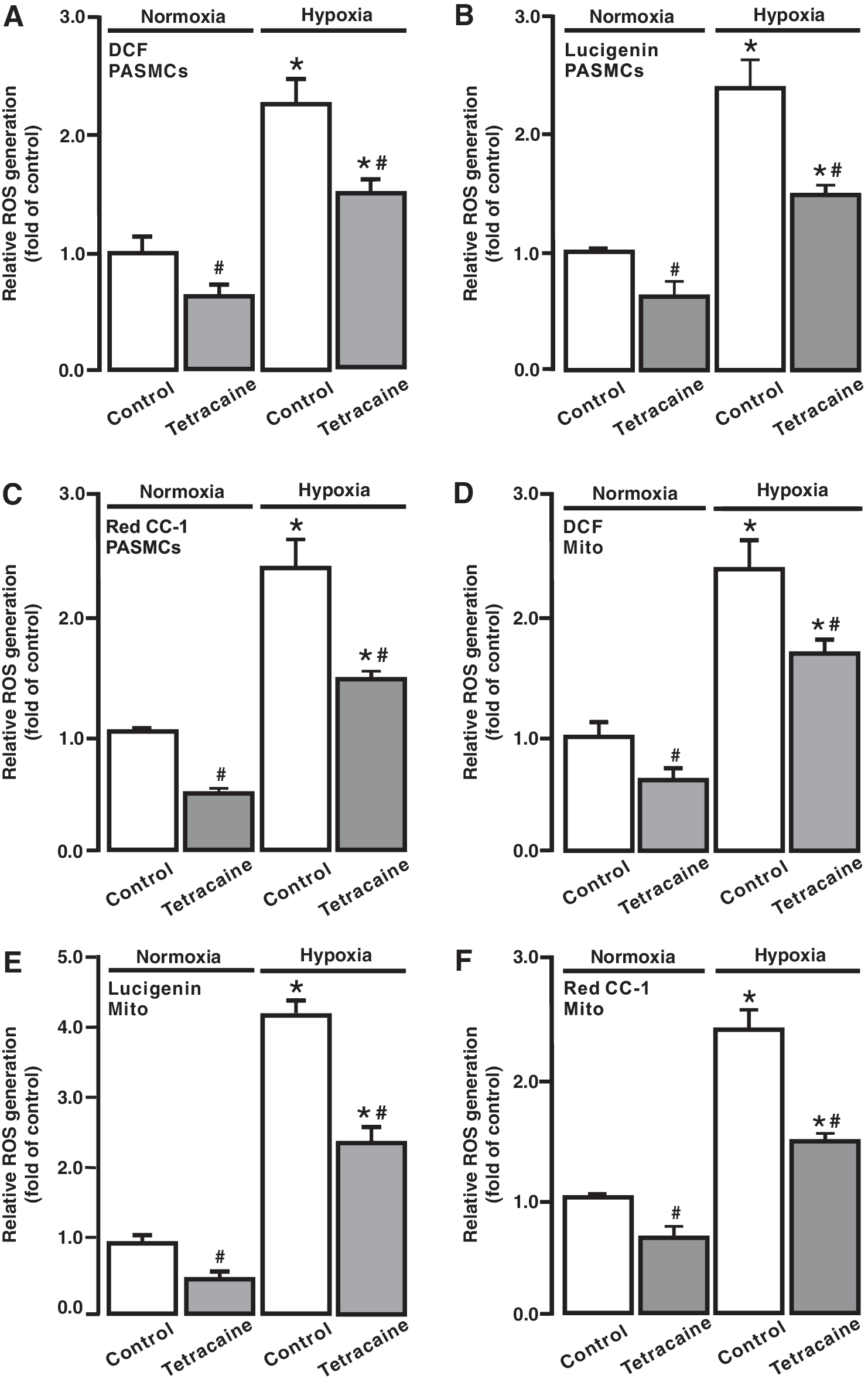
We next assessed the effect of tetracaine on [ROS]m. To achieve this, cells were treated with tetracaine, mitochondria were isolated, and ROS generation was measured. Hypoxia-induced and resting [ROS]m were attenuated in cells pretreated with tetracaine (Fig. 7D–F).
