
Abstract
Significance:
Pulmonary hypertension (PH) is a progressive disease characterized by pulmonary vascular remodeling and lung vasculopathy. The disease displays progressive dyspnea, pulmonary artery uncoupling and right ventricular (RV) dysfunction. The overall survival rate is ranging from 28–72%.
Recent Advances:
The molecular events that promote the development of PH are complex and incompletely understood. Metabolic impairment has been proposed to contribute to the pathophysiology of PH with evidence for mitochondrial dysfunction involving the electron transport chain proteins, antioxidant enzymes, apoptosis regulators, and mitochondrial quality control.
Critical Issues:
It is vital to characterize the mechanisms by which mitochondrial dysfunction contribute to PH pathogenesis. This review focuses on the currently available publications that supports mitochondrial mechanisms in PH pathophysiology.
Future Directions:
Further studies of these metabolic mitochondrial alterations in PH could be viable targets of diagnostic and therapeutic intervention.
Introduction
Pulmonary hypertension (PH) is a complex disease defined by persistent increases in pulmonary vascular resistance, severe remodeling of the pulmonary arteries, lung vasculopathy, right ventricular (RV) dysfunction, and death (141). Metabolic changes, similar to those observed in cancer, have been reported in smooth muscle cells (SMCs) (84), endothelial cells (44), and fibroblasts (163) of PH patients (16, 27, 44, 98, 125, 141). The Warburg metabolism observed in cancer cells was also described in PH whereby mitochondrial and cytosolic alterations drive a metabolic adaptation characterized by a switch to glycolysis (104). Thus, cellular reprogramming of metabolism may result in functional and molecular abnormalities seen in pulmonary artery cells, including excessive proliferation, apoptosis resistance, and inflammation (44). Multiple mechanisms of mitochondrial dysfunction have been proposed to contribute to PH pathogenesis, including electron transport chain (ETC) dysfunction (110), altered mitochondrial membrane potential (96), mitochondrial DNA (mtDNA) damage (32), increased production of mitochondrial reactive oxygen species (mROS) (1, 145), impaired mitochondrial biogenesis and mitophagy (2, 155), and improper mitochondrial dynamic via fission/fusion (59, 110, 114). Most of those mechanisms are linked such that, for example, increased mROS and mtDNA damage could initiate apoptosis, impair biogenesis, or change mitochondrial structure or phenotype. The ETC dysfunction could lead to disruption of ATP productions and to an increase in mROS. The ensued oxidative stress strongly contributes to PH pathogenesis via its ability to induce DNA and protein damage. Recently, disturbed mitochondrial function and/or biogenesis have emerged to play a vital role in cellular mechanisms such as SMC proliferation and trans-differentiation in PH (157). Decreased mitochondrial biogenesis would interrupt mitochondrial oxidative capacity and energy production. Recent evidence supports the potential for mitochondria to drive pulmonary smooth muscle cell (PASMC) and pulmonary artery endothelial cells (PAECs) to an apoptosis-resistant phenotype (59). Dysregulated angiogenesis (142), growth factor induction, and apoptosis resistance in PASMCs (136) have been cited as key pathogenetic mechanisms. In this review, the evidence for mitochondrial dysfunction in PH pathologies will be discussed in the context of whether such mechanisms may represent novel therapeutic perspectives in PH.
Mitochondrial Dysfunction
ETC dysfunction
Mitochondrial respiratory chain dysfunction and altered mitochondrial metabolism associated with a glycolytic shift are described in PH (4, 7, 114, 154). At the molecular level, the mechanisms underlying the metabolic shift toward increased glycolysis observed in PASMC during the pathogenesis of PH are not fully understood. Recent studies showed that the glycolytic enzyme α-enolase (ENO1) regulates the metabolic reprogramming of PASMC (30). In addition, ENO1 levels were elevated in PH patients and in animal models of chronic hypoxic pulmonary hypertension. Inhibition of ENO1 decreases PASMC proliferation and de-differentiation, and induces PASMC apoptosis, whereas the overexpression of ENO1 promotes de-differentiation, and apoptotic-resistant PASMC phenotype via the 5′ AMP-activated protein kinase (AMPK)-thymoma viral proto-oncogene (Akt) pathway (30).
