
Abstract
Aims:
A subpopulation of cancer cells, termed cancer stem cells (CSCs), has stemness properties, such as self-renewal and differentiation, which drive cancer recurrence and tumor resistance. CSCs possess enhanced protection capabilities to maintain reduced intracellular levels of reactive oxygen species (ROS) compared with nonstem-like cancer cells. This study investigated whether reductive stress could regulate self-renewal activity in breast CSCs.
Results:
We found that manifestation of stemness in breast cancer stem-like cells was associated with an elevated production of reduced glutathione (GSH) maintained by upregulation of glutamate cysteine ligase catalytic subunit (GCLC) and consequently, lowered ROS levels. This was accompanied by upregulation of phospho-AMP-activated protein kinase, FoxO3a, and Bmi-1. Notably, expression of nuclear factor erythroid-derived 2-like 2 (Nrf2) protein was substantially increased in cells undergoing sphere formation. We noticed that expression of Bmi-1 was inhibited after introduction of Nrf2 short interfering RNA into MCF-7 mammosphere cells. Silencing of Nrf2 expression suppressed the xenograft growth of subcutaneously or orthotopically injected human breast cancer cells.
Innovation:
Association between Nrf2 and self-renewal signaling in CSCs has been reported, but the underlying molecular mechanism remains largely unresolved. This study demonstrates the Nrf2-mediated signaling pathway in maintenance of reductive stress in breast CSCs.
Conclusion:
Nrf2 overactivation in breast CSCs upregulates GCLC expression and consequently enhances GSH biosynthesis with concurrent reduction in intracellular ROS accumulation, thereby provoking the reductive stress. The consequent upregulation of nuclear FoxO3a and its binding to the promoter of the gene encoding Bmi-1 account for the self-renewal activity of breast cancer stem-like cells and their growth in a xenograft mouse model.
Innovation
Cancer stem cells (CSCs) possess enhanced protection capabilities against oxidative stress induced by reactive oxygen species (ROS) compared with nonstem-like cancer cells. In this study, we investigated whether reductive stress could regulate self-renewal activity in breast CSCs. The manifestation of stemness in CSC-like breast cancer cells was associated with an elevated ratio of reduced/oxidized glutathione and consequently, lowered ROS levels. Therefore, activation of nuclear factor erythroid-derived 2-like 2-mediated FoxO3a-Bmi1 signaling accounts for self-renewal activity, and regulation of redox status may provide therapeutic strategies in breast CSCs.
Introduction
C
It is noteworthy that CSCs maintain relatively low levels of reactive oxygen species (ROS) (32). Diehn et al. have reported that lower ROS levels in mammary CSCs might confer their resistance to ionizing radiation (9). It is hence speculated that low ROS content in CSCs is accompanied by disturbances in cell cycle regulation, which may account for their dormancy (42). Chen et al. found that treatment of hematopoietic stem cells with the antioxidant N-acetylcysteine resulted in the acquisition of stemness characteristics (5).
In general, expression of cellular antioxidant defense proteins is mainly regulated by the redox-sensitive transcription factor, nuclear factor erythroid-derived 2-like 2 (Nrf2) (29). Reduced glutathione (GSH) is a principal antioxidant molecule responsible for the elimination of ROS. GSH production is catalyzed by the glutamate cysteine ligase complex modifier subunit and glutamate cysteine ligase catalytic subunit (GCLC), under the control of Nrf2 (44). Recently, association between Nrf2 and self-renewal signaling in CSCs has been reported (38, 46, 49), but the underlying molecular mechanism remains largely unresolved.
Forkhead box O (FoxO) family transcription factors function as cellular redox sensors. Their transcriptional activities are affected by the alteration of intracellular and extracellular redox status (21). FoxOs play a critical role in enhancing self-renewal capability of stem cells (45). Bmi-1 is a key molecule involved in the maintenance of stem cells as well as cancer development (41). Here, we report that breast CSCs maintain low levels of intracellular ROS by upregulating Nrf2-induced expression of GCLC, thereby maintaining self-renewal activity via the FoxO3a-Bmi1 axis.