RyR2 gene KO blocked the hypoxia-induced [ROS]m in PASMCs
Our previous studies have shown that RyR1, RyR2, and RyR3 all are expressed and mediate acute hypoxic Ca2+ and contractile responses in PASMCs; however, the role of RyR2 dominates over that of RyR1 and RyR3 (17, 18, 51). In complement to the effect of pharmacological inhibition of RyRs, we examined the effect of RyR2 gene KO on the hypoxic ROS generation in PASMCs. As shown in Figure 8A–C, acute hypoxic exposure for 5 min caused a much smaller ROS generation in PASMCs isolated from RyR2 KO mice than control (wild type [WT]) animals. The resting ROS generation was also reduced in RyR2 KO PASMCs.

The hypoxic increase in ROS generation was significantly suppressed in isolated mitochondria from RyR2 KO PASMCs, and the resting [ROS]m was decreased as well (Fig. 8D–F).
Discussion
One of the major novel findings resulting from this study is that the application of the classic RyR agonist caffeine to activate RyRs on the SR can cause a significant [ROS]m (i.e., CIRG) in isolated PASMCs. This discovery complements our previous reports, in which increasing [ROS]i (e.g., application of H2O2) opens RyRs and induces SR Ca2+ release (i.e., RICR), leading to an increase in [Ca2+]i in PASMCs (18, 28, 29, 37). ROS-mediated increase in [Ca2+]i plays an important role in hypoxia-induced pulmonary vasoconstriction, vasoremodeling, and hypertension (1, 41, 53). Presumably, the crosstalk between Ca2+ and ROS signaling may enhance the hypoxia-induced ROS generation in PASMCs, which provides a positive feedback mechanism to mediate the hypoxic ROS generation and associated cellular responses.
In this study, we have also found that the application of norepinephrine, a major neurotransmitter in vascular SMCs to activate α-adrenergic receptors and increase [Ca2+]i (21, 24, 50, 51), can result in a large increase in ROS generation as well in PASMCs. It is well known that the activation of α-adrenergic receptors induces SR Ca2+ release by IP3Rs (21, 24, 50, 51). Ca2+ release via IP3Rs may activate adjacent RyRs to induce further SR Ca2+ release, that is, a local Ca2+-induced Ca2+ release (CICR) process (51), in PASMCs. Thus, the role of norepinephrine to induce ROS generation is likely to be, at least in part, implemented by RyR-dependent Ca2+ release as a result of local IP3R/RyR interaction-mediated CICR (19).
We and other investigators have reported that mitochondria are a major source for ROS generation in PASMCs, and mitochondrial ROS are primarily generated at complex III (18, 28, 29, 37, 42, 43, 46, 47). Consistent with these previous reports, the current study has shown that ROS generation is largely increased in isolated mitochondria from PASMCs pretreated with caffeine to induce Ca2+ release due to activation of RyRs. Likewise, ROS generation is also augmented in isolated mitochondria from PASMCs after treatment with norepinephrine to induce Ca2+ release from the SR as a result of the opening of IP3Rs (19, 51).
Moreover, by using mitochondria-targeted Ca2+ sensor, we successfully measured and quantified the changes in [Ca2+]m in cells following exposure of different concentrations of caffeine or norepinephrine. In complement to caffeine- and norepinephrine-induced responses, different concentrations of exogenous Ca2+ also significantly increase ROS generation in isolated mitochondria and complex III from PASMCs. These data not only reveal that Ca2+ release from the SR may cause [ROS]i (CIRG) primarily at mitochondrial complex III in PASMCs but also further support our novel concept that the SR can locally communicate with closely neighboring mitochondria in the format of CIRG in PASMCs.
Our recent work has unveiled that RISP KD at complex III blocks the hypoxic ROS generation in isolated PASMCs, mitochondria, and complex III, whereas its gene overexpression produces the opposite effects; thus, RISP is a crucial molecule for the hypoxic ROS generation in PASMCs (16, 48). In agreement, we have also shown that RISP plays an important role in hypoxia-induced increase in [Ca2+]i in PASMCs and vasoconstriction in PAs (16, 48). Supporting our findings, Waypa et al. have also found that RISP KO abolishes the hypoxia-induced increase in [Ca2+]i in PAs and right ventricular systolic pressure (45). In line with the important role of RISP, in the current study, we have discovered that RISP KD inhibits Ca2+- and caffeine-induced ROS generation in isolated mitochondria and/or complex III from PASMCs. Further experiments are needed to determine whether Ca2+ may cause [ROS]m by producing a direct or an indirect effect on RISP in PASMCs.