Disruption of the mitochondrial ETC is an important cause of ROS dysregulation. Flow of electrons from Complex I to Complex IV generates a gradient of protons in the inner membrane. This gradient is used by ATP synthase to produce ATP or by uncoupling proteins to produce heat. The enzymatic activity of the mitochondrial respiratory chain complexes showed a decrease in Complex I and III activity in muscle of PH patients (41, 108, 110). Mitochondrial respiratory complexes require iron-sulfur clusters to transfer electrons that require the iron-sulfur chaperon gene NFU1 (NFU1 iron-sulfur cluster scaffold, mitochondrial). Mutations in NFU1 gene resulted in decreased activities of mitochondrial respiratory Complexes I, II, and III and have been associated with PH development (4, 90). There is general agreement that the mitochondrion is likely the acute oxygen sensor and a source of the signaling mediator ROS. Blockade of electron flux results in decreased hydrogen peroxide (H2O2) production (24, 144, 148, 152). Decreased mROS production in hypoxia has been attributed to reduced electron flux. On the other hand, an alternative hypothesis that hypoxia increases mROS is based on the assumption that in the absence of O2, there is a block of the ETC and a retrograde accumulation of electrons leading to autoxidation of the chain and superoxide production (38, 55, 149). ETC function is disrupted by inhibition of any of its complexes. Notably, inhibition of Complex III results in maximal ROS production and maximal mitochondrial damage (24). Inhibition of Complex III can enhance superoxide production in both the intermembrane space and the mitochondrial matrix (88). Superoxide radical is scavenged by the mitochondrial superoxide dismutase—superoxide dismutase 2 (SOD2) and transformed into less reactive H2O2 (144). However, nitric oxide (NO) produced in the vasculature can also scavenge superoxide to form peroxynitrite, a highly potent oxidant that could inhibit other ROS scavengers by induction of protein nitration and nitrosylation, including SOD2 (109), and initiate many pathophysiological processes (46, 134). Recent investigation showed that hypermethylation of SOD2 gene could decrease its expression and contribute to the observed increase in mROS in PASMCs from PH patients (8). This epigenetic regulation appears to be specific to the pulmonary arteries as a result of DNA methyltransferases 1 and 3 upregulation in pulmonary, but not systemic, arteries (7 –9). Specific mitochondrial abnormalities related to clinical PH and experimental PH include induction of pseudohypoxia via activation of hypoxia-inducible factor 1α (HIF-1α), impaired mitochondrial–endoplasmic reticulum interaction, and mitochondrial fission (44, 155). Further, HIF-1α overexpression decreased nuclear respiratory factor 1 (NRF1) and thus modulated mitochondrial biogenesis (101, 127). In addition, patients with HIF-2α overexpression mutations develop PH (45, 47). On the contrary, heterozygous HIF-2α-knockout mice were protected from hypoxic PH, and endothelial prolyl hydroxylase 2 knockdown promoted HIF-2α-dependent pulmonary vascular remodeling and PH in mice (31). Though true that there has been significant debate as to the nature of the mitochondria's role with respect to hypoxic signaling, it is now generally accepted that increases in mROS are triggered in this setting and that these drive subsequent activation of both HIF-dependent transcriptional mechanisms and carotid body hypoxia responses (19, 20, 49, 149, 151). Although additional studies are vital to elucidate the unique effects of mitochondria in PH, current evidence suggests that there may be therapeutic benefits targeting HIFs in the pulmonary vasculature.
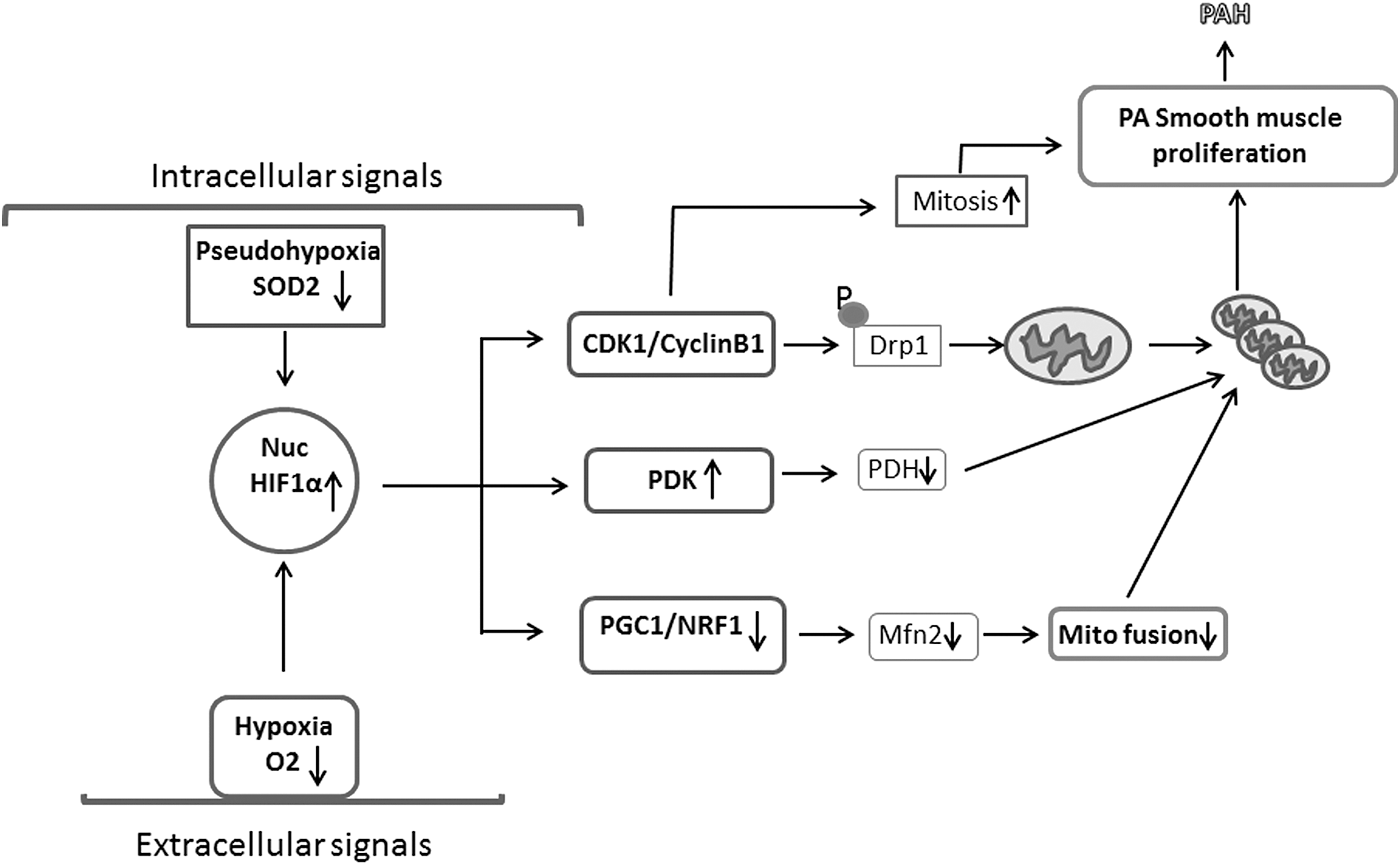
Mitochondria with defective ETC also exhibit low respiratory chain coupling and inefficient use of oxygen (155). These abnormalities were linked to the chronic inflammatory process in PH (131) and to the progressive proliferation of SMCs that obliterate the vascular lumen (137). Mitochondria integrate many molecular signals in PH (34). Mitochondria produce signaling mediators that control multiple critical mechanisms of cellular function (Fig. 1) (34). Mitochondria signals through ROS or diffusible metabolites such as α ketoglutarate (αKG) can regulate transcription factors such as nuclear factor of activated T cells and HIF-1α that play key roles in the vascular remodeling of PH. Mitochondria can modulate epigenetic signaling through its products such as αKG and citrate, major regulators of epigenetic mechanisms, including histone methylation and acetylation, respectively, that have been shown to contribute to PH pathogenesis (36). Mitochondria are critical regulators of apoptosis, and solid evidence support that mitochondria-dependent apoptosis is suppressed in PH (34, 36). Although inflammatory cytokines can suppress mitochondrial function, mitochondria themselves can activate the NLPR3 (NACHT, LRR, and PYD domains-containing protein 3) inflammasome (66), leading to a cascade that results in increased levels of lungs and circulation inflammatory cytokines in patients with PH (36). These data support that mitochondrial dysfunction is central to the pathogenesis of PH (36, 84, 131).
