
Abstract
Aims:
Excessive reactive oxygen species (ROS) are detrimental to immune cellular functions that control pathogenic microbes; however, the mechanisms are poorly understood. Our aim was to determine the immunological consequences of increased ROS levels during acute bacterial infection.
Results:
We used a model of Streptococcus pneumoniae (Spn) lung infection and superoxide dismutase 3-deficient (SOD3−/−) mice, as SOD3 is a major antioxidant enzyme that catalyses the dismutation of superoxide radicals. First, we observed that in vitro, macrophages from SOD3−/− mice generated excessive phagosomal ROS during acute bacterial infection. In vivo, there was a significant reduction in infiltrating neutrophils in the bronchoalveolar lavage fluid and reduced peribronchial and alveoli inflammation in SOD3−/− mice 2 days after Spn infection. Annexin V/propidium iodide staining revealed enhanced apoptosis in neutrophils from Spn-infected SOD3−/− mice. In addition, SOD3−/− mice showed an altered macrophage phenotypic profile, with markedly diminished recruitment of monocytes (CD11clo, CD11bhi) in the airways. Further investigation revealed significantly lower levels of the monocyte chemokine CCL-2, and cytokines IL-23, IL-1β, and IL-17A in Spn-infected SOD3−/− mice. There were also significantly fewer IL-17A-expressing gamma-delta T cells (γδ T cells) in the lungs of Spn-infected SOD3−/− mice.
Innovation:
Our data demonstrate that SOD3 deficiency leads to an accumulation of phagosomal ROS levels that initiate early neutrophil apoptosis during pneumococcal infection. Consequent to these events, there was a failure to initiate innate γδ T cell responses.
Conclusion:
These studies offer new cellular and mechanistic insights into how excessive ROS can regulate innate immune responses to bacterial infection.
Introduction
Extracellular superoxide dismutase (EC-SOD or SOD3) is one of the most abundantly expressed antioxidant enzymes found in the airways where it acts as a scavenger of potentially damaging superoxide radicals (O2˙−). SOD3 has been detected in endothelial cells, vascular smooth muscle cells, alveolar type 2 epithelial cells, fibroblasts, and within intracellular phagocytic vesicles of leukocytes and myeloid-derived cells recruited to sites of inflammation and infection (15, 17). Found anchored to heparin-sulfate proteoglycans on the cell membrane and collagen in the extracellular matrix (ECM), SOD3 is involved in regulating extracellular reactive oxygen species (ROS) homeostasis. Unlike its related family enzymes (SOD1-2), SOD3 preferentially regulates ROS levels in the extracellular spaces, and SOD metabolites such as H2O2 have been shown to regulate the production of cytokines and chemokines (12, 18). Although the mechanisms are not fully understood, dysfunctional SOD3 or excessive ROS are strongly associated with enhanced inflammation, lung injury, and a deficiency to control bacterial infection. Leukocytes including macrophages and neutrophils express SOD3 that is enriched within membrane-bound vesicles of phagocytes, and macrophages lacking SOD3 produce more ROS and show significantly impaired phagocytosis and killing of Escherichia coli (20).
Several studies have also demonstrated the importance of SOD3 in protecting the airways from ROS-induced ECM degradation, tissue damage, and inflammation. SOD3−/− mice display more severe lung injury (edema) and accelerated mortality compared with wild-type (WT) mice under hyperoxia conditions (5). Consistent with these findings, mice overexpressing human SOD3 in the airways attenuated the hyperoxic lung injury response by reducing edema and neutrophil infiltration (11). In addition, lung injury and fibrosis caused by bleomycin treatment were reduced in transgenic mice overexpressing SOD3 (3). Furthermore, when SOD3−/− mice are exposed to chronic cigarette smoke, they develop greater airspace enlargement and more advanced emphysema in comparison to WT mice. The overexpression of SOD3 was shown to be protective in the same chronic cigarette smoke exposure model (40). Interestingly, there are contradictory data on the role of SOD3 during acute bacterial infections. SOD3−/− mice infected with E. coli in the lungs demonstrated greater pulmonary inflammation and reduced clearance of bacteria to suggest that SOD3 facilitates clearance of bacteria and limits inflammation by promoting phagocytosis (20). Consistent with the response to E. coli, SOD3−/− mice inoculated with heat-killed bacteria (Klebsiella pneumoniae) showed a trend toward increased neutrophilic inflammation compared with WT mice (29). In contrast, the overexpression of SOD3 in mice during Listeria monocytogenes infection had detrimental impacts on mortality and bacterial clearance and suggests that excessive SOD3 activity reduces protective innate immune responses of this intracellular bacterial pathogen (4).
Innovation
Chronic diseases such as chronic obstructive pulmonary (COPD) are associated with excessive reactive oxygen species (ROS) production or oxidative stress (7, 24), and macrophages are functionally compromised as they display reduced phagocytosis of bacteria (30). Consistent with this defect, COPD patients are more susceptible to common respiratory pathogens, such as Streptococcus pneumoniae (27). However, it is not known whether excessive ROS/oxidative stress can actually drive increased susceptibility to pneumococcal infection at a molecular and cellular level. Our study discovered a critical role for superoxide dismutase 3 (SOD3) in regulating inflammatory responses to infection including monocyte recruitment and expansion of pulmonary IL-17A+γδ T cells. Our data show that a deficiency in SOD3 signaling causes early neutrophil apoptosis, which initiates a potent anti-inflammatory signal to prematurely switch off secondary innate cellular responses.
What remains unresolved and the focus of this study is whether SOD3 regulates innate immunity to Streptococcus pneumoniae (Spn or pneumococcus). Spn is a leading cause of bacterial pneumonia and invasive disease, and there are multiple cellular and molecular processes that facilitate clearance of pneumococcus. Spn entering the lungs are first phagocytosed by resident patrolling alveolar macrophages (AMs), which release chemokines to promote a rapid wave of neutrophil recruitment into the alveolar space. This coincides with activation of innate mucosal cells including gamma-delta T cells (γδ T cells) by inflammatory cytokines released by activated AMs including IL-23 and IL-1β. Pulmonary γδ T cells are distributed in the lungs and play an important role in bacterial clearance during pneumococcal infection (23). γδ T cells coordinate a new wave of neutrophil and monocyte recruitment, where monocytes mature into macrophages to complete bacterial clearance and initiate the resolution/repair phase of infection. The γδ T cell-mediated recruitment of monocytes and neutrophils is dependent on the inflammatory cytokine IL-17A, as depletion of IL-17A during pneumococcal colonization prevents recruitment of monocytes and neutrophils (42). The aim of this study was to determine the effects of SOD3 deficiency on mucosal immunity during Spn-induced lung inflammation. Our data demonstrate that high extracellular and phagosomal ROS due to SOD3 deficiency enhance neutrophil apoptosis and reduce the inflammatory response during Spn-induced acute inflammation, identifying a novel pathway by which SOD3 regulates innate immunity.