Results
The intracellular redox status is critical for the manifestation of stemness in breast cancer stem-like cells
It has been reported that CSCs, in general, exhibit lower ROS levels than nonstem cells (9, 39), which is maintained by upregulation of antioxidant gene expression. The correlation between recurrent breast cancer and antioxidant gene expression was assessed by gene set enrichment analysis (GSEA) of clinical data deposited from GSE2034. Through the GSEA analysis, we found that the ROS-related gene set was significantly enriched in breast tumor from nonrelapsed patients, whereas biosynthetic and metabolic processes of ROS were downregulated in the relapsed breast cancer (Fig. 1A). When MCF-7 cells were grown as mammospheres for 5 days, the cellular ROS level was found to be decreased compared with that in parent cells as measured by fluorescence-activated cell sorting (FACS) analysis (Fig. 1B and Supplementary Fig. S1A).

We isolated CD24low/CD44high breast cancer stem-like cells from MCF-7 mammospheres by use of the FACSAria™ cell sorter, and the intracellular accumulation of ROS was measured by 2′,7′-dichlorofluorescein diacetate (DCF-DA) staining. We observed that CD24low/CD44high breast cancer stem-like cells contain low levels of ROS when compared with parent MCF-7 cells (Fig. 1C and Supplementary Fig. S1B). In general, superoxides in the cytoplasm and mitochondria are detected by using dihydroethidium (DHE) and MitoSox dye, respectively (33). We observed that mammospheres derived from MCF-7 cells exhibited a decreased ROS level detected by DHE staining, whereas MitoSox staining did not show any significant changes (Fig. 1D).
GSH is the most important hydrophilic antioxidant that protects cells from exogenous and endogenous toxicants, including ROS (4). The ratio of GSH to an oxidized form (GSSG) has been considered as an indicator of cellular redox status. Thus, the reduced ratio of GSH/GSSG is an indication of oxidative stress, whereas an increased ratio is indicative of reductive stress (4). We noted that CD24low/CD44high breast cancer stem-like cells exhibit the higher GSH/GSSG ratio as well as total intracellular GSH than CD24high/CD44low cells sorted from MCF-7 mammospheres (Fig. 1E). In addition, there was a concurrent increase of CD44, GCLC, and Bmi-1 protein expression in CD24low/CD44high breast cancer stem-like cells (Fig. 1F and Supplementary Fig. S1C).
To further verify that the redox status in mammosphere formation is essential for regulation of Bmi-1-mediated self-renewal signaling, we performed the immunoblot analysis using anti-Bmi1 antibody after the treatment of MCF-7 mammospheres with buthionine sulfoximine (BSO), an inhibitor of GCLC, and also with hydrogen peroxide (H2O2). As shown in Figure 1G and H, treatment of mammosphere cells with these compounds provoking prooxidant status resulted in the decreased Bmi-1 protein expression (Supplementary Fig. S1D, E), suggesting that the ROS production might render these cells incompetent for self-renewal activity. Thus, we intended to determine whether GCLC could regulate the self-renewal activity in the process of generating the third-generation mammospheres. Depletion of GSH by BSO treatment led to a diminution of the number and the size of mammospheres (Fig. 1I). In contrast, treatment of MCF-7 mammospheres with glutathione-reduced ethyl ester (GSH-MEE), a membrane permeable derivative of GSH, resulted in the increased size and the number of mammospheres (Fig. 1J). Notably, the GSH-mediated increase in mammospheres was abrogated by BSO treatment (Fig. 1K).
Furthermore, we examined the GCLC mRNA expression levels in different CD24/CD44 phenotypes using a published database (GSE2034). As shown in Supplementary Figure 2A, the CD24low/CD44high group expressed higher amount of GCLC than did the CD24high/CD44low group in breast cancer patients. Further, GCLC expression correlated with poor clinical outcome. In the GCLC-amplified group, the probability of relapse-free survival was significantly higher than that in the low GCLC expression group (Supplementary Fig. S2B). To determine whether CD44 could be regulating the expression of GCLC transcripts in the patient tumor, we evaluated trends in transcript abundance of each gene in the breast cancer patients with relapse. Particularly, the correlation between CD44 and GCLC significantly increased in relapse patient samples of GSE1456 (Supplementary Fig. S2C).
