
Abstract
Aims:
Interventions to inhibit oxidative stress and apoptosis, important pathogenic contributors toward the progression of chronic kidney disease (CKD), are not well established. Here, we investigated the role of a transforming growth factor beta (TGFβ) superfamily neutralizing protein, follistatin (FST), in the regulation of apoptosis and oxidative stress in glomerular mesangial cells (MCs) and in the progression of CKD.
Results:
The endoplasmic reticulum (ER) stress inducer thapsigargin (Tg), known to cause MC apoptosis, led to a post-translational increase in the expression of FST. Recombinant FST protected, whereas FST downregulation augmented, Tg-induced apoptosis without affecting Ca2+ release or ER stress induction. Although activins are the primary ligands neutralized by FST, their inhibition with neutralizing antibodies did not affect Tg-induced apoptosis. Instead, FST protected against Tg-induced apoptosis through neutralization of reactive oxygen species (ROS) independently of its ability to neutralize activins. Importantly, administration of FST to mice with CKD protected against renal cell apoptosis and oxidative stress. This was associated with improved kidney function, reduced albuminuria, and attenuation of fibrosis.
Innovation and Conclusion:
Independent of its activin neutralizing ability, FST protected against Tg-induced apoptosis through neutralization of ROS and consequent suppression of oxidative stress, seen both in vitro and in vivo. Importantly, FST also ameliorated fibrosis and improved kidney function in CKD. FST is, thus, a novel potential therapeutic agent for delaying the progression of CKD. Antioxid. Redox Signal. 31, 551–571.
Introduction
Chronic kidney disease (CKD) is a major cause of morbidity and mortality, affecting more than 10% of the world's population (24). It is characterized by an excess of pro-fibrotic and inflammatory cytokines, most notably transforming growth factor beta (TGFβ) and its family members, that lead to cellular stresses, including endoplasmic reticulum (ER) stress and oxidative stress. These result in renal cell injury and fibrosis that lead to progressively worsening renal dysfunction and eventually kidney failure (10, 44, 64, 72). Although oxidative stress and apoptosis occur in several renal cell types, these processes in glomerular mesangial cells (MCs) play a major role in glomerular fibrosis and consequent loss of renal function in CKD of varying etiology (57, 63, 64).
Reactive oxygen species (ROS) generation and apoptosis are key pathophysiologic contributors to chronic kidney disease (CKD) that are not as yet effectively targeted with current therapies. We present novel data demonstrating that the glycoprotein follistatin (FST), best known for its ability to neutralize transforming growth factor beta (TGFβ) family members called activins, has potent antiapoptotic and antioxidant effects in CKD through its ability to scavenge ROS and, in turn, therapeutically protects against the progression of CKD through improving kidney function and reducing renal fibrosis.
Oxidative stress is an important pathogenic contributor to CKD. The accumulation of excessive reactive oxygen species (ROS) occurs from impaired balance of free radical production and clearance (11, 23, 60, 66). ROS arise from the metabolism of oxygen and include small reactive molecules such as superoxide (SO), hydrogen peroxide (H2O2), peroxynitrite (ONOO−), hydroxyl radicals (HO), nitric oxide (NO), and the peroxy radical (ROO). Under physiologic conditions, ROS maintain redox homeostasis, act as vital intracellular second messengers, and mediate the activation of genes that are important in processes ranging from cell proliferation to differentiation and growth (11, 66). Excess oxidative stress, however, has been shown to induce apoptosis in several renal cell types, including glomerular MC, and is recognized as an important factor in the pathogenesis of CKD of varying etiology (23, 26, 63).
ER stress is also an established contributor to CKD pathogenesis, with its inhibition reducing the progression of CKD in several studies (10, 44, 72). ER stress was shown to induce apoptosis in renal cells, including MC, through chronic activation of the unfolded protein response (10, 40, 44, 55, 72). It also induces cellular death by promoting oxidative stress (31, 68). Thus, both oxidative stress and apoptosis are important contributors to the progression of CKD to end-stage renal disease, and interventions that can protect against these stressors would provide important therapeutic benefit (45, 56).
Recent work has shown a protective therapeutic role for the secreted glycoprotein follistatin (FST) against ROS production and apoptosis in nonrenal cells (73). FST is an endogenous inhibitor of members of the TGFβ superfamily, with the greatest potency against activins. Once activins are bound by FST, they are cleared by internalization of the complex followed by lysosomal degradation (19, 27). Activin A has been shown to induce ROS production and mediate apoptosis in several cell types (7, 49, 67), and interestingly, elevated serum and kidney activin A was recently found in a mouse CKD model (1). Although the use of a ligand trap to inhibit activin A signaling attenuated fibrosis in this model, neither ROS nor apoptosis were studied (1).
In this study, we thus sought to determine whether FST can protect against oxidative stress and apoptosis induced by ER stress in renal MC, and inhibit the progression of CKD in vivo. We show for the first time that FST protects against both oxidative stress and apoptosis, and that this occurs through neutralization of ROS in a manner that is independent of activins. In vivo, FST alleviated renal oxidative stress, protected against the development of renal fibrosis, and improved kidney function in a mouse model of CKD.
Results
The ER-stress inducer thapsigargin causes MC apoptosis and post-translationally increases the expression of FST
The ER-stress inducer thapsigargin (Tg), a noncompetitive inhibitor of the sarco/endoplasmic reticulum Ca2+ Mg2+-ATPase (SERCA), is a potent activator of apoptosis in several cell types, including MC (31, 39, 40, 55, 68). We first confirmed that Tg promoted apoptosis in cultured primary mouse MCs. Apoptosis was assessed by quantifying the exposure of phosphatidylserine (PS) on the outer leaflet of the plasma membrane by using a luminescent Annexin V binding assay. Tg significantly increased PS-Annexin V binding as compared with vehicle-treated cells (Fig. 1A). Effector caspases 3 and 7 are members of the cysteine (cys) aspartic acid-specific protease family that, once activated through cleavage, irreversibly mark the cell's entry into the apoptotic signaling pathway (56). Tg significantly increased the enzymatic activity of caspase 3 and 7 (Fig. 1B) and led to increased cleavage of caspase 3 (Fig. 1C). These results confirm that Tg promotes MC apoptosis.
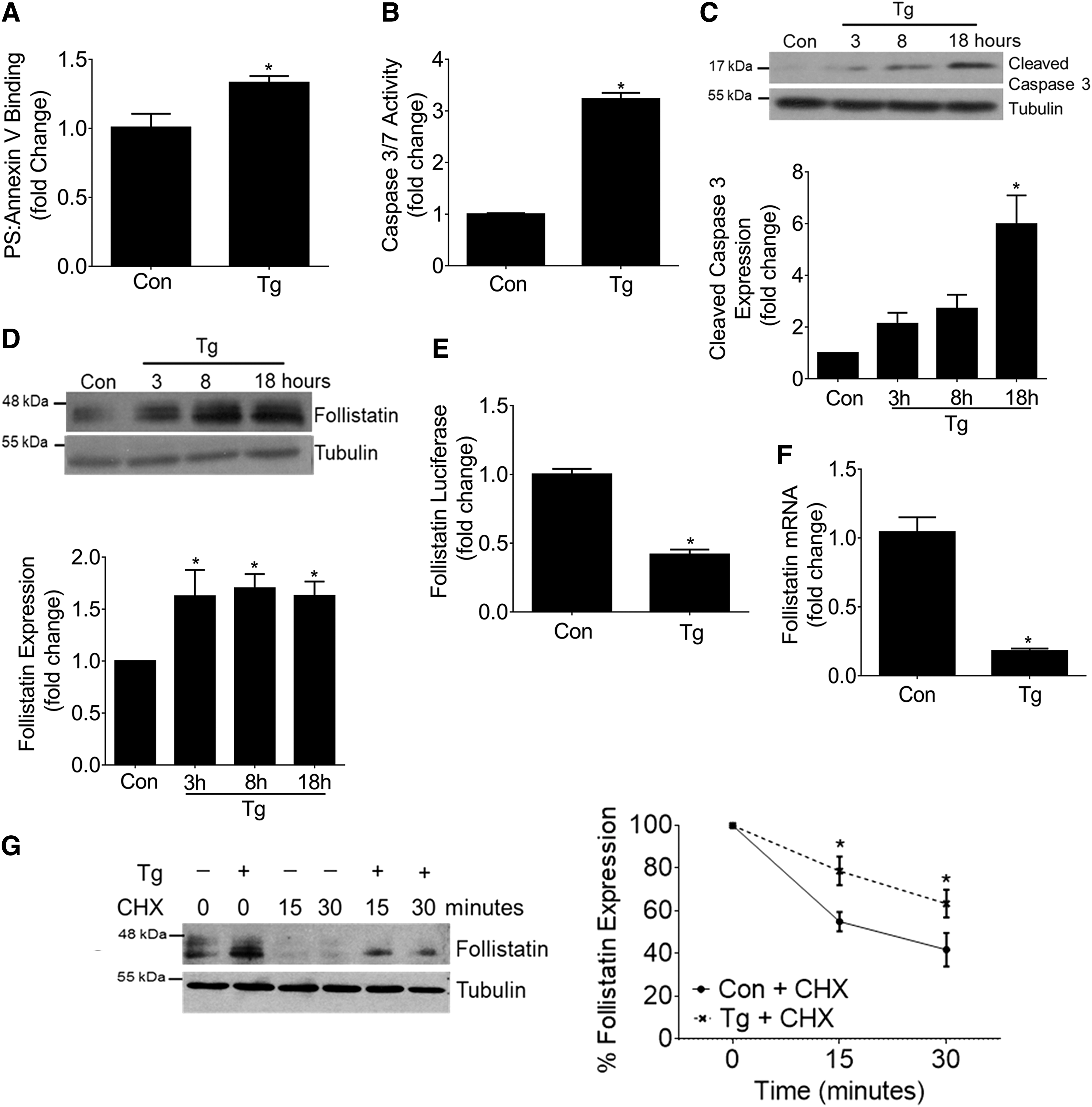
Although FST has been described to serve as a protective stress-responsive protein under several stressors (8, 13, 73), whether it is regulated by ER stress is unknown. Thus, we first examined the effect of Tg on the expression of FST. Figure 1D shows that Tg time dependently increased FST protein expression. To determine whether this was mediated by increased transcription, the effects of Tg on a full-length mouse FST promoter luciferase construct were assessed. Contrary to expectations, FST promoter activity was significantly repressed by Tg (Fig. 1E). FST messenger RNA (mRNA) levels, as assessed by quantitative real-time polymerase chain reaction (qRT-PCR), were also decreased by Tg (Fig. 1F). These results show that the increase in FST seen in response to Tg is not mediated by increased transcriptional activity. We next studied the effects of Tg on FST post-translational regulation. To assess whether Tg affects FST protein stability, cycloheximide (CHX) was used to block de novo protein synthesis after Tg treatment for 8 h. Figure 1G shows that FST protein is usually rapidly degraded, with Tg leading to significant stabilization. Thus, Tg post-translationally increases FST expression through enhancing its protein stability.