Results
SOD3-deficicent macrophages have increased phagosomal superoxide but similar Spn killing capacity
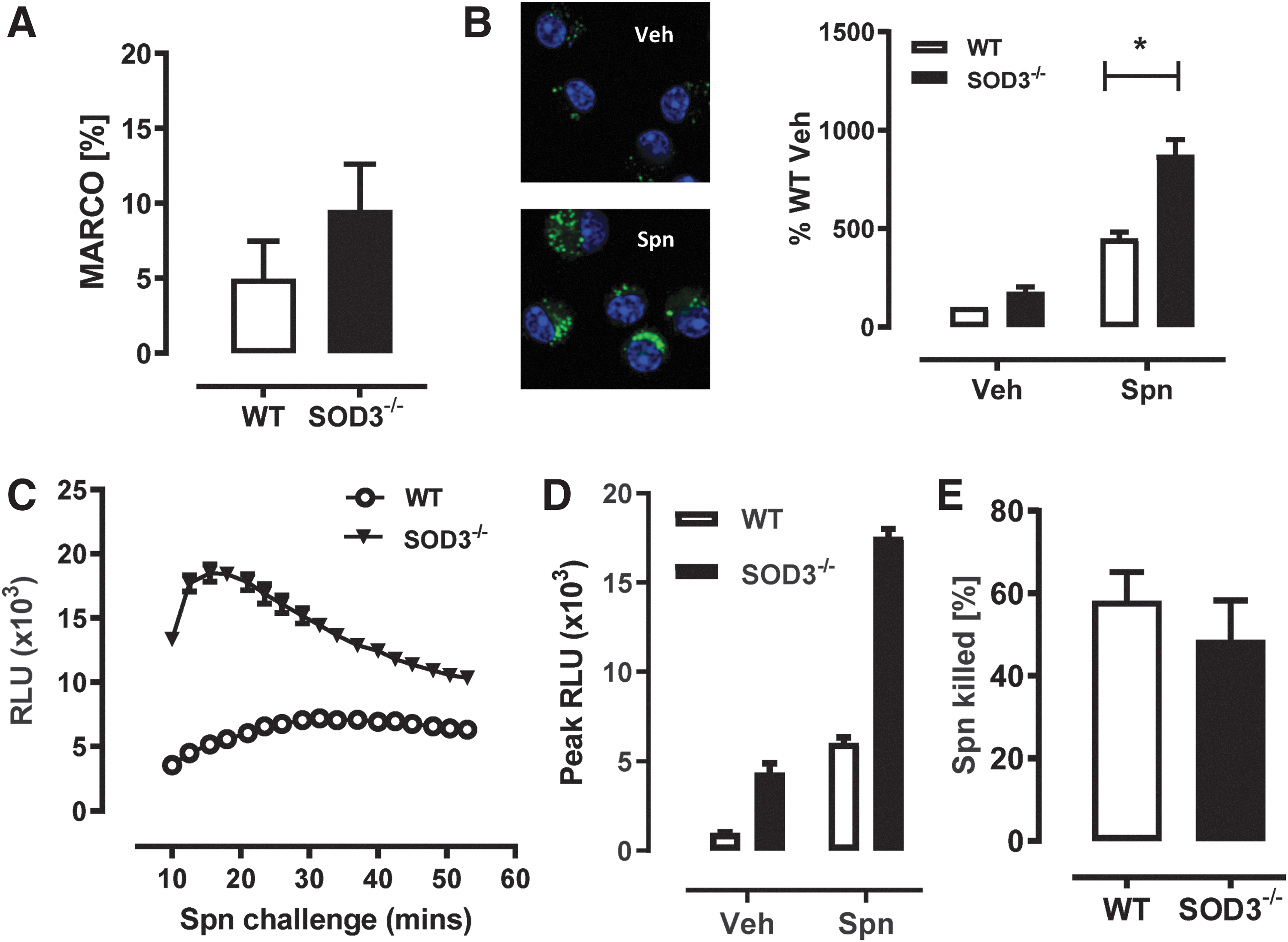
Since it is not known whether SOD3 can regulate phagosomal ROS levels during pneumococcal infection, we investigated responses to acute bacterial infection under in vitro conditions in AM isolated from SOD3−/− and WT mice. First, flow cytometry was used to measure the levels of macrophage receptor with collagenous structure (MARCO), one of the dominant Spn receptors expressed on myeloid cells such as AM described as CD45+, F4/80+, CD11chi, and CD11blo (Fig. 1A). We observed no significant differences in the level of MARCO expressed on the surface of AM from both groups of mice (∼5%–10%) indicative of comparable surrogate Spn binding potential. We next measured AM superoxide levels in response to the live bacteria and observed that SOD3−/− AMs had significantly higher levels of phagosomal superoxide (an approximate doubling above that of WT vehicle) compared with AMs isolated from WT mice (Fig. 1B). In addition, using a L-012 chemiluminescence assay, increased ROS levels were detected from bone marrow-derived macrophages (BMDMs) isolated from SOD3−/− mice compared with WT mice over a 60-min Spn challenge (Fig. 1C, D). However, despite this increased phagosomal ROS, we did not observe enhanced bactericidal capacity in SOD3−/− macrophages compared with WT control AMs (Fig. 1E). These in vitro data demonstrate that in response to Spn, excessive superoxide accumulates in the absence of SOD3, resulting in a significant increase in intracellular phagosomal superoxide in SOD3−/− phagocytes. It also demonstrated that enhanced ROS levels in macrophages did not improve bacterial killing capacity.