We have surmised that the increased [Ca2+]i may give rise to an increase in mitochondrial Ca2+ uptake, leading to ROS generation in mitochondria and at complex III. Consistent with our conjecture, treatment with Ru360, a specific mitochondrial Ca2+ uptake inhibitor (1), prevents caffeine from inducing ROS generation in PASMCs. Likewise, caffeine-induced ROS generation is also blocked in mitochondria from PASMCs after treatment with Ru360. More interestingly, Ru360 inhibits hypoxia-induced [ROS]m. These results reveal that mitochondrial Ca2+ uptake serves as a critical step for Ca2+-dependent [ROS]m, thereby playing an important role in hypoxic cellular responses in PASMCs.
Previous investigations from our laboratory and others have demonstrated that RyRs play an important role in hypoxia-induced increase in [Ca2+]i in PASMCs and pulmonary vasoconstriction in PAs (17, 18, 25 –27, 36, 51). Hypoxia may inhibit voltage-dependent K+ (Kv) channels and activate store-operated Ca2+ (SOC) channels, leading to Ca2+ and contractile responses in PASMCs; however, the hypoxic inhibition of Kv channels and the activation of SOC channels may be secondary to SR Ca2+ release, presumably via RyRs (25 –27). These data reinforce the importance of RyRs in hypoxic cellular responses in PASMCs.
Our laboratory and others have demonstrated that both pharmacological and genetic inhibition of [ROS]m almost completely inhibit hypoxia-induced Ca2+ and contractile responses in PASMCs (8, 16, 18, 28, 29, 37, 42 –46, 48). As such, we have assumed that hypoxia-induced, ROS-initiated RICR may cause further ROS generation (CIRG), providing a positive feedback mechanism to contribute to hypoxia-evoked physiological and pathological functions in PASMCs. Indeed, in this study, we have observed that treatment with tetracaine to block RyRs remarkably inhibits the hypoxic ROS generation in PASMCs. The hypoxic response is also reduced in isolated mitochondria from PASMCs after pretreatment with tetracaine.
Our previous investigations have demonstrated that RyR1 or RyR3 KO can partially inhibit, whereas RyR2 KO completely inhibits, the hypoxia-induced increase in [Ca2+]i in PASMCs and pulmonary vasoconstriction in PAs (17, 18, 51). Thus, all three known RyR subtypes are involved in hypoxic Ca2+ and contractile responses in PASMCs; however, RyR2 is the most valuable player. In agreement with this notion, we have unveiled that hypoxia causes a much smaller increase in ROS generation in PASMCs from RyR2 KO mice than control (WT) mice. Furthermore, the hypoxic ROS generation is significantly reduced in isolated mitochondria from RyR2 KO mice. These pharmacological and genetic findings for the first time demonstrate that RyR2 functions as a key molecule to implement the focal communication from the SR to mitochondria, leading to CIRG in PASMCs during hypoxic stimulation.
In summary, our current study proves a concept that Ca2+ release from the SR via RyR2 can increase [Ca2+]i, mitochondrial Ca2+ uptake, and RISP-dependent mitochondrial CIRG. Previously, we have discovered that hypoxia first causes ROS generation and then the opening of RyRs to induce Ca2+ release from the SR in PASMCs, leading to HPV and associated pulmonary hypertension (16, 18, 28, 29, 37, 48, 49). Similar findings have been reported by Waypa et al. (43, 45, 46). Here, we present evidence that hypoxia-induced SR Ca2+ release is uptaken by MCUs, which in turn increases [Ca2+]m and subsequently leads to RISP-dependent [ROS]m, providing a positive feedback mechanism to contribute to the hypoxia-induced increase in [ROS]i in PASMCs. This novel communication from the SR to mitochondria in the form of CIRG may play an important role in the overall hypoxia-induced Ca2+ and contractile responses in PASMCs.