FST protects against Tg-induced MC apoptosis
Since FST has been shown to function as an antiapoptotic protein, we hypothesized that the increase in FST in response to Tg may also serve to protect against apoptosis (8, 13, 73). We first assessed whether FST downregulation using small interfering RNA (siRNA) augments Tg-induced apoptosis. Figure 2A confirms downregulation of FST and shows that this exacerbates Tg-induced apoptosis as measured by cleavage of caspase 3, as well as by caspase 3/7 enzymatic activity (Fig. 2B). We next assessed the efficacy of exogenous recombinant FST in protecting against Tg-induced apoptosis. Figure 2C shows that FST significantly reduced Tg-induced apoptosis, as assessed by PS-Annexin V binding, caspase 3/7 enzymatic activity (Fig. 2D), and caspase 3 cleavage (Fig. 2E).
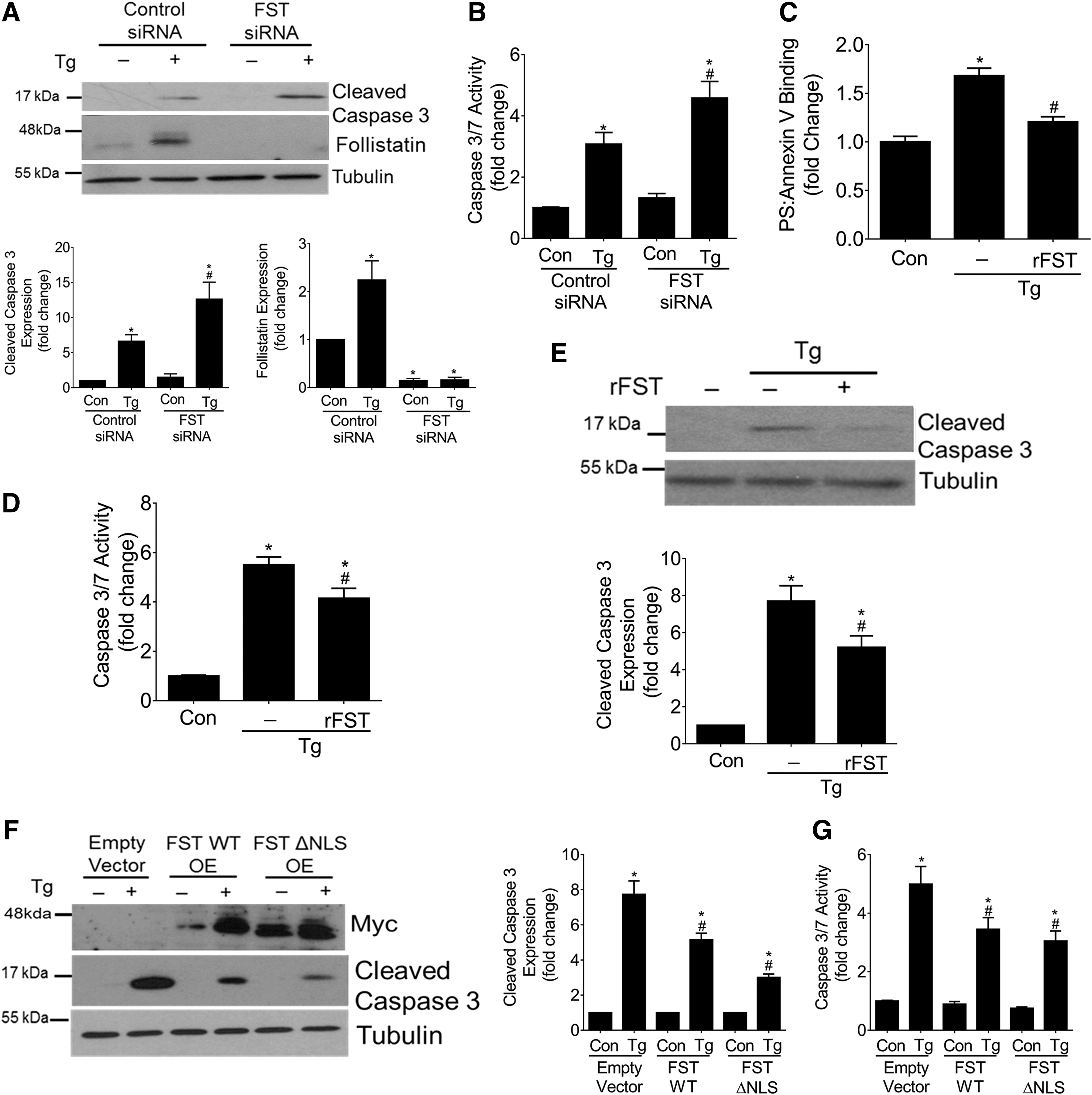
Finally, we assessed the effects of FST overexpression on Tg-induced apoptosis. We transfected MC with myc-tagged wild-type FST or the FSTΔNLS mutant (13). The FSTΔNLS vector encodes a mutated FST protein that lacks a nuclear localization signal (NLS) (13), with FST nuclear import shown to be important for the protection of cancer cells against apoptosis (13). We first confirmed that Tg upregulated the expression of both transfected proteins (Fig. 2F). We next determined whether Tg promotes nuclear FST localization by using immunoblotting of cytosolic/nuclear preparations. Although we did find basal nuclear expression of wild-type FST, its localization within the nucleus was not increased by Tg. Nuclear exclusion of the FSTΔNLS mutant protein was also confirmed (Supplementary Fig. S1A). Further, our results show that both wild-type FST and FSTΔNLS were equally effective against reducing Tg-induced apoptosis, as measured by caspase 3 cleavage (Fig. 2F) and caspase 3/7 enzymatic activity (Fig. 2G). These results show that FST protects against Tg-induced apoptosis, and that this occurs independently of its ability to localize within the nucleus.
Finally, FST is canonically known to be a secreted protein (19, 73). Based on our findings that the addition of exogenous FST was effective in decreasing Tg-induced apoptosis (Fig. 2C–E), we questioned whether wild-type FST and FSTΔNLS were both secreted into the medium in MCs in response to Tg. Supplementary Figure S1B shows the basal secretion of both forms of overexpressed FST proteins without augmentation by Tg. Taken together, these data show that FST protects against Tg-induced apoptosis and that this protection is independent of its ability to localize to the nucleus.
FST does not inhibit Tg-induced Ca2+ release or ER stress
We next sought to determine how FST attenuates Tg-induced MC apoptosis. Since Tg promotes apoptosis through depletion of ER Ca2+ with concurrent accumulation of cytosolic Ca2+ (55), we examined whether FST alters Tg-induced Ca2+ release. Cytosolic Ca2+ levels were assessed by using the ratiometric intracellular calcium indicator, Fura-2 and spectrofluorometry. We found that the addition of recombinant FST did not affect Tg-induced cytosolic Ca2+ influx (Fig. 3A).