SOD3 deficiency accelerates neutrophil cell death and significantly reduces airway neutrophils
Our in vitro findings (Fig. 1) demonstrated that increased phagosomal ROS production did not impair Spn killing by SOD3−/− macrophages. To investigate these findings in vivo, WT and SOD3−/− mice were infected with Spn intranasally and the bacterial burden in the upper and lower airways was determined by quantifying colony-forming units (CFU) in the nasal tissue and bronchoalveolar lavage fluid (BALF) (Fig. 2A). Following Spn infection, similar CFUs in nasal tissue of WT and SOD3−/− mice were detected at day 1, ∼103 CFU/mL, which increased to 104 CFU/mL by day 2. Pneumococcal burden in the lower airways (BALF) was also not significantly altered in SOD3−/− mice at either day 1 or day 2 compared with infected WT mice; however, there was a trend toward more rapid clearance in SOD3−/− mice (Fig. 2A).
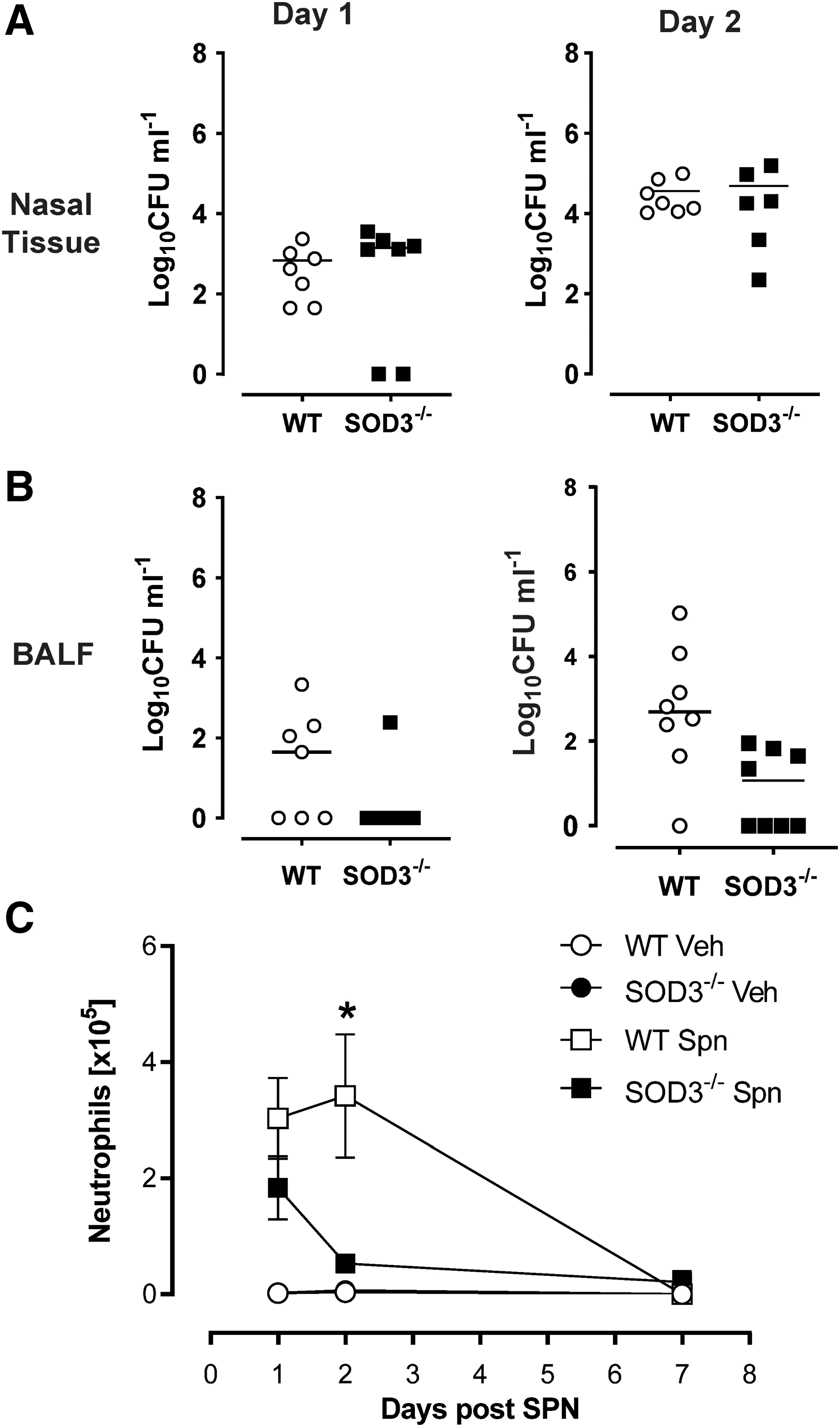
To determine whether the innate immunological response was perturbed by altered ROS homeostasis in SOD3−/− mice, we evaluated neutrophil recruitment in response to Spn lung infection over a 7-day period in SOD3−/− and WT mice. At day 1, there was increased neutrophil trafficking into the airways (bronchoalveolar lavage [BAL]) in response to infection, which was not significantly altered in SOD3−/− mice compared with Spn-infected WT mice. However, by day 2, there were significantly (p = 0.009) fewer neutrophils in SOD3−/− (53,252.26 ± 11,908.24) mice compared with WT mice (341,934.95 ± 106,166.30) (Fig. 2B). By day 7, neutrophils were no longer detected in the BALF of WT or SOD3−/− mice. These results indicated that the impairment in neutrophil numbers at day 2 was not due to an initial airway recruitment defect but rather a defect in other pathways/mechanisms.
To further explore whether the reduction in BAL neutrophil numbers at day 2 was reflective of the inflammation or injury in lung tissue following pneumococcal infection, histology was performed on Spn-infected WT and SOD3−/− mice. Representative images of the whole lung lobe demonstrate the presence of peribronchial inflammation and alveolitis (inflammation of parenchyma) during the acute phase (day 2) of infection in WT mice (Fig. 3A). The degree of inflammation and injury was blindly scored and revealed a significant reduction in peribronchial inflammation in SOD3−/− mice, which is consistent with BAL neutrophil numbers (Fig. 3B). There was also a significant reduction in alveolitis in SOD3−/− mice compared with WT mice (Fig. 3C). By day 7, the resolution of inflammation had occurred in both WT and SOD3−/− mice, where the peribronchiolitis and alveolitis scores were graded as mild and were not significantly different between the genotypes (Fig. 3D–F). Consistent with the resolution of inflammation at day 7, total respiratory compliance was not altered by pneumococcal infection in WT (vehicle: 0.032 ± 0.002 vs. Spn: 0.032 ± 0.002 mL/cmH2O) or SOD3−/− mice (vehicle: 0.039 ± 0.002 vs. Spn: 0.037 ± 0.003 mL/cmH2O).