mtDNA damage
mtDNA has a significant role in the regulation of mitochondrial functioning.
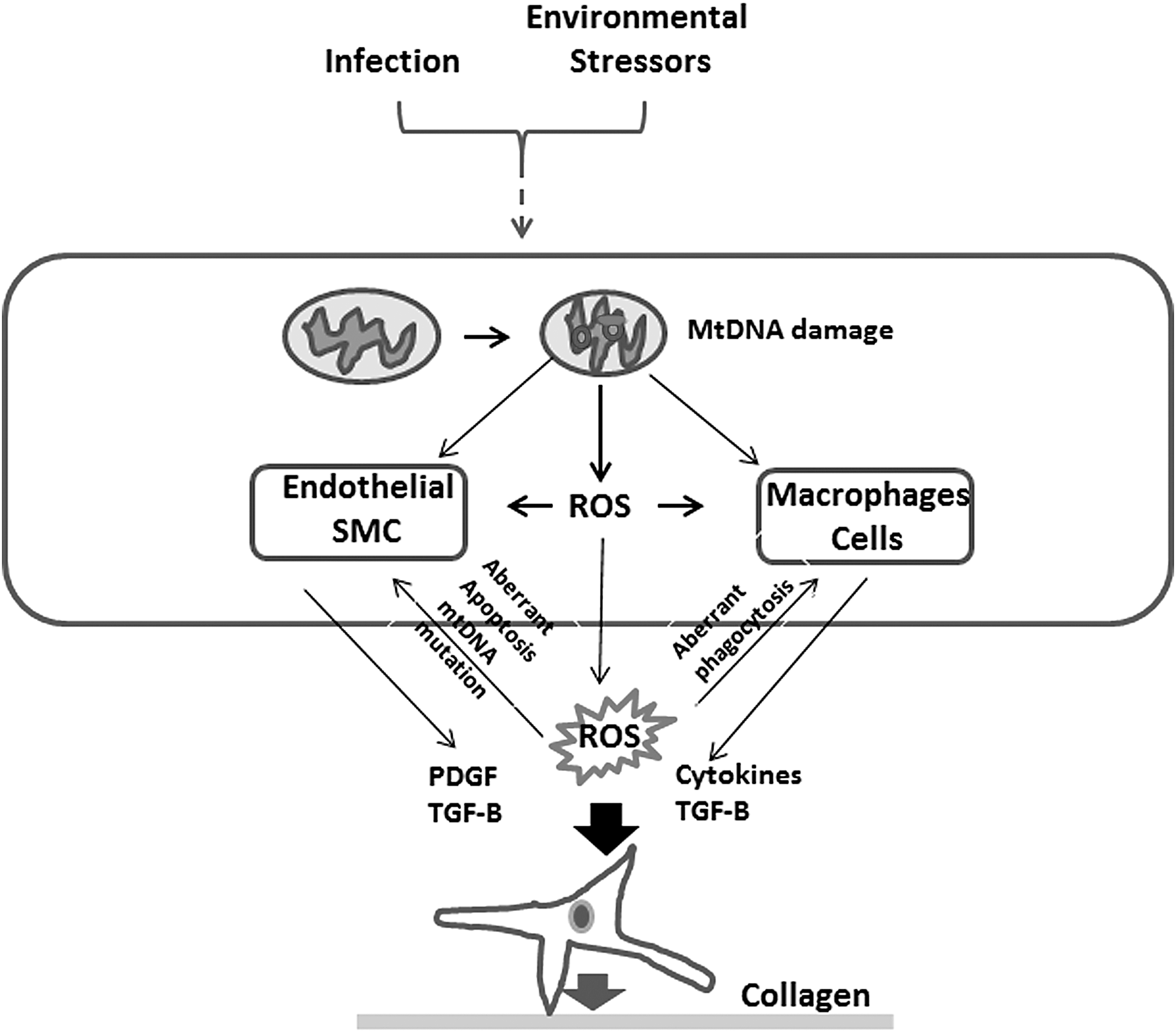
mtDNA is more sensitive to oxidative damage than is nuclear DNA, especially when considering mitochondrial-derived ROS (26, 82). Interestingly, decreased mitochondrial oxidative repair enzymes resulted in increased cytotoxicity and subsequent apoptosis. In contrast, overexpression of these enzymes seems to have a protective effect on the cell (118). Oxidation of mtDNA can lead to increased mitochondrial dysfunction and subsequent increased apoptosis, which is rescued by overexpression of 8-oxoguanine DNA glycosylase (Ogg1; 112). These protective effects of Ogg1 were reproduced in other forms of oxidant injury, including ventilator-induced and hyperoxia-induced lung injury (58). Further studies also demonstrated that Ogg1 has a protective effect on lung injury and PH (26).