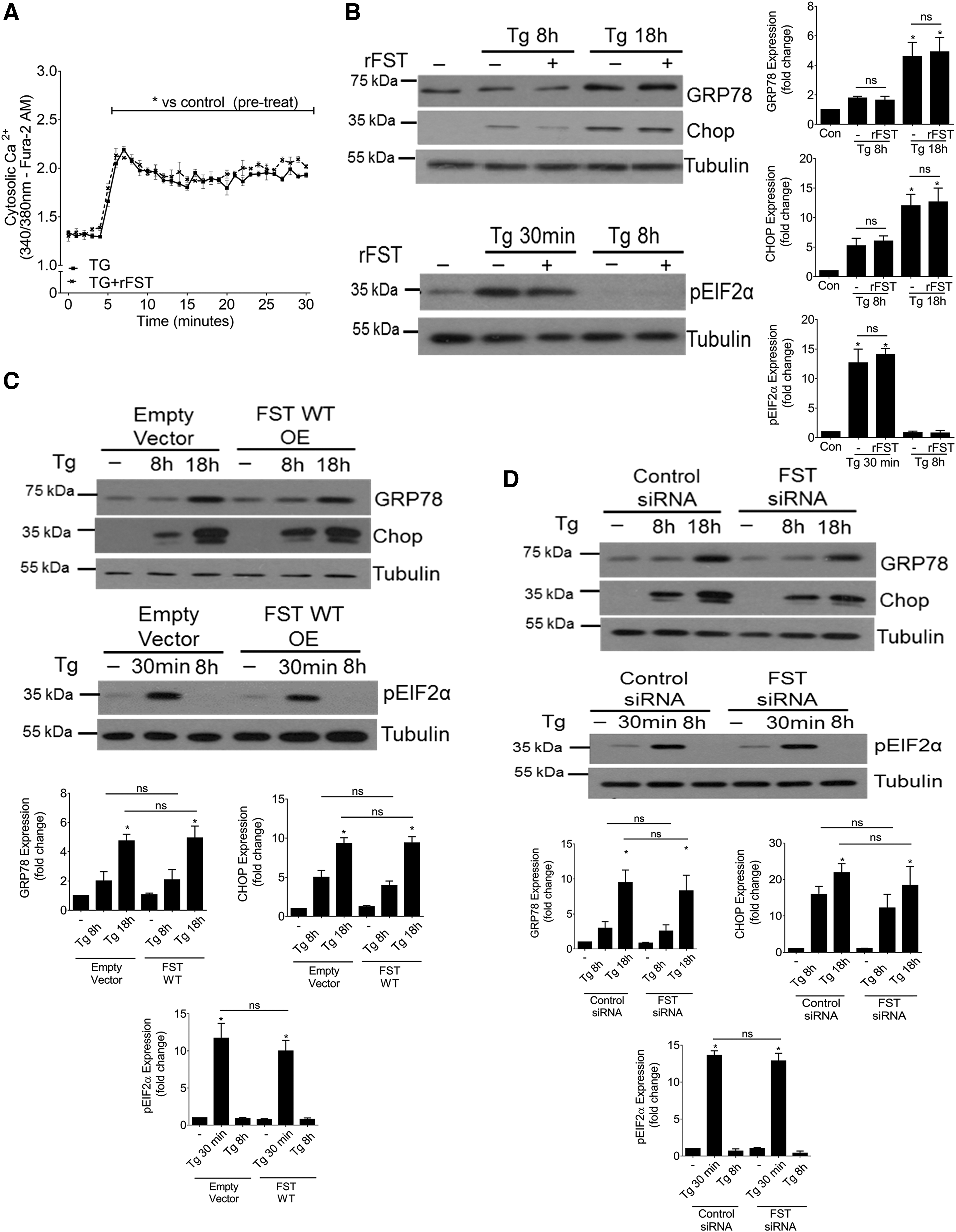
Based on its ability to deplete ER Ca2+ stores, Tg also leads to ER stress and activation of the ER stress-mediated apoptotic pathway (10, 47). Thus, we investigated whether FST inhibits ER stress. We found that Tg-induced ER stress, as measured by the upregulation of GRP78 and CHOP and the phosphorylation of eIF2α, was not affected by the addition of recombinant FST (Fig. 3B). Similarly, overexpression of wild-type FST (Fig. 3C) or siRNA-mediated downregulation of FST (Fig. 3D) did not affect Tg-induced ER stress. These results suggest that FST protects against Tg-induced MC apoptosis independently of Ca2+ regulation and ER stress induction.
FST protects against Tg-induced MC apoptosis by inhibiting ROS
Tg has been shown to induce oxidative stress in numerous cell types by promoting ectopic cytosolic Ca2+ accumulation (31, 68). Further, ROS are known to play an important role in the induction of apoptosis, including that effected by Tg (61, 68). Interestingly, FST was shown to be an oxidative stress-responsive protein that is effective at inhibiting ROS in several cell types (34, 74). We, thus, sought to determine whether FST inhibits Tg-induced apoptosis through attenuation of ROS production. A proprietary ROS/Superoxide detection cocktail (ROS-ID Total ROS/Superoxide Kit) was used to concurrently assess and differentiate SO from other intracellular ROS species, including H2O2, ONOO−, HO, NO, and ROO. In this study, superoxides are abbreviated as “SO,” whereas ROS collectively refers to species including H2O2, ONOO−, HO, NO, and the ROO, unless otherwise specified.
We first assessed ROS and SO production after Tg exposure to confirm that this can be seen in MC. Figure 4A shows that Tg led to an accumulation of ROS in MCs that was attenuated by the H2O2 scavenger PEGylated-catalase. Tg did not affect SO production. Spectrofluorometric quantification confirmed the inhibitory effects of PEGylated-catalase on Tg-induced ROS production (Fig. 4B, C). Catalase also significantly blocked Tg-induced apoptosis, as assessed by PS-Annexin V binding (Fig. 4D) and caspase 3/7 enzymatic activity (Fig. 4E). We next assessed whether FST can inhibit Tg-induced ROS production. The addition of recombinant FST drastically reduced the accumulation of intracellular ROS (Fig. 4F), which was confirmed with spectrofluorometric quantification (Fig. 4G, H). Interestingly, incubation of MC with the SO scavengers superoxide dismutase (SOD) and TEMPOL effectively blunted Tg-induced apoptosis in MC (Fig. 4I). Since Tg did not increase intracellular SO, we questioned whether SO scavengers could deplete basal SO levels in MC, which could explain these protective antiapoptotic effects. Indeed, basal depletion of SO was confirmed (Supplementary Fig. S2).
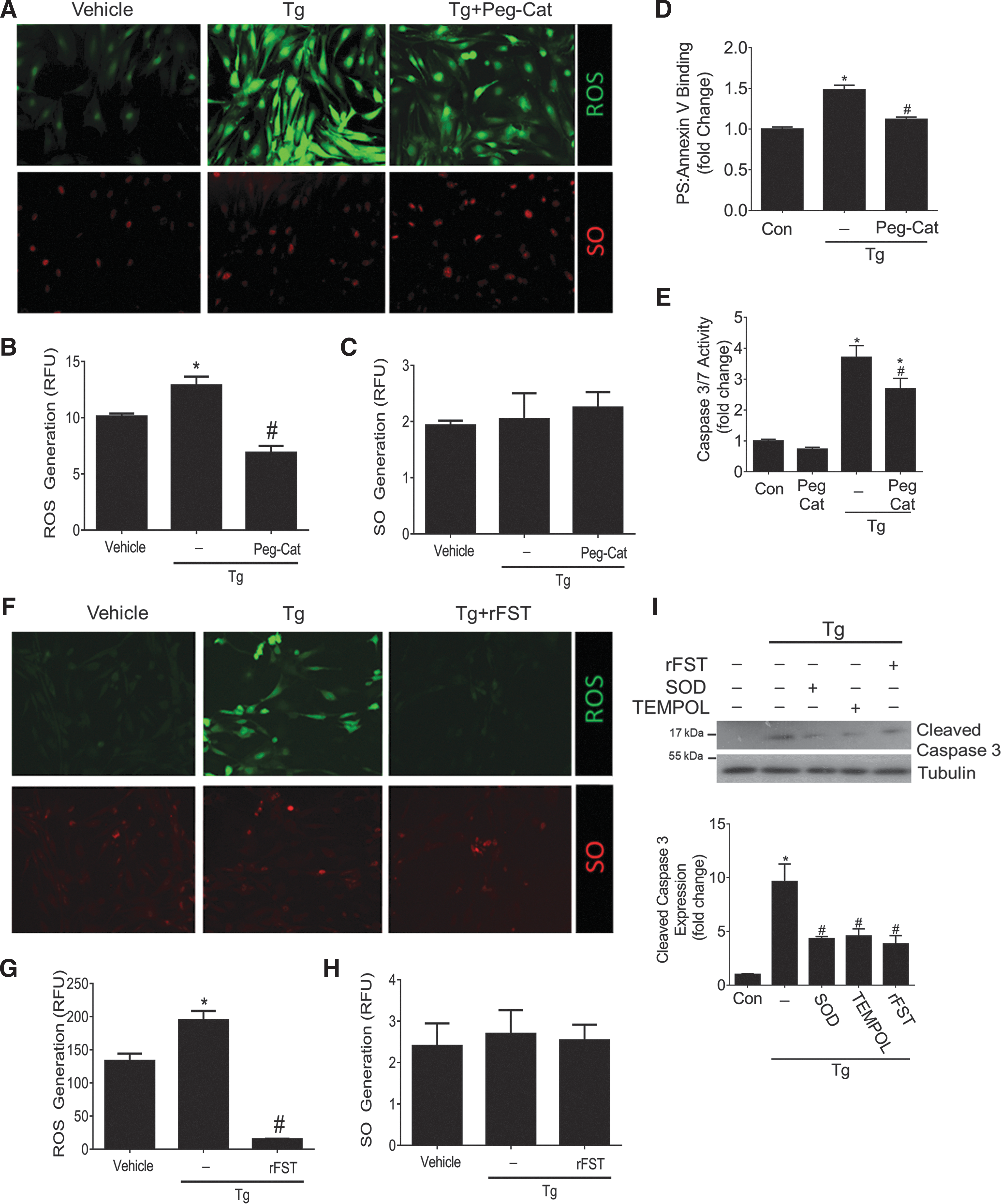
These data collectively illustrate that FST inhibits intracellular ROS, and that inhibition of basal SO or Tg-induced ROS production protects against Tg-induced apoptosis. This raised the question of the mechanism by which FST can inhibit intracellular ROS.
Activins A and B do not mediate Tg-induced apoptosis and ROS production
FST has the greatest neutralizing potency against activins (19, 27). Secreted activins act in an autocrine manner through cell surface receptor binding and activation of downstream signaling events (17). The role of activin B is unclear, whereas activin A has been shown to induce ROS production and initiate apoptosis in several cell types (7, 9, 17, 35, 49, 67). We, thus, investigated whether activin production and secretion is regulated by Tg in MC. Figure 5A shows that Tg did not increase the cellular protein expression of activins A or B. The secretion of activin A into the medium was also not increased, as assessed by enzyme-linked immunosorbent assay (ELISA), although the expected decrease with FST was seen (Fig. 5B). Thus, Tg does not increase activin A or B.