We hypothesized that cell death due to enhanced ROS levels may be a contributing factor to the significant decrease in neutrophils in SOD3−/− mice observed at day 2. To investigate this, we assessed neutrophil cell death ex vivo in the BALF of Spn-infected mice at day 1 (Fig. 3A, B) by Annexin V/propidium iodide (PI) staining on cells positive for Ly6G and CD45. We observed significantly increased (p = 0.02) proportions of apoptotic neutrophils at day 1 in Spn-infected SOD3−/− (74.55% ± 5.25%) versus WT (53.21% ± 4.41%) mice (Fig. 4A, B). This was also demonstrated in immature neutrophils from the whole bone marrow stimulated with Spn where we observed a significant doubling in the percentage of immature neutrophils (Ly6G+, CD11c−ve) undergoing cell death 3 h after Spn stimulation (Fig. 4C) in cells deficient of SOD3 (WT: 13.44 ± 2.24 vs. SOD3−/−: 34.17% ± 3.18%). After this time (6 h), the lack of SOD3 did not influence neutrophil cell death. These data indicated that deficiency in the extracellular and phagosomal antioxidant SOD3 significantly enhanced neutrophil apoptosis, thereby reducing the numbers of airway neutrophils present at day 2 after infection. To confirm that SOD3 also regulates ROS levels in neutrophils infected with Spn, bone marrow-derived neutrophils were isolated and superoxide production was quantified using the L-102 chemiluminescence assay. ROS levels gradually increased in response to Spn inoculation over a 2-h incubation period (Fig. 4D), and peak levels were significantly increased in neutrophils isolated from SOD3−/− mice compared with WT mice (Fig. 4E).

In vivo SOD3 deficiency reduces CCL-2 and subsequent monocyte recruitment
Since monocytes also traffic into the airways in response to infection, a previously described flow cytometry approach was used to define three airway macrophage subsets including monocytes (8). CD11b (MAC-1) and CD11c (Integrin alpha X) within the total F4/80+ve pool of macrophages were used to further identify other macrophage subsets including resident alveolar (CD11chi, CD11blo), intermediate (CD11chi, CD11bhi) airway macrophages, as well as monocytes (CD11clo, CD11bhi) by flow cytometry. In the absence of infection, most macrophages are resident AMs (CD11chi, CD11blo) (Fig. 5A, B). However, 2 days after Spn infection, two other macrophage subsets emerged including intermediate macrophages (CD11chi, CD11bhi) and monocytes (CD11clo, CD11bhi) in WT mice. Intriguingly, these two macrophage/monocytic subsets were not observed in SOD3 mice after infection. SOD3−/− mice had significantly less monocyte subset (CD11clo, CD11bhi) in the BAL (5,844.70 ± 3,710.37) compared with WT mice (96,661.96 ± 40,595) (Fig. 5B). This occurred independent of cell death as WT and SOD3−/− macrophages at day 1 showed almost identical proportions of cell death in the whole F4/80 pool of BAL cells (Fig. 5C). These data indicate that high oxidative stress alters the macrophage profile in response to Spn and suggests a possible failure to recruit monocytes into the airways. Therefore, we measured gene expression and protein secretion of CCL-2 (monocyte chemotactic protein-1). In response to Spn, we observed a large induction of CCL-2 in WT mice, which did not occur in SOD3−/− mice. There was significantly less CCL-2 in SOD3 mice compared with WT mice with a twofold decrease in gene expression (WT: 8.35 ± 1.98 vs. SOD3−/−: 4.32 ± 0.92) in the lungs. Secretion of CCL2 protein into the BALF was also significantly reduced in SOD3−/− mice (WT: 2,744.03 ± 534.77 vs. SOD3−/−: 1,018.76 ± 366.79) (Fig. 5D).

Inflammatory cytokines are significantly reduced in SOD3−/− mice in vivo
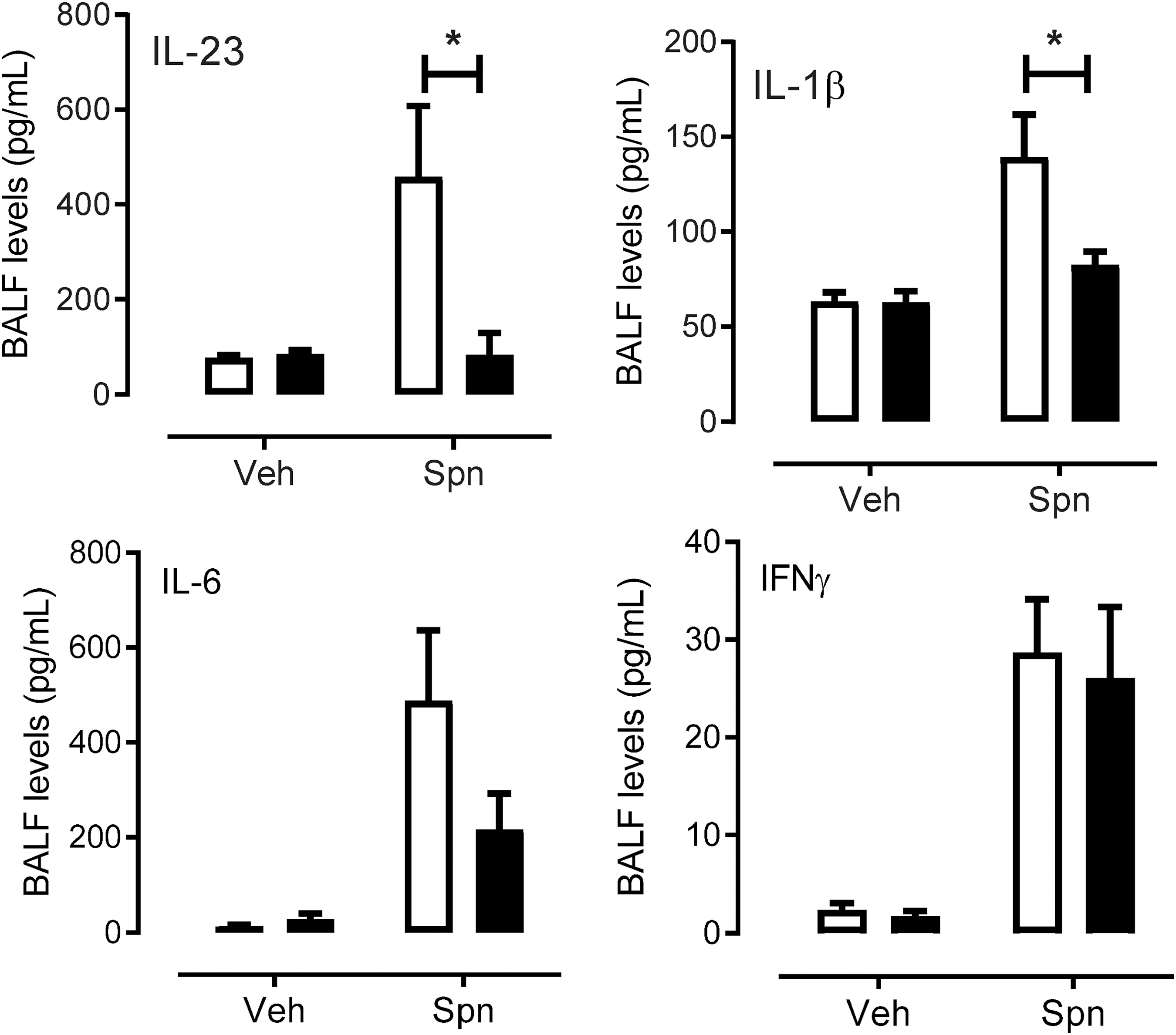