mtDNA can be released from cells after apoptosis as fragments (118, 153). These fragments act as damage-associated molecular patterns (DAMPs), which play a role in innate immunity, and inflammation-mediated endothelial remodeling (153). DAMPs contribute to activation of toll-like receptors (153), including Toll receptor 4 (TLR4) and Toll receptor 9 (TLR9) (74). Recent data showed that TLR4 activation of platelets is associated with development of PH (13), and in sickle cell disease, DAMPs are linked to development of the vasculopathy that leads to PH (105). TLR9 activation may lead to further damage and fragmentation of mtDNA, suggesting a feed-forward mechanism to amplify mitochondrial injury (70). DAMPs are also recognized by NOD-like receptors (NLRs) that augment responses by immune cells. Particularly, the NLRP3 inflammasome is activated by mitochondrial-associated DAMPs, and it has been linked with the pathogenesis of PH (89). Suppression of the inflammasome cascade inhibited the development of PH in animal models (158). Oxidative stress due to hypoxia/reoxygenation also induced mtDNA damage, increased caspase-3/7 activity in PAECs, and impaired reversal of PH (18).
Impaired mitochondrial quality control
Mitochondrial number, or mass, regulates cellular bioenergetics capacity and is controlled by mitochondrial quality control (MQC), a process that couples the genetic programming for mitochondrial biogenesis and mitophagy. Mitochondrial biogenesis involves a bi-genomic program of nuclear- and mitochondrial-encoded genes that are rapidly regulated to adjust mitochondrial mass, distribution, and phenotype (101, 127). The program requires key transcription factors, including NRF1 and NRF2 and the coactivator peroxisome proliferator-activated receptor gamma (PPARγ) (PPARγ coactivator 1-α [PGC1α]) that regulate mitochondrial transcription factor A (TFAM) to promote mtDNA genes (100). Several experimental models of PH demonstrate severe reduction in mitochondrial mass and biogenesis gene expression (73, 123, 156), though a mechanistic understanding of mitochondrial biogenesis regulation in PH is lacking. Several mediators affect mitochondrial biogenesis in PH, including NO/cGMP signaling and heme oxygenase 1 (HO-1)/carbon monoxide (CO) signaling (93, 101, 127). In a fetal lamb experimental model of PH, the decreased PGC1α, ETC complex expression, and mtDNA copy number were reversed by an NO donor (28, 119). Thus, NO and NO derivatives could restore mitochondrial biogenesis and are currently under clinical investigation as therapies in PH (40, 122). Further, in vivo enhancement of HO-1 using agonists of HO-1 prevented the development of hypoxic PH and inhibited the structural remodeling of the pulmonary vessels (25, 146). In experimental PH in rats, the expression of biogenesis markers PGC1α, NRF1, and TFAM mRNA was decreased in the RV (147). Recently, the histone deacetylase sirtuin 1 (SIRT1) was implicated as a vital regulator of mitochondrial function in the heart and vascular smooth muscle (76, 77). SIRT1 and SIRT3 effect mitochondrial biogenesis via post-translational modification of PGC-1α (85). Knockdown of Sirt3 decreased PGC-1α-dependent gene expression of subunits I and II (mitochondrial encoded genes) as well as subunit VIIa (nuclear encoded genes) from cytochrome c oxidase (68). Further, a loss-of-function SIRT3 polymorphism is associated with PH in human and animal studies (150). SIRT1 and SIRT3 also regulate mitochondrial phenotype and function in arterial SMCs (76). Also, SIRT1/SIRT3 mediates deacetylation of Cyclophilin D, a modulator of mitochondrial membrane permeability that regulates mitochondrial delta psi membrane potential and mitochondrial function during hemorrhagic shock (74, 76).