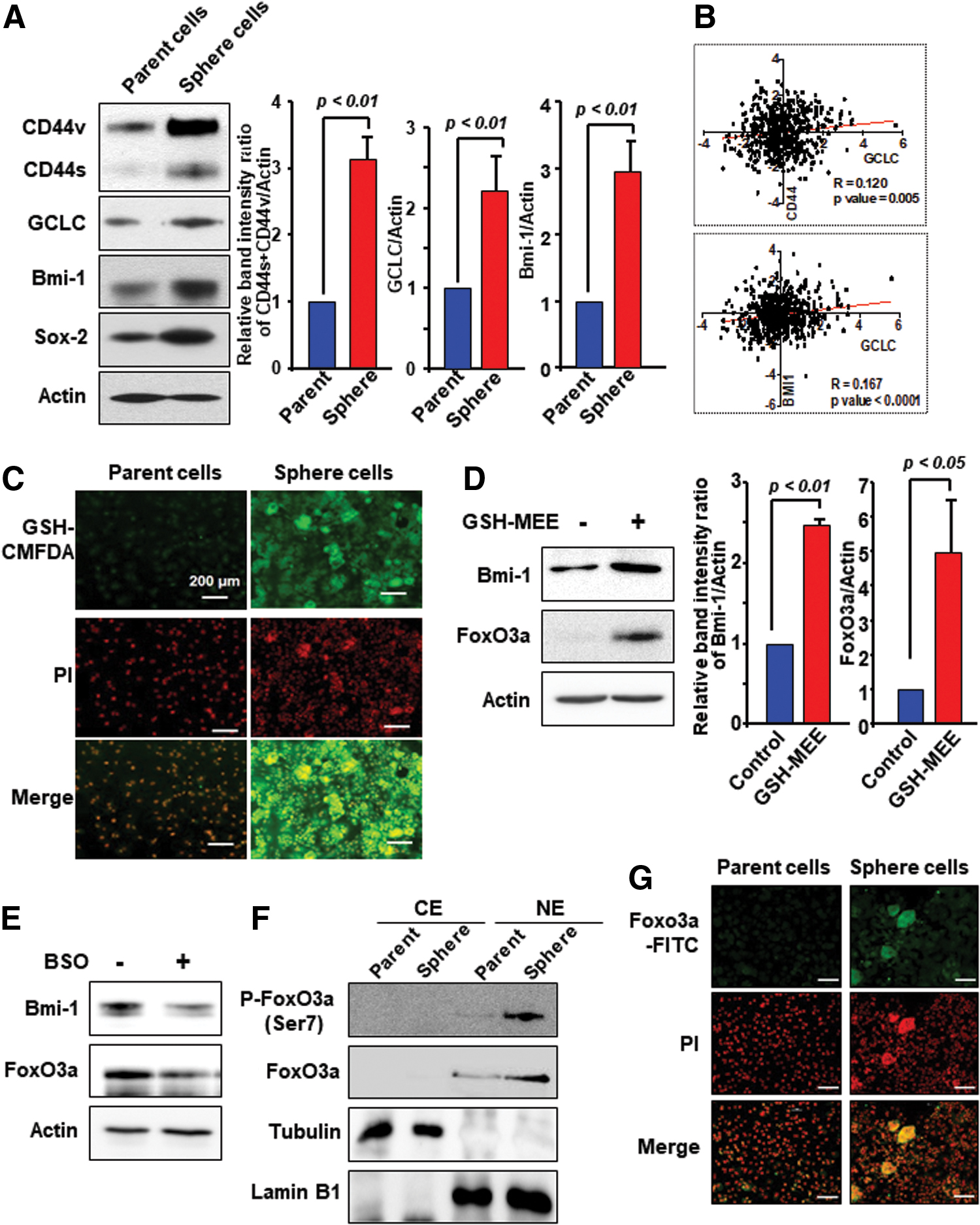
GCLC-mediated GSH production upregulates expression of FoxO3a and Bmi-1 in mammosphere cells
Recently, Hu et al. have reported that heterogeneity of CSCs expressing different CD44 isoforms in breast cancer is associated with a CSC subpopulation with enhanced lung metastasis capacity (15). In line with this notion, there is an increase in the expression of CD44s and CD44v types, GCLC and Bmi-1 in third-generation mammospheres as compared with parent cells (Fig. 2A and Supplementary Fig. S3A). Sox-2 was used as a sphere cell marker. To determine the possible involvement of GCLC in regulating the expression of CD44 and Bmi-1 transcripts in patient tumor samples, we evaluated trends in transcript abundance of each gene in the breast cancer samples of GSE1456. As shown in Figure 2B, GCLC expression was well correlated with the levels of CD44 and Bmi-1 gene expression.