Cytokines produced by monocyte-derived macrophages are crucial for maintaining an inflammatory response during infection and were measured due to the significant decrease in monocyte numbers (Fig. 6). We measured several inflammatory cytokines by cytometry bead array and flow cytometry in the BALF of mice infected with Spn. We observed a significant induction of IL-23 and IL-1β in Spn-infected WT mice compared with WT vehicle-treated mice (Fig. 6). However, in SOD3−/− mice there was a failure to induce both cytokines above vehicle treated mice (Fig. 6). IL-23 was significantly lower (p < 0.05) in SOD3−/− mice compared with WT mice treated with Spn (SOD3−/−: 84.122 ± 45.40 vs. WT: 458.21 ± 149.413 pg/mL). There was also a significantly lower concentration of IL-1β observed in SOD3−/− (82.52 ± 7.02 pg/mL) compared with WT (139.27 ± 22.56 pg/mL) mice treated with Spn (Fig. 6). There were no significant changes observed for other measured cytokines, such as IL-6 and IFNγ (Fig. 6).
Increased extracellular superoxide reduces the proportion of IL-17A+ γδ T cells and lung IL-17A levels
Since IL-1β and IL-23 are known to promote induction of IL-17A from innate γδ T cells and we observed a reduction in secretion of these cytokines, this prompted us to investigate the regulation of γδ T cells after Spn infection in our model. Lungs from Spn-infected mice were harvested, and single-cell suspensions were treated with phorbol 12-myristate 13-acetate (PMA) and ionomycin in the presence of Brefeldin-A before intracellular staining was carried out for IL-17A. Absolute lung γδ T cell numbers were increased during Spn infection at day 2 in WT mice, and this response was significantly (p < 0.05) reduced in SOD3−/− mice (5279.52 ± 1996.03) compared with WT mice (12,279.75 ± 2169.810) (Fig. 7A). Additionally, Spn-infected SOD3−/− mice (expressed as a frequency of the live CD45+ve fraction) had significantly (p < 0.0108) fewer IL-17A+ γδ T cells compared with WT mice (∼40% less) (Fig. 7B, C). To determine whether this reduction influenced total IL-17A levels in the lungs following pneumococcal infection, IL-17A expression was quantified using RT-quantitative PCR. Relative to vehicle-treated mice, Spn lung infection resulted in a 75-fold increase in IL-17A transcript levels at day 2, and this response was significantly reduced to 11-fold in SOD3−/− mice (Fig. 7D, p < 0.05). In addition, IL-17A protein levels in lung homogenate were determined by ELISA, where Spn-induced IL-17A levels were reduced to uninfected control levels in SOD3−/− mice (Fig. 7E, p < 0.05). These data demonstrate that in the absence of SOD3, there is a failure to expand IL-17A+γδ T, which contributes to a significant reduction in IL-17A in the whole lung.

Discussion
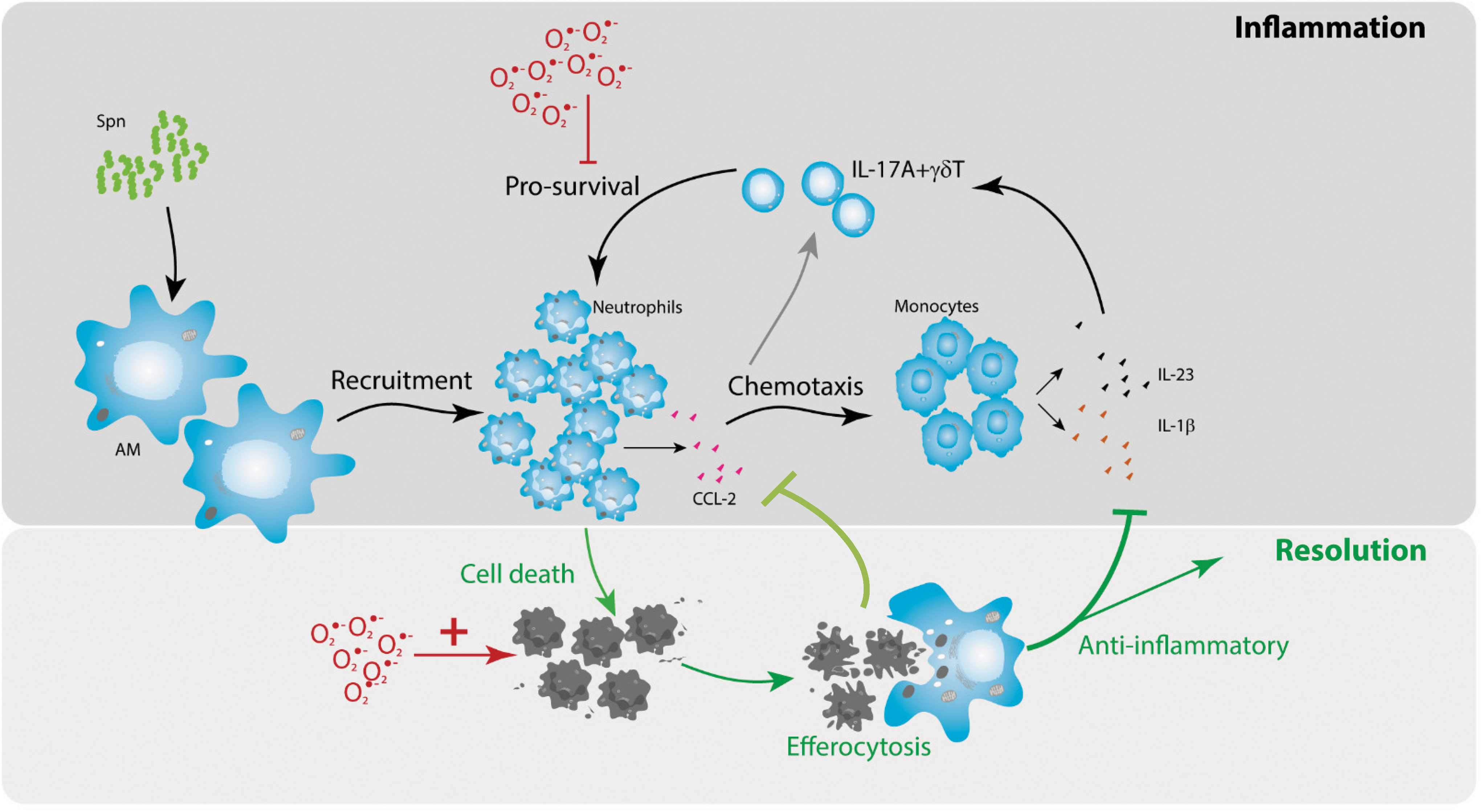
Our data demonstrate for the first time that SOD3 regulates the redox state of phagocytic vesicles during pneumococcal infection. We also show that the lack of SOD3 (EC-SOD) has a dramatic impact on the IL-17A+γδ T cells and survival of neutrophils in the airways after pneumococcal infection. The early emergence of apoptotic neutrophils will stimulate their removal by efferocytosis, the process by which phagocytic macrophages engulf apoptotic cells. This can have profound impacts during acute inflammatory responses. Efferocytosis is not only essential for the removal of apoptotic/necrotic cells during the resolution phase but also promotes the active suppression of inflammatory mediators produced by macrophages (9). Indeed, this negative feedback loop has been previously postulated; however, we are the first to demonstrate the involvement of SOD3 signaling. Using adhesion molecule-deficient mice (CD18−/− mice), Stark et al. showed that phagocytosis of apoptotic cells (neutrophils in particular) induces a very strong anti-inflammatory signal, which suppressed the secretion of macrophage/dendritic cell-derived IL-23. This in turn dampened the IL-17A response from “neutrophil regulatory” T cells, such as γδ T cells (28). We observed a similar diminished IL-23 and IL-1β response in SOD3−/− mice, which led to inhibition of IL-17A+γδ T proliferation and lung IL-17A levels during pneumococcal lung infection.