Since FST functions as a potent activin antagonist, we next investigated whether FST protects against Tg-induced ROS production and apoptosis through neutralization of basally present activins (19, 73). We used activin A and/or B neutralizing antibodies. To first confirm their efficacy in neutralizing activins, we tested Smad3 activation downstream of activins by using the CAGA12 reporter luciferase assay, which contains 12 repeats of the Smad3 consensus binding site. Supplementary Figure S3 confirms that both antibodies inhibit signaling of their targeted activin. However, neither antibody alone or in combination inhibited Tg-induced ROS production in MCs (Fig. 5C), with spectrofluorometric quantification shown in Figure 5D and E. Tg-induced apoptosis, as assessed by caspase 3/7 enzymatic activity, was similarly unaffected by activin A and/or B neutralization (Fig. 5F). These results suggest that the ability of FST to protect against Tg-induced ROS production and apoptosis in MCs is independent of its neutralization of activins A/B.
To determine whether activins can exacerbate Tg-induced ROS accumulation and apoptosis in MCs, we treated cells with Tg in combination with either activin. The addition of recombinant activin A or B did not promote intracellular ROS or SO accumulation, nor did it augment ROS production in response to Tg (Fig. 5G). Spectrofluorometric quantification is shown in Figure 5H and I. Similarly, activin A or B did not promote apoptosis, as assessed by caspase 3/7 enzymatic activity (Fig. 5J). These data demonstrate that activins do not contribute to Tg-induced ROS production and apoptosis in MCs, and that FST is protective independently of its activin neutralizing activity.
FST acts as an ROS scavenger
Since we observed that FST attenuates ROS detection, we next sought to determine whether FST could act as an ROS scavenger. Here, we evaluated the effects of FST on oxidative stress induced by using a general oxidative stress agent, pyocyanin, a cytotoxic pigment secreted by Pseudomonas aeruginosa that induces production of various ROS species (2). As expected, pyocyanin led to the accumulation of both ROS and SO in MCs after 1 h, and this was effectively inhibited by FST (Fig. 6A). Notably, FST inhibited the production of both general ROS species and SO, with effects on SO production similar to those seen by SOD, an enzyme that is well established to play a role in the breakdown and degradation of SO (Fig. 6A). Spectrofluorometric quantification confirmed the inhibitory effects of FST and SOD on pyocyanin-induced ROS and SO production (Fig. 6B, C). It should also be noted here that we observed a small, but significant decrease in ROS by SOD. This is consistent with the known spontaneous conversion of SO to more reactive species such as ONOO−, such that SOD reduction of SO would also reduce these other ROS species (70). This effect might also contribute to the antiapoptotic effect of SOD seen in Figure 4I, despite the absence of significant SO induction by Tg.

Next, we tested the effects of FST on H2O2-induced DCF oxidation by using MC loaded with 2′,7′-dichlorofluorescin diacetate (H2DCFDA). Here, MCs treated with H2O2 were co-incubated with either recombinant FST or catalase, an enzyme that catalyzes the decomposition of H2O2. Figure 6D shows that FST significantly suppressed H2O2-induced DCF oxidation, with potency somewhat greater than that seen with catalase. The specificity of FST in reducing ROS and SO was confirmed by using immunoglobulin G (IgG), one of the most abundant proteins found in serum. Mouse IgG, unlike FST, was unable to neutralize pyocyanin-induced ROS and SO production (Supplementary Fig. S4A–C).
Last, we utilized cell-free assays to determine whether FST can directly scavenge ROS. First, to assess the ability of FST to neutralize H2O2 in a cell-free system and to assess whether FST has peroxidase activity, we utilized a nonfluorescent 10-Acetyl-3,7-dihydroxyphenoxazine (ADHP) probe that reacts with H2O2 and is oxidized in the presence of peroxidase activity to form a highly fluorescent compound, resorufin. Using this assay, we first found that recombinant FST did not possess endogenous peroxidase activity when compared with the positive control horseradish peroxidase (HRP) (Fig. 6E). However, FST incubation with H2O2 significantly decreased H2O2 availability to HRP, indicating its ability to scavenge this ROS species (Fig. 6F). In MC treated with H2O2, this scavenging effect was dose dependent, seen to increase from 25 to 1000 ng/mL of FST (Supplementary Fig. S5A). Further, FST scavenging of H2O2 protected MC against H2O2-induced apoptosis (Fig. 6G) and dose dependently against Tg-induced apoptosis (Supplementary Fig. S5B). In a separate cell-free assay, we examined the effects of FST on xanthine oxidase-generated SO. Here, we found that FST decreased the presence of detectable SO by 20% (0.5 μg FST) and 29% (1 μg FST), in comparison to an 81% percent decrease with SOD (Fig. 6H). Thus, FST has the ability to dose dependently scavenge ROS, which would, at least in part, contribute to the effects seen on intracellular ROS and its antiapoptotic effects.
To address the mechanism by which exogenous FST administration could attenuate intracellular ROS detection (Figs. 4F and 6A), we wished to determine whether recombinant FST could be internalized in the short timeframe we used for treatment in our studies. We, thus, incubated MC with FST for 30 min, and assessed intracellular levels by immunoblotting total cell lysate. Supplementary Figure S5C shows that FST indeed dose dependently enters cells. Since FST internalization is known to occur after its binding to activins (18), we then assessed whether neutralization of activin A would attenuate its internalization. As seen in Supplementary Fig. S5C, FST internalization was only prevented at low doses of FST (25 ng). Interestingly, at higher doses of FST (1000 ng), which are required for FST to fully exert its protective antiapoptotic effects, internalization was not blunted by activin A neutralization. Thus, exogenous FST is rapidly internalized, partially via activin A and partially via an alternate and as-yet-undescribed mechanism, to attenuate intracellular oxidative species.
FST inhibits NOX4 upregulation
With longer-term incubation, FST was shown to inhibit upregulation of NAD(P)H oxidase (Nox) subunits Nox1/5 by silica particles in human lung epithelial cells (34). We, thus, sought to determine whether Tg upregulated the expression of Nox subunits and whether these responses were regulated by FST. Nox4 specifically has been shown to be an important generator of intracellular H2O2 in numerous cell types, including MC (15, 28). Figure 7A and B show that Tg led to a pronounced and transient increase in NOX4 transcript at 4 h, which was diminished by 8 h. Nox1 and Nox2, which are primarily SO-generating enzymes, were not affected by Tg (Fig. 7A). Nox3 transcript was not detected in MC (data not shown). Next, since Nox4 is constitutively active, we assessed whether FST could inhibit Tg-induced Nox4 production and, in turn, activity. Here, we found that Tg-induced Nox4 protein expression was prevented by the addition of exogenous recombinant FST (Fig. 7C). Since oxidative stress and the ERK pathway were shown to be important regulators of Nox4 expression (16), we also tested whether Tg-induced Nox4 induction and its repression by FST is mediated through effects on ERK activation and reduction in oxidative stress. As seen in Figure 7D, ERK activation, assessed by its phosphorylation, was inhibited by FST and the ROS scavenger catalase, and as expected by inhibition of its upstream kinase MEK with U0126. These all also effectively blocked the induction of Nox4 expression by Tg (Fig. 7E), suggesting that FST inhibits Nox4 induction by Tg through attenuation of ROS-induced ERK activation.
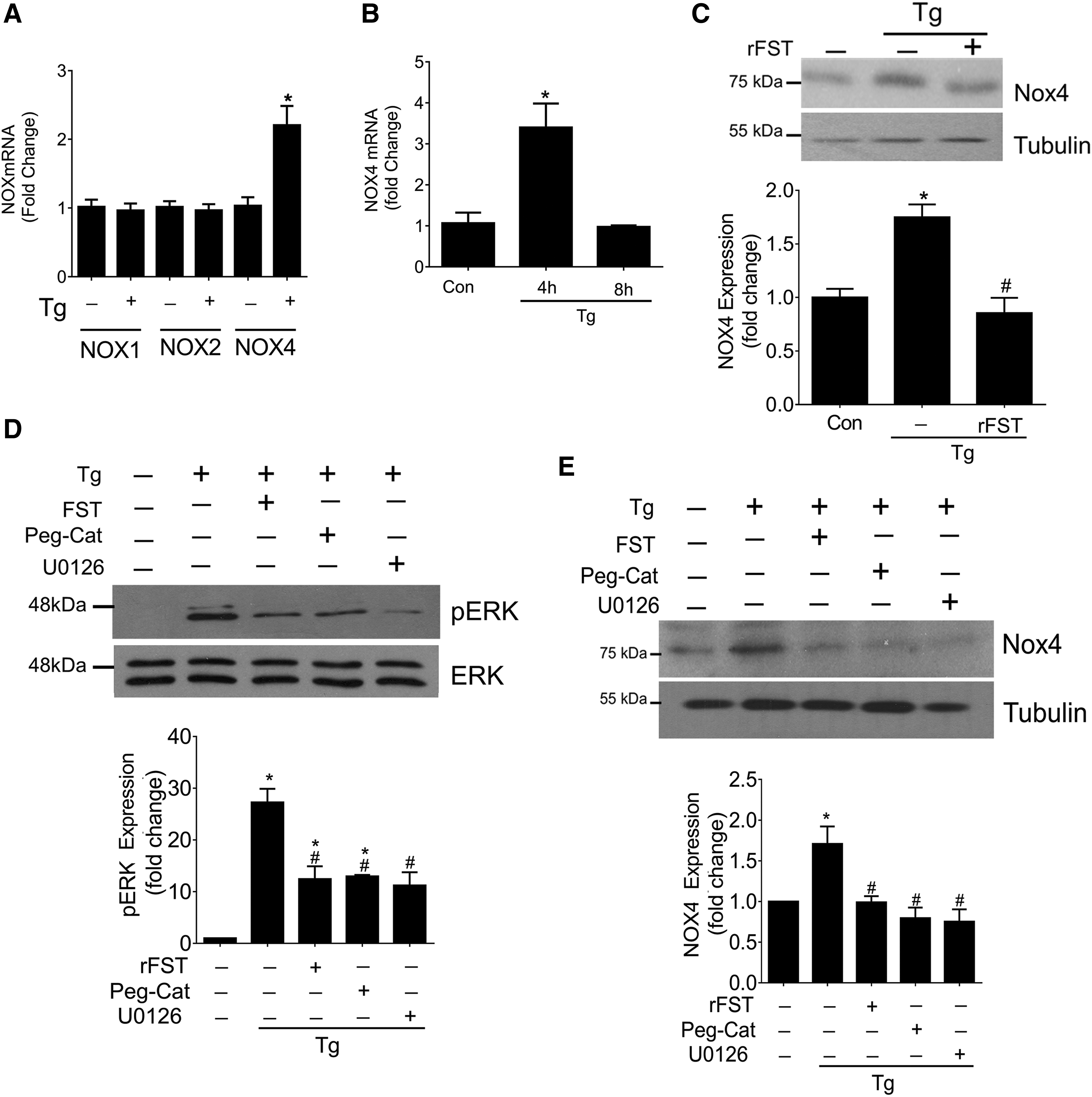
Collectively, these results demonstrate that Tg-induced apoptosis is mediated, at least in part, through oxidative species and that the protective antiapoptotic effects of FST may be attributed to its potent ability to inhibit oxidative stress through both direct and indirect mechanisms.