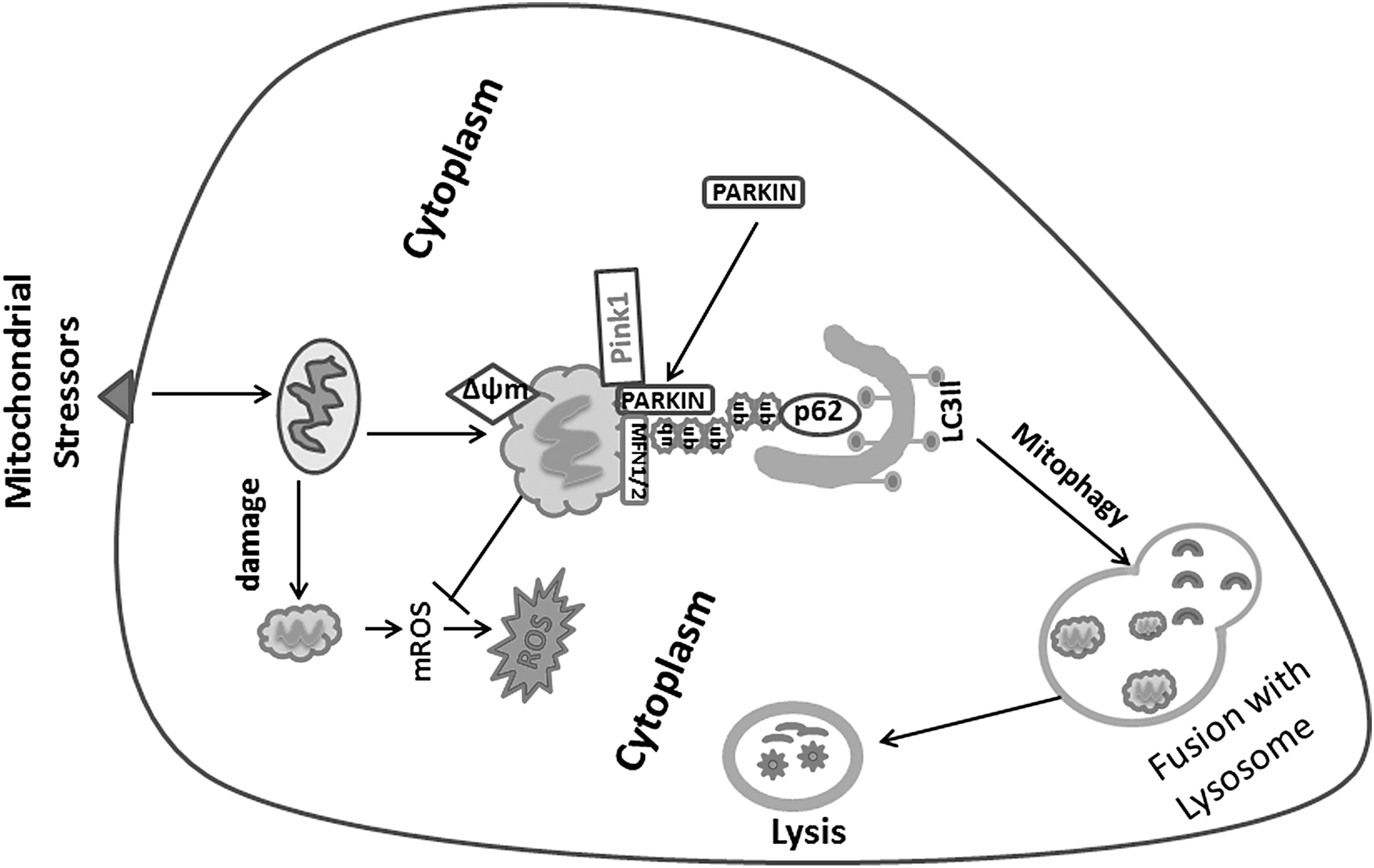
Mitophagy is the selective autophagy of mitochondria, and it is an important quality control mechanism that eliminates damaged mitochondria and recycles damaged mtDNA. Defects in mitophagy have been implicated in certain cancers and a number of pulmonary diseases (3, 71, 115). Specifically, the imbalance between mitochondrial biogenesis and mitochondrial turnover leads to functional impairment of the cell. This relationship has been demonstrated in various pathological processes, including aging (89). There is evidence of increased mitophagy and decreased mitochondrial biogenesis in human patients with PH as well as in experimental mouse models of the PH (59, 113). Mitophagy is initiated by a change in mitochondrial membrane potential that results in accumulation of PTEN-induced kinase 1 (Pink1) on the mitochondrial outer membrane, leading to the recruitment of cytoplasmic Parkin (E3 ubiquitin-protein ligase parkin), and subsequent ubiquitination of damaged mitochondria (52). Oxidative injury enhances mitophagy, which is an early response to promote survival (3, 161). However, excessive mitophagy to lung endothelium can lead to cell death (10, 135, 160). It is increasingly reported that dysregulated endothelial metabolism and mitochondrial dysfunction can cause PH and right heart dysfunction. The regulation of autophagy and mitophagy involves complex interaction and signal transduction networks (Fig. 2). This process is initiated by a change in mitochondrial membrane potential with accumulation of Pink1 on the outer membrane, leading to recruitment of cytoplasmic Parkin and facilitating ubiquitination of damaged mitochondria (52). Uncoupling protein 2 (UCP2) is a calcium uniporter that maintains endopalsmic reticulum-dependent Ca2+ influx to the mitochondria, thus regulating calcium-dependent proteins such as pyruvate dehydrogenase, ROS production, and mitochondrial-mediated apoptosis (82). Endothelial cells deficient in UCP2 gene showed increased mitophagy and reduced mitochondrial mass (95). Loss of UCP2 increased levels of mitophagy proteins Pink1 and Parkin to increased mitophagy, and it resulted in the development of spontaneous PH (59), emphasizing the role of endothelial mitophagy and the UCP2 pathway in the development of pulmonary vascular remodeling. Increased mitophagy and inadequate mitochondrial biosynthesis was shown to be associated with increased apoptosis in the endothelium. These changes were associated with physiologic evidence of PH in mice, such as increased RV systolic pressure and RV hypertrophy (82). In addition, mitochondrial dysfunction and higher rate of mitochondrial fragmentation were reported in the PASMCs of patients with PH, suggesting inadequate clearance of dysfunctional mitochondria by mitophagy (81). LC3B knockout mice had exacerbated PH in response to hypoxia, suggesting that autophagy is protective against chronic hypoxia-induced PH (72). Also, in a mouse PH model induced by pulmonary artery constriction, increased microtubule-associated proteins 1A/1B light chain 3 (LC3) and sequestosome 1 (P62) were associated with RV failure (107). Imbalances in mitophagy may affect mitochondrial mass, but the role of mitophagy and autophagy in PH is incompletely understood.