We also observed a profound alteration in macrophage subsets present during pneumococcal infection in SOD3−/− mice, demonstrating for the first time that this enzyme can regulate the phenotype of airway macrophages during bacterial lung infections. The failure to recruit monocytes is most likely to be driven by a failure to stimulate CCL2 production. Monocytes can produce a suite of inflammatory cytokines, and their migration to the site of infection is dependent on chemokines such as CCL-2 (39). It is important to note that lung CCL-2-CCR2 axis is crucial for protection against S. pneumoniae infection in vivo (38). CCL-2 can also act as a chemotactic factor for the recruitment of dendritic cells and activated memory T cells (6). Multiple cell types including monocytes/macrophages, epithelial and endothelial cells, as well as neutrophils can secrete CCL-2. It has been proposed that neutrophils secrete CCL2 and this induced chemotaxis of IL-17-expressing T cells (25). We propose that the early emergence of apoptotic neutrophils in SOD3−/− mice and their clearance by efferocytosis result in a strong anti-inflammatory effect, exemplified by the markedly reduced secretion of CCL2. SOD3 will likely protect neutrophils from early apoptosis by either preventing ROS-induced cell death or promoting neutrophil survival. It has previously been shown that overexpression of SOD3 leads to phosphorylation of Akt and FoxO3a, with translocation of FoxO3a to the cytoplasm, causing the simultaneous downregulation of Bim but enhancement of antiapoptotic miRNA miR21 (16). In this previous study, SOD3 played a dual-function role as both prosurvival and antiapoptotic mediator. Future work delineating the mechanism by which SOD3 deficiency and excessive phagosomal ROS levels drive early neutrophil cell death will need to be carried out.
Intriguingly, the failure to expand IL-17A+ γδ T cells in SOD3−/− mice was not associated with a failure to clear the pathogen in the upper or lower airways. This is consistent with our observation that SOD3 deficiency does not impair the pneumococcal killing capacity of macrophages. Our data suggest that resident lung and AMs and the early preserved neutrophil response at day 1 are sufficient to contain and eradicate the 19F pneumococcus serotype during the early phase of infection. We chose to investigate the 19F serotype as it readily colonizes the airways without causing invasive disease in mice, and it is frequently detected in human populations, particularly in developing countries where there is poor vaccine coverage (33). The 19F serotype can, however, cause severe pneumonia if there is a defective immune response (1). There are also conflicting data regarding the role of neutrophils during experimental pneumococcal lung infections, where serotype appears to dictate whether neutrophils are indispensable or redundant for bacterial clearance. In one study, neutrophil depletion proved beneficial during infection with the invasive serotype 8 as the degree of pneumonia and septicemia was reduced, which resulted in prolonged survival (21). In contrast, neutrophil depletion resulted in markedly higher bacterial loads in the lungs and blood of mice infected with the invasive serotype 2 pneumococci (13). Hence, there is a fine balance between neutrophil function and pathogen clearance, where neutrophils contribute to pathogen clearance, but an excessive response can cause tissue damage. Our data herein identify a previously unappreciated role for SOD3, which plays a central role during pneumococcal infection by coordinating IL-17A-dependent neutrophil trafficking.