It has been demonstrated that overexpression of antiapoptotic proteins, such as Bcl-2, causes redistribution of GSH to the nucleus, thereby altering the nuclear redox status (24). Notably, nuclear GSH plays a role in regulating cellular proliferation, and influences both telomerase activity and histone function (12). To investigate the subcellular distribution of GSH in breast cancer stem-like cells, mammospheres were incubated with the 5-chloromethylfluorescein diacetate fluorescent probe for 30 min. Next, the images were taken by light microscopy to capture red fluorescence of nuclei and green fluorescence for GSH. As illustrated in Figure 2C, GSH was detected in the nucleus as well as cytoplasm of mammosphere cells.
It has been reported that FoxO activity is regulated by intracellular GSH and ROS (13, 21). To determine whether intracellular GSH can induce expression of FoxO3a, thereby regulating Bmi-1, MCF-7 cells were treated with GSH-MEE for 48 h. As shown in Figure 2D, exogenous supply of GSH resulted in upregulation of FoxO3a and Bmi-1 (Supplementary Fig. S3B). In contrast, expression of Bmi-1 and FoxO3a was decreased in BSO-treated MCF-7 mammosphere cells (Fig. 2E and Supplementary Fig. S3C). Moreover, mammosphere formation was accompanied by FoxO3a phosphorylation at the Ser 7 residue, and subsequently nuclear accumulation in MCF-7 cells (Fig. 2F, G and Supplementary Fig. S3D).
GSH induces phosphorylation of AMP-activated protein kinase and FoxO3a for self-renewal activity of breast cancer stem-like cells
It has been reported that the crosstalk between AMP-activated protein kinase (AMPK) and FoxO under metabolic stress is essential for redox maintenance (48). We found that there was a robust increase in phospho-AMPK (Thr172) and P-FoxO3a (Ser7) in mammospheres compared with those in parent MCF-7 cells (Fig. 3A, B and Supplementary Fig. S4A). Ho et al. have reported that the Ser7 residue of FoxO3a is the only phosphorylation site associated with its nuclear enrichment (14). We found that levels of Bmi-1 protein expression and FoxO3a phosphorylation were decreased in AMPK-knockdown MCF-7 mammosphere cells (Fig. 3C and Supplementary Fig. S4B). Further, phosphorylation of AMPK and FoxO3a was markedly enhanced in MCF-7 mammosphere cells treated with GSH-MEE (Fig. 3D and Supplementary Fig. S4C). Conversely, treatment of GSH-enriched mammospheres with the prooxidant H2O2 abrogated the AMPK activation (Fig. 3E and Supplementary Fig. S5A). Moreover, treatment of mammosphere cells with H2O2 resulted in decreased expression of both total and phosphorylated FoxO3a proteins (Fig. 3F and Supplementary Fig. S5B). These findings suggest that a redox shift toward GSH accumulation can trigger phosphorylation of AMPK and FoxO3a, which accounts for self-renewal activity of breast cancer stem-like cells.

FoxO3a induces Bmi-1 transcription by directly binding to its proximal promoter
After revealing the FoxO3a upregulation by GSH, we determined whether FoxO3a could regulate the expression of Bmi-1. We noticed that expression levels of Bmi-1 protein and its mRNA transcript were decreased in FoxO3a knockdown MCF-7 mammosphere cells (Fig. 4A, B and Supplementary Fig. S6A). To understand how the Bmi-1 gene expression is regulated by FoxO3a, we analyzed the Bmi-1 promoter region and identified a single putative FoxO3a binding site in the upstream of the transcription start site. The information on the promoter region (promoter ID: BMI1_2) was obtained from a Eukaryotic promoter database. FoxO3a binding to the Bmi-1 promoter was verified by a chromatin immunoprecipitation (ChIP) assay (Fig. 4C).