Materials and Methods
Preparation of PASMCs
PASMCs were prepared from mouse resistance PAs as we have described previously (16 –18, 28, 29, 37, 48, 51). All experiments using mice were conducted in compliance with the National Institutes of Health guidelines and approved by the Animal Care and Use Committee of Albany Medical College. Resistance (third and smaller intralobar) PAs were dissected, with removal of the endothelium and connective tissues, from male Swiss Webster mice at 2 months old. PASMCs were isolated from the dissected PAs using the two-step enzymatic digestion method, in which the arteries were digested with papain and then collagenase in physiological saline solution (PSS). The harvested PASMCs were cultured in modified Dulbecco's minimal essential medium for three passages and used in experiments.
PASMCs from smooth muscle-conditional RyR2 KO mice (C57/B6 background) were obtained using the same method described above. RyR2 KO mice were generated by crossbreeding RyR2 flox/flox mice with smooth muscle-specific myosin heavy-chain Cre recombinase mice. The KO mice were genotyped by polymerase chain reaction analysis of tail tip DNAs. Western blot analysis revealed that these mice showed the absence of RyR2 expression in PAs.
Detection of ROS generation
ROS generation in PASMCs was first measured using CM-H2DCF/DA (Molecular Probes), as this fluorescent dye has been most widely used to determine [ROS]i (mainly H2O2) in living cells (5, 32, 37, 41). The measurement was conducted, as described in our previous publications (16, 28, 29, 37). A suspension of PASMCs was incubated with CM-H2DCF/DA (5 μM) at 37°C for 30 min, followed by washing for 10 min. The dye was excited at 485 nm, and the emitted fluorescence at 532 nm was collected using a FlexStation-III spectrophotometer (Molecular Devices). Fluorescent intensity was normalized to control.
There are the criticisms of the use of H2DCF due to its oxidation and cytotoxicity; however, this probe at low concentrations (1–10 μM) in serum-free media is suitable for the ROS measurement in almost all cell types (9, 15, 23, 41). Despite this fact, we utilized the chemiluminescent probe lucigenin (mainly for measuring O2 −) as a supplementary approach to examine [ROS]i in PASMCs (37). Lucigenin (20 μM; Molecular Probes) was added to one well of a 96-well plate containing 1.5 × 105 PASMCs. The emitted chemiluminescence derived by lucigenin was detected. The initial (maximal) values of lucigenin-derived chemiluminescence were normalized to the control group. The fluorescent Redox Sensor Red CC-1 was also used to measure both O2 − and H2O2 generation (37). Cells were loaded with Red CC-1 (1 μM; Molecular Probes) at 37°C for 30 min. Fluorescence derived from Red CC-1 was measured using an excitation wavelength of 544 nm and emission wavelength of 612 nm. [ROS]i was determined by the difference in fluorescence between the wells containing the assay buffer with and without cells.
To detect ROS generation in isolated mitochondria, we utilized the same method reported in our previous publication (16). Isolated mitochondrial samples (50 μg protein) were added in microplate wells containing 5 mM pyruvate, 2.5 mM malate, 10 μM H2DCF/DA, and 5 μM horseradish peroxidase (HRP). After 10 min of incubation, different concentrations of caffeine, norepinephrine, and free Ca2+ were added to medium. Fluorescence was measured at 37°C using the FlexStation-III spectrophotometer with an excitation wavelength of 485 nm and emission wavelength of 532 nm. ROS generation was determined by subtracting the fluorescence intensity measured in control wells that contained assay buffer without mitochondria, H2DCF/DA, and HRP. Lucigenin was also used in detecting ROS generation in mitochondria. Fifty micrograms of isolated mitochondrial sample was incubated with lucigenin (20 μM), 5 mM pyruvate, and 2.5 mM malate for 10 min. Lucigenin-derived chemiluminescence was detected in an absorbance of 550 nm after the addition of different concentrations of free Ca2+ to the medium.