FST protects against apoptosis and oxidative stress in vivo in CKD
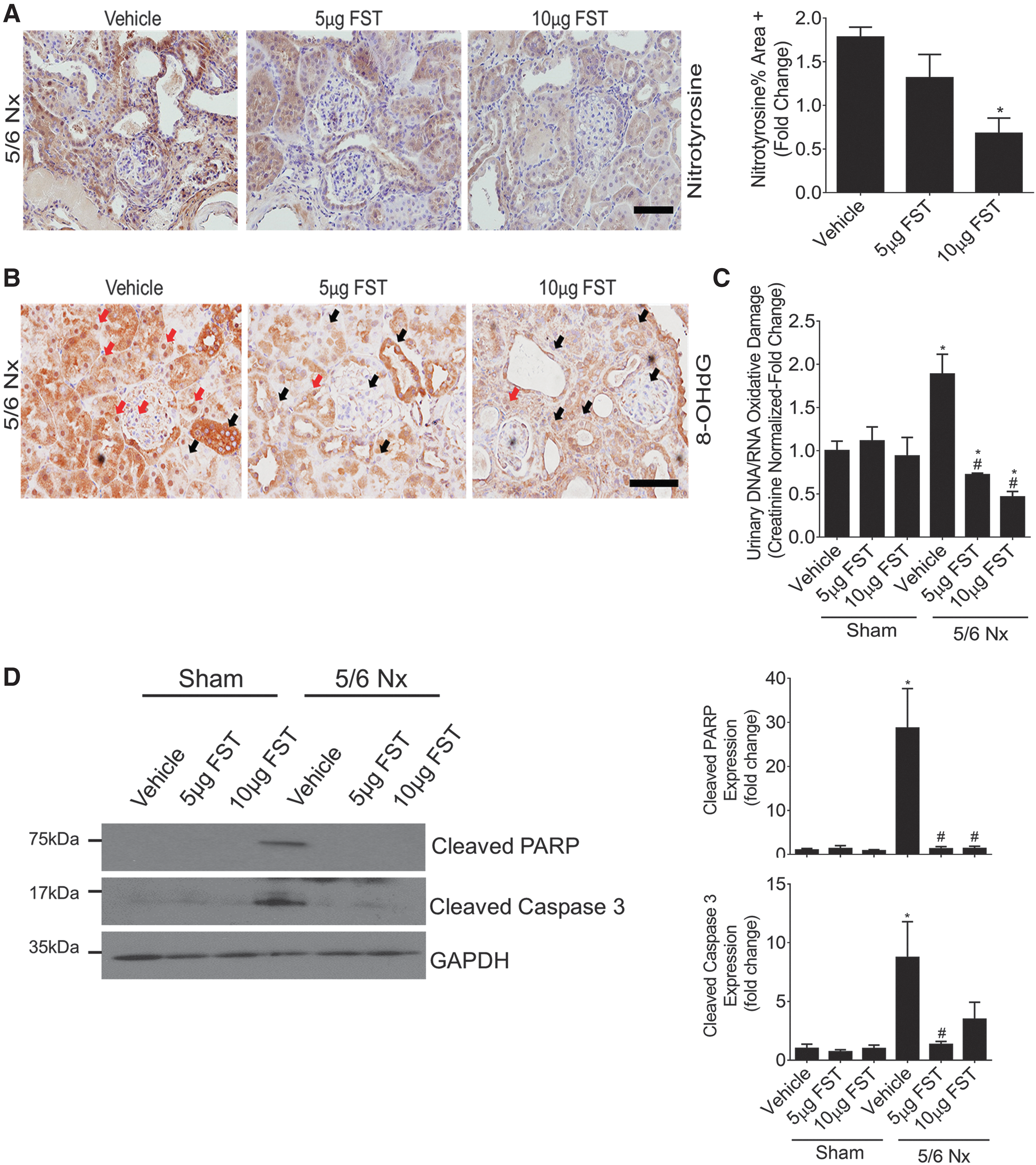
ER stress is a known important contributor to the progression of fibrosis and kidney dysfunction in rodent and human CKD (36, 44). The production of ROS has also been implicated in the pathogenesis of CKD (23, 50, 64). Further, oxidative stress and apoptosis have been correlated with worsening kidney function (56), with several studies showing beneficial effects of inhibiting apoptosis and ROS on limiting CKD progression (6, 52, 53). Since we have shown that FST is effective at inhibiting ER stress-induced apoptosis and ROS accumulation in cultured MCs, we next examined whether FST administration is protective in vivo. We used a well-established 5/6 nephrectomy (5/6 Nx) model of CKD due to renal mass reduction in which ER stress, ROS production, and renal cell apoptosis are known to occur (29, 36, 42, 44). Mice were treated with one of two doses of recombinant FST for 9 weeks before analysis of renal oxidative stress. Immunohistochemistry (IHC) demonstrated a dose-dependent increase in kidney FST after treatment, suggesting its successful targeting to the kidney (Supplementary Fig. S6).
We first analyzed mouse kidneys for the expression of nitrotyrosine, a commonly used marker for ONOO− formation and oxidative stress in vivo. As expected, global protein nitrotyrosination within the glomeruli and tubules of CKD mice was dose dependently inhibited by FST (Fig. 8A). Second, within nuclear and mitochondrial DNA, the presence of oxidized deoxyguanosine molecules, or 8-hydroxy-2′-deoxyguanosine (8-OHdG) is a well-established biomarker of oxidative stress. As expected, we observed prominent expression of 8-OHdG within the nuclei of tubular and glomerular cells in CKD mice treated with vehicle, with staining significantly reduced by FST (Fig. 8B). The release of multiple oxidized guanine species into the urine also serves as a strong indicator of renal oxidative stress in vivo. We assessed this by ELISA, which measures the major oxidative damage DNA/RNA markers 8-OHdG, 8-hydroxyguanosine, and 8-hydroxyguanine (8-OHG). This also demonstrated increased oxidative stress in CKD, which was inhibited by FST in a dose-dependent manner (Fig. 8C). Last, we assessed apoptosis by cleavage of caspase 3 and PARP. The increased apoptosis clearly seen in kidneys of mice with CKD was inhibited by FST (Fig. 8D). Thus, FST is also effective at inhibiting oxidative stress and apoptosis in vivo in mice with CKD.
FST preserves kidney function and protects against renal fibrosis in mice with CKD
Having seen a prominent reduction in oxidative stress and apoptosis in CKD mice treated with FST, we next assessed whether these mice are protected against the progression of CKD. The 5/6 Nx mouse model of CKD is characterized by progressive decline in kidney function, albuminuria, and glomerular and tubulointerstitial fibrosis (14, 29). We found that CKD mice treated with 5 μg FST had a higher glomerular filtration rate (GFR), showing protection against the decline in kidney function (Fig. 9A). FST (5 μg) also reduced albuminuria in these mice (Fig. 9B).