Mitochondrial Dynamics
Mitochondrial fission
Mitochondrial dynamics reflect the connectivity of the mitochondrial network and the shape of individual mitochondria (Fig. 3). Mitochondria exist in dynamic networks that can rapidly divide (fission) or join together (fusion) and move about the cell (trafficking) (159). Besides metabolic changes (132), the mitochondria in PH display phenotypic and structural changes affecting the mitochondrial dynamics that are linked to cell cycle regulation (81) and ROS production (139). Dynamin-related protein 1 (Drp1) is post-translationally modified and activated by dephosphorylation at Serine 637 or phosphorylation at Serine 616 (140). The activated Drp1 translocates to the outer mitochondrial membrane where it is bound to fission protein 1 (Fis1), mitochondrial fission factor, and mitochondrial dynamics proteins of 49 and 51 kDa (MiD49 and MiD51) (140). Drp1 and its binding partners form a ring-like structure that constricts and divides the mitochondrion, leading to mitochondrial fission (5). Mitochondria from human PH SMCs or fibroblast cells are fragmented and characterized by decreased levels of fusion proteins (mitofusin-2 [Mfn2], optic atrophy-1 [Opa1]) and upregulated levels of fission proteins (Drp1, Fis1) (104, 113). In addition, increased Drp1 phosphorylation and activation was observed in PH mitochondria, whereas the mitofusion protein, Mfn2 was deregulated (113). Increased mitochondrial fission could result in increased fragmented mitochondria that promote a hyperproliferative, apoptosis-resistant mitochondrial phenotype seen in PASMC in PH (81). Moreover, excessive mitochondrial fission in RV myocytes in PH leads to increased mitochondrial-derived ROS production (Fig. 4) and impaired RV function (139), but not changes in cell proliferation. Recent reports showed that inhibiting mitochondrial fission by targeting Drp1 has therapeutic potential in PH. The selective Drp1 GTPase inhibitor, mitochondrial division inhibitor 1 (Mdivi-1), was found to decrease proliferation of PASMC from PH patients and improve hemodynamics in animal models of PH (81). Further, Mdivi-1 inhibits mitochondrial fission in left ventricular (LV) myocytes and improves LV function in the ischemia-reperfusion injury model and in the cardiac arrest model in rodent models (120, 121). Recently, P110, a seven-amino-acid peptide representing a homology sequence between Drp1 and Fis1 linked to TAT47–57 carrier peptide, was found to selectively inhibit pathological, but not physiological mitochondrial fission (54, 106). Though FIS1 is not important to the increased fission observed in PASMC from PH patients (21), blocking the interaction between Drp1 and Fis1 using P110 preserves mitochondrial morphology and cellular function in a rat model of PH (54). Although further studies are required, results to date seem to support a role for mitochondrial fission in promoting altered bioenergetics, increased proliferation, and resistance to apoptosis in PH-fibroblasts cells, an observation that may highlight potential therapeutic targets in the future.
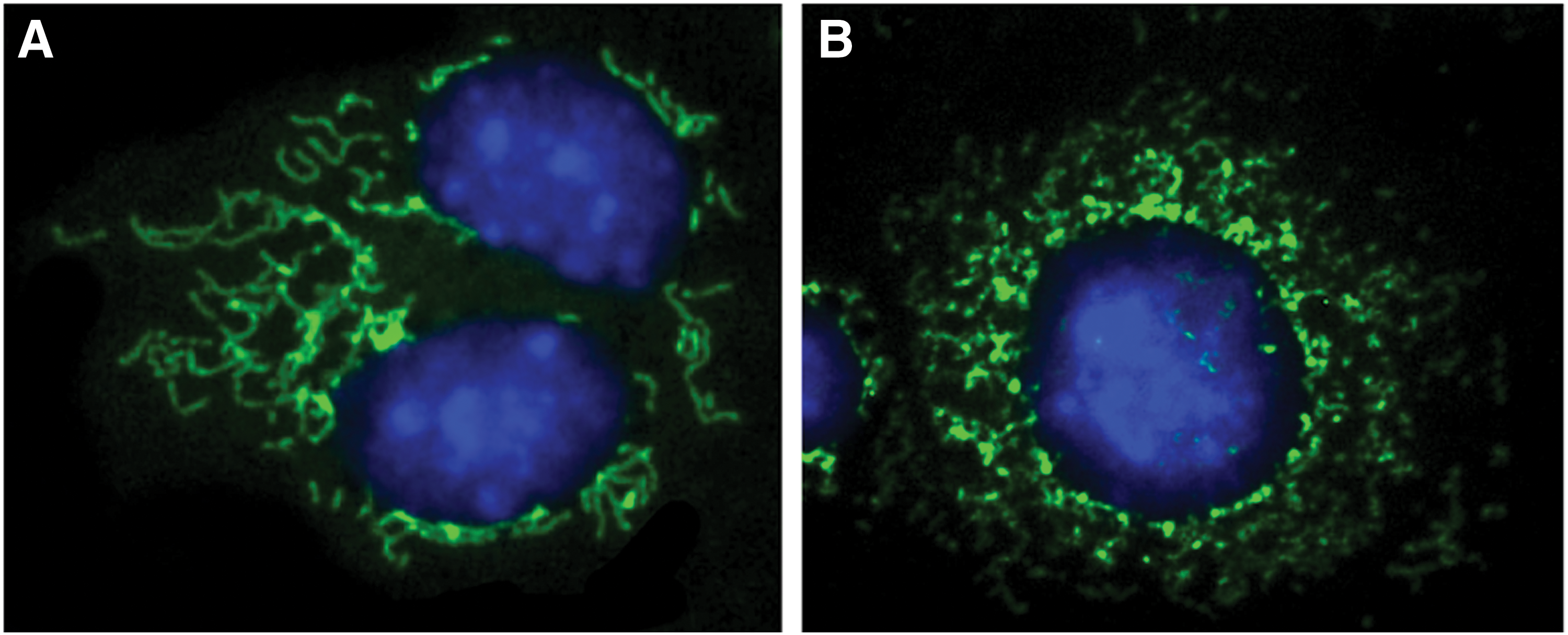
Fusion and mitofusin-2 in PH
The GTPase, mitofusin-1 (MFN1) facilitates mitochondrial fusion, which permits redistribution of proteins/genes and may alleviate the accumulation of mutations in the mtDNA (116). In addition, fusion can modify rates of cell proliferation. Due to its antiproliferative effect, MFN2 was initially referred to as hyperplasia suppressor gene (22). In addition to MFN1 and 2, mitochondrial fusions require annealing of the inner mitochondrial membrane to the OPA1.
