
Abstract
Significance:
Oxidative stress in the cell is characterized by excessive generation of reactive oxygen species (ROS). Superoxide (O2 −) and hydrogen peroxide (H2O2) are the main ROS involved in the regulation of cellular metabolism. As our fundamental understanding of the underlying causes of lung disease has increased it has become evident that oxidative stress plays a critical role.
Recent Advances:
A number of cells in the lung both produce, and respond to, ROS. These include vascular endothelial and smooth muscle cells, fibroblasts, and epithelial cells as well as the cells involved in the inflammatory response, including macrophages, neutrophils, eosinophils. The redox system is involved in multiple aspects of cell metabolism and cell homeostasis.
Critical Issues:
Dysregulation of the cellular redox system has consequential effects on cell signaling pathways that are intimately involved in disease progression. The lung is exposed to biomechanical forces (fluid shear stress, cyclic stretch, and pressure) due to the passage of blood through the pulmonary vessels and the distension of the lungs during the breathing cycle. Cells within the lung respond to these forces by activating signal transduction pathways that alter their redox state with both physiologic and pathologic consequences.
Future Directions:
Here, we will discuss the intimate relationship between biomechanical forces and redox signaling and its role in the development of pulmonary disease. An understanding of the molecular mechanisms induced by biomechanical forces in the pulmonary vasculature is necessary for the development of new therapeutic strategies.
Introduction
The entire pulmonary vasculature is exposed to biomechanical forces that can have profound physiological and pathological effects. In the vasculature, biomechanical forces are realized via two types of hemodynamic loads: tensile wall shear stress (WSS) caused by blood flow on the vessel and compressive circumferential stress caused by pressure loading. Flowing blood constantly exerts hemodynamic loads on the endothelium lining the blood vessels once the heart begins to produce a fetal circulation (75). As blood flow passes over the vessel luminal surface, it produces a frictional force known as shear stress (SS) or WSS, which acts tangentially to the vessel (75) (Fig. 1A).
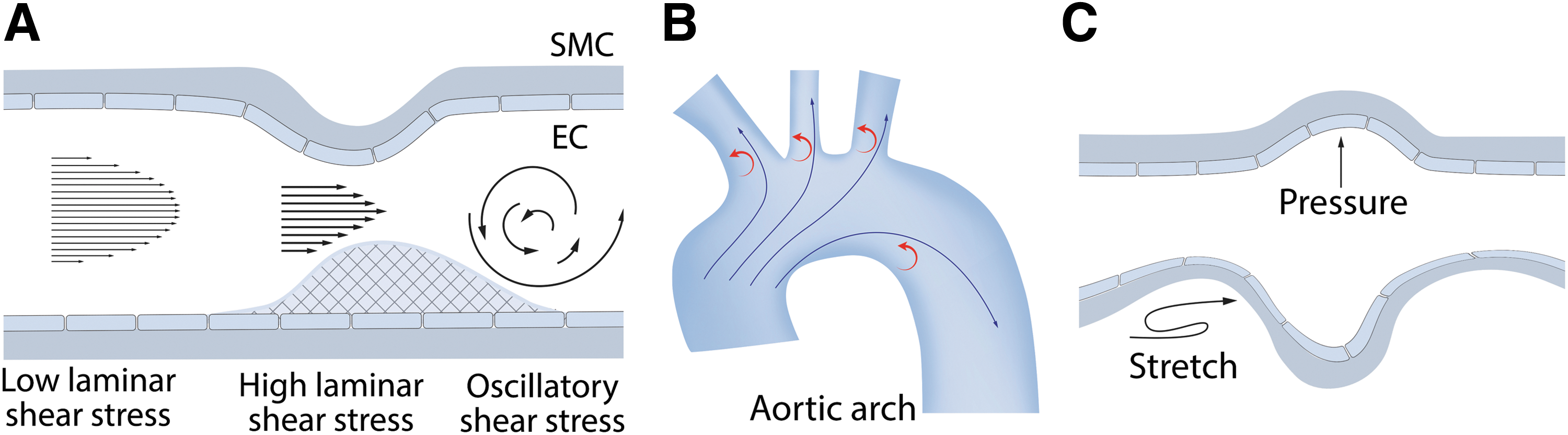
In vitro, many effects of physiological WSS can be reproduced by laminar shear stress (LSS), induced by steady laminar flow, and pulsatile shear stress, induced by periodic flow with a positive mean flow rate, stimulating a physiological response that maintains normal endothelial functions (Fig. 1A). LSS causes the alignment of the endothelial cells (ECs) in the direction of the flow (231). LSS globally affects EC homeostasis via multiple cell signaling cascades, the activation of specific transcription factors, and mechanosensitive gene expression. Blood vessels also contain athero-prone sites where wall geometry, afterload, and distal conditions combine to create areas of nonuniform flow such as turbulent or oscillatory flow as well as areas with modulated physiological SS (Fig. 1A, B). These increases or decreases in LSS (low and high SS) can have pathological consequences.
While SS acts tangentially to the vessel luminal surface (75) (Fig. 1A), the concomitant blood pressure exerts a load that acts perpendicularly to the cell surface, creating a compressive stress on the pulmonary vessel (75). As the blood pressure within the pulmonary system rises and falls depending on the cardiac cycle, this results in a circumferential stress and this is transmitted circumferentially to cells in the lung through contacts with the extracellular matrix (75) (Fig. 1C). The alveolar-capillary unit present in the lung is also exposed to mechanical forces as a result of the respiratory cycle (20), resulting in lung capillary strain (20).
Under certain conditions (such as high tidal volume lung mechanical ventilation or high blood pressure), excessive circumferential or compressive loading can induce pathological changes in the challenged cells. In vitro, an excessive circumferential loading can be reproduced by special devices designed to apply physiological (5% elongation) or excessive (15%–20% elongation) cyclic stretch (CS) to the cell monolayers. The following sections discuss our most up-to-date understanding of the effects of biomechanical forces on the lung and the role played by redox pathways in transducing these signals into both physiological and pathological cellular responses.
EC Surface Proteins as Mechanosensors
Integrins
Integrins are heterodimeric transmembrane adhesion receptors responsible for cell focal adhesions (FAs) that function by linking cytoskeletal structures to the extracellular matrix (130). Integrins are also involved in cell signaling events via scaffolding specific signaling macromolecules (128). Integrins can also serve as mechanosensors, providing “outside-in” signaling in response to increased blood pressure, SS, or circumferential tensile stress (242) (Fig. 2). Low SS signaling via integrins has been linked to the activation of multiple proinflammatory pathways (60 –62), whereas an excessive CS-dependent in vitro stimulation of β3-subunit expression has been shown to be protective for CS-challenged cells through cellular reorientation (257).

Immunofluorescence microscopy has identified a rapid reorganization of FA contacts and the activation of focal adhesion kinase, and the depletion of paxillin, an FA protein scaffold, delays the cell orientation changes indicating the importance of integrin-mediated signaling (127). Exposing smooth muscle cells (SMCs) to an excessive CS also induces both αv- and β3-integrin expression, Src activity, talin degradation, and binding and processing of prothrombin (173).
The integrin β4 has been shown to be involved in the anti-inflammatory response in EC (56) and in mouse models of acute lung injury (ALI) (57). Interestingly, the tyrosine phosphorylation in the C-terminal intracellular domain of integrin β4 is activated by CS-mediated mechanical stress, leading to the loss of its anti-inflammatory property in ECs (55). Mechanical forces appear to regulate integrin(s) via phosphorylation and this has been shown to be critical for proinflammatory cytokine expression (IL-6, IL-8, MCP-1, and RANTES) (55). Oscillatory SS- or high-pressure-dependent release of angiotensin II (Ang II), endothelin-1 (ET-1), vascular endothelial growth factor (VEGF), and other vasoactive factors can, in turn, activate integrin functions (204, 271).