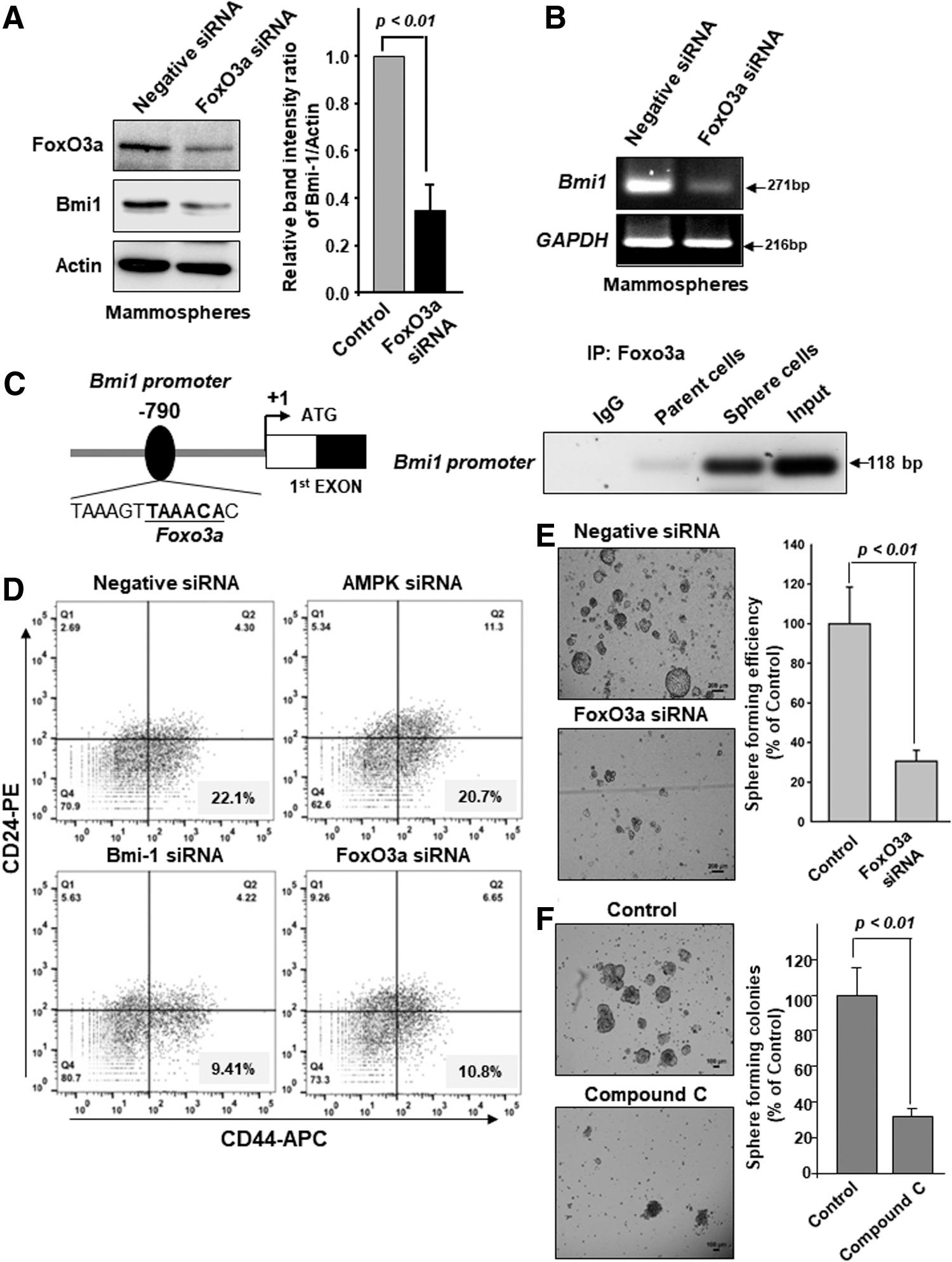
In addition, the proportion of the CD44high/CD24low cell population in MCF-7 mammospheres was decreased by knockdown of AMPK, Bmi-1, and FoxO3a using target-specific short interfering RNA (siRNA) (Fig. 4D). Use of unstained and singly dye-stained cells as compensation controls is common, so we set the standard for flow cytometry gating (Supplementary Fig. S6B). We also noticed that knockdown of FoxO3a expression in MCF-7 cells abrogated the formation of secondary mammospheres (Fig. 4E). Treatment with compound C, a well-known inhibitor of AMPK, led to a diminution of the number and the size of mammospheres (Fig. 4F). Furthermore, we performed the correlation study with the breast cancer cohort of early breast cancer patients (GSE1456, n = 159). The moderate correlation between Foxo3a and Bmi-1 was found (Supplementary Fig. S6C). Taken together, these findings indicate that FoxO3a transcriptionally regulates Bmi-1 to promote self-renewal activity of breast cancer stem-like cells.
Nrf2 activation contributes to self-renewal activity of breast mammosphere cells
Based on the upregulated expression of GCLC, we examined whether the Nrf2 signaling pathway could be activated in breast cancer mammospheres. We observed that expression of Nrf2 protein was substantially increased in cells growing under sphere-forming conditions (Fig. 5A and Supplementary Fig. S7A). In addition, the nuclear accumulation of Nrf2 was evident in the mammospheres derived from MCF-7 cells (Fig. 5B and Supplementary Fig. S7B), which was further verified by immunocytochemical analysis (Fig. 5C). Further, the sphere formation was abrogated by Nrf2 silencing (Fig. 5C). As there was a concomitant increase in the expression of CD44 and Nrf2 in third-generation mammospheres, we explored the involvement of CD44 in self-renewal activity via the Nrf2 signaling.
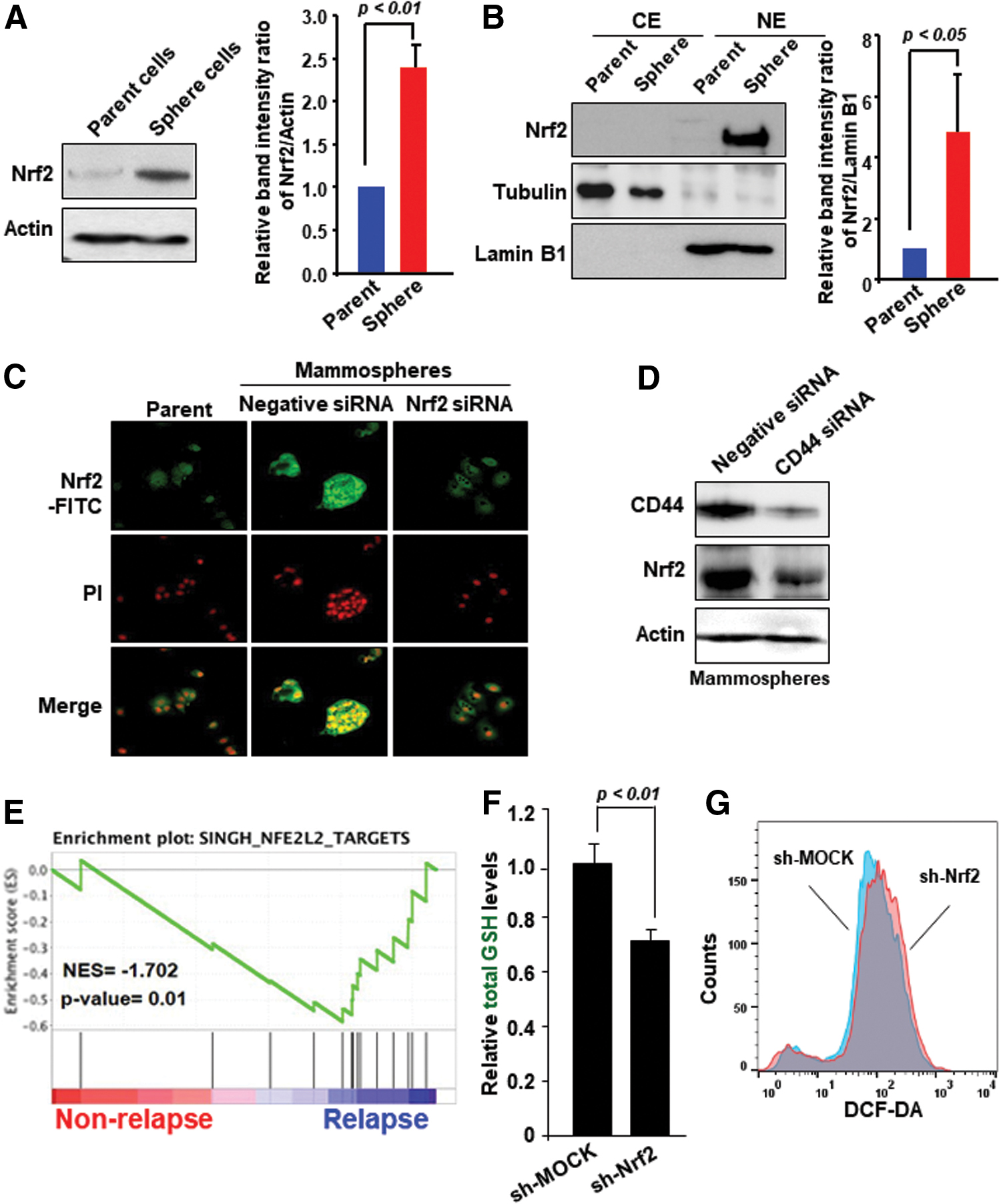
Variant forms of CD44 are likely to be involved in activation of Nrf2-mediated signaling (18). The expression of Nrf2 was markedly inhibited in MCF-7 mammosphere cells silenced for CD44 expression (Fig. 5D and Supplementary Fig. S7C). Notably, the high levels of Nrf2 are significantly related to relapsed breast cancer, according to the GSE2034 dataset (Fig. 5E). Collectively, these findings suggest that activation of Nrf2 responsible for the GCLC upregulation in breast cancer stem-like cells is attributable, at least in part, to CD44. We found the decreased intracellular levels of GSH and increased ROS production in Nrf2-knockdown breast cancer (MDA-MB-231) cells. It is likely that reduced production of GSH may provoke inefficient cellular detoxification of ROS in Nrf2-silenced MDA-MB-231 cells (Fig. 5F, G).
Nrf2 plays a role in the sphere-forming capability of breast cancer cells through upregulation of GCLC and Bmi-1
Next, we examined the expression of Bmi-1 in Nrf2-knockdown or overexpressing breast cancer cells. In general, MDA-MB-231 cells exhibit a higher Nrf2 expression compared with MCF-7 cells (47). As shown in Figure 6A, silencing Nrf2 caused downregulation of the Bmi-1 expression in MDA-MB-231 cells (Supplementary Fig. S8A). Conversely, overexpression of Nrf2 enhanced the mRNA and protein levels of Bmi-1 in MCF-7 cells (Fig. 6B and Supplementary Fig. S8B). We also noticed that knockdown of Nrf2 expression in MCF-7 cells abrogated the formation of secondary mammospheres (Fig. 6C). The proportion of CD44high/CD24low cell population in MCF-7 mammospheres was decreased by knockdown of Nrf2 using target-specific siRNA, whereas Nrf2 overexpression induced stemness properties (Fig. 6D).

The human GCLC promoter contains the antioxidant response element, which is the cis-acting consensus sequence responsible for Nrf2 binding (26). We investigated whether Nrf2 could upregulate GCLC expression in breast cancer stem-like cells, necessary for their self-renewal activity mediated by Bmi-1 expression. For this purpose, two different short hairpin RNAs (shRNAs) targeting Nrf2 were transfected into MDA-MB-231 cells that constitutively express Nrf2. Of these, the sh-Nrf2 (#2) efficiently lowered the levels of Nrf2, GCLC, and Bmi-1 mRNA and protein in MDA-MB-231 cells (Fig. 6E and Supplementary Fig. S8C). Likewise, mammosphere formation was abolished in Nrf2-silenced MDA-MB-231 cells (Fig. 6F). Moreover, expression of GCLC and Bmi-1 as well as phosphorylation of AMPK and FoxO3a was decreased in Nrf2-knockdown MDA-MB-231 cells (Fig. 6G and Supplementary Fig. S9). These results suggest that Nrf2 overactivation upregulates GCLC, and subsequently Bmi-1 responsible for self-renewal capability of breast cancer stem-like cells.
Knockdown of Nrf2 attenuates the tumorigenicity of breast cancer cells and mammospheres in vivo
To assess the oncogenic role of Nrf2 in the mammary cancer progression, athymic nude mice were subcutaneously injected with MDA-MD-231 cells transfected with either the control or Nrf2-specific shRNA. In MDA-MB-231 xenografts, Nrf2-shRNA introduction suppressed the tumor growth (Fig. 7A). Knockdown of Nrf2 significantly reduced the size of tumors derived from MDA-MB-231 cells in the xenograft mouse model (Fig. 7B). We confirmed reduced Nrf2 expression by immunohistochemical (IHC) staining of tumor tissues, and quantification of IHC image was performed by ImageJ analysis (Fig. 7C and Supplementary Fig. S10). It was accompanied by decreased expression of GCLC, FoxO3a, and Bmi-1 proteins (Fig. 7D and Supplementary Fig. S11).
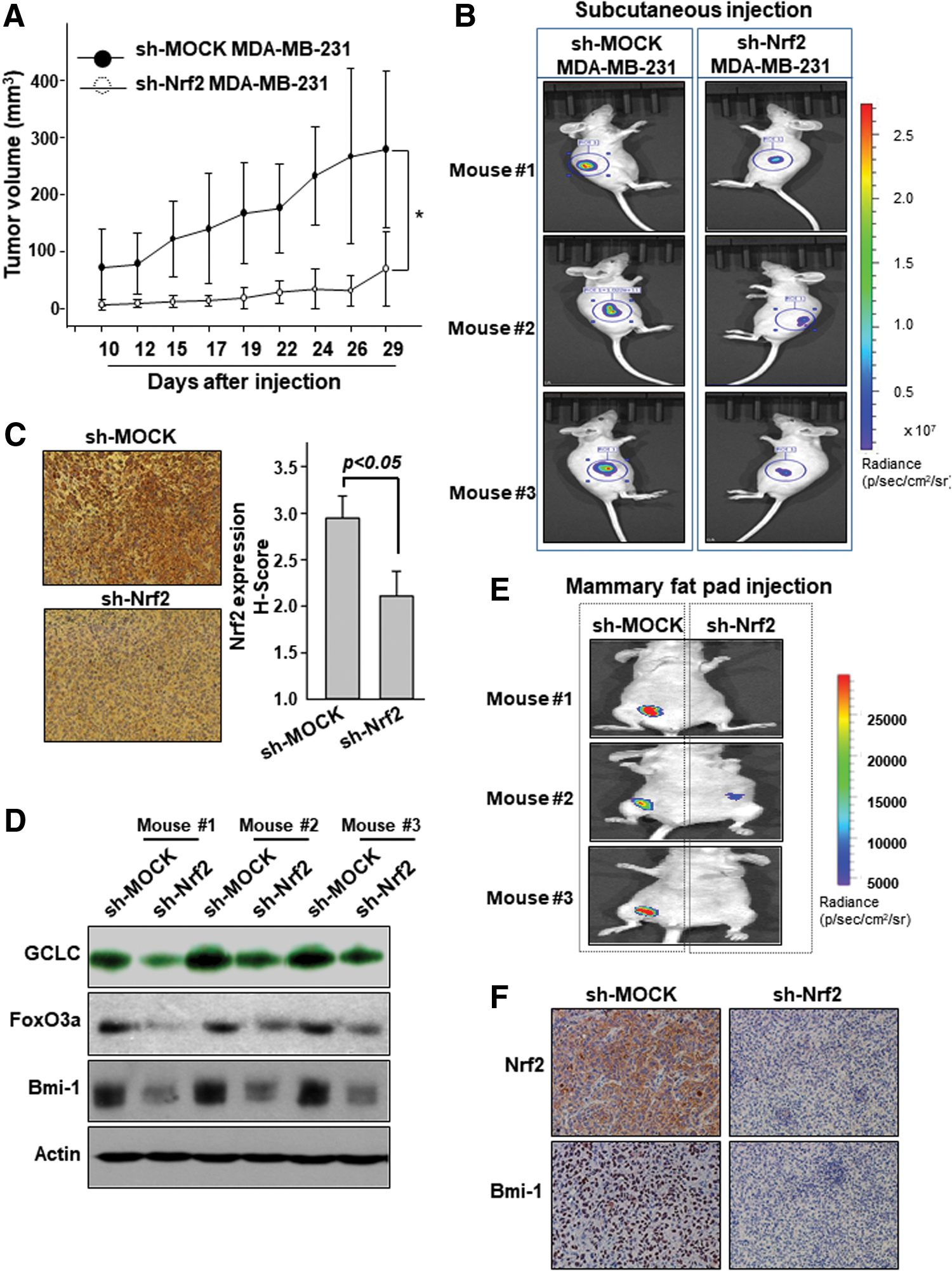
In another experiment, MDA-MB-231 sphere-forming cells transfected with lentivirus encoding Nrf2 shRNA or control shRNA were transplanted orthotopically into the fourth inguinal mammary fat pads of female athymic nude mice. After 10 weeks of injection, the mice were anesthetized using isoflurane and in vivo tumor green fluorescence protein (GFP) fluorescence was measured by the in vivo imaging system (IVIS) spectrum computed tomography (CT). The mice injected with control shRNA cells were able to form tumors more rapidly than those with Nrf2-knockdown cells (Fig. 7E). Overall morphology of the tumor was visualized with hematoxylin and eosin staining (Supplementary Fig. S12). IHC analysis showed a relatively weak staining of Bmi-1 in tumor tissues of mice transplanted with Nrf2-knockdown sphere-forming cells (Fig. 7F and Supplementary Fig. S13).
Discussion
There has been increasing evidence for a causal relationship between intracellular redox status and manifestation of stemness (9, 39). It has been known that CSCs maintain reduced levels of ROS compared with nonstem cells (30, 39). CSCs retaining a low level of ROS exhibit elevated expression of stemness-associated molecules such as Notch-1 and telomerase, which confers a self-renewal potential (16, 32).
Paul et al. have identified complex crosstalk between dynamic intracellular ROS flux and Nrf2 in the airway basal stem cells, whereby Nrf2 activates the