PH patient and experimental PH models showed decreased MFN2 expression (114). Genetic inhibition by siRNA of MFN2 recapitulates a PH phenotype in PASMC, leading to a fragmented mitochondrial network and enhanced proliferation. Several possible mechanisms may contribute to the downregulation of MFN2 in PH patients, including activation of platelet-derived growth factor (117) and endothelin 1 (126); both can also reduce MFN2 expression in arterial SMC (22). Moreover, decreased levels of NRF-1/PGC-1α, major transcription factors regulating MFN2, can downregulate transcripts of MFN2 in PH (114). PGC-1α enhances the activity of the MFN2 promoter through estrogen-related receptor-alpha-binding elements that regulate MFN2 transcription (124). Adenoviral overexpression of MFN2, using Ad-MFN2, recovers mitochondrial networks, lessens proliferation, and restores normal rates of apoptosis in PH PASMC (99).
Mitochondrial Dysfunction in the Right Ventricle
RV failure is a common pathway for mortality in PH due to decreased perfusion pressure and insufficient capillary networks with energy failure, altered metabolism, and resultant ischemia and cardiac failure (6). Mitochondrial dysfunction is established as an important factor in the metabolic shift reported in the failing left ventricle (91). In the RV in PH, mitochondrial metabolic abnormalities include an increase in uncoupled glycolysis (102, 130), disorders of fatty acid oxidation (FAO) (43), and induction of glutaminolysis (103, 113). Electron microscopy revealed decreased number of mitochondria and substantial abnormalities in size and shape of mitochondria in failing RV (103). The relevance of these findings in the RV requires validation. Moreover, decreased expression of PGC-1α in failing RV decreases mitochondrial gene expression and gene expression of proteins required for fatty acid metabolism (103). Mitochondrial dynamics are critical in the heart for cardiac bioenergetics, yet the role of mitochondrial dynamics in RV failure is unclear. Embryonic deletion of DRP1 or a tandem deletion of MFN1 and MFN2 is lethal in mice (72). However, an inducible, cardiac-specific DRP-1 knockout model led to reduced ejection fraction, hypertrophy, fibrosis, and death of most animals within 4 months (61). In contrast, cardiac deletion of a single mitofusin isoform appears to protect against ischemia-reperfusion injury and results in only mild mitochondrial dysfunction (97).
Mitochondrial-Targeted Therapies
Targeting ROS
Besides acting as a significant source of cellular ROS, mitochondria are also targets of cellular ROS that damage mtDNA and proteins, particularly within the respiratory complexes. ROS modify mitochondrial proteins such as inactivation of SOD2 and aconitase, or alter their function as in cytochrome c (17, 80). Oxidative macromolecular damage in vascular cell mitochondria is known to affect mitochondrial function (12). However, the regulation of mROS generation and its pathophysiological role in PH is much less understood. Several lines of evidence suggest that mitochondrial-directed therapies may modulate lung remodeling. Impaired ETC Coenzyme Q reductase (complex I) would result in enhanced production in mtROS (111). To protect mitochondria from oxidative damage, mitochondria-targeted antioxidants such as mitoubiquinone mesylate (MitoQ) were developed (79). MitoQ acts to quench ROS, including superoxide and peroxynitrite, and protects mitochondria from oxidative damage (79). Recent studies showed that a mitochondria-targeted SOD mimetic, mitoTEMPO, significantly attenuates angiotensin II-induced hypertension in mice (33). mitoTEMPO treatment was reported to inhibit vascular oxidative stress and completely restored endothelial-dependent relaxation (33). Also, mitoTEMPO was shown to decrease the activity of transforming growth factor β and provide efficacy in a mouse model of asthma (63). Another mitochondrial-targeted antioxidant Szeto-Schiller (SS) compound SS-31 was found to reduce oxidative stress, improve cardiac function, and attenuate cardiac hypertrophy by scavenging H2O2 and peroxynitrite (29, 133, 162). The antioxidant transcription factor nuclear factor erythroid derived 2, like 2 (Nrf2) has emerged as a potential target for pharmacological attenuation of mitochondrial oxidative stress in the cardiovascular system (143). Naturally occurring Nrf2-activating compounds (14, 51) were found to restore endothelial function and decrease vascular oxidative stress in rodents. Collectively, these studies demonstrate that antioxidant strategies targeting mitochondria ROS could have potential therapeutic benefits in PH diseases.