Thus, integrins are implicated in downstream cell signaling events stimulated by other receptors including mechanosensors. Integrin signaling is also important in regulating reactive oxygen species (ROS) generation and oxidative stress. For example, superoxide release is induced in mouse neutrophils by α4-integrin-dependent adhesion on vascular cell adhesion molecule 1 (VCAM-1) (211), whereas tumor necrosis factor α (TNFα) has been shown to cause the redistribution of β2-integrins and NADPH oxidase (NOX) subunits (gp91phox, p22phox, p47phox, and p67phox) to a Triton X-100-insoluble fraction human neutrophils (299), suggesting an integrin-dependent activation of NOX. Ligation of β1-integrins has also been linked to p47phox membrane redistribution and hydrogen peroxide (H2O2) generation in human neutrophils (272). Thus, integrin-dependent signaling is intimately involved in the cellular response to biomechanical forces and the vascular damage induced by excessive ROS production.
Endothelial receptors and ion channels
The membrane microdomain, caveolae, is critically involved in mechanotransduction in EC. Caveolae serve as a platform that allows the assembly of cell signaling complexes, including receptors and ion channels, and components of cell–cell, and cell–matrix, contacts. Thus, caveolae can integrate “outside-in” signaling by functionally linking various mechanoreceptors with their downstream effectors. Caveolae microdomains are also important in assembling endothelial junctions and FAs into mechanosensitive signaling units. Therefore, perturbing the caveolae structure can produce an abnormal response to biomechanical forces applied to the endothelium.
Depleting the major structural protein of caveolae, caveolin-1 (cav-1) decreases the sensitivity to WSS in cav-1−/− mice that includes an attenuated increase in [Ca2+]i (45). Caveolae also support the ion channels involved in the EC hyperpolarization and Ca2+-dependent cell signaling that occurs in response to WSS. Studies in cav-1 knockout (KO) mice revealed that the impaired Ca2+-dependent signaling is linked to a decreased activity of the TRPV4 Ca2+ channel that normally colocalizes with cav-1 on the plasma membrane (226).
The TRPV4-dependent [Ca2+]i increase is essential for Ca2+-activated K+ channels (KCa), which induce endothelium-dependent hyperpolarization (EDH) and regulate vascular tone (109, 165). In EC, TRPV4 and KCa receptors are colocalized in caveolae (109). In human lung microvascular EC, under static conditions, TRPV4 colocalizes with small conductance KCa2.3 channel in caveolae, whereas SS stimulation also recruited intermediate conductance KCa3.1 channel to the complexes in caveolae (109), suggesting an importance of these channel complexes for vascular cell hyperpolarization (Fig. 2). Thus, mechanosensitive ion channels localized in caveolae are important players in the fine regulation of vascular tone and blood pressure.
The secretion of vasoactive factors (ET-1, Ang II, VEGF, PDGF, TNFα, etc.) is also regulated by biomechanical forces and these can be indirectly involved in mechanosensing via their respective endothelial or SMC receptors (Fig. 2). For example, mechanosensitive release of ATP (27, 278) can further stimulate P2X and P2Y purinoceptors, such as P2X4 (an ATP-dependent Ca2+ channel) (232, 297, 298) and P2Y1/P2Y2 (G-protein-coupled receptors [GPCRs]) (37, 38), followed by activation of respective cell signaling pathways (Fig. 2). All have been linked to ROS generation (Fig. 2).
Regulation of Vasoactive Molecules by Biomechanical Forces
Vasodilators
Nitric oxide (NO) is a vasorelaxant produced by NO synthase isoforms converting
In blood vessels, NO is synthesized in ECs and diffuses to the adjacent SMCs, where it activates soluble guanylate cyclases (sGCs) (67). This leads to activation of cGMP-dependent PKG (cGMP-dependent protein kinase) and other effector proteins, including ion channels, ion pumps, and phosphodiesterases (PDEs) (43). In addition, NO in SMCs promotes the activation of cAMP-dependent protein kinase/protein kinase A (PKA), inhibiting SMC proliferation (136). NO is also involved in preventing platelet and leukocyte activation and adhesion to the vessel wall (147).
SS increases NO production via endothelial nitric oxide synthase (eNOS) phosphorylation and by stimulating EC receptors that increase intracellular Ca2+ (269). Exposing ECs to LSS can also suppress ROS levels (190, 289). In contrast, exposing EC to SS using an irregular flow pattern leads to higher levels of ROS and less available NO (166). NO generation attenuates insulin-like growth factor 1 (IGF-1) and the insulin-induced elevation in H2O2 levels via a cGMP-dependent event in SMC (313). As eNOS promoter activity and protein levels in ECs are suppressed by SMC-derived H2O2, this suggests that a feedback mechanism exists that may contribute to the NO signaling (287).
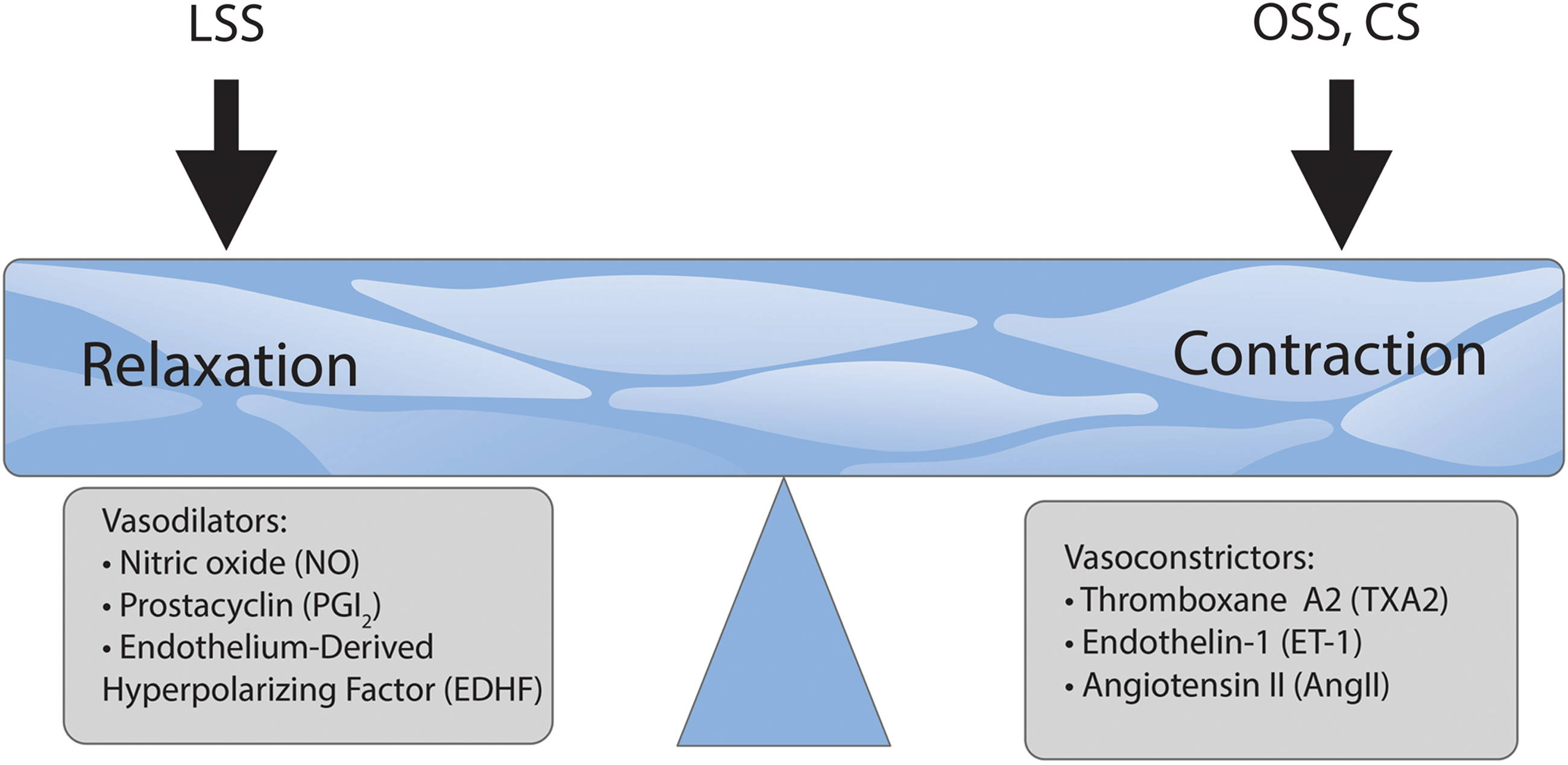
Derived from arachidonic acid via the action of cyclooxygenase-2 (COX-2) and prostaglandin I synthase (PGIS), prostacyclin (PGI2) is another vasodilator with a broad range of effects in the vasculature (Fig. 3).