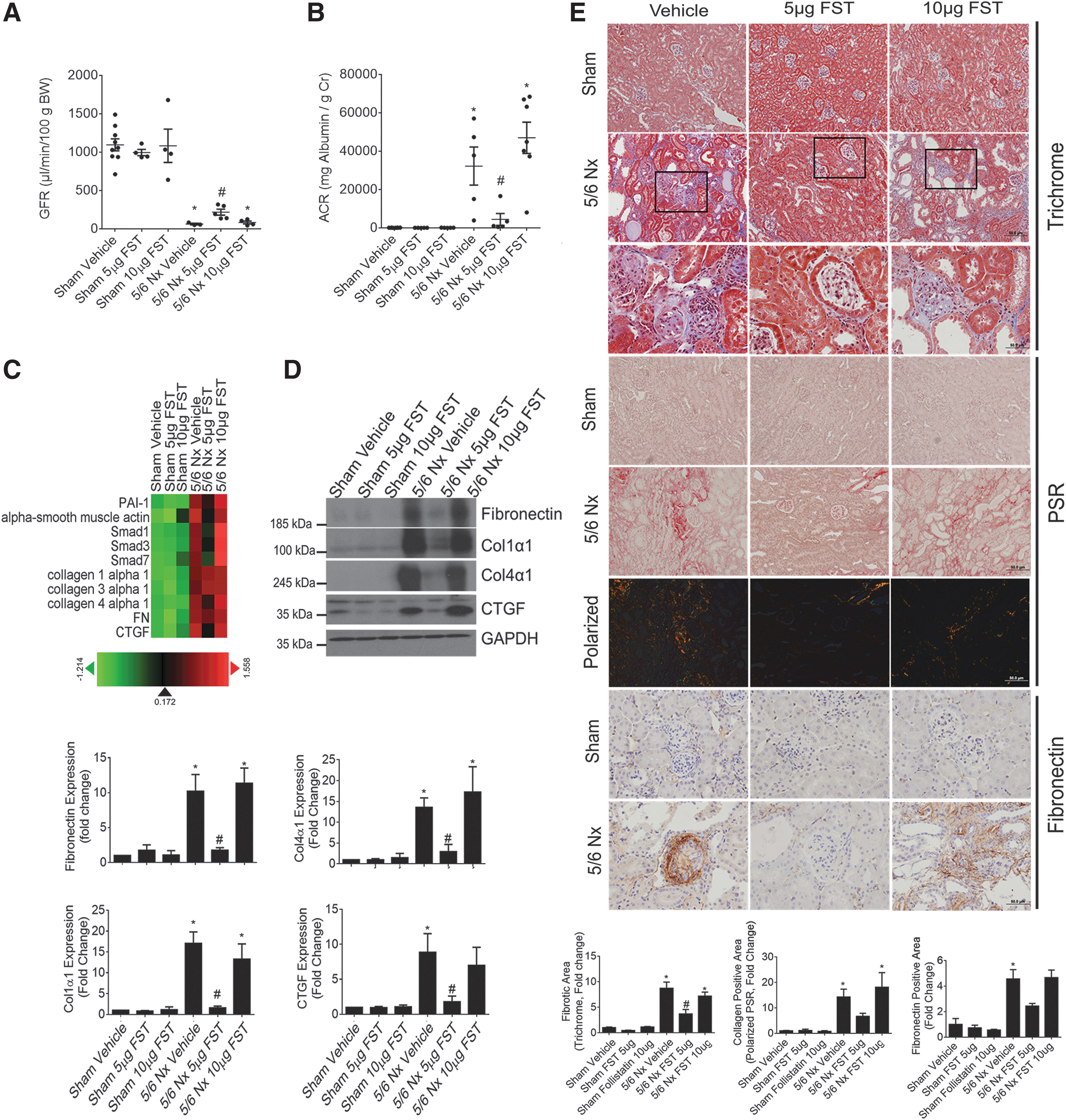
Next, we carried out an RNA screen by using Nanostring to assess for changes in the renal expression of profibrotic and extracellular matrix (ECM) genes involved in fibrosis in CKD. Figure 9C shows an agglomerative clustered heat-map of kidney RNA expression profiles from CKD mice treated with FST. These data demonstrate a reduction of profibrotic and ECM markers by 5 μg of FST, which was confirmed by immunoblotting as shown in Figure 9D. Glomerular and tubulointerstitial fibrosis was also histologically assessed by using picrosirius red (PSR) and trichrome staining, both of which mark the deposition of collagens, as well as with IHC staining for fibronectin. Figure 9E shows both glomerular and tubulointerstitial fibrosis in vehicle-treated CKD mice that were significantly attenuated by 5 μg FST. Thus, in addition to protecting against renal oxidative stress, apoptosis, and kidney function, FST also reduced renal fibrosis in CKD. Interestingly, our data illustrate that although a higher dose of FST (10 μg) provides a greater reduction in renal oxidative stress, this does not directly correlate with a further improvement in kidney function and renal fibrosis. Potential mechanisms are discussed in the Discussion section.
Discussion
Interventions to inhibit chronic oxidative stress and renal cell apoptosis are essential toward halting the progression of CKD to kidney failure. We investigated a potential protective role of FST, a TGFβ superfamily neutralizing protein that primarily neutralizes activins, in the regulation of renal cell apoptosis and oxidative stress. Recent work has shown that FST protects against ROS production and apoptosis in several settings, both in vitro and in vivo (73). This has been believed to be due to neutralization of activin A in some cases, although in others the role of activins has been less clear. For example, FST protection against glucose deprivation-induced apoptosis in cancer cells required its nuclear localization and attenuation of ribosomal RNA (rRNA) synthesis (13). Herein, we found that FST functioned as a cellular stress-responsive protein. Importantly, in cultured MC and when administered systemically to mice with CKD, FST protected against apoptosis, reduced renal fibrosis, and improved kidney function. This was associated with its direct and potent ability to inhibit ROS as a scavenger and thereby to repress oxidative stress, independent of its ability to neutralize activins. Our data, thus, identify a novel, activin-independent, role for FST in attenuating oxidative stress and apoptosis. Together with its antifibrotic properties, FST may be a highly effective novel therapeutic agent for renal protection in a CKD setting.
In keeping with our findings, a protective role of FST against apoptosis has also been demonstrated in several other settings. For example, in pulmonary epithelial cells, FST protected against silica-induced oxidative stress and cell death (34). Conversely, siRNA-mediated downregulation of FST significantly augmented apoptosis in bovine granulosa cells (8). Although these and our studies show an increase in FST expression by various cell stressors, the mechanism by which this increase occurs markedly differs between settings. For example, silica nanoparticles in epithelial cells induce the transcriptional activation of FST (34), whereas glucose starvation in cancer cells induces FST mRNA stabilization (12). In our studies, the increased expression of FST in response to Tg was specifically attributed to a post-translational increase in protein stability. How FST is post-translationally stabilized in response to MC stress requires further investigation.
ER stress is an important pathogenic contributor not only to CKD but also to numerous other chronic diseases (10, 43). Our data show a novel protective role for FST against ER-stress induced apoptosis, effected in our studies by Tg. This is a noncompetitive inhibitor of the SERCA (31, 39, 40, 68). Interestingly, FST attenuated apoptosis without affecting the induction of ER stress by Tg. This may be through its effects on inhibition of oxidative stress, since ROS scavengers inhibited Tg-induced apoptosis in our studies. Indeed, apoptotic induction by Tg in other cells such as cortical neurons and hepatocytes could be alleviated by inhibition of oxidative stress (31, 68).
How FST may function to prevent oxidative stress is not as yet completely understood. Alternate splicing and processing of the FST gene results in extracellular secretion of two major isoforms containing 288 and 315 amino acids (27). These differ in their localization. FST-288 is bound to the cell surface via heparan sulfates (27). In FST-315, an acidic tail blocks this cell-surface binding site, causing its release into the circulation (27). FST most potently not only neutralizes activins but can also inhibit several other TGFβ family members, including myostatin, GDF9, TGFβ3, and BMPs 2, 4, 6, and 7, although with much lower affinity (27). Interestingly, FST-targeted cytokines, through Smad3-dependent and -independent signaling pathways, have been shown to promote ROS production and apoptosis in numerous cell types (7, 49, 62, 67). The role of the primary FST-targeted cytokine, activin, in promoting oxidative stress and apoptosis in MC has not been previously examined. Although activins were shown to induce apoptosis in other cells such as hepatic stellate cells, hepatocytes, and myeloma cells (7, 20, 46, 58, 75), our data show that neither activin A nor B promotes oxidative stress or apoptosis, and that their inhibition is not protective against Tg-induced apoptosis and ROS induction in MC. However, other ligands targeted by FST, namely myostatin and BMP4, have also been shown to promote apoptosis and/or oxidative stress in other settings (25, 62, 74). Whether these other ligands neutralized by FST could contribute to its protective effects against ROS induction and apoptosis in MC remains to be determined.
Reactive species can be produced intracellularly by the NADPH oxidase (Nox) enzyme system. A recent study showed that FST inhibits expression of the subunits Nox1 and Nox5 in human pulmonary epithelial cells (34). It was also shown to reduce activin A-induced Nox2 upregulation in endothelial cells (33). We, thus, assessed the effects of FST on the expression of Nox4, a constitutively active enzyme expressed by MC that is involved in the intracellular production of H2O2 and free radicals such as SO (28, 38). We found that Tg-induced Nox4 upregulation was suppressed by FST. However, it should be noted that the role of Nox4 in CKD is not entirely clear. In some studies, Nox4 is shown to be protective, whereas in others, it promotes cell apoptosis, inflammation, and fibrosis (38, 71). Nonetheless, these and our findings support the notion that FST protects against oxidative stress by affecting the expression and/or activity of intracellular Noxs.
How FST might regulate the expression of Nox4 is as yet unknown. FST was shown to localize to the nucleus and regulate RNA synthesis in glucose-deprived cancer cells (13). Our studies with a mutant FST protein that is unable to enter the nucleus, however, clearly demonstrated that nuclear localization is not required for its ability to protect against ROS generation and apoptosis. Although endogenous nuclear FST in MC may have contributed, this role would likely be minor. Our data also do not support a role for activin-mediated Nox4 induction by Tg given that we did not find any effect of activin neutralization on ROS production. Further, although Smad3 activation by TGFβ1 regulated Nox4 expression in kidney myofibroblasts (5), we also did not observe Smad3 activation by Tg in MC (not shown). Similarly, activation of Smad1, a mediator of BMP signaling, was also unaffected by FST (not shown), suggesting that secretion of TGFβ1 family ligands is not involved in the induction of Nox4 by Tg. Interestingly, however, as found in endothelial cells (16), we identified that Nox4 induction by Tg was mediated by ROS-dependent activation of the ERK1/2 kinases. The ability of FST to scavenge ROS appears critical to its inhibition of ERK1/2-Nox4 induction.
Oxidative species, including H2O2 and SO, can also be generated and regulated extracellularly through cell-surface bound Noxs and SODs, where they have been shown to exert deleterious effects in CKD (65, 70). SO are generally short lived and not readily permeable through cell membranes (70). On the other hand, H2O2 is uncharged, highly stable, and cell-membrane permeant (70). Thus, we questioned whether FST selectively inhibits ROS intra- or extra-cellularly. FST is a secreted glycoprotein that functions extracellularly to neutralize its ligands (27), after which the ligand-FST complex is internalized by endocytosis and cleared by the lysosomal degradation pathway (18). Using cell-free assays, our data show that FST is able to directly scavenge ROS in cell-free conditions, suggesting its ability to scavenge both intra- and extracellularly located ROS. These results are the first to show that FST is able to directly neutralize SO and H2O2. However, treatment with FST quite rapidly also decreased intracellular ROS generation, as assessed by ROS-specific immunofluorescent dyes, in a dose-dependent manner. We thus tested whether this could be explained by internalization of the exogenously administered FST. Surprisingly, we found that MC are able to rapidly internalize extracellular FST, and that this occurs in the presence of activin A. However, when FST is administered at higher doses (≥100 ng), internalization was also found to be independent of activin A. How this occurs is not understood and requires further research.
Since we found that FST is able to scavenge ROS, but does not have endogenous peroxidase activity, we examined its amino acid composition for characteristic features of an ROS scavenger. Either free reduced cys or methionine (met) residues, such as the free reduced Cys34 in human serum albumin, can be readily oxidized and thus act as an endogenous antioxidant (30, 37, 51). FST is highly enriched in cys (n = 36, 11.3%) and met (n = 5, 1.5%) residues (59). However, all of the 36 cys residues in FST are disulfide bonded and thus lacking in free reactive sites that can be readily oxidized (69). On the other hand, the met residues in FST are available for oxidation by H2O2 and met modification does not affect the ability of FST to bind and neutralize activins (59). Interestingly, however, oxidation of tryptophan (trp) in FST was shown to almost completely inhibit activin binding (59). Thus, it is likely that FST can act as a sink for ROS through oxidation of its met and trp residues, and with the latter this is associated with loss of activin neutralizing activity. Further studies are needed to test this hypothesis.
Oxidative stress and apoptosis are critical contributors to the pathogenesis of CKD of varying etiology (22, 23, 56). These stressors induce renal fibrosis, and therapies that reduce renal oxidative stress have been shown to reduce fibrosis and improve renal function (21, 54). ER stress has also been identified in both human and rodent CKD, including in mice with 5/6 Nx as used in our studies (29, 36, 42, 44). Our in vivo data confirm the relevance of our in vitro findings. Indeed, in our mice with experimentally induced CKD, FST was highly protective against both oxidative stress and renal cell apoptosis. Our data are also consistent with the reported attenuation of hepatocyte apoptosis by FST in a model of liver fibrosis (although oxidative stress was not examined here) (48). At the 5 μg FST dose, these protective effects were associated with improvement in renal function, a decrease in urinary albumin excretion, and a significant reduction in renal fibrosis. Indeed, FST has been shown to act as an antifibrotic agent in various organs (3, 41, 48). Interestingly, this antifibrotic effect was believed to be predominantly due to attenuation of activin A signaling. Our data support an additional and potent antioxidative property of FST that contributes to its in vivo efficacy.
Why the higher dose of FST (10 μg), while further reducing oxidative stress, did not improve kidney function and renal fibrosis in our studies is not yet clearly understood. Interestingly, administration of heavily oxidized albumin or tyrosine to animals at high doses over time was shown to induce kidney or liver fibrosis (4, 32). These findings may explain why a higher dose of FST in our in vivo study, while reducing global renal oxidative stress, did not linearly result in improvements in kidney function and fibrosis when compared with the lower efficacious dose. Alternatively, it is also plausible that excessive reduction of ROS such as H2O2, which at physiologic basal levels serve as important homeostatic cell signaling regulators, contributes to worsening disease progression. Future studies will aim at a better understanding of this finding.
Taken together, our data support a novel role for the antifibrotic protein FST as an ROS scavenger with protective effects both in vitro and in vivo in CKD. Future studies will focus on understanding the pharmacokinetic and pharmacodynamic profile of FST as a step toward therapeutic implementation.
Materials and Methods
Cell culture
Primary mouse MCs were isolated from B6129SF1/J mice (Jackson Laboratory) by using Dynabeads (Invitrogen). They were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 20% fetal bovine serum (FBS; Invitrogen), penicillin (100 μg/mL), and streptomycin (100 μg/mL) at 37°C in 95% O2, 5% CO2. Passages 7–14 were used. MCs were serum deprived in 0.5% FBS 24 h before treatment unless otherwise stated. Drugs/reagents used in the study are provided in Supplementary Table S1.
Transfection
Transient expression of plasmids was achieved by using electroporation with the ECM 830 Square Wave Electroporation System (Harvard Bioscience). Briefly, MCs resuspended in electroporation buffer containing the appropriate plasmids (0.5 μg luciferase plasmid with 0.05 μg β-Galactosidase or 10 μg protein expression plasmid) were electroporated by using a single square pulse set at 200 V for 35 ms. siRNA-mediated (50 nM) knockdown was achieved by using RNAiMAX (Thermo Fisher Scientific) as per the manufacturer's recommendation. MCs were serum deprived 24 h after transfection before treatment and harvest. Plasmids and siRNA used in the study are provided in Supplementary Tables S2 and S3.
Luciferase assay
MC lysis was achieved by using Reporter Lysis Buffer (Promega) as per the manufacturer's recommendation. Luciferase activity was measured on clarified cell lysate by using the Luciferase Assay System (Promega) with a luminometer (Junior LB 9509; Berthold). β-galactosidase activity, used to normalize for transfection efficiency, was measured in clarified cell lysates by using the β-Galactosidase Enzyme Assay System (Promega) with a plate reader absorbance set at 420 nm (SpectraMax Plus 384 Microplate Reader; Molecular Devices).
Protein extraction and immunoblotting
MC cell lysis and total cellular protein extraction was carried out by using a buffer containing 20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1% Triton X-100, 1 mM EDTA, 1 mM EGTA, 2.5 mM sodium pyrophosphate, 1 mM/β-glycerophosphate, 2 mM DTT, 1 mM sodium vanadate, 1 mM phenylmethylsulfonyl fluoride, 1 μg/mL leupeptin, and 2 μg/mL aprotinin. Cell lysates were centrifuged (15,000 rpm, 10 min, 4°C), supernatant was collected, protein concentration was quantified and sample boiled in sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) sample loading buffer containing 250 mM Tris HCl (pH 6.8), 10% SDS, 30% (v/v) glycerol, 10 mM DTT, and 0.05% (w/v) bromophenol blue (5 min, 100°C).
Secreted proteins were isolated and concentrated from cell culture media by using trichloroacetic acid and acetone precipitation. Briefly, 1 volume of trichloroacetic acid was added to 4 volumes of cell culture media and incubated (10 min, 4°C). Samples were centrifuged (15,000 rpm, 10 min, 4°C); the resulting pellet was washed three times in cold acetone, air-dried, resuspended, and boiled in SDS-PAGE sample loading buffer.
MC cell lysis and cytoplasmic/nucleus protein extraction was carried out by using hypotonic lysis buffer containing 20 mM HEPES (pH 7.6), 20% glycerol, 10 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 0.1% NP40, 2 mM DTT, 1 mM sodium vanadate, 1 mM phenylmethylsulfonyl fluoride, 1 μg/mL leupeptin, and 2 μg/mL aprotinin. Cell lysates were centrifuged (500 rpm, 10 min, 4°C); the pellet containing the nucleus was suspended in buffer and sonicated; and supernatant (cytoplasmic extract) was collected, protein concentration quantified, and boiled in SDS-PAGE sample loading buffer.
Total, nuclear and/or cytoplasmic protein lysates (10–50 μg) and secreted protein lysates (total yield) were separated on SDS-PAGE for subsequent immunoblotting. Densitometric analysis was carried out by using ImageJ (
Quantitative RT-PCR
RNA from MCs was extracted by using Ribozol RNA Extraction Reagent (Amresco) as per the manufacturer's recommendation, with 1 μg of RNA reverse transcribed into complementary DNA (cDNA) by using qScript cDNA SuperMix Reagent (Quanta Biosciences). qRT-PCR was carried out by using the Power SYBR Green PCR Master Mix (Thermo Fisher Scientific) on the Applied Biosystems ViiA 7 Real-Time PCR System (Thermo Fisher Scientific). mRNA expression and fold changes were calculated by using the ΔΔCT method, where 18S was used as the endogenous control. Primers sequences used in the study are provided in Supplementary Table S5.
Activin A ELISA
At endpoint, secreted activin A was quantified from clarified (15,000 rpm, 10 min, 4°C) MC culture media by using the activin A Quantikine ELISA Kit (R&D Systems).
Apoptosis assays
Caspase-Glo 3/7 Assay (Promega) was used to assess the enzymatic activity of executioner caspases 3 and 7. Briefly, at endpoint, MCs seeded in an opaque-walled clear-bottom 96-well plate were washed in phosphate buffered saline (PBS) and incubated with freshly prepared Caspase-Glo 3/7 reagent (45 min, room temperature). After incubation, luminescence readings, indicative of caspase 3/7 enzymatic activity, were obtained with a microplate reader (LUMIstar Galaxy; BMG Labtech). RealTime-Glo Annexin V Apoptosis Assay (Promega) was used to measure apoptosis in real time in live cells. Briefly, MCs seeded in an opaque white-walled clear-bottom 96-well plate were loaded with freshly prepared Annexin V Detection Reagent in DMEM supplemented with 1% bovine serum albumin (1 h, 37°C). Immediately after loading, treatment was initiated (18 h, 37°C). At endpoint, luminescence readings, indicative of cell surface PS-annexin V binding, were obtained with a microplate reader (LUMIstar Galaxy; BMG Labtech).
Intracellular calcium assessment
Cell-permeant Fura-2 AM (Thermo Fisher Scientific) was used to measure the intracellular concentration of Ca2+ in real time in live cells. Briefly, MCs seeded in an opaque black-walled, clear-bottom 96-well plate were loaded with Fura-2 AM in Ca2+ free HBSS (5 μM, 45 min, room temperature, dark). Immediately after loading, baseline fluorescence readings (ex340/em510 nm and ex380/em510 nm) were taken every minute for 5 min by using a temperature-controlled fluorescent microplate reader at the indicated time points (Gemini EM Spectra Max; Molecular Devices). Experimental drugs/treatments were then introduced, and fluorescence readings were taken every minute thereafter for 30 min (37°C). Intracellular Ca2+ concentrations were calculated by quantifying the ratio of fluorescence signal obtained at 340 and 380 nm (F340nm/F380nm).
ROS and SO detection
ROS-ID Total ROS/Superoxide detection kit (Enzo Life Sciences) and H2DCFDA (Thermo Fisher Scientific) were used for the assessment of oxidative stress. To assess ROS generation through DCF oxidation, MCs seeded in an opaque black-walled, clear-bottom 96-well plate were loaded with H2DCFDA in the dark (20 μM, 45 min, 37°C). Immediately after loading, cells were treated and ROS generation was assessed by measuring fluorescence emissions using a temperature-controlled fluorescent microplate reader set at 37°C (ex490 nm/em525 nm, Gemini EM Spectra Max; Molecular Devices). To simultaneously assess and differentiate between specific ROS species (H2O2, ONOO−, HO, NO, and ROO) and SO, MCs seeded in an opaque black-walled, clear-bottom 96-well plate were loaded with a cocktail of Oxidative Stress Detection Reagent and Superoxide Detection Reagent (ROS/SO cocktail, 5 μM each, 45 min, 37°C, dark). Immediately after loading, cells were treated and ROS and SO generation were assessed by measuring fluorescence emissions using a temperature-controlled fluorescent microplate reader set at 37°C (ex490 nm/em525 nm for ROS and ex550 nm/em620 nm for SO, Gemini EM Spectra Max; Molecular Devices). For some experiments, ROS-ID Total ROS/Superoxide detection kit (Enzo Life Sciences) was used for fluorescence imaging of intracellular ROS and SO accumulation. Briefly, MCs seeded in an eight-well chamber slide were treated and then loaded with the ROS/SO cocktail (5 μM each, 45 min, 37°C, dark). Immediately after loading, slides were cover-slipped and images were taken by using a fluorescein filter set (ex490 nm/em525 nm) for ROS detection and a rhodamine filter set (ex550 nm/em620 nm) for SO detection (EVOS FL Cell Imaging System; Thermo Fisher Scientific).
ROS and SO scavenging assays
A Colorimetric OxiSelect Superoxide Dismutase Activity Assay Kit (Cell Biolabs) was used to measure the ability of FST to neutralize SO. Briefly, SO was generated by using the xanthine/xanthine oxidase complex in an opaque-walled clear-bottom 96-well plate. A chromogen that produces a water-soluble formazan dye on reduction by SO anions was used to quantify the amount of SO. The chromogenic reaction was measured by using a plate reader absorbance set at 490 nm (SpectraMax Plus 384 Microplate Reader; Molecular Devices).
A Fluorometric OxiSelect Hydrogen Peroxide/Peroxidase Assay Kit (Cell Biolabs) was used to measure the ability of FST to neutralize H2O2 and to assess whether FST has peroxidase activity. Nonfluorescent ADHP was added and allowed to react with H2O2, which is oxidized in the presence of peroxidase activity to form a highly florescent compound, resorufin. Fluorescent emissions are proportional to the amount of H2O2. Fluoresce emissions were measured by using a plate reader (ex530 nm/em590 nm, Gemini EM Spectra Max; Molecular Devices).
Animal studies
Animal studies were carried out in accordance with the principles of laboratory animal care and McMaster University and Canadian Council on Animal Care guidelines. Male CD1 mice were obtained from Charles River Laboratories. CKD was achieved by using the 5/6 Nx renal mass reduction model. Briefly, at 6–7 weeks of age, anesthetized mice underwent resection of the upper and lower poles of the left kidney. After a 1-week recovery period, anesthetized mice underwent a right nephrectomy. Sham mice were anesthetized and the kidney was manipulated without resection. Resected kidney weights were divided by the nephrectomized right kidney weight (Nx ratio) and mice were placed into six groups, with the Nx groups containing mice with roughly equal Nx ratios: Sham-Vehicle (n = 5), Sham-FST- 5 μg (n = 5), Sham-FST-10 μg (n = 5), 5/6 Nx-Vehicle (n = 5), 5/6 Nx-FST-5 μg (n = 5), and 5/6 Nx-FST-10 μg (n = 7). After completion of the 5/6 Nx, vehicle-treated mice were injected (IP) every other day with vehicle (20 mM NaPO4, 500 mM NaCl, pH 7). FST-treated mice were injected (IP) every other day with human recombinant FST-288, provided by Paranta Biosciences Limited and followed for 9 weeks.
At study endpoint, urine was collected and albumin-to-creatinine ratio was measured according to the manufacturer's instructions (Albuwell M, Exocell for urine albumin and Crystal Chem for creatinine). To assess DNA/RNA oxidative damage by ELISA, urine samples were centrifuged (3000 rpm, 5 min, 4°C), diluted 500-fold in ELISA buffer, and assessed by using a DNA/RNA Oxidative Damage ELISA kit (Cayman Chemicals). This kit measures major oxidative damage markers 8-OHdG, 8-hydroxyguanosine, and 8-OHG. Obtained concentrations (pg/mL) were normalized against urinary creatinine values, and they were measured by using a kit (Exocell).
GFR was assessed in conscious mice by measuring the clearance of fluorescein isothiocyanate (FITC)-labeled sinistrin (Fresenius Kabi Linz). Briefly, a 5% FITC-sinistrin solution was injected retro-orbitally, after which blood was collected from the saphenous vein at 7, 15, 30, 60, 90, and 120 min. Plasma fluorescence was assessed by using a fluorometer (Gemini EM; Molecular Devices) at 485 nm excitation and 538 nm emission. After GFR assessment, mice were perfused with cold PBS. Kidney portions (renal cortex) were snap-frozen in liquid nitrogen for RNA or protein analysis or fixed in formalin for IHC.
RNA was isolated by using Trizol (Invitrogen). Nanostring analysis on the extracted RNA was carried out at the Farncombe Metagenomics Facility at McMaster University. Data were analyzed by using nSolver 4.0. Samples were normalized by using background subtraction with the negative control, and the geometric means of the housekeeping genes (actin, gusb, pgk1, and pp1a). For protein analysis, the cortex was sonicated in lysis buffer, centrifuged (15 min, 13,000 rpm, 4°C) and the supernatant was separated on a SDS-PAGE for subsequent immunoblotting. Protein expression was normalized against GAPDH.
Formalin-fixed sections (4 μm) were stained with trichrome or PSR. For fibronectin IHC, 4 μm FFPE kidney sections were deparaffinized; endogenous peroxidase activity was blocked; tissues were blocked with 5% horse serum and incubated in primary antibody (overnight, 4°C). Tissues were incubated with biotinylated secondary antibodies (Vector Labs; 30 min, room temperature) and then with streptavidin/peroxidase (30 min, room temperature; Vector Labs). Chromogenic color development was carried out by using Nova Red (Vector Labs), followed by counterstaining using Gill's hematoxylin (Sigma) and mounting (Permount; Thermo Scientific). Images were quantified by measuring the percentage of positive area examined under transmitted light by using ImageJ. All micrographs were captured at × 200 and × 400 magnification by using the BX41 Olympus microscope.
Statistical analysis
Statistical analyses were performed by using GraphPad Prism 6. A Student's t-test or one-way analysis of variance (ANOVA) was used to determine statistical significance between two or more groups of data, respectively. Post hoc significance of pairwise comparisons was assessed by using Tukey's HSD. Statistical significance between two categorical independent variables was assessed by using a two-way ANOVA, with Bonferroni's multiple-comparisons test. A p value <0.05 (two-tailed) was considered significant. Data are presented as mean ± standard error of the mean. The number of experimental repetitions (n) is indicated in the figure captions.
Footnotes
Acknowledgments
The authors acknowledge the support of St. Joseph's Healthcare for nephrology research. They thank Dr. M. Bilandzic (Prince Henry's Institute, Australia) for providing pGL3-CAGA12-luc, Dr. J. Yoon (Maine Medical Center Research Institute) for providing mFST-luc, and Dr. X. Gao (Institute of Environmental Medicine, China) for providing the WT-FST and ΔNLS-FST over-expression constructs. They are also grateful to Paranta Biosciences Limited (Australia) for providing human recombinant FST-288 for animal administration. Finally, they thank Richard van Krieken for his assistance in blood collection for GFR measurements. This work was supported by the Canadian Institutes of Health Research (CIHR; J.C.K.), MOP 136868 and Kidney Foundation of Canada (J.C.K.), KFOC160011. N.M. is the recipient of a studentship award from the Research Institute of St. Joe's Hamilton.
Authors' Contributions
N.M. and A.L.G. performed experiments and analyzed data; D.Z. performed experiments; N.M. wrote the article; A.L.G., N.M., and J.C.K. conceived the ideas; B.G. assisted with animal studies; and all authors read and approved the final article.
Author Disclosure Statement
The authors declare that they have no competing financial interests.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
Supplementary Table S5
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.