Our study demonstrates that in SOD3−/− mice, early neutrophil recruitment (day 1) is not significantly altered, but there was a rapid decline in neutrophil numbers at day 2 following acute pneumococcal lung infection. Consistent with the decline in neutrophil numbers as determined by flow cytometry, analysis of the degree of lung inflammation by histology demonstrated a significant reduction in peribronchial and alveoli inflammation in SOD3−/− mice infected with Spn. This is somewhat contradictory to previous studies where SOD3−/− mice displayed an increased lung neutrophil response to heat-killed bacteria at day 1 after inoculation (29). This differential neutrophil response may reflect our use of viable bacteria and our analysis of the later day 2 time point, where neutrophil apoptosis only resulted in a significant decline at day 2. Our study is also somewhat contradictory to the observation that transgenic mice overexpressing SOD3 displayed increased neutrophil apoptosis in response to intravenous inoculation with L. monocytogenes (4). This may reflect that Spn is an extracellular pathogen, whereas L. monocytogenes is an intracellular pathogen that can replicate in leukocytes. However, our findings are consistent with the observation that gp91phox−/− mice, which cannot generate Nox-dependent superoxide, display greater lung neutrophilic inflammation during pneumococcal infection (22). The failure to produce superoxide during pneumococcal infection resulted in a significant decrease in neutrophil apoptosis (22). This finding is very consistent with our study as the absence of SOD3 resulted in increased superoxide levels during pneumococcal infection, and this resulted in greater neutrophil apoptosis. This has intriguing therapeutic implications as Nox-dependent superoxide is not required for killing of Spn (22); hence, the use of SOD mimetics may restore host immunity without compromising pneumococcal killing and clearance.
In summary (Fig. 8), a lack of SOD3 increases phagosomal ROS levels in leukocytes during acute pneumococcal infection without altering macrophage-mediated bacterial killing. However, neutrophils were particularly susceptible to the lack of SOD3 levels, as they entered apoptosis much more rapidly. We propose that the clearance of apoptotic neutrophils by efferocytosis stimulates a potent anti-inflammatory effect mediated by markedly lower CCL2 levels during bacterial lung infection. This idea has been previously proposed and proven by others (9, 28). Indeed, the clearance of apoptotic cells by phagocytic cells plays an important role in the resolution of inflammation where apoptotic cell engulfment activates signals that suppress the release of proinflammatory cytokines, as reviewed in Refs (19, 26). The failure to recruit circulating inflammatory monocytes due to reduced CCL2 levels will in turn result in a reduction in cytokines including IL-23 and IL-1β. As a consequence of reduced CCL2 and IL-23/IL-1β levels, there is a failure to stimulate γδ T cells to proliferate and produce IL-17A. The attenuated signal leads to further reduced neutrophil survival due to an increase in apoptosis. Herein, we identify a new role for SOD3 during pneumococcal infection, whereby SOD3 is required for innate T cell responses.
Materials and Methods
Animals and pneumococcal infection protocol
All animal experiments described in this article were approved by the Animal Ethics Committee of RMIT and conducted in accordance with the guidelines of the National Health and Medical Research Council of Australia. The SOD3−/− mice were originally generated in the laboratory of Dr. Stefan Marklund (5). The SOD3−/− mice used in our experiments were a gift from the University of Pittsburgh, where SOD3−/− mice were backcrossed to C57BL/6J mice as previously described (14). Tail clippings from pups generated from breeding pairs were genotyped by TransnetYX (US). Wild-type C57BL/6J mice were used as control animals in this study and purchased from the Animal Resource Centre Pty. Ltd. (Perth, Australia). Six- to 12-week-old male mice were used in all experiments and housed at 20°C on a 12-h day/night cycle in sterile micro-isolators and fed a standard sterile diet of Purina mouse chow with water allowed ad libitum. S. pneumoniae (EF3030- capsular type 19F) (Spn) was used in all the described experiments. Mice were infected intranasally with 105 CFU Spn in 35 μL saline under light isoflurane anesthesia. On day 1, 2, or 7 after infection, mice were culled by intraperitoneal overdose of sodium pentobarbitone. BAL was performed via tracheotomy, and total/differential BAL cell counts were determined as previously described (35).
BAL cell harvest and lung single-cell suspensions
The lungs of anesthetized mice were lavaged in situ with a final volume of 1.2 mL of ice-cold PBS, as previously described (34). Total number of viable cells in the BALF was determined using bromide/acridine orange exclusion. Lung single-cell suspension was prepared as previously described (2). Briefly, lung lobes were minced with scissors and digested in a solution of PBS containing Liberase™ Research Grade (Sigma–Aldrich) for 45 min at 37°C with shaking. The digest was washed with FACS buffer (PBS containing 2% FBS) and cells were harvested by centrifugation (400 g at 4°C for 10 min). Red blood cells were lysed using red blood cell lysis buffer (1.55 M NH4Cl, 0.1 M KHCO3, 10 mM disodium EDTA, pH 7.4) for 5 min at room temperature. Cells were filtered through a 40-μm nylon net strainer, washed, and resuspended in FACS buffer before surface staining. Cell counts were carried out using ethidium bromide/acridine orange exclusion.
Antibodies and flow cytometry
All antibodies were purchased from Biolegend unless otherwise stated. Anti-mouse antibodies: fluorescein isothiocyanate-conjugated CD45, γδ TCR, phycoerythrin (PE)-conjugated IL-17A, and relevant Rat IgG1k isotype control, MHCII (Mouse I-A[d]), MARCO, and relevant isotype control mouse IgG1 (R&D Systems); allophycocyanin (APC)-conjugated TCRβ, CD11b, PE-Cy™7-conjugated CD11c, IL-17A, and relevant Rat IgG2ak isotype control (Thermo Fisher Scientific); CD45, CD45 and Pacific Blue-conjugated CD4, F4/80, APC-eFluor 780-conjugated Ly-6G (Gr-1; Thermo Fisher Scientific); and Biotinylated CD49b. A secondary streptavidin APC-Cy™7 was also used according to the manufacturer's instructions (Thermo Fisher Scientific). To avoid nonspecific binding, anti-mouse CD16/32 mAb (Mouse BD Fc Block™) (2.4G2; BD) was used. Up to 4 × 106 cells in FACS buffer were typically stained with primary antibody at 4°C for up to 1 h on ice in the dark. Secondary antibodies were stained for 20 min under the same conditions and harvested by centrifugation (described above). Data from cells were collected using the LSRFortessa or Aria Fusion (BD), and FlowJo software (BD) was used for data analysis. Cells were gated to exclude doublets and nonviable cells (either by PI or by DAPI exclusion), all immune cells were gated as CD45+ve cells, and lymphocytes were determined to be low SSC-A/FSC-A and myeloid cells were determined to be high SSC-A/FSC-A before more specific markers/antibodies (listed above) were used to define different populations.
For intracellular staining, up to 5 × 106 single cells from the lungs of WT or SOD3−/− mice treated with vehicle or Spn for 2 days were cultured in a 24-well plate in DMEM containing 2.5% FCS and 10 mg/mL Brefeldin A (Thermo Fisher) with/without 50 ng/mL PMA and 1 μg/mL ionomycin for 4 h at 37°C in 5% CO2. Cells were washed twice in ice-cold PBS and harvested by centrifugation (400 g at 4°C for 10 min) before surface staining using the above list of antibodies to determine each cellular subset. Intracellular staining was carried out using the Fixation and Permeabilization Kit (Thermo Fisher Scientific) according to the manufacturer's instructions. IL-17A was stained using anti-mouse IL-17A (as listed above). Cells were gated to exclude doublets and aggregates by SSC-W/H and/FSC-H/W, all immune cells were gated as CD45+ve cells, and lymphocytes were determined to be low SSC-A/FSC-A and myeloid cells were determined to be high SSC-A/FSC-A before more specific markers/antibodies (listed above) were used to define different populations. The CBA Flex Systems Kit (BD Biosciences) was used to measure the described cytokines in the BALF according to the manufacturer's instructions and as previously described in Ref (2). FlowJo software (Treestar, Inc., Ashland, OR) was used for final analysis, and positive IL-17A signaling was determined relative to the relevant isotype controls (listed above).
CFU assay, ELISA, and RT-quantitative PCR
BALF and nasopharyngeal tissue were collected, and serial dilutions were cultured on gentamicin (5 μg/mL) horse blood agar plates, as previously described (35, 37). CFUs were counted after overnight incubation at 37°C.
RNA was purified from snap-frozen lung tissue using RNeasy Kit according to the manufacturer's instructions, and cDNA was prepared as previously described (36). RT-quantitative PCR was performed using bioinformatically validated TaqMan primer/probes. All threshold cycle values (Ct) were normalized to a reference gene (18S), and the relative fold change was determined by the ΔΔCt method, as previously described (36).
For lung tissue, protein lysate isolation tissue was resuspended in a lysis buffer (10 μL/mg, 150 mM NaCl, 50 mM Tris-HCl, pH 7.4; 1% NP-40, protease inhibitor cocktail). The Murine IL-17A ELISA Kit (eBioscience) was used to measure IL-17A concentration in the protein lysate according to the manufacturer's instructions. Protein content was assessed by BCA (Peirce, Thermo Fisher), and IL-17A concentration was normalized to total protein for each sample.
ROS measurement and bacterial killing assay
Phagosomal ROS were measured as previously described (32). Briefly, SOD3−/− and WT AMs were isolated from the BAL of naive animals and seeded (1 × 105 cells per well/24-well plates) onto glass coverslips for 24 h to allow for adherence in serum-free RPMI medium before 5 min pretreatment with OxyBURST Green H2HFF (100 μM). Cells were infected with Spn (multiplicity of infection [MOI] 10) or PBS for 60 min, then washed with PBS, and fixed with 4% PFA for 15 min. After three PBS washes, cells were mounted on coverslips onto microscope slides with 10–20 μL of DAPI, then analyzed, and photographed on a Nikon upright confocal fluorescence microscope (Nikon D-eclipse C1). BMDMs from WT and SOD3 mice were also generated by culturing mouse bone marrow cells with 10 ng/mL of recombinant mouse GM-CSF for 10 days. On day 10, nonadherent macrophages were seeded at a density of 50,000 cells per 96 well in a 96-well Optiview plate and incubated overnight at 37°C, 5% CO2, and 95% humidity in a CO2 incubator. In addition, neutrophils were isolated from the bone marrow of WT and SOD3−/− mice through negative selection, as previously described (41). Briefly, bone marrow cells were collected from the femur and tibia of WT and SOD3−/− mice, and neutrophils were isolated using the MojoSort™ Mouse Neutrophil Isolation Kit (Biolegend, CA) with the MidiMACS™ magnetic separator (Miltenyi Biotec, Bergisch Gladbach, Germany) according to the manufacturer's instructions.
ROS production was quantified using L-012-enhanced chemiluminescence from isolated macrophages and neutrophils that were infected with Spn (MOI 10), as previously published (32). Briefly, Krebs-HEPES buffer containing L-0-12 (100 μM) was added into a 96-well Optiview plate, and photon emission (relative light units per second) was detected using the Chameleon luminescence detector (model 425105; Hidex, Finland) and recorded from each well for 120 min. For the bacterial killing assay, macrophages were incubated with S. pneumoniae at an MOI of 10 for 1 h at 37°C with rotation to allow for phagocytosis of bacteria by macrophages. The supernatant was then discarded to remove extracellular bacteria, and at selected time points (0, 30, 60, 90, and 120 min), the cells were lysed and intracellular bacteria plated on horse blood agar plates using serial dilution.
Histological examination of lung injury and assessment of lung function
For histology, the left lung was dissected, fixed in neutral buffered formalin (10%), and processed in paraffin wax. Sections (5 μm thick) were cut longitudinally, stained with hematoxylin and eosin, and whole slide scans were generated by light microscopy using the Olympus VS120 microscope scanner. The lung injury score was determined, as previously published (31). Briefly, a grading system that combined assessments of peribronchiolar inflammation and alveolitis by two independent assessors was used, where a score of 0 was indicative of healthy lungs (i.e., no damage); 1-very mild damage; 2-mild damage; 3-moderate damage, 4-severe damage, and 5-extremely severe histological changes.
Baseline lung function measurements were carried out using a low-frequency forced oscillation technique (FOT) and a small-animal ventilator (FlexiVent; Scireq, Montreal, QC, Canada), as previously published (10). Briefly, mice were ventilated, and single frequency FOT was performed to measure total respiratory system resistance, elastance, and compliance, where compliance describes the ease with which the respiratory system can be extended.
Statistical analyses
Data were normally distributed and expressed as mean ± standard error of the mean (SEM); n represents the number of mice. Differences were determined by two-way ANOVA, followed by Bonferroni post hoc test for multiple comparisons, where appropriate. All statistical analyses were performed using GraphPad Prism™. Statistical significance was taken to indicate the probability levels less than 0.05 (p < 0.05).
Footnotes
Author Disclosure Statement
No competing financial interests exist.