PGI2 binds to the PGI2 receptors (IP) (66) located on both platelets and SMCs (199), inhibiting platelet aggregation (63). Acting via Gs GPCR prostaglandin receptors, PGI2 induces cAMP synthesis and the well-described PKA-dependent pathway of the cytoskeletal reorganization and relaxation (263). The effects of PGI2 are tightly related to NO effects since PGI2 potentiates NO release and, in turn, NO potentiates the effect of PGI2 on SMCs (248).
PGI2 also exerts protective effects in the vasculature by inhibiting SMCs hypertrophy, migration, and proliferation (187). In patients with hypertension, production of vasoactive prostanoids is selectively impaired and this may contribute to the increased systemic vascular resistance and increased incidence of thrombosis (188). PGI2 exerts protective cardiovascular effects that counterbalance the harmful effects of thromboxane A2 (TxA2) (187). Disturbance of the balance between PGI2 and TxA2 has been associated with vascular disorders such as pulmonary hypertension (PH). ROS can activate COX-2 expression, enabling the production of both PGI2 and TxA2 (182). PGI2 has been shown to inhibit the activity of NOX, whereas peroxynitrite induces tyrosine nitration in PGIS, inactivating the enzyme (12, 314) and increasing the levels of TxA2 (2).
Endothelium-derived hyperpolarizing factor (EDHF) produced by the EC is a vasodilator of unknown nature that is shown to be important for vascular tone in smaller arteries, although a number of publications established its compensatory role for some pathological states, leading to an impairment of eNOS activity (300) (Fig. 3).
Vasorelaxation can also occur after endothelial stimulation through a non-NO nonprostanoid pathway originally ascribed to the actions of endothelium-derived hyperpolarizing factor (265) (Fig. 3). EDHF involves hyperpolarization, generated in the endothelium, which spreads via myoendothelial gap junctions to the SMCs, and it is this hyperpolarization that results in relaxation of SMCs (65, 85, 92, 228). Flow-induced vasodilation that is independent of endothelium-derived NO (EDNO) and PGI2 is typically due to EDH of the underlying SMCs (86).
EDHF initiates SMC hyperpolarization directly after its release from the endothelium (40, 84). The endothelial hyperpolarization is initiated by the activation of KCa channels (92). H2O2 is believed to be an EDHF that acts primarily on the prearterioles and arterioles where EDH-mediated relaxation becomes more important than EDNO (181, 243, 244). SS can induce the release of H2O2 from ECs that acts as an EDHF that contributes to flow-induced vasodilation in coronary arterioles (189). H2O2 can induce this hyperpolarization by several mechanisms, including cGMP or cAMP-meditated pathway, activation of PKA/PLA2, or the direct activation of various K+ channels (245).
Vasoconstrictors
The opposite effect on vascular tone and blood pressure occurs via vasoconstrictors (Fig. 3). Another arachidonic acid derivative, TxA2, secreted by platelets, acts via Gq GPCR thromboxane receptors (TP), inducing platelet aggregation and blood clot formation and reducing blood flow. TxA2 is a functional antagonist of PGI2 and their balance supports vascular homeostasis. TxA2 promotes platelet aggregation and expresses adhesive cofactors for platelets such as von Willebrand factor, fibronectin and thrombospondin, and procoagulant factors (262).
TxA2 exerts its biological activity through its cognate TP GPCR receptor (194). TxA2 receptor also promotes cell migration and proliferation of SMCs (133, 205, 301). TxA2 is a functional antagonist of PGI2 and their balance supports vascular homeostasis. ROS have been shown to induce the release of TxA2 in different tissues (1, 113, 114). ROS can enhance arteriolar tone by diminishing endothelium-derived NO responses, generate a COX-2-dependent endothelial-derived contracting factor (EDCF) that activates TP, and enhance vascular SMCs reactivity (182). In the vasculature, O2 •− elicits constriction through activation of TP-dependent mechanisms (141, 266). Thus, ROS through the release of TxA2, a vasoconstrictor prostanoid, can also mediate vascular contraction.
ET-1 is a potent peptide vasoconstrictor produced by the EC. The product of the EDN1 gene, preproendothelin-1 (ppET-1), is proteolytically processed to an active 21-amino acid peptide ET-1 secreted from the EC into the circulation (72) (Fig. 3). ET-1 is a GPCR agonist inducing Ca2+ elevation in affected cells. In the vasculature, ET-1 has pleiotropic effects producing SMC constriction via ETA receptors and inducing relaxation via endothelial ETB receptors (72). ETA and ETB receptors promote the proliferation of pulmonary artery SMCs (PASMCs) (74). Increased ROS production caused by ET-1 promotes vasoconstriction and vascular remodeling via the suppression of NO activity (77). ET-1 messenger RNA (mRNA) and peptide expression are significantly upregulated in both PH models and patients (107, 274).
ET-1 receptor A and B antagonists have been used as pulmonary arterial hypertension (PAH) drugs with potent antiproliferative, anti-inflammatory, and endothelium-protective properties (48). Physiological levels of SS have a negative effect on the expression of ppET-1 and ET-1-converting enzyme (ECE-1) in the EC (178, 191). This downregulation of the ET-1 system depends on eNOS activation and oxidative stress (179, 191). ET-1 promotes a vascular and interstitial remodeling, stimulates the proliferation of SMCs, fibroblast activation, and proliferation (241) via increases in NOX-derived ROS (287). SS and NO are potent inhibitors of ET-1 gene expression (217, 222, 253, 255). Recently, it has been shown that mitochondria-targeted antioxidant, mitoTEMPO, can inhibit ET-1-induced constriction of rat mesenteric arteries (50), confirming a link between ET-1 and mitochondria-derived ROS that had been shown in EC (255).
Ang II is produced from angiotensin I in the lung by angiotensin-converting enzyme (ACE). Ang II is a potent vasoconstrictor acting via GPCR Ang II type 1 and type 2 receptors (AT1R and AT2R) (Fig. 3). LSS (10 dyn/cm2, 24 h) upregulates ACE expression in SMCs (111) and Ang II promotes SMC remodeling, cell growth, fibrosis, collagen deposition, and contractility (268, 313). AT1R is likely a redox-coupled mechanosensor that regulates oxidative stress as studies have demonstrated AT1R is closely associated with ROS production (25, 163, 282) via Nox-4-dependent oxidative stress pathways (312). LSS can also induce ROS levels by an AT1R-mediated downregulation of eNOS expression mediated via Akt1 and Erk activity (49). Ang II is also a proinflammatory mediator that stimulates the production of inflammatory cytokines and causes oxidative stress via AT1Rs to promote hypertension (18, 137, 308).
Regulation of ROS Generation by Biomechanical Forces
NADPH oxidase
The NOX family consists of seven isoforms (NOX-1–5 and DUOX-1 and DUOX-2) that act as transmembrane catalytic subunits and require additional proteins to assemble large functionally active complexes. NOX complexes produce ROS (superoxide anion and H2O2) using NADPH and molecular oxygen as substrates (152).