Targeting mitochondrial biogenesis
Therapeutic modalities targeting mitochondrial biogenesis and reversing glycolytic shift via NRF-1/HO-1 activation were successful in several experimental models (100, 101, 127). In addition, activation of the CO/HO-1 pathway is known to induce mitochondrial biogenesis and improve MQC (11, 100, 127).The ability of endothelial-derived NO to activate the transcriptional machinery driving mitochondrial biogenesis is well established (92). Several drugs with NO-releasing properties currently used in clinical practice, mainly for cardiovascular diseases, may also affect nitric oxide synthase mRNA, protein, or activity levels with special modulation of endothelial nitric oxide synthase (37). Arginine or citrulline supplements have been proposed for the treatment of mitochondrial disorders associated with NO deficiency (39). However, the rationale for simple NO-based therapy depends on the capacity of the patient to increase the rate of proliferation of mitochondria with intact oxidative phosphorylation systems, a limitation in patients with hereditary mtDNA mutations and a low background heteroplasmy. Recently, nitrite, recognized as an NO donor specifically in hypoxic/acidic conditions, has been proposed to mediate cytoprotection without substantially altering normal tissue. In addition, there are a number of AMPK activators known to induce mitochondrial biogenesis (56). Numerous studies have documented a role for AMPK in regulating mitochondrial function and biogenesis and promoting anti-inflammatory effects (94). Further, mitochondrial transfer via cell therapy emerged as a potential therapeutic alternative to restore the dysfunctional pool of mitochondrial persisting in acute lung injury (62) and remodeled lungs of cigarette smoke-exposed mice (78). Exogenous mesenchymal stromal cells (MSCs) transfer functioning mitochondria to the lung epithelium and enhance the pool of functional mitochondria to restore ATP production in several experimental models. MSCs transfer technology was reported to have anti-inflammatory and anti-fibrotic therapeutic effects (111). At present, data collected at pre-clinical and clinical levels support allogeneic MSC treatment as a potential therapy for lung diseases (111). Therefore, MSC therapy could present a promising therapeutic approach for hosts with lung/heart diseases; mainly due to mitochondrial mechanisms that lead to correction of mitochondrial defects and restoration of cell bioenergetics. In conclusion, the area of mitochondrial-based therapy to support mitochondrial biogenesis, MQC, and anti-inflammation is still in its infancy. The recent discovery of a group of novel redox-regulated molecular pathways that protect cell viability through the support MQC offers a plethora of new targets and the opportunity for unique small-molecule treatments for diseases in which persistent mitochondrial dysfunction is a contributing factor.
Targeting metabolic reprograming
The metabolic changes in pulmonary arterial hypertension (PAH) cells resemble the cancer-associated Warburg effect, involving incomplete glucose oxidation (GO) through aerobic glycolysis with depressed mitochondrial catabolism, enabling the fueling of anabolic reactions with amino acids, nucleotides, and lipids to sustain proliferation (16, 69, 75, 104). Besides augmented glycolysis, cancer and PAH cells depend on other metabolic fuels, including increased fatty acid synthesis and enhanced glutamine metabolism to sustain elevated proliferation rates and resistance to cell death signals (60, 138). This metabolic reprograming has been shown to provide a selective pro-survival advantage to distal pulmonary artery cells, regardless of oxygen availability (16, 35, 104, 131). It has been acceptable that mitochondrial dysfunction and decreased GO is associated with increased FAO and that partial inhibition of FAO may represent a therapeutic target in PH.. Recently, trimetazidine and ranolazine, FAO inhibitors were found to reverse the glycolytic shift back toward GO (57, 65). In pulmonary artery banding and monocrotaline-induced models, the use of trimetazidine and ranolazine reversed the metabolic changes in RV hypertrophy, enhanced GO, and improved RV function (43, 53). Accordingly, multiple clinical trials are currently underway or have been completed by using FAO inhibitors (43, 65, 67).
Further, recent studies showed an association between mitochondrial dysfunction and the induction of reductive carboxylation, an alternative pathway for glutamine catabolism that supports biosynthesis of lipids (15, 23, 64, 86, 87, 128). The ability of reductive glutamine carboxylation to supply nicotinamide adenine dinucleotide may enable cells to maintain a high rate of glycolytic flux in the setting of reduced tricarboxylic acid cycle activity (50). Though the contribution of reductive carboxylation to lipid formation from glutamine is controversial due to the possibility of isotope exchange (42), studies suggest that reductive carboxylation occurs in vivo and can support lipogenesis for tumor growth and progression (48, 129) and can also control the levels of mROS (64). In addition, glutamine enhances upregulation of reduced glutathione and may regulate oxidative stress (83) (Fig. 5).

Conclusions and Future Perspective
It is clear that dysregulated vasculature metabolism and mitochondrial dysfunction drive important aspects of PH. Nevertheless the molecular aspects of this pathway are not fully defined. Mitochondria may participate in PH through modifications of glycolysis, oxidative phosphorylation, and anaplerosis to provide substrates for proliferation and generation of signaling messengers such as ROS, Ca2+ to modulate vasculature function. The current standard care for PH patients is mainly dependent on treating physiological symptoms such as vasoconstriction; however, the growing consensus is that new therapeutic modalities targeting the remodeling process may provide disease modification and endless clinical benefit to patients with PH (111). Despite the complex and diverse pathophysiology of PH, cumulative data on metabolic reprogramming and perturbations in mitochondrial function and dynamics in PH are evident. The renaissance in mitochondrial biology research focuses on the emerging evidence of mitochondrial dysfunction in human diseases. Apart from targeting the mitochondria to alter ROS production, the proteins regulating mitochondrial dynamics, particularly Drp1, may hold therapeutic potential. Pharmacological modalities such as Mdivi-1 could be of great potential in restoring mitochondrial dysfunction in lung diseases. Inhibiting Drp1 in preclinical studies has resulted in decreased fission and cell proliferation in human PASMC from PH patients as well as in rodent PASMC (81), suggesting that regulators of the mitochondrial network are viable therapeutic targets for diseases of impaired oxygen sensing. Future work will require further definition of the molecular signals and regulatory checkpoints of these processes. It is also anticipated that the novel research outcomes could be converted rapidly into clinical applications for establishing a new era of metabolic diagnostics and therapeutics for PH and its fatal consequences.