To detect ROS generation in complex III, immunocapture kit specific for complex III (Abcam) was used to isolate according to the manufacturer's instruction, as we have described previously (16). Freshly isolated complex III (3 μg) was incubated in the respiration buffer containing 40 μM reduced decylubiquinone, 2 mM potassium cyanide, 50 μM cytochrome c, 10 μM H2DCF/DA, and 5 μM HRP for 10 min. Different concentrations of free Ca2+ or the hypoxic gas mixture were applied to respiration buffer, and ROS were detected at 37°C using the FlexStation-III spectrophotometer. Lucigenin was also used in detecting ROS generation in complex III. Three micrograms of isolated complex III was incubated with buffer containing 40 μM reduced decylubiquinone, 2 mM potassium cyanide, 50 μM cytochrome c, and 20 μM lucigenin for 10 min. Different concentrations of free Ca2+ were added to medium, and lucigenin-derived chemiluminescence was detected by luminometer in an absorbance of 550 nm.
ROS generation in complex I was assessed using a similar procedure to that used in the aforementioned complex III experiments. Complex I was isolated using its specific immunocapture kit (Abcam), and ROS were measured in buffer containing 8 mM MgCl2, 2.5 mg/mL bovine serum albumin, 0.15 mM NADH, and 1 mM potassium cyanide (16, 20).
Intracellular ROS were also assessed using the specific ROS biosensor pHyPer-cyto (2, 16). Primary PASMCs were transfected with pHyPer-cyto plasmid vector. After transfection for 48 h, caffeine or norepinephrine was added to induce ROS generation. HyPer was alternatively excited at 420 and 500 nm. Emitted fluorescence at 510 nm was measured at 37°C using the FlexStation-III spectrophotometer.
[Ca2+]m detection with mutant aequorin
Mitochondria-targeted Ca2+ sensor pcDNA3.1+/mit-2mutAEQ (Asp119 to Ala, Asn28 to Leu points mutations on aequorin) was a gift from Javier Alvarez-Martin (plasmid No. 45539; Addgene, Watertown, MA). Following the procedure provided by Addgene (6), pcDNA3.1+/mit-2mutAEQ was purified and transfected to primary cultured mouse PASMCs for 48 h. After transfection, PASMCs were incubated for 2 h at room temperature in standard medium (145 mM NaCl, 5 mM KCl, 1 mM MgCl2, 1 mM CaCl2, 10 mM glucose, and 10 mM HEPES [pH 7.4]) with 2 μM coelenterazine w (Biotium).
After incubation, PASMCs were treated with different concentrations of caffeine and norepinephrine for 5 min. Emitted luminescence was recorded by a luminometer and then calibrated for the Ca2+ concentration by following a previous publication (6).
Rieske iron–sulfur protein KD
Lentiviral shRNAs specific for RISP were used to KD its expression in PASMCs, as described in our previous publications (16, 48). Lentiviruses containing RISP shRNAs and nonsilence shRNAs were purchased from Thermo Scientific OpenBiosystems and then packaged using pCMV-dR8.2 dvpr and pCMV-VSV-G packing vectors. The efficiency of RISP KD was determined using Western blot analysis.
Hypoxia
To induce hypoxic responses, cell, mitochondrion, or complex preparations were treated with a normoxic PSS (110 mM NaCl, 3.4 mM KCl, 2.4 mM CaCl2, 0.8 mM MgSO4, 25.8 mM NaHCO3, 1.2 mM KH2PO4, and 5.6 mM glucose [pH 7.4]) that was aerated with a 21% O2, 5% CO2, and 74% N2 mixture for 10 min and then hypoxic PSS gassed with a 1% O2, 5% CO2, and 94% N2 mixture for 5 min, as we have reported previously (16 –18, 38, 48, 51). As a control, preparations were treated with normoxic PSS alone.
Statistical analysis
Levels of statistical significance was evaluated with data from at less three independent experiments using one- or two-way analysis of variance with an appropriate post hoc test. Data are shown as means ± standard error of the mean. The differences between the means of data at p < 0.05 were considered statistically significant.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by the American Heart Association (AHA) Scientist Development Grant 0630236N (Y.-M.Z.) and Established Investigator Award 0340160N (Y.-X.W.), National Natural Science Foundation of China 81160011 (Z.Y.), and National Institutes of Health R01HL108232 and R01HL122865 (Y.-X.W.).