The regulation of NOX isoforms is diverse, including rather simple Ca2+-dependent activation of NOX-5 (267) and complex modulation of NOX-1/NOX-2 activities via association with various effector proteins such as Rac-1/2, NoxA1, p47phox, and p67phox that, in turn, can be regulated by a number of cell signaling pathways. In addition, NOX-4 is constitutively active and is mainly regulated by gene expression (152). NOX isoforms function in normal physiological processes and in the development/progression of vascular pathologies (261).
Owing to their complex regulation, NOX isoforms can be stimulated by biomechanical forces. In cell culture models, long-term LSS (30 dyn/cm2, 24 h) downregulates mRNA and protein expression of NOX-2 and p47phox in an eNOS-dependent manner (82). LSS also downregulates the expression of NOX-4 via antioxidant response element (ARE), Oct-1-binding site, and NF-E2-related factor 2 (Nrf2) (110), whereas oscillatory SS can stimulate expression/activity of NOX isoforms, p47phox-dependent superoxide generation, and monocyte adhesion (129).
More detailed studies of LSS and oscillatory SS have identified different roles of activated NOXs with LSS activating an NOX-2–p47phox complex that stimulates eNOS phosphorylation and NO production, and oscillatory SS leading to eNOS uncoupling via an NOX-1–NOXO1 complex (247). Disturbed flow (low and oscillatory SS) studied in vivo using a model of partial ligation of the mouse carotid artery identified a p47phox-dependent endothelial dysfunction, leucocyte recruitment, and infiltration (185), leading to the development of atherosclerosis (196. 197). In EC, NOX-4-derived superoxide has also been shown to interfere with PGI2 bioactivity (193).
Xanthine oxidase
Xanthine oxidoreductase is the enzyme that catalyzes the oxidation of hypoxanthine to xanthine and uric acid during purine metabolism (250, 286). The enzyme exists in two forms: xanthine dehydrogenase and xanthine oxidase (XO). XO is one of the major sources of ROS in the vasculature (183) producing superoxide and H2O2 and can be induced by TNFα (99). XO activity and superoxide generation are stimulated by oscillatory SS (183). A number of studies have identified a role for XO in the pathogenesis of ventilator-induced lung injury (VILI) via a p38-dependent mechanism (81), and p38/XO inhibition attenuates VILI pathogenesis (153, 258). Increased XO activity also impairs shear-dependent and endothelium-dependent vasodilation (80, 151).
Endothelial NO synthetase
A number of studies have established a regulatory role of post-translational modifications (PTMs) of eNOS. Multiple phosphorylation sites implicate several protein kinases in the modulation of eNOS activity.
Tyrosine phosphorylation of eNOS induced by H2O2 in EC increases the association of eNOS with caveolin-1 (104). Phosphorylation by Akt1 at Ser1177 increases NO synthesis (79), and LSS or pulsatile SS induces this PI3K/Akt1-dependent phosphorylation in a Ca2+-independent manner (94, 161).
Several reports describe the phosphorylation of the same site, Ser1177, by protein kinases A and G (PKA and PKG), AMP-dependent protein kinase (AMPK), and Ca2+-calmodulin-dependent protein kinase II (CaMKII); PKA also phosphorylates Ser633 and Ser615 [reviewed in Boo and Jo (31)]. LSS (15 dyn/cm2) induces PKA-dependent phosphorylation of eNOS at Ser633, which positively regulates its activity (30, 32).
eNOS-mediated NO signaling can also be inhibited by asymmetric dimethylarginine (ADMA), a product of cellular protein degradation (29). Increased levels of ADMA have been shown to be associated with PH (91, 229). ADMA levels have been shown to be stimulated by increased pulmonary blood flow (PBF) and pressure in vivo (254) and this leads to the uncoupling of eNOS and the peroxynitrite-mediated nitration and activation of Akt1 (216). This, in turn, induces the mitochondrial redistribution of eNOS that causes mitochondrial dysfunction and increases mitochondrial ROS generation and further increase in cellular oxidative stress (256). The ADMA degrading enzymes, dimethylaminohydrolases (DDAH), are now considered key regulators of eNOS-produced NO (93, 162).
In ALI models, the ADMA/DDAH balance is critical for the endothelial barrier disruption and disease progression, and DDAH II overexpression reduces lipopolysaccharide (LPS)-mediated increases in oxidative/nitrosative stress in vivo (3). DDAH II is inhibited via an Src-dependent phosphorylation (149, 238). As Src activity is stimulated by biomechanical forces (39, 76, 173), this could be a common mechanism for increasing cellular ADMA levels.
eNOS is also susceptible to a protein kinase C (PKC)-dependent phosphorylation at Thr495 (96, 186), this correlated with increases in NOS-derived superoxide and decreased NO levels (51). A similar Ang II-mediated increase in eNOS uncoupling was also recently identified in LPS-challenged EC that is mediated via a NOX-2-induced glutathionylation of eNOS (103, 292). Regulation of both eNOS gene expression and eNOS mRNA stability is also sensitive to various biomechanical stimuli, including LSS and oscillatory SS, LPS, and oxidative stress. The literature data regarding the regulation of eNOS gene expression have been extensively summarized by Searles (235).
Mitochondrial function, biogenesis, and network dynamics
Mitochondrial generation of ATP requires the activity of the electron transport complexes (ETCs) I–IV acting in concert with ATP synthase (Fig. 4A).
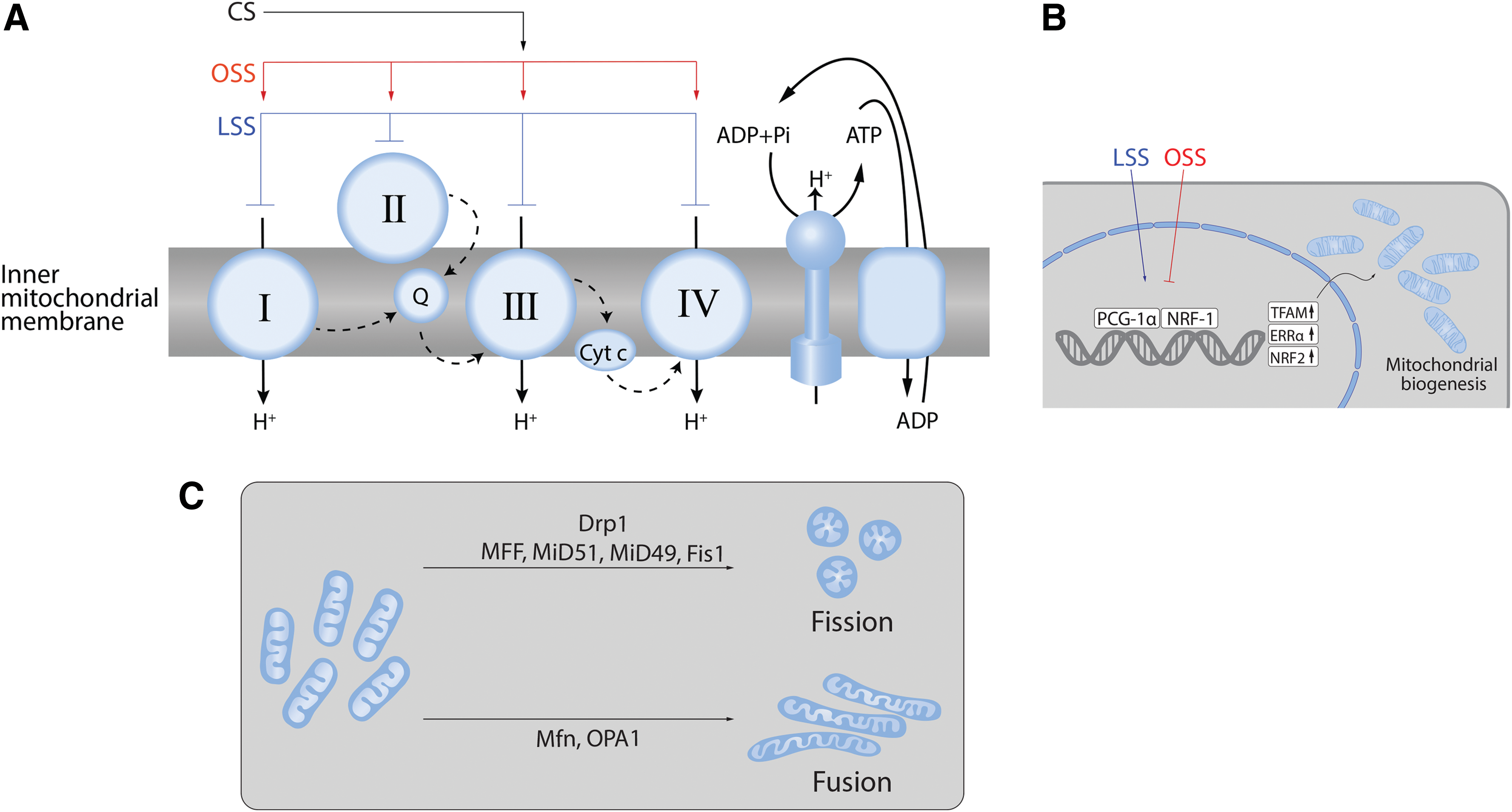
Biomechanical forces have been shown to modulate ETC activity (Fig. 4A). For example, LSS-induced NO production mediates a sustained suppression of ETC I, II/III, and IV (116). Mitochondrial ROS generation is also regulated by SS due to the eNOS-derived NO and reactive nitrogen species (RNS)-mediated inhibition of mitochondrial electron transport (116). Increased PBF and pressure also attenuate mitochondrial function via the nitration-mediated inhibition of carnitine acetyl transferase (CrAT) and the reduction in CrAT, carnitine palmitoyltransferase type 1 (CPT1), and carnitine palmitoyltransferase type 2 (CPT2) expression (239, 256). The resulting disruption of β-oxidation leads to increased mitochondrial ROS generation.
The reduction in CrAT, CPT1, and CPT2 expression appears to be caused by a loss of peroxisome proliferator-activated receptor γ (PPARγ) signaling via increased WSS and/or increased pressure (240). PPARγ antagonists also induce mitochondrial ROS in the lung (237). Oscillatory shear stress also increases mitochondrial superoxide production via an NOX-c-Jun N-terminal kinase signaling pathway (260). At present the effect of CS on mitochondrial-mediated ROS in vascular cells is limited. One study in SMCs has shown that CS (15% elongation, 24 h) stimulates NOX-4 activity via a mechanism that requires CIII activity (288). How CS modulates mitochondrial-mediated ROS in EC is unresolved.
Mitochondrial biogenesis is a complex process involving the replication of mitochondrial DNA (mtDNA) that contains 37 genes encoding 13 subunits of electron transport chain complexes I, III, IV, and V (139). Again, biomechanical forces have been shown to regulate this process (Fig. 4B).
LSS has been shown to activate the AMPK pathway in EC (83, 200). As a result, AMPK stimulates DNMT1, RBBP7, and HAT1 signaling pathways (175) and stimulates mitochondrial biogenesis via peroxisome proliferation and the activated receptor gamma coactivator-1α (PGC-1α), which, in turn, activates nuclear respiratory factor (NRF)-1, NRF-2, transcription factor A mitochondrial, and transcription factor B mitochondrial (41, 277) (Fig. 4B). LSS can also stimulate mitochondrial biogenesis through Sirtuin 1, an NAD+-dependent deacetylase (59, 143). This laminar flow-enhanced mitochondrial biogenesis may also protect ECs against oxidative stress by stimulating PGC-1α-induced ROS-detoxifying enzymes (59). Thus, mitochondrial biogenesis is also involved in controlling the redox state of the endothelium.
A common misconception is that the mitochondria are present as static individual organelles, within the cell. In reality, mitochondria are dynamic: constantly forming elongated tubes, through the process of fusion and, through fission, splitting into small less connected mitochondria (44, 52, 131, 195) (Fig. 4C). This process has been termed “mitochondrial network remodeling.”
The correct balance of fission and fusion is critical for mitochondrial homeostasis. Mitochondrial fragmentation (fission) has been linked to increased apoptotic cell death (36, 158). However, the seminal work of Archer's group has shown that in PH, the increase in fission is associated with a hyperproliferative antiapoptotic SMC phenotype (10, 54, 223, 224).
Fusion permits the mixing of the contents between mitochondria and may be a pathway that protects the mitochondria (115) (Fig. 4C). Three mitochondrial guanosine triphosphatases (GTPases) regulate mitochondrial fusion: the mitofusins (Mfn)-1 and -2 and the optic atrophy 1 protein (OPA-1). Fusion is also an underappreciated regulator of cell proliferation as the initial term for Mfn-2 was “hyperplasia suppressor gene” due to its antiproliferative effect when overexpressed (46, 52).
Fission is mediated through the GTPase activity of dynamin-related protein 1 (Drp1) (Fig. 4C). Drp1 is present in the cytosol and translocates to the mitochondria when activated. On the mitochondrion, it assembles into oligomeric structures that mechanically constrict and fragment the mitochondria (170).
Drp1 is regulated by a complex array of PTMs, including S-nitrosylation, ubiquitination, sumoylation, O-GlcNAcylation, and phosphorylation (115, 203). The best studied PTM with respect to Drp1 is its phosphorylation that occurs at Ser616 and Ser637. Phosphorylation at Ser616 activates Drp1 to promote mitochondrial fission (135, 230). Cyclin-dependent kinase 1 (Cdk1) phosphorylates Drp1 at Ser616. Phosphorylation at Ser637 has been shown to occur through PKA, Cam kinase, and Pim1 (115). Phosphorylation at Ser637 inhibits Drp1 oligomerization, sequesters Drp1 in the cytosol, and can, therefore, suppress mitochondrial fission (135, 230). Ser637 can be dephosphorylated by calcineurin that enhances Drp1 mitochondrial translocation and so stimulates fission. Rho kinase (ROCK) has also been shown to phosphorylate Drp1 (34).
ROCK exists as two isoforms 1 and 2 and is known to be a major player in the pulmonary vascular disease (PVD) through its ability to reorganize the actin cytoskeleton. One of the major upstream activators of ROCK is RhoA (Ras homologous GTP-binding protein A) (275, 290). The canonical activation of RhoA GTPase involves the activation of GPCRs and/or tyrosine kinases, resulting in the activation of guanine nucleotide exchange factors (GEFs) that enhance the exchange of GDP for GTP and translocation of GTP-RhoA to the plasma membrane. Upon translocation to the plasma membrane, GTP-RhoA is able to activate ROCK.
A new mechanism of RhoA activation has been recently identified in which post-translational (PTM) nitration events can directly stimulate RhoA nucleotide exchange, independent of GEF activation (215). Thus, there could be a link between nitrosative stress and mitochondrial fission, although this has not been explored.
The effects of biomechanical forces on mitochondrial network remodeling are also still far from resolved as the limited published data are conflicting. For example, LSS has been shown to both increase mitochondrial fission and apoptosis (234) and increase mitochondrial fusion (293). As already described, the transient receptor potential cation channels are important players in mechanotransduction pathways (148). Increased calcium uptake is associated with LSS and is essential for the initiation of mitochondrial fission (35).
Nrf2 and Krüppel-like factor 2
Biomechanical forces can also regulate the removal of ROS. For example, LSS-dependent activation of Erk5 induces the activity of Nrf2 (144). Nrf2 acts via the ARE and stimulates expression of a number of antioxidant enzymes, including NAD(P)H:quinone oxidoreductase 1, glutathione reductase, glutathione peroxidase (GPx), and catalase (138). Nrf2-dependent upregulation of these enzymes has been shown to protect cardiac fibroblasts, macrophages, and cardiomyocytes against oxidative/nitrosative stress (42, 310, 311).
HO-1, the downstream target gene of Nrf2, is also capable of suppressing atherosclerotic lesion formation by reducing the oxLDL-induced transmigration of monocytes (132) and protecting against oxidative stress and inflammation, two of the predominant mechanisms in atherosclerosis.
The activation of transcription factor, Krüppel-like factor 2 (KLF2), is also stimulated by LSS (12 dyn/cm2, 16–24 h) (144). VCAM-1 mRNA levels are decreased in ECs exposed to LSS (209), whereas KLF2 inhibits the expression of vascular cell adhesion protein 1 (VCAM-1) as well as E-selectin (78, 281). This suggests a link between LSS-mediated increase in KLF-2 and a decrease in monocyte attachment to the endothelium. In human umbilical vein endothelial cell (HUVEC), KLF2 activity is also associated with SS-induced extracellular ATP release followed by P2X4 Ca2+-channel activation (232), suggesting a functional link between calcium-mediated signaling, antioxidant and antiatherogenic gene expression, and vasorelaxation.
KLF-2 also suppresses inflammatory gene expression via the inhibition of NFκB and AP-1 (33). KLF2 also improves the nuclear localization of Nrf2, and the combined actions of these two factors are thought to constitute the majority of the LSS-induced endothelial gene expression (95).
Mechanosensitive MicroRNAs and Cell Homeostasis
MicroRNAs (miRNAs) are single-stranded noncoding small RNAs that play an important role in the regulation of gene expression via binding targeted mRNA and suppressing their translation or inducing their degradation (157) (Fig. 5A).

In ECs, miRNA profiling has revealed 21 miRs that are differentially expressed (8 up- and 13 downregulated) after 24 h pulsatile SS (283) (Fig. 5B). Multiple miRNAs have been shown to be regulated by biomechanical forces (Fig. 5B). Fluid SS, like other physiological stimuli, can both induce and suppress gene expression, including expression of miRNA genes. Thus, miRNA expression patterns depend on specific transcription factors activated by different types of SS.
Pulsatile SS activates miR-10a expression via a retinoic acid receptor RARα/KLF2-dependent mechanism, and this miRNA downregulates VCAM-1 expression. In contrast, oscillatory SS induces histone deacetylase-dependent suppression of miR-10a expression (154). RARα/RXRα agonists rescue miR-10a expression in oscillatory SS regions in vivo. Moreover, induction of miR-10a by RARα/RXRα agonists protects ApoE−/− mice against atherosclerosis, inhibiting VCAM-1 expression and inflammatory cell infiltration (156). In addition, miR-23b, induced by pulsatile SS via KLF2-dependent transcription, possesses antiproliferative properties, repressing cyclin H and cell cycle progression. Oscillatory SS has no effect on miR-23b, however, and, therefore, does not induce cell cycle arrest (14). In HUVECs, induction of miR-19a by laminar flow leads to cyclin D1 downregulation and cell cycle arrest (214), whereas under the same conditions, miR-101 has an antiproliferative effect, suppressing mTOR expression (53).
Biomechanical forces affect the expression of several miRNAs that are either directly or indirectly involved in cellular redox balance [reviewed in Marin et al. (174)]. Among the mechanosensitive miRNAs, directly targeting ROS-regulating enzymes are miR-221/222 (252) and miR-92a (90, 294), which inhibit eNOS and miR-17* [inhibits superoxide dismutase (SOD) 2] (296) that are all downregulated by LSS. Conversely, miR-21 (252), miR-25 (100), and miR-23b (283) are upregulated by LSS and have been shown to inhibit NOX-4. miR-30b is also upregulated by LSS and inhibits catalase expression (117). Oscillatory SS upregulates miR-221/222 (252), miR-92a (90, 294), and miR-663, all of which inhibit eNOS expression. Oscillatory SS also upregulates miR-21 (304), which inhibits SOD3 expression.
Mechanosensitive miRNAs can also indirectly regulate oxidative stress by affecting ICAM-1/VCAM-1 expression and, therefore, adhesion and activation of neutrophils on the endothelium and subsequent ROS generation. LSS upregulates miR-10a (154) and miR-126 (118, 119), both of which inhibit VCAM-1 expression, whereas oscillatory SS downregulates miR-10a (154), which increases VCAM-1 expression. Several other miRs upregulated by oscillatory SS [miR-21 (309), miR-34a (89), and miR-663 (198)] have also been described as ICAM-1/VCAM-1-inhibitory miRs, suggesting a complex interplay in the regulation of these adhesion receptors by mechanical forces.
miR-486-5p, which is downregulated by oscillatory SS, may also be another indirect regulator of cellular redox balance as it can inhibit the expression of the phosphatase, PTEN, leading to increased Akt1 activity (125, 279). As Akt1 can phosphorylate eNOS, this could regulate NO levels.
Sirtuin-1 expression, a positive regulator of mitochondrial biogenesis, is inhibited by miR-92a (90, 164) and miR-217 (184), both of which are upregulated by oscillatory SS. LSS also upregulates miR-20a that inhibits VEGF expression (283).
Expression of PPARγ, a nuclear hormone receptor, is negatively regulated by miR-21 under oscillatory SS conditions (309). Detailed studies of PPARγ functions have identified its role in maintaining endothelial function and its loss has been shown to enhance the development of atherosclerosis, hypertension, and PH (6, 17, 126, 192, 237, 240). Experiments with pulmonary artery endothelial cells (PAECs) obtained from PH patients and mouse model of endothelial PPARγ loss-of-function showed that high levels of ET-1 correlated with the downregulation of PPARγ and miR-98 (140). Another miRNA family, miR-130/301, also negatively regulates the expression of PPARγ under conditions of excessive blood flow and pressure (15). miR-130/301-driven downregulation of PPARγ induces two downstream pathways that results in the decreased expression of miR-424/503 (in PAEC) and miR-204 (in PASMC); both pathways stimulate cell proliferation and are critical for PH promotion (16).
The exposure of ECs to CS (15% elongation, 24 h) also induces a dramatic change in the miRNA expression profile. Intriguingly, several miRs that inhibit NOX-4 expression are regulated with miR-4428, miR-1273 being downregulated and miR-17-5p, miR-93-5p, miR-23a-3p being upregulated. This divergent regulation is suggestive of a complex regulatory mechanism of NOX-4 expression via miRNAs. CS also downregulates miR-4763-3p that is a negative regulator of eNOS expression and miR-551b-5p that inhibits ICAM-1 expression (307). Taken together, these data demonstrate complex multileveled regulation of pathological pathways in vasculature by miRNAs that are responsive to biomechanical forces to either enhance vasoprotective effects or support excessive ROS generation.
Vascular Diseases/Pathologies Resulting from Altered Biomechanical Forces
Pulmonary hypertension
PH is biomechanically characterized as an increase in the resistive and reactive components of pulmonary vascular impedance (201, 285). In severe forms of PH, a progressive increase in the pulmonary vascular resistance leads to right heart pressure overload and right heart failure (102). Thus, changes in biomechanical forces are likely important in PH development. Increased levels of oxidative stress markers have been detected in PH patients (303) underpinned by multiple molecular, genetic, and epigenetic abnormalities, which cause endothelial dysfunction, pathological vascular remodeling, and mitochondrial metabolic abnormalities (4). WSS-dependent endothelial alterations within the complex pathobiology of PH play a very important role in blood clotting, inflammation, vascular tone, metabolism, angiogenesis, and repair. WSS is required for the development and maintenance of severe occlusive vascular lesions after Sugen-induced pulmonary vascular injury (280).
As was shown in healthy volunteers, a relationship between vascular WSS and flow-dependent vascular dilation can be directly accessed by phase contrast magnetic resonance imaging (MRI) (246), and in vivo data collected using MRI demonstrated site-specific WSS magnitudes in arterial system (206). Furthermore, using MRI, an occurrence of disturbed blood flow in pulmonary artery was directly demonstrated in PH patients. Vortex blood flow patterns and early systolic retrograde flow in main pulmonary artery were detected in all PH patients studied and were absent in healthy individuals; PA flow velocities and WSS were lower than those in control group (13, 202). Vortical blood flow duration in main pulmonary artery correlates with PH progression (220).
Disturbed blood flow is considered to be a critical trigger of PH development, since it stimulates numerous signaling pathways leading to oxidative stress, endothelial dysfunction, and expression of atherogenic factors. Experimental models of PH demonstrate dysregulation of oxidative signaling, with elevated ROS/RNS, reduced SOD, GPx, and catalase (4).
In the chronic hypoxia model of PH, pulmonary vascular remodeling is primarily mediated by NOX-2- and NOX-4-dependent ROS production (14, 134, 159). In PASMC, transforming growth factor (TGF)-β1 treatment stimulates increased NOX-4 levels, resulting in increased ROS that drives cellular proliferation, suggesting that NOX-4 mediates TGF-β1-dependent pulmonary vascular remodeling (4, 180, 251). NOX-4 also mediates the effects and hypoxia-induced factor-1α (HIF-1α) (28, 97, 227), which is critical to the pathogenesis of PAH.
Mitochondrial metabolic abnormalities are emerging as key players in the pathobiology of PAH (224, 239). Activation of HIF-1α causes the switch to a glycolytic phenotype, thereby suppressing oxidative phosphorylation, with multiple downstream consequences including mitochondrial depolarization (7). The underlying mechanism has been studied in animal models and is usually considered to be multifactorial through changes in eNOS production and uncoupling (256, 306), alteration in
Evidence of mitochondrial fragmentation has also been identified due to a decrease in the expression of MFN2, and MFN2 overexpression attenuates the severity of PH (225). Several miRNAs are dysregulated in PH patients (19), including miR-204, which in healthy pulmonary artery SMCs (PASMCs) inhibits the STAT3/HIF-1α pathway (68).
Recent studies demonstrated that endothelial-to-mesenchymal transition (EndMT) could contribute to PH development and complexity. In PH models, various insults (such as hemodynamic stress and hypoxia) applied to the endothelium induce a loss of cell–cell and cell–matrix contacts, decrease of endothelial marker expression (VE-cadherin, PECAM-1, and von Willebrand factor), and increase of SMC- and fibroblast-specific proteins (α-SM-actin, fibronectin, SM-myosin, and calponin) (8, 98). TGF-β1-activated signaling was shown to contribute to EndMT (9, 98). Hypoxia-induced EndMT occurs via HIF-1α-mediated transcription followed by Twist1 activation (302), which, in turn, may lead to upregulation of TGF-β receptor 2 and Smad2 phosphorylation (172) linking hypoxia and TGF-β signaling.
Recently, HIF-2α-mediated transcription network was also demonstrated as critical for EndMT and PH development: siRNA-directed depletion of HIF-2α downregulated expression of Snai1/2 and EndMT in lung EC from idiopathic PAH patients (264). Also, HIF-2α-mediated upregulation of endothelial arginase II may contribute to an impairment of NO signaling in hypoxia-challenged EC and development of PH, since arginase II and eNOS utilize the same substrate,
The diverse and complex mechanisms underlying the pathogenesis of PH offer the potential for new therapies. Specific therapies that have been developed for PH patients include the endothelin receptor antagonists, phosphodiesterase 5 (PDE5) inhibitors, prostanoids, sGC stimulators, and calcium channel blockers. New therapeutic targets have arisen since the emergence of the recent data that mitochondrial abnormalities and the presence of a hypoxic state are key to PH pathogenesis
Targeting various pathways (e.g., STAT3, mTORC, Akt1, PI3K, FoxO, and NFκB) in addition to dysregulated metabolic and mitochondrial signaling networks may help to reverse disease. Drugs aimed at blocking apoptosis might prevent the development of vascular remodeling in PAH, whereas promoting apoptosis in end-stage PAH might improve it (259). The treatment of PH could also benefit from advancements in precision medicine, by applying treatments that already exist in other areas. Combining two or more therapeutic approaches may be a strategy for the treatment of PH (101).
Congenital heart disease
In the United States, congenital heart disease (CHD) occurs in at least 8 of every 1000 live births and accounts for >24% of birth defect-related infant deaths (108). All congenital heart defects, in which a large intra- or extracardiac communication allows unrestricted pressure and volume overload of the pulmonary circulation, can lead to the development of PH (105, 221). The resulting shunt increases the pressure in the pulmonary arteries, leading to increased SS, circumferential wall stretching, and endothelial dysfunction. However, the classification of the PVD associated with CHD belies the complexity and varying physiology of predisposing cardiac lesions—from the classic example of unrestrictive ventricular septal defect to complex single ventricle lesions (Table 1).
Risk of Pulmonary Vascular Disease in Differing Lesions Associated with Congenital Heart Disease and Increased Pulmonary Blood Flow
Defects in bold represent high flow/direct high-pressure lesions; defects in italics represent high flow/variable direct high pressure.
ASD, atrial septal defect; CHD, congenital heart disease; PVD, pulmonary vascular disease.
The natural history of PVD associated with systemic-to-pulmonary shunt reveals the differential, or perhaps incremental, effects of increased PBF and increased pulmonary arterial pressure. In patients with increased blood flow alone—pretricuspid valve lesions such as atrial septal defects—the development of PVD is uncommon and presents late, among 5%–15% of patients by the fourth decade of life (249). In stark contrast, in patients with increased blood flow and a direct pressure stimulus from the systemic ventricle—post-tricuspid lesions—the development of PVD is common, and develops early in life. Thus, the progression of PVD in these lesions reflects the severity of the hemodynamic insults to the pulmonary vasculature with lesions that exert only increased shear forces, from increases in blood flow alone, having a slower progression than those that have both flow and direct pressure stimuli (1, 120, 150).
Altered expression of vasoactive mediators, such as ET-1, PGI2, and NO, in CHD results in vasoconstriction, whereas aberrant expression of VEGF and fibroblast growth factor promotes vascular remodeling (1). These changes contribute to a progressive increase in pressures in the right ventricle (1). Compared with patients with other PH etiologies, the increases in pulmonary pressure seen in patients with PH–CHD occur early (during infancy rather than during adulthood), and this seems to provide PH–CHD patients with a prognostic advantage. More than 50% of patients with large unrestrictive ventricular septal defect will develop PH and cyanosis due to a reversal of left-to-right shunting, known as Eisenmenger syndrome (142).
Treatment of PH–CHD has evolved in recent years with options for either late repair in some patients (surgical) or PH disease-targeting therapy (87, 121). The use of PH-specific therapies in CHD significantly lowers the rate of cumulative mortality when compared with no therapy. Clinical studies evaluating oxidative stress and antioxidant status in children with CHD have revealed significant elevation of the oxidative stress biomarkers, malondialdehyde (MDA) and protein carbonyl, in patient plasma samples, as well as proinflammatory cytokines, such as IL-6 and TNFα (212). Superoxide and H2O2 levels have also been shown to increase (24). A comparison of total oxidant system (TOS) and total antioxidant system (TAS) in plasma collected from cyanotic acyanotic CHD patients and age-matched control individuals revealed a significant TOS increase and TAS decrease in cyanotic CHD patients (88).
Acute lung injury
ALI and its more severe form, acute respiratory distress syndrome (ARDS), are pathological states of lung dysfunction of various etiology such as Gram-positive or Gram-negative respiratory infection, sepsis, trauma, acid aspiration, or toxic gas inhalation (219, 270). Although ALI/ARDS is not necessarily a pathology induced by biomechanical forces, it can be associated with VILI in patients. Therefore, studies of so-called two-hit models (bacterial toxin challenge or any other ALI-related stimuli plus in vitro CS or in vivo lung mechanical ventilation) are considered to be more clinically relevant (21 –23) (Fig. 6). Published data demonstrate that the toxin pretreatment dramatically potentiates effects of excessive CS (18% elongation) inducing cell signaling pathways that lead to the barrier-disruptive cytoskeleton remodeling, cell–cell contact loss, expression of proinflammatory cytokines, and adhesive molecules (21 –23, 291).

The most studied models of ALI are cultured pulmonary cell monolayers or animals challenged with either Gram-negative (LPS) or Gram-positive (pneumolysin, PLY; listeriolysin, LLO) bacterial toxins. In these models, the pivotal role of oxidative/nitrosative stress in the endothelial dysfunction is well documented. On molecular level, ALI is characterized by an excessive ROS generation and mitochondrial dysfunction. The major sources of ROS generation are NOX isoforms, which are activated by LPS in neutrophils (NOX-1) or EC (NOX-2) (171, 233). In ECs and SMCs, LPS induces oxidative stress and AP-1-/NFκB-mediated proinflammatory responses via NOX-4 activation via TLR4 (207, 208, 210). Uncoupled eNOS is another critical source of ROS in ALI/ARDS models (112, 238) that leads to peroxynitrite generation followed by protein tyrosine nitration and that plays an important role in ALI/ARDS pathogenesis (e.g., nitrated RhoA activation) (112, 149, 215).
Gram-positive bacterial toxins are pore-forming proteins that may rapidly induce oxidative stress via perturbance of [Ca2+]i homeostasis and mitochondrial dysfunction. PLY increases mito-ROS and decreases mitochondrial O2 consumption (160). In vitro, PLY or LLO shows dose-dependent negative effects on the endothelial barrier and, due to robust [Ca2+]i elevation, the stimulation of PKC and CaMKII activities (71). The activation of PKCα has been shown to trigger pulmonary endothelial dysfunction (168). PKC-dependent phosphorylation of eNOS at T495 leads to eNOS uncoupling (51, 255).
Actin stress fiber formation and contraction in PLY-challenged EC are the result of RhoA activation, Rac1 inhibition, myosin light chain (MLC), and filamin A phosphorylation causing the barrier disruption (70). Oxidative stress-mediated activation of protein tyrosine kinases followed by VE-cadherin tyrosine phosphorylation can also impair adherens junctions (167). In animal models, Gram-positive toxins induce endothelial dysfunction causing vascular leakage and pulmonary edema (71, 168).
Ventilator-induced lung injury
Mechanical ventilation of lungs is one of few clinical approaches effective for ARDS patients. However, excessive mechanical stress induced by ventilation may also cause lung tissue damage. This mechanical force, which is difficult to control in individual patients, may result in abnormal cyclic strain of the lung tissue (11). Such prolonged abnormal strain affects the lung vasculature inducing endothelial dysfunction and an inflammatory response [reviewed in Wang et al. (284)].
Experimental data obtained in EC subjected to excessive CS in specially designed devices or in animals subjected to mechanical ventilation demonstrate dramatic changes in cell signaling and cell metabolism affecting virtually all levels of EC and SMC homeostasis, including cytoskeletal structures and cell–cell contacts (adherens and tight junctions), protein modifications, gene expression, and cytokine/chemokine secretion.
Numerous studies have implicated ROS-modulating enzymes (NOX, NOS, and XO) as well as mitochondrial-derived ROS in VILI pathology (176, 177, 218, 276). Uncoupling of eNOS due to a functional BH4 shortage also exists in VILI (276). Exposing EC to various levels of CS has highlighted the critical role of the RhoA/ROCK signaling pathway in the development of endothelial dysfunction (21). Activation of ROCK appears to be due to a specific Rho-GEF, GEF-H1 (21, 47). GEF-H1 activation has been linked to microtubule disassembly (145) that occurs under excessive mechanical force (106). Importantly, GEF-H1 inhibition results in a decrease of proinflammatory factor levels in excessive CS-challenged EC. Therefore, a link between RhoA/ROCK pathway and NFκB-dependent proinflammatory response has been demonstrated (145).
Protective Approaches
Since the generation of excessive ROS/RNS and the resulting oxidative/nitrosative stress in the lung play a central role in the development and progression of a number of lung pathologies, antioxidant therapies have been tested as a general approach to protect the lung against the effects produced by abnormal biomechanical forces. However, such a general antioxidant approach has demonstrated either very modest or no therapeutic effect in humans due to inability to distinguish between harmful effects of excessive ROS and physiological ROS-mediated processes (213, 273). These failures have led to new ideas regarding antioxidant therapies that are designed to specifically target individual ROS/RNS-generating enzyme(s) or specific intracellular sites of ROS generation (e.g., dysfunctional mitochondria), which have been defined as critical for a particular disease.
Studies have focused on the specific inhibition of distinct NOX isoforms. Small-molecule inhibitors of NOX were tested in atherosclerosis mouse model (streptozotocin-treated ApoE−/− mice) and NOX-1/NOX-4 inhibitors (such as GKT137831) application showed decreased lesions and macrophage infiltration [reviewed in Altenhöfer et al. (5)]. NOX-inhibitory peptides have had success in inhibiting p22phox function, p47phox phosphorylation, and translocation and superoxide generation [reviewed in Cifuentes-Pagano et al. (64)].
Another protective approach being tested is the use of modulators of the downstream effectors regulated by excessive biomechanical forces and oxidative stress. For example, a number of publications have shown that ROCK inhibitors can be efficient in animal models of PH (58, 305). A more precise approach by preventing RhoA activation by Y34 tyrosine nitration using a specific RhoA-shielding peptide significantly protects the pulmonary vasculature against LPS-mediated damage in vivo (215). This type of precise targeting of ROS/RNS-dependent activation or inhibition of key regulators based on a fundamental understanding of a disease pathology could lead to more targeted and effective antioxidant therapies.
Another interesting therapeutic direction is the development of disintegrins, peptides originated from snake venoms that specifically interact and inhibit particular integrins [reviewed in Daavid et al. (73)]. This approach is aimed at preventing thrombocyte aggregation and leukocyte adhesion and activation and, therefore, has the potential to exert antioxidative and anti-inflammatory effects in the lung.
Recent preclinical and/or clinical studies in other complex pathologies (such as cancer) have demonstrated an increased therapeutic efficiency using drug combinations. Such combined approaches may be successful in the therapy of pulmonary diseases. In this regard,
Concluding Remarks
The exposure of the pulmonary vasculature to biomechanical forces affects the lung in a number of important ways, allowing cells in the vasculature to respond to a changing external environment via alterations in the production/secretion of vasoactive factors, gene expression changes, ROS generation, and mitochondrial bioenergetics/biogenesis/network dynamics. Lung injury and disease can alter the normal patterns of these forces, resulting in pathological signaling events that are intimately involved in disease progression. However, despite substantial investigations, there are still many unresolved issues surrounding how vascular cells respond to mechanical stress. This limitation is based on both the different types of forces to which the lung is exposed and the complexity of the lung itself.
Indeed, most of our data come from single cell types exposed to a single mechanical force. Thus, more sophisticated experimental systems that will allow the analysis of multiple cell types exposed to both SS and pressure/stretch will be necessary to more accurately determine how the pulmonary vasculature responds to a changing mechanical environment and how this is subverted in pathological conditions to drive the disease progression.
Footnotes
Acknowledgment
This research was supported, in part, by HL60190 (S.M.B.), HL137282 (S.M.B.), HL134610 (S.M.B.), HL142212 (S.M.B. and E.A.Z.), HL136603 (A.A.D.), HD072455 (E.M.), HL115014 (J.X.-J.Y.), and HL061284 (J.R.F.), all from the National Institutes of Health.