
Abstract
Significance:
Pulmonary hypertension (PH) is characterized by elevated vascular resistance due to vasoconstriction and remodeling of the normally low-pressure pulmonary vasculature. Redox stress contributes to the pathophysiology of this disease by altering the regulation and activity of membrane receptors, K+ channels, and intracellular Ca2+ homeostasis.
Recent Advances:
Antioxidant therapies have had limited success in treating PH, leading to a growing appreciation that reductive stress, in addition to oxidative stress, plays a role in metabolic and cell signaling dysfunction in pulmonary vascular cells. Reactive oxygen species generation from mitochondria and NADPH oxidases has substantial effects on K+ conductance and membrane potential, and both receptor-operated and store-operated Ca2+ entry.
Critical Issues:
Some specific redox changes resulting from oxidation, S-nitrosylation, and S-glutathionylation are known to modulate membrane receptor and ion channel activity in PH. However, many sites of regulation that have been elucidated in nonpulmonary cell types have not been tested in the pulmonary vasculature, and context-specific molecular mechanisms are lacking.
Future Directions:
Here, we review what is known about redox regulation of membrane receptors and ion channels in PH. Further investigation of the mechanisms involved is needed to better understand the etiology of PH and develop better targeted treatment strategies.
Introduction
Normally, the pulmonary circulation is maintained in a low-pressure, low-resistance state with little resting tone. Pulmonary arteries are thin-walled, and rely heavily on pulmonary arterial distension and recruitment for reducing pulmonary vascular resistance when cardiac output is elevated. Under pathophysiological conditions, however, active vasoconstriction and vascular remodeling lead to enhanced pulmonary vascular resistance and subsequent pulmonary hypertension (PH) (Fig. 1). The World Health Organization (WHO) classifies five categories of PH, including PH that is secondary to left heart disease (WHO Group 2), thromboembolic disease (Group 4), or blood disorders and other less common causes (Group 5). However, for the purposes of this review, we focus on PH due to hypoxia (WHO Group 3), such as that resulting from chronic obstructive pulmonary disease, interstitial lung diseases, chronic high-altitude exposure, and sleep apnea. We also discuss redox signaling in pulmonary arterial hypertension (PAH) (WHO Group 1), which includes heritable and idiopathic forms of the disease and is characterized by narrowing of the small pulmonary arteries.

A rise in both basal and stimulated pulmonary arterial smooth muscle cell (PASMC) intracellular free Ca2+ concentration ([Ca2+]i) is a major trigger for enhanced pulmonary vascular resistance in PH, and can be attributed to persistent PASMC depolarization, receptor stimulation, mechanical stimuli, and alveolar hypoxia. Anomalies in cellular redox potential and alterations in reactive oxygen species (ROS) homeostasis can largely influence the expression, function, and regulation of receptors, ion channels, and transporters involved in PASMC Ca2+ homeostasis and ultimately play an important role in the regulation of vascular tone and development of PH.
Oxidative stress is considered a hallmark of PH. In this context, ROS, including hydrogen peroxide (H2O2) and superoxide (•O2 −), mediate changes in vascular reactivity and vascular remodeling (Fig. 1) by enhancing responses to vasoconstrictor stimuli and promoting increased cellular proliferation and migration, respectively. Reactive nitrogen species, such as nitric oxide (NO) and peroxynitrite (ONOO−), can also contribute to redox signaling through electron transfer (167). Although ROS are a natural by-product of oxygen metabolism, abnormally high levels of these free radicals can overwhelm cellular antioxidant capacity. This oxidative stress—characterized by lipid peroxidation, DNA oxidation, and post-translational oxidative protein modifications—can create an environment of inflammation, damaged cell membranes, autoimmunity, and cell death.
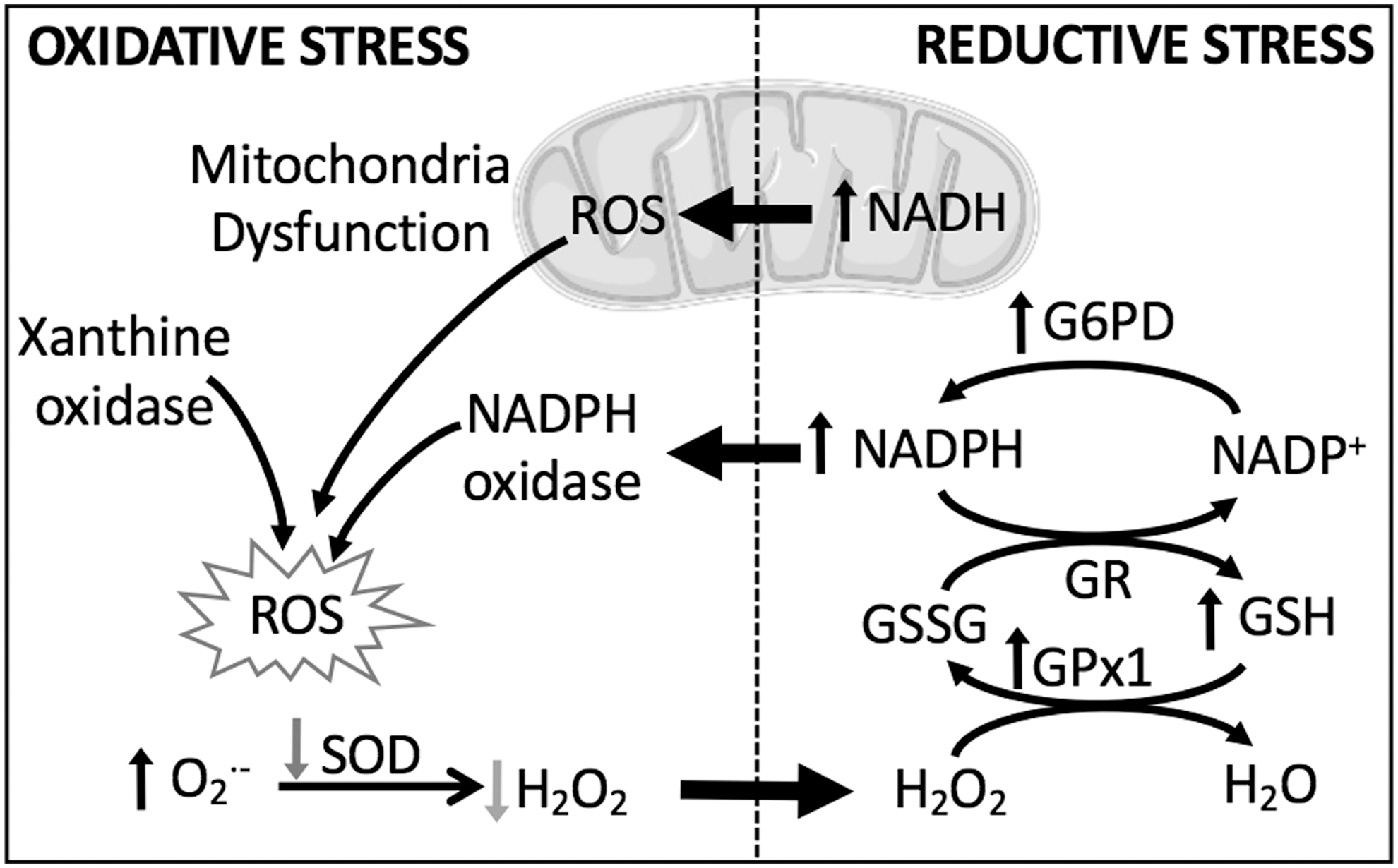
Although it is understandable why antioxidant strategies and oxidant inhibitors would pose an attractive treatment option for PH, these strategies have not always proven to be beneficial in treating the disease. Accordingly, there is a growing appreciation that reductive stress, defined by aberrantly high levels of intracellular reducing equivalents and a loss of ROS, is equally detrimental. Reductive and oxidative stress are closely intertwined processes, and we consider how prolonged reductive stress may cause cellular dysfunction, in part by increasing ROS (Fig. 2). We discuss how the interplay between reductive and oxidative stress leads to changes in membrane receptor function and ion channel activity, enhanced vasoconstriction and increased vascular remodeling associated with PH.
A Central Role of Reductive and Oxidative Stress in Hypoxic PH
Oxidative stress is an important pathogenic mechanism in PH, as demonstrated in both patients with the disease and an overwhelming number of experimental models of PH (67, 122, 152). This fact can create the improper assumption that an increased production of ROS is always detrimental. However, ROS play a critical role as second messengers in normal physiology, and an improper depletion of oxidant signaling and/or overproduction of reducing equivalents can be equally damaging by altering protein function (particularly with respect to disulfide bonds), reducing mitochondrial efficiency, and shifting cellular metabolism [reviewed in Pérez-Torres et al. (136)]. Furthermore, chronic reductive stress, indicated by high levels of reducing equivalents such as nicotinamide adenine dinucleotide (phosphate) reduced/oxidized (NAD(P)H/NAD(P)+) and GSH/GSSG, can paradoxically induce ROS production by feedback regulation (Fig. 2). In addition, during reductive stress, when electron acceptors are less readily available, some redox proteins have been shown to donate electrons directly to O2, thus generating ROS in the form of •O2 − (82). One can appreciate the necessity for redox equilibrium, held in check by balanced redox couples, sufficient antioxidants, and moderate levels of ROS.
Types of ROS: •O2 − and H2O2
The main ROS of interest in PH, •O2 − and H2O2, can be generated from multiple sources, including the NADPH oxidase (NOX) family of enzymes (which will be discussed in more detail below), and as a by-product of the mitochondrial electron transport chain (ETC). At complex IV of the ETC, O2 serves as the terminal acceptor of electrons, yet a fraction of unpaired electrons can leak from the ETC to yield •O2 − radicals, commonly at complex I and complex III (112). To remove this highly reactive ROS, mitochondria express a unique scavenging enzyme, superoxide dismutase 2 (SOD2), which transforms •O2 − radicals into the less injurious H2O2. Various models of PH and patients with PAH exhibit increased •O2 − generation and low levels of SOD expression and activity (8, 15, 138, 141), indicating the importance of this antioxidant system in the pathogenesis of PH.
The role of H2O2 in PH pathology is less clear. In a mouse hypoxic PH model, increased expression of SOD2 and subsequent high levels of H2O2 exacerbated right ventricular systolic pressure (RVSP), right ventricular hypertrophy, and pulmonary vascular remodeling (2). However, we and others have observed decreased H2O2 in response to hypoxia that is due to dysregulation of SOD and increased glutathione peroxidase (GPx) expression (15, 138, 141).
Sources of ROS
In addition to NOX isozymes and the ETC mentioned above, the pulmonary vasculature contains several other sources of ROS generation, such as arachidonic acid oxygenases, xanthine oxidase, and uncoupled endothelial nitric oxide synthase (eNOS). Arachidonic acid oxygenases insert O2 into arachidonic acid and produce •O2 − as a by-product, but there is little evidence that ROS from these sources contribute to PH (152). Xanthine oxidase oxidizes hypoxanthine to xanthine and xanthine to uric acid, generating NADH and •O2 − in the process, and this pathway is upregulated in response to hypoxia (135) (Fig. 2). eNOS normally synthesizes the vasodilator NO, but when its cofactor BH4 (tetrahydrobiopterin) is depleted, as in the case of chronic hypoxia (CH), eNOS produces •O2 − instead. eNOS downregulation and dysfunction are both associated with PH (37, 51).
Nicotinamide adenine dinucleotide (phosphate) in reductive stress
In the context of hypoxic PH, arterial cells undergo metabolic changes that can create an environment of reductive stress [reviewed in Stenmark et al. (160)]. Indeed, like in cancer, PH is characterized by a Warburg-like shift to aerobic glycolysis, high rates of proliferation and migration, and resistance to apoptosis (137). These mechanisms will be discussed in more detail below, but we first examine how hypoxia leads to reductive stress. Hypoxia-inducible factors (HIFs), such as HIF-1α, regulate oxygen-dependent expression of hundreds of genes in many cell types, including the pulmonary vasculature. Upregulation of HIF-1 has been implicated in decreased apoptosis, increased proliferation and migration, and enhanced contractility of pulmonary arteries [reviewed in Shimoda and Laurie (154)].
One of the many effects of HIF signaling is to acutely stimulate increased glucose transport into pulmonary vascular cells (Fig. 3). Under normal conditions, hexokinase phosphorylates glucose to glucose-6-phosphate (G6P), which generates electron donors that reduce oxygen and provide energy through the mitochondrial ETC. However, hypoxia inhibits the respiratory chain due to the lack of available O2, leading to accumulation of reduced nicotinamide adenine dinucleotide (NADH) (123). Since complex I of the ETC is the entry point for electrons from NADH, buildup of NADH then stimulates complex I to oxidize more NADH to NAD+. This leads to an increase in electron leakage at complex I, allowing for the reduction of O2 to •O2 − [reviewed in Murphy (112)] (Fig. 3). In this scenario, a high level of the reducing equivalent NADH can lead to oxidative stress by the increased formation of •O2 −.
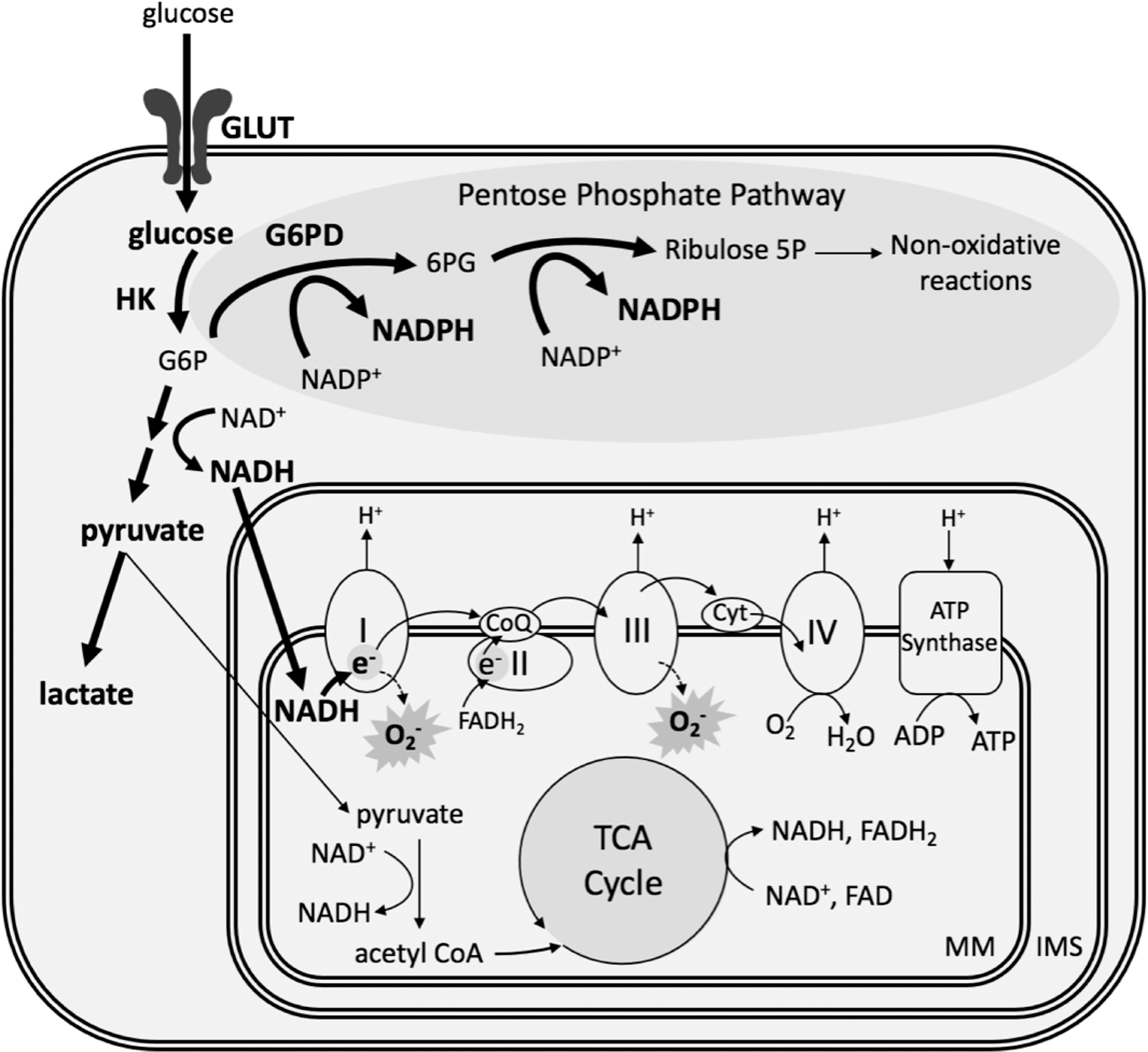
In the cytoplasm, increased glucose uptake in response to hypoxia also shunts some G6P to the pentose phosphate pathway via G6P dehydrogenase (G6PD), where it is used to generate high levels of reduced nicotinamide adenine dinucleotide phosphate (NADPH), relative to its oxidized counterpart (NADP+) (Fig. 3). Hypoxia increases G6PD activity and promotes PASMC proliferation (56), while inhibition of G6PD prevents PASMC conversion to the synthetic phenotype by reducing mitochondrial •O2 − generation (22).
NADPH is a critical reducing equivalent for the H2O2 scavenging systems of the mitochondrial matrix. First, it functions as a substrate for glutathione reductase to regenerate reduced glutathione (GSH) from its oxidized counterpart (GSSG). GSH is the main source of reducing equivalents for the GPx family of enzymes. Glutathione peroxidase isoform 1 (GPx1) inhibits the intracellular accumulation of H2O2 by reducing it to H2O (Fig. 2). Indeed, models of CH-induced PH have observed increased GPx1 expression and activity, and accordingly low levels of H2O2 (138).
L-2-hydroxyglutarate in reductive stress
Hypoxia limits carbon flux through the tricarboxylic acid (TCA) cycle, and shifts energy metabolism through glycolysis and the pentose phosphate pathway. Accordingly, most TCA metabolites are reduced in PASMCs following hypoxia, with the notable exception of α-ketoglutarate (αKG) and its reduced metabolite 2-hydroxyglutarate (2HG) (123). Both
2HG competitively inhibits αKG-dependent enzymes, which can have profound effects on chromatin remodeling, and L-2HG stabilizes HIF-1α signaling following hypoxia (176). L-2-hydroxyglutarate dehydrogenase (L2HGDH) normally metabolizes L-2HG back to αKG in a redox reaction via the oxidation of NADH and reduction of flavin adenine dinucleotide (FAD); yet, L2HGDH mRNA and protein expression are reduced following hypoxia (123). Oldham et al. (123) provide evidence that L2HGDH knockdown enhances hypoxia-mediated increases in NADH/NAD+ in pulmonary cells, while overexpression of L2HGDH has the opposite effect. Altogether, these data suggest that L-2HG metabolism plays an important role in regulating both energy pathways and cellular redox homeostasis following hypoxia in the pulmonary vasculature.
NOXs in oxidative stress
NADPH also functions as a substrate for the NOX family of enzymes, which are an important source of ROS in the vasculature (Fig. 2). NOX activation results in the transfer of electrons from NADPH through FAD to oxygen, generating •O2 −. Of the seven NOX isoforms that have been identified in mammals, four isoforms (NOX1, NOX2, NOX4, and NOX5) are commonly expressed in the vasculature (84), although less is known about NOX5 since it has only been identified in primates and not in rodents (58). Upregulation and activation of NOX increase ROS production in vascular tissues, which can have multiple downstream effects. NOX-derived ROS directly inhibit KV channel activity (107) in PASMCs, as well as increase intracellular Ca2+ concentration by targeting both store-operated Ca2+ entry (SOCE) and activity of ryanodine receptors (RyRs) (185). Both mechanisms have been linked to enhanced PASMC contraction. In addition, NOX-derived ROS can further enhance ROS production by affecting other enzymes, such as eNOS or xanthine oxidase (83).
NOX1 and NOX2 primarily generate •O2 −, which can be rapidly converted to H2O2 by SOD. NOX4 has uniquely been shown to produce H2O2 directly (36, 168). NOX1 contributes to PASMC migration and proliferation in a rat model of monocrotaline-induced PAH (178); yet, it does not play a substantial role in hypoxic PH. NOX2 and NOX4 have both been implicated in hypoxia-induced PH (44, 98, 108, 118), although the relative importance of each isoform to disease progression is somewhat in dispute. NOX2 activation contributes to hypoxia-induced endothelial dysfunction by reducing NO bioavailability (48, 97) and to depolarization-induced vasoconstriction in pulmonary arteries (118). The importance of NOX2 in PH is illustrated by the fact that Nox2−/− mice show reduced pulmonary vascular resistance and lower RVSP (98).
NOX4 expression is increased in PASMCs following hypoxia (35, 108), in patients with idiopathic pulmonary arterial hypertension (IPAH) (53) and in monocrotaline-induced PAH (12). NOX4-derived ROS have been shown to promote vascular remodeling by both PASMC (55, 161) and adventitial fibroblast proliferation (92). However, Veith et al. provided evidence that Nox4−/− mice were not protected against CH-induced indices of PH, including elevated RVSP, right ventricular hypertrophy, and pulmonary arterial vascular remodeling (179). These results contrast with those obtained from experiments in isolated cells, indicating that the in vivo and in vitro cellular environment may drastically affect the contributions of NOX isoforms to PASMC dysfunction.
Redox Regulation of Membrane Receptors
Both G protein-coupled receptors (GPCRs) and receptor tyrosine kinases (RTKs) are expressed in PASMCs and pulmonary arterial endothelial cells (PAECs), control multiple cell functions, and are implicated in the development of PH [reviewed in Refs. (19, 68)]. In general, little is known regarding specific redox regulation of GPCRs. However, evidence from pulmonary arteries demonstrates that S-nitrosothiols (SNO) can directly modify GPCRs through S-nitrosylation, an important post-translational modification, leading to inhibition of vasoconstriction (121) (Fig. 4). Consistent with this notion, a recent study found that chronic administration of sodium nitrite increased lung S-nitrosylation and physiological NO signaling in rat pups, effectively reversing CH-induced PH (70). Therefore, SNO and protein S-nitrosylation may serve to mitigate GPCR activation.
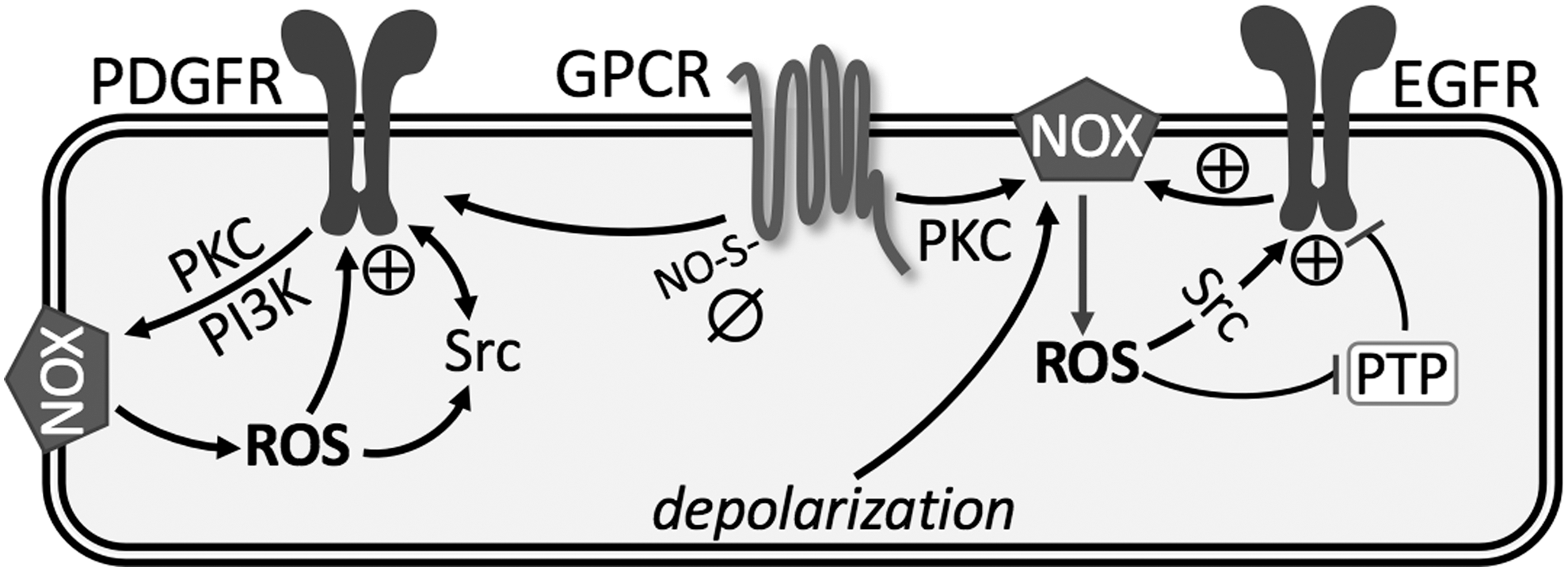
More notable in the pulmonary vasculature are the various signaling pathways linking GPCR to activation of NOX-derived ROS, which augment pulmonary vasoconstriction and development of PH (73, 98, 118, 153, 186). In addition, GPCR-induced ROS target parallel signaling mechanisms, including RTKs, protein tyrosine phosphatases (PTPs), protein kinase C (PKC), mitogen-activated protein kinases, and transcription factors [reviewed in Ushio-Fukai (177)]. Accumulating evidence suggests that ROS are the primary signaling intermediates through which GPCRs transactivate RTKs, such as epidermal growth factor receptor (EGFR) and platelet-derived growth factor receptor (PDGFR) (18, 40).
Epidermal growth factor receptor
Activation of EGFR promotes receptor homo- or heterodimerization, resulting in autophosphorylation of intracellular tyrosine residues, thereby recruiting various adapter molecules and activating an array of downstream signaling cascades involved in growth, proliferation, and cell contraction. In addition to canonical activation by ligands such as epidermal growth factor (EGF), transforming growth factor-α (TGF-α), and amphiregulin, EGFR can be transactivated by a variety of stimuli, including GPCR stimulation, depolarization, mechanical deformation of cell membranes, and redox-dependent mechanisms [reviewed in Refs. (61, 87)] (Fig. 4). EGFR therefore serves to integrate information from distinct upstream pathways to downstream cellular responses.
Although lung EGFR levels are not altered in either human PAH or rodent models of PH, EGFR signaling is enhanced in these models, independent of changes in expression (31, 118, 140). EGFR contributes to PH in mice, which overexpresses the EGFR ligand TGF-α (29), and chronic EGFR inhibition improves survival and attenuates arterial remodeling in rats with monocrotaline-induced PH (140). EGFR activation associated with monocrotaline exposure is thought to result from oxidative dimerization of EGFR through a Src- and ligand-independent mechanism (140).
Oxidative modifications of EGFR occur via ROS produced by NOX isoforms. In vascular smooth muscle cells, c-Src acts as an upstream regulator of EGFR transactivation and is also required for ROS generation (Fig. 4) (151). In aortic vascular smooth muscle, binding of angiotensin II to AT1 receptor triggers NOX-dependent •O2 − generation, which results in phosphorylation and activation of EGFR (46). In PASMC, H2O2-mediated oxidation stimulates intrinsic intracellular protein-tyrosine kinase activity and formation of covalently modified dimeric EGFR (140). This H2O2-induced EGFR transactivation has also been observed in systemic vascular smooth muscle, but seems to require metalloprotease-dependent shedding of heparin-binding EGF-like growth factor (47, 69, 106). Regardless, this oxidant-induced dimerization plays an important role in stimulating contraction and proliferation of PASMC observed in PH (118, 140).
In addition, several studies illustrate that ROS production by NOX inactivates PTPs due to oxidation of a conserved catalytic cysteine residue that is essential for phosphotyrosine hydrolysis, thereby enhancing and prolonging EGFR activation (61) (Fig. 4). However, whether similar inactivation of PTPs accounts for enhanced EGFR-dependent vasoconstrictor reactivity in PH remains to be determined. Furthermore, we have identified a unique effect of CH to couple depolarization-induced EGFR activation to NOX2-mediated pulmonary arterial constriction without a change in EGFR protein levels, a response not present in control vessels (118). These data suggest that there may be reciprocal activation of EGFR and NOX, leading to progression of pathogenesis of PH.
Platelet-derived growth factor receptor
Transactivation of PDGFR by GPCR also involves NOX-derived ROS. Similar to EGFR, PDGFR promotes the proliferation and migration of PASMC in the progression of PH. In addition, PDGF plays an important role in promoting apoptosis resistance of PAEC and development of endothelial plexiform lesions (90). Using genetic approaches, the activated PDGFR-β isoform has been found to be a key contributor to pulmonary vascular remodeling in experimental models of PH (32, 172). Selective disruption of PDGF-dependent phosphatidyl-inositol-3-kinase (PI3K) and phospholipase Cγ (PLCγ) activity is sufficient to abolish these pathogenic responses in vivo (172). Interestingly, redox regulation of PDGFR tyrosine autophosphorylation and its signaling is related to NOX activity through PKC and PI3K. Through its receptors, PDGF activates c-Src kinase, PI3K and PKC, leading to activation of NOX and increased H2O2, which in turn contributes to the full activation of both PDGFR and c-Src tyrosine phosphorylation (17) (Fig. 4).
Redox Regulation of K+ Channels
Abnormal expression and activity of K+ channels in PASMCs have been implicated in increased vasoconstriction in both human PAH and animal models of PH (63, 196). Located at the cell membrane, these channels are very selective for K+ and play an important role in regulating cell membrane potential. Four main types of K+ channels have been identified in the vasculature and will be discussed in more detail: voltage-gated (Kv) channels, calcium-activated (KCa) channels, tandem-pore domain (K2P) channels, and inwardly rectifying, ATP-sensitive (KATP) channels (Fig. 5).
Voltage-gated (KV) channels
KV channels are the largest subgroup of K+ channels, and they are involved in several fundamental cellular functions related to the sensing and controlling of membrane potential. These ion channel protein complexes open upon membrane depolarization and allow efflux of K+ down its electrochemical gradient. Therefore, in excitable vascular smooth muscle cells, increased K+ channel expression and/or activity leads to hyperpolarization and subsequent vasodilation. In contrast, low expression and/or closure of K+ channels increases the net influx of K+ ions, which depolarizes the cell, activates voltage-gated Ca2+ channels (VGCCs), and enhances vascular smooth muscle contraction (197).
The intracellular redox environment can directly affect KV channels by altering function, or indirectly through mechanisms that change KV channel expression at the cell membrane. KV1.1, KV1.2, KV1.5, KV2.1, KV3.1, and KV9.3 subtypes have been found in PASMCs (9). Among these, the expression of KV1.2, KV1.5, and KV2.1 is reduced in PH (105, 139) and in PASMCs following hypoxia (64, 182, 188). Although there is some controversy as to whether increased or decreased ROS mediate redox regulation of KV channels, there is little doubt that KV channel currents are decreased in response to hypoxia.
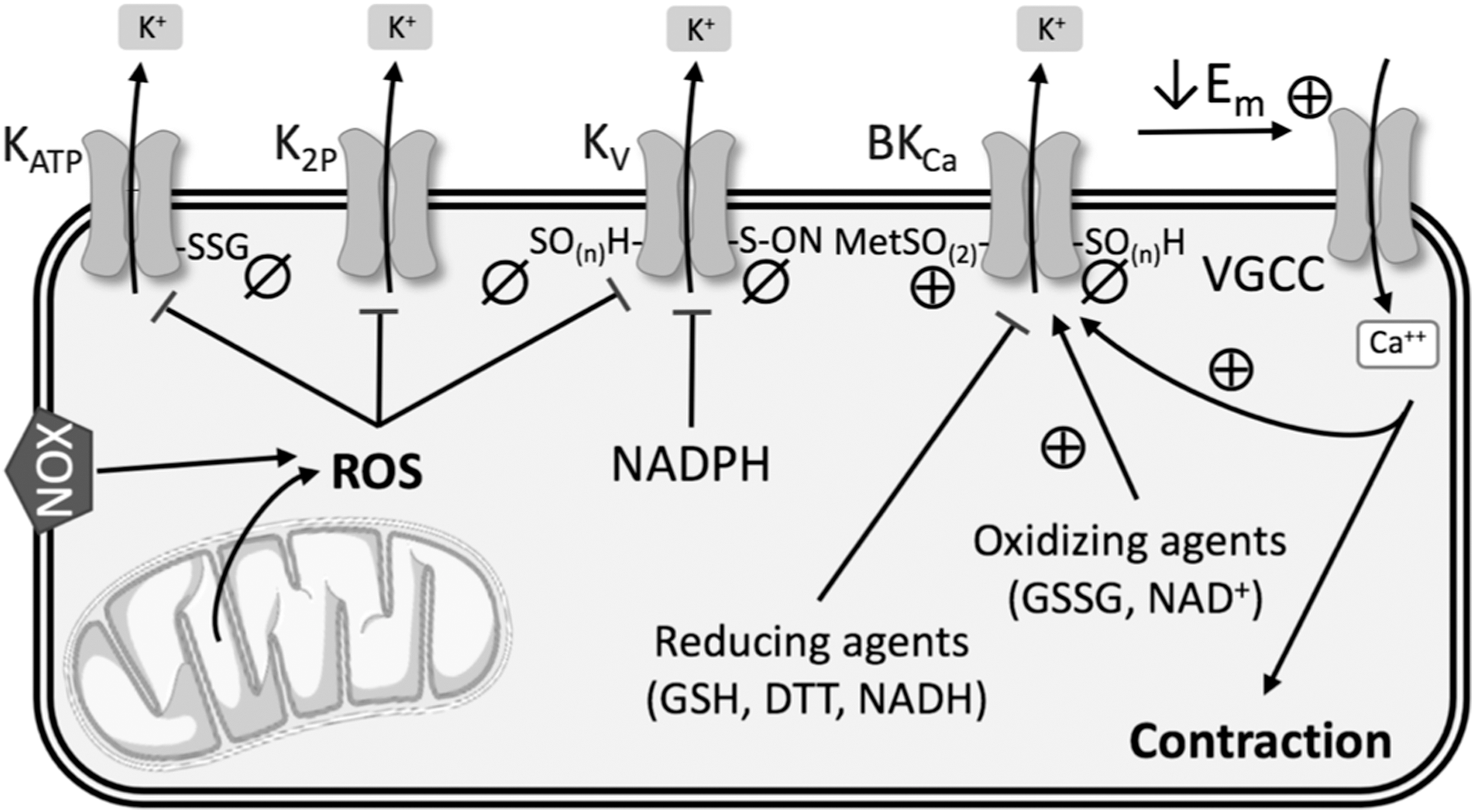
Cysteine residues in the KV channel complex are of interest since the oxidation state of their thiol groups has a substantial effect on channel functionality in cardiac, neural, and other tissues (71, 126, 148). A few studies have suggested this kind of direct regulation of KV channel function by NOX-derived ROS in the vasculature in the context of PH. Activation of NOX and the subsequent production of H2O2 inhibited KV channels in rat pulmonary arteries (27), which was corroborated by evidence that hypoxia increased NOX4-derived ROS in PASMCs, resulting in KV1.5 cysteine oxidation and reduced KV currents (107) (Fig. 5). Svoboda et al. indicated that in atrial myocytes, chronic oxidative stress led to sulfenic acid modulation of KV1.5 cysteine residues and subsequent internalization of this channel (166). They suggest that a similar mechanism may occur in the vasculature in hypoxic PH, but direct evidence is lacking.
KV channel activity may also be modified by the redox state of pyridine nucleotides (Fig. 5). KV channels consist of transmembrane α-subunits that conduct K+ and cytoplasmic β-subunits that modify channel activity. The N-terminus of the β-subunits forms a “ball-and-chain” N-type inactivation that obstructs ion flow through the channel. Binding of NADP+ to the C-terminus of KV channel β-subunits removes N-type inactivation, whereas binding by NADPH stabilizes channel inactivation and reduces KV channel currents (175). However, further work is needed to confirm this regulatory role in the pulmonary vasculature.
Deficiencies in mitochondrial metabolism modify KV channel activity via more indirect mechanisms. Mitochondrial dysfunction increases HIF-1α translocation to the nucleus, where it reduces KV channel expression at the transcriptional level (15). A decline in mitochondrial oxidative phosphorylation via complex I inhibition also reduces KV channel currents (142, 187), and Moral-Sanz et al. (110) suggest that this may be in part due to AMPK activation. In some cell types, AMPK activation leads to increased endocytosis of KV channels via Nedd4-2 ubiquitination (6), but whether AMPK reduces KV channel membrane expression in the context of PH has not been directly tested. On the contrary, several studies have indicated that activation of AMPK may instead be protective against hypoxia-induced pulmonary vascular remodeling (125, 190, 199).
Most of what we know about the role of KV channels in PH has focused on KV1.5 and KV2.1 subtypes. However, KV7 (KCNQ) channels have also become a target of interest in hypoxic PH (150). KV7 channel blockers promote depolarization, vasoconstriction, and increase arterial pressure in rat pulmonary arteries, while KV7 channel activators have opposite effects in PASMCs (76, 77, 150). In neurons, H2O2 and •O2 − oxidatively modify a triple cysteine pocket to inhibit KV7 channel currents (49, 95, 126), and S-nitrosylation of Cys445 in the C-terminus of KCNQ1 also reduces channel functionality (10, 162) (Fig. 5). However, redox regulation of these channels has not been extensively studied in hypoxic PH.
Calcium-activated (KCa) channels
KCa channels, which are activated by both membrane potential and intracellular Ca2+, are subdivided into three categories according to their conductance: big (BKCa), intermediate (IKCa), and small (SKCa). The large conductance BKCa channels are inhibited in hypoxic (34, 39) and monocrotaline-induced PH (143). The relative contribution of BKCa channels to PASMC K+ currents varies depending upon species, age, and pulmonary artery branch order. Some have suggested that BKCa channels may play a larger role in fetal or neonatal pulmonary vasculature than in adult PASMCs (28, 144). In addition, proximal pulmonary artery segments exhibit higher expression of BKCa channels, while distal segments contain more KV-enriched PASMCs (7).
Direct regulation of BKCa channel activity by ROS has produced conflicting results. H2O2 has been shown to both activate and inhibit BKCa channels, although through different mechanisms. Cysteine oxidation decreases channel activity, whereas methionine oxidation increases K+ conductance through BKCa channels (157, 171, 174) (Fig. 5). In PASMCs, reducing equivalents such as GSH, dithiothreitol, and NADH inhibit BKCa conductance, whereas oxidizing agents such as NAD+ and GSSG increase channel open probability (85, 133) (Fig. 5). ONOO− (16, 100) and hydrogen sulfide (91) also inhibit BKCa channel activity in nonpulmonary SMCs, likely by redox modifications to cysteine residues; yet more work is needed to confirm this effect in the pulmonary vasculature.
Tandem-pore domain (K2P) channels
Six subgroups, based upon structure and function, make up the K2P family of channels: TASK, TWIK, TREK, TALK, THIK, and TRESK. These channels are usually open at negative membrane potentials, so they play a major role in establishing K+ conductance (57). Regulation of the activity of these channels can be quite complex because they respond to a multitude of stimuli, including pH, stretch, and post-translational modifications (169).
TASK-1 (KCNK3) is expressed in PASMCs and plays an important role in regulating resting membrane potential (50, 54, 57). TASK-1 knockdown causes depolarization in PASMCs (57, 124), and both hypoxia and the vasoconstrictor endothelin inhibit TASK-1 function (114, 170). Furthermore, loss-of-function mutations in the pH-sensitive K2P channel TASK-1 have been implicated in patients with PAH (102). Hypoxia-induced decreases in TASK-1 K+ currents are dependent upon NOX4 in HEK 293 cells (86, 134) (Fig. 5), but little is known about redox regulation of TASK-1 in the pulmonary vasculature. Studies are complicated by the fact that TASK-1 does not appear to play much of a role in PASMCs in mice, limiting the availability of animal models (80). However, TWIK-2 (KCNK6) knockout mice exhibit elevated RVSP and pulmonary vascular remodeling at 20 weeks of age, and thus may be a better animal model for studying the role of K2P channels in PH (131).
Inwardly rectifying, ATP-sensitive (KATP) channels
KATP channels are a subgroup of inwardly rectifying K+ (Kir) channels that are insensitive to voltage and usually closed under normal conditions (104). The Kir6.1 and 6.2 subunits have been detected in PASMCs (30), and KATP channel openers have been shown to ameliorate both monocrotaline- and hypoxia-induced models of PH (88, 132, 181). Excessive ROS inhibit KATP channel activity in cerebral and coronary arteries (42, 109, 146), and vascular KATP channels are inhibited by S-glutathionylation of a cysteine residue of the Kir6.1 subunit (194) (Fig. 5). However, this mechanism of regulation has yet to be confirmed in the pulmonary circulation.
Redox Regulation of Ca2+ Homeostasis
The intracellular free Ca2+ concentration ([Ca2+]i) plays an important role in the regulation of vascular tone, cell proliferation, and activation of genes via Ca2+-sensitive transcription factors. Ca2+ homeostasis requires the coordination of several fundamental systems, including release of Ca2+ from sarco-/endoplasmic reticulum (SR/ER), Ca2+ entry through plasma membrane channels, Ca2+ sequestration into cytoplasmic organelles, and Ca2+ extrusion from the cell. Dysregulation of these systems leads to a rise in both resting and stimulated PASMCs [Ca2+] i in patients with IPAH as well as in various animal models of PH (72, 94, 155) (Fig. 6). The crucial role of increased [Ca2+] i in a wide array of cell functions underlines the importance of understanding the redox mechanisms regulating [Ca2+] i in PH.
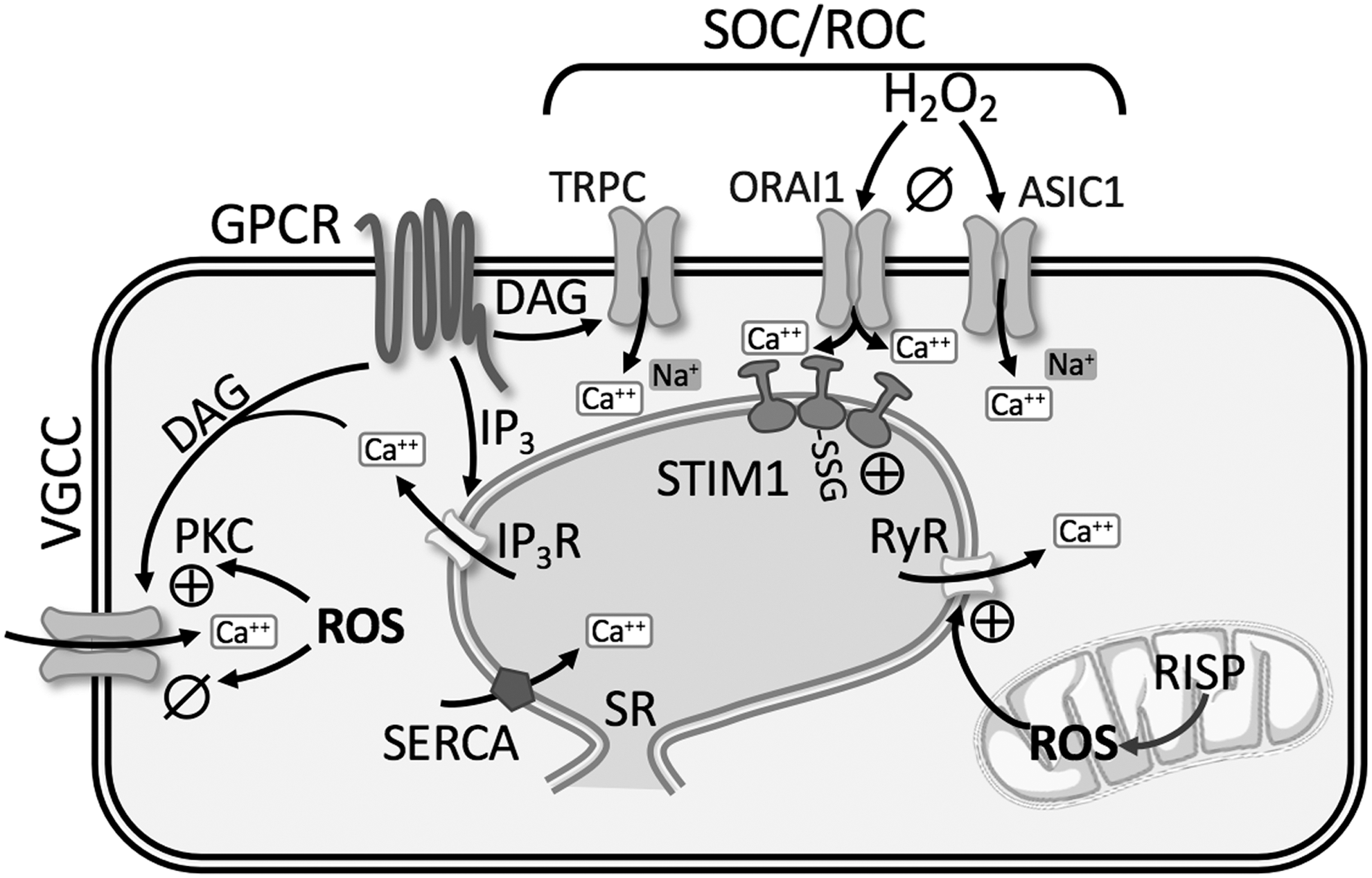
Redox modulation of intracellular Ca2+ stores
Ca2+ is released through channels in the SR membrane known as RyRs and inositol 1,4,5-trisphosphate receptors (IP3R), whereas uptake of Ca2+ into the SR/ER occurs via the sarco-/endoplasmic Ca2+-ATPase (SERCA2a and SERCA2b). RyR and IP3R each have three subtypes (RyR1–3; IP3R1–3) that form homotetrameric channels, and all subtypes are expressed in PASMCs (201, 203).
Ryanodine receptor
The classical activation of RyR occurs through Ca2+-induced Ca2+ release (CICR), whereby local elevations of [Ca2+] i stimulate further Ca2+ release from RyR. In monocrotaline-induced pulmonary hypertensive rats, PASMC stretch results in a greater CICR response mediated by RyR, which is not present under control conditions (52). Studies on RyR1, RyR2, and RyR3 null mice revealed that RyR channel activity and Ca2+ release are largely increased in PASMCs from CH-induced pulmonary hypertensive mice (203). Furthermore, pulmonary artery constriction, remodeling, and hypertension following CH were prevented in RyR2 null mice, and partially reduced in RyR1 and RyR3 mice (203).
The endogenous RyR regulator, FK506 binding protein 12.6 (FKBP12.6), stabilizes RyR in a closed state (93, 202). There is greater dissociation of FKBP12.6 from RyR2 in PASMC from CH-induced pulmonary hypertensive mice leading to SR Ca2+ leak (93, 203). Evidence suggests that ROS production from the Rieske iron-sulfur protein (RISP) of mitochondrial complex III subsequently leads to FKBP12.6/RyR2 dissociation in PASMCs, as lentiviral RISP shRNA reduces CH-induced PH in mice (93, 203) (Fig. 6). Collectively, these studies demonstrate that redox regulation of RyR leads to augmented intracellular Ca2+ release in PH.
Inositol 1,4,5-trisphosphate receptor
Activation of GPCR increases the synthesis of IP3 and diacylglycerol (DAG) via PLC, which subsequently binds to IP3R on SR/ER causing release of Ca2+. Various exogenous oxidants can stimulate IP3R-mediated Ca2+ release [reviewed in Joseph (75)]. Thiol-oxidizing agents increase the sensitivity of the IP3R through formation of protein-glutathionylation leading to greater Ca2+ release (11) (Fig. 6). Although redox regulation of IP3R may represent a fundamental mechanism for regulating Ca2+ homeostasis during pathological oxidative stress, little is known regarding IP3R involvement in PH.
Sarco-/endoplasmic reticulum calcium ATPase
Intracellular Ca2+ homeostasis can also be dysregulated at the level of SERCA, a transporter that sequesters Ca2+ into the SR/ER after its depletion during excitation–contraction coupling (52). SERCA2 expression is downregulated in small pulmonary arteries from IPAH patients, as well as in the monocrotaline-induced rat model of PH (59). Gene transfer of SERCA2a in both a monocrotaline-induced rat PH model and a pulmonary vein banding porcine model demonstrated decreased pulmonary artery pressure, and improved right ventricular function (3, 59). The redox state of cysteine residues on SERCA is important for enzymatic function; however, modifications of different cysteine residues have been shown to result in both inhibition and activation of SERCA (1, 38). Whether there is redox modulation of SERCA2, in addition to the downregulation observed in PH, is currently unknown and requires further investigation of this emerging field.
Redox modulation of plasma membrane Ca2+ channels
Chelation and removal of extracellular Ca2+ reduce PASMC [Ca2+] i similar to control levels, indicating that a constant Ca2+ influx is required for maintenance of elevated resting PASMC [Ca2+] i in PH (72, 94, 155). Ca2+ influx occurs through both L- and T-type VGCCs, and voltage-independent receptor-operated channels (ROCs) and store-operated channels (SOCs). Although the majority of these Ca2+-permeable channels are known to be redox regulated, the overall implications toward the pathogenesis of PH are not well defined.
Voltage-gated Ca2+ channels
Alterations in membrane potential regulate the open probability of VGCC. VGCCs are classified into six different types (L, T, N, P, Q, and R). L-type VGCCs are abundantly expressed in PASMCs and are characterized by their high voltage of activation, large single channel conductance, and slow voltage-dependent inactivation (119). T-type VGCCs are present in both PASMC and PAEC (111, 117, 128, 189), and are activated at much more negative membrane potentials (low voltage activated). They are activated rapidly, deactivated slowly, exhibit small single channel conductance, and are resistant to dihydropyridine antagonist (119).
Pulmonary arteries from CH-induced pulmonary hypertensive neonatal piglets and adult rats and mice have increased expression of L-type (Cav1.2) and T-type (Cav3.1/3.2) VGCC (62, 180). Furthermore, in pulmonary hypertensive patients and animal models of PH, PASMC resting membrane potential is persistently depolarized (156, 165, 196), which presumably leads to greater activity of L- and T-type VGCCs (111, 180). Although it has been demonstrated that exogenous and endogenous redox compounds affect the activity of both L- and T-type VGCCs in the pulmonary circulation, there is not a general agreement on the effects of oxidation on channel activity. Here, we focus on redox modulation of L-type VGCC.
L-type VGCC
The α1C subunit contains multiple redox-sensitive cysteine residues, but the specific effect of redox changes on VGCC in smooth muscle seems to be variable (23, 67). Consistent with several reports that antioxidants and NOX inhibition reduce pulmonary vasoconstriction (73, 118, 153, 186), NOX-derived ROS have been shown to augment VGCC activity (20, 65, 153) (Fig. 6). In bovine PASMC, the thromboxane mimetic, U-46619, leads to activation of VGCC and Ca2+ influx through NOX-derived ROS in a PKC-dependent manner (20). Sphingosylphosphorylcholine-induced rat pulmonary vasoconstriction is coupled to NOX1-mediated increase in ROS, PKCɛ, and consequent enhancement of VGCC-dependent pulmonary vasoconstriction (153).
It is important to note that in these cases, ROS only potentiate VGCC activity, which is likely mediated through PKC-mediated phosphorylation. However, ROS do not seem to directly initiate Ca2+ influx. In fact, the opposite effect is observed when thiol-specific reducing or oxidizing compounds are applied directly. In cloned rabbit smooth muscle cells, L-type VGCC currents are inhibited by 2,2-dithiodipyridine, a specific lipophilic oxidizer of sulfhydryl groups, and dithiothreitol reverses this effect (23). Similarly, ROS produced by xanthine oxidase and hypoxanthine, as well as direct application of H2O2, attenuate PASMC Ca2+ influx through VGCC, decrease [Ca2+] i , and cause relaxation in pulmonary arteries in mice (81) (Fig. 6). Taken together, these data suggest that ROS and oxidizing agents directly inhibit VGCC, but may indirectly enhance L-type VGCC activity via agonist-induced signaling through PKC.
Disunity in the redox regulation of L-type calcium channels might be due to the extensive phosphorylation of this channel by different kinases, which are activated by ROS and might at least partially counterbalance the inhibitory effects of direct oxidation of this channel (147). Despite evidence that VGCC antagonists attenuate PH in animal models (43, 62, 79), the clinical use of the classical dihydropyridine Ca2+ channel blockers is not therapeutically effective for a majority of patients with PH and may even cause deterioration of the disease (173). Consistent with this notion, inhibition of VGCC has little effect on resting [Ca2+] i or enhanced agonist-induced Ca2+ influx in experimental models of PH (72, 94, 183).
Even with a persistent depolarization in PASMC from hypertensive models, VGCCs appear to be less active, possibly due to direct effect of ROS to inhibit VGCC that over-rides depolarization-mediated activation of the channel. Indeed, it has been shown that VGCC activity is independent of changes in membrane potential in pulmonary hypertensive animals (101). Together, these studies suggest a minimal role of L-type VGCC in development and progression of PH, and are consistent with a greater role for a direct effect of oxidative stress to inhibit L-type VGCC function.
Store- and receptor-operated Ca2+ channels
Nonselective cation channels play a pivotal role in increasing [Ca2+] i in PASMCs. Activation of GPCRs or RTKs increases the synthesis of IP3 and DAG via PLC. IP3-mediated release and depletion of Ca2+ from the SR induce Ca2+ influx through SOC (Fig. 6). DAG directly activates Ca2+ influx through ROC. SOCE is enhanced in PASMC from pulmonary hypertensive patients (158, 195) and various animal models of PH (72, 94, 99, 184), while PAEC SOCE is decreased in CH-induced pulmonary hypertensive rats (127, 200). SOCE involves a dynamic and highly regulated process mediated by several proteins, second messengers, and ion channels that likely assemble into a macromolecular complex. Due to the numerous molecular components involved, the relationship between redox state, ROS, and SOCE is not completely understood.
Patch clamp studies reveal two different types of ionic currents evoked by store depletion: (i) a current mediated by a Ca2+ release-activated channel (CRAC) displaying high selectivity for Ca2+ (89) and (ii) a nonselective cation channel current (192). Many agree that stromal interaction molecule 1 (STIM1) is the endoplasmic/sarcoplasmic reticulum Ca2+ sensor relaying the signal to plasma membrane Ca2+-permeable ion channels (96, 145). In PASMCs, STIM1 and/or STIM2 has been shown to interact with both the CRAC-related channel, ORAI1, ORAI2, ORAI3, and the classical/canonical transient receptor potential channel (TRPC1, TRPC3, TRPC4, and TRPC6) (26, 116, 158, 184). In addition, our laboratory has recently shown a novel role for the Ca2+-permeable cation channel, acid-sensing ion channel 1 (ASIC1) to mediate enhanced SOCE following CH (72, 74).
The effect of ROS on SOCE can vary from tissue to tissue. In vascular smooth muscle and endothelial cells, H2O2 seems to cause a profound inhibition of SOCE (45, 130, 138, 149) (Fig. 6). Our laboratory has recently shown that H2O2 inhibits ASIC1-dependent SOCE in rat PASMC (138). Furthermore, removing H2O2 through the addition of catalase or the glutathione peroxidase mimetic, ebselen, increased SOCE in rat PASMC. We also found that in pulmonary arteries from SOD1−/− mice, with reduced levels of H2O2 production, arterial wall Ca2+ is increased through an ASIC1-dependent SOCE mechanism (138). The enhanced SOCE in PASMC from CH animals is inhibited by exogenous H2O2, suggesting that the lower endogenous levels of H2O2 in PASMC from CH animals facilitate the activation of SOCE (138).
ASIC1 is a known redox-sensitive ion channel, where reducing agents potentiate ASIC1 activity, plasma membrane depolarization, and Ca2+ influx, while oxidizing agents have the opposite effect (24, 25, 198). Similarly, pretreatment with H2O2 attenuates ORAI1- and ORAI2-mediated Ca2+ influx, whereas ORAI3-mediated Ca2+ influx is resistant to H2O2-mediated inhibition (14). In contrast to these findings that oxidizing agents limit SOCE, S-glutathionylation of STIM1 mimics the effects of luminal Ca2+ depletion, promotes the oligomerization of STIM1, and increases basal [Ca2+] i in DT40 cells (60). Whether S-glutathionylation of STIM1 occurs in the setting of PH is unknown. Overall, the change in redox associated with PH leads to enhanced SOCE.
Transient receptor potential vanilloid 4
As mentioned above, several members of the TRPC subfamily of ion channels show increased sensitivity to agonists in PASMCs from PAH patients and animal models of PH. Although all TRP channels members of the melastatin-related and vallinoid-related (TRPV) subfamilies are present in pulmonary arteries, TRPV4 is the only other TRP channel shown to be upregulated in PH (193). TRPV4 is a nonselective cation channel permeable to Ca2+ that is expressed in both PASMC and PAEC. In PASMC, TRPV4 plays a role in basal and stimulated pulmonary vascular tone in PH (33, 191). In PAEC, activation of TRPV4 elicits NO-dependent vasodilation (103), as well as increasing pulmonary microvascular endothelial cells (PMVECs) permeability (5). TRPV4 plays a key role in H2O2-induced increases in [Ca2+] i and barrier dysfunction in PMVECs (164). Similarly, in an animal model of PH, TRPV4 was not upregulated in PMVEC, but elevated production of mitochondrial-derived ROS activated TRPV4 Ca2+ influx, leading to enhanced migratory and proliferative capacity of PMVEC from pulmonary hypertensive animals (163).
Although TRPV4 is known to be redox-sensitive, these studies do not illustrate a direct effect of ROS/H2O2 on TRPV4. On the contrary, TRPV4 in systemic endothelial cells is sulfhydrated by the reducing agent, H2S, which correlates with H2S-mediated endothelial- and TRPV4-dependent vasodilation in mesenteric arteries (115). Together, these results indicate the importance of TRPV4 in PH and provide some insight into potential redox regulation; however, further studies are needed to elucidate the direct effect of redox modification on TRPV4 channel function.
Targeting Oxidative Stress in PH with Antioxidants
The strong evidence that oxidative stress associated with PH results from increased oxidant production and decreased antioxidant capacity leads to studies testing the efficacy of antioxidant therapeutic strategies in the setting of PH. N-acetylcysteine (NAC), a derivative of the amino acid
A more direct antioxidant approach utilizing overexpression of SOD reduces PH resulting from CH (120), monocrotaline (78), and in lambs with prenatal ligation of the ductus arteriosus (159). Similarly, Elmedal et al. (41) demonstrated that TEMPOL, a synthetic compound that mimics SOD enzyme activity, normalizes RVSP and slightly reduces right ventricular hypertrophy in CH rats. A more recent study from our laboratory provided corroborating evidence that TEMPOL reduced RVSP and vasoconstrictor reactivity in both CH- and CH/SU5416-treated rats. However, TEMPOL did not reduce right ventricular hypertrophy, and was instead associated with an unexpected increase in medial hypertrophy and adventitial fibrotic lesion formation in CH/SU5416 rats (73).
The reason TEMPOL increased the severity of arterial remodeling in the CH/SU5416-treated rats is not clear, but may result from phenotypic changes in cellular responsiveness to ROS in angioproliferative PAH. Together, these data suggest that despite protective effects of TEMPOL in limiting vasoconstrictor reactivity and elevated RVSP, the exacerbation of arterial remodeling may pose therapeutic limitations of using an SOD mimetic in severe PAH. Furthermore, the dietary supplement Protandim, which increases expression of antioxidant enzymes such as SOD and heme-oxygenase-1, neither reduced RVSP nor pulmonary vascular remodeling in CH/SU5416-treated rats, but prevented right ventricular hypertrophy and preserved right ventricular function (13).
While such findings complicate the ability to interpret antioxidant efficacy, they suggest that different antioxidants may preferentially target different ROS, and these ROS species may, in turn, have divergent effects related to the development of PH. An additional point of consideration is that these antioxidants may target ROS in different cellular compartments. To this point, pulmonary vascular remodeling was elevated with overexpression of the mitochondrial SOD2, but attenuated by overexpression of mitochondria-targeted catalase, suggesting that increased mitochondrial H2O2 contributes to CH-induced PH hypertension (2). Further studies examining the mitochondrial-targeted antioxidant, MitoQ, show inhibition of the acute hypoxia-induced increase in •O2 − but not CH-induced PH (129). These findings indicate a differential role of ROS in pulmonary vascular responses to acute and CH, and suggest that the type of ROS as well as their intracellular compartmentalization may vary over the course of disease progression, making effective treatment of PH with antioxidants difficult.
Conclusions
The development and progression of PH are widely associated with disruption of intracellular redox homeostasis leading to overall “redox stress” that includes both oxidative stress and reductive stress. Oxidative stress is accompanied by increased production of free radicals and failure of antioxidant defense mechanisms leading to oxidation of protein, fatty acids, and DNA damage. Reductive stress is characterized by an excess of reducing equivalents, such as NADH, NADPH, and GSH, that cause mitochondrial dysfunction, and alters transcription, translation, and post-translational modifications. In addition, chronic reductive stress can lead to feedback regulation that further induces oxidative stress.
Although this redox imbalance either directly or indirectly impacts the function of various ion channels and membrane receptors in the pulmonary vasculature, the outcome of this redox stress fundamentally alters both vasoconstrictor and arterial remodeling components of PH by mediating downstream proproliferative, promigratory, and antiapoptotic effects. Many questions remain unanswered regarding (i) the dynamic inter-relationship between oxidants and reductants, and how increases in both can lead to the same outcome; (ii) mechanisms of intracellular compartmentalization or cell-type-specific alterations in redox stress; and (iii) whether antioxidants and/or antireductants could provide a therapeutic option for treating PH.
Although oxidative stress has long been a hallmark of PH, antioxidant strategies and oxidant inhibitors have not always proven to be beneficial in treating this disease, and in some cases the outcomes have been detrimental. The scientific interest in reductive stress has increased over recent years; however, very little is known about potential antireductive therapy. This review highlights our current understanding of specific membrane receptor and ion channel regulation by redox, and underscores the importance of considering the dynamic events related to both oxidative and reductive stress. While critical advances have been made in characterizing the ROS/redox-specific cellular signaling pathways involved in enhanced vasoconstriction and vascular remodeling in PH, a greater appreciation of the functional relationships between these pathways is needed to better understand the etiology of PH and to develop more effective treatments and preventative measures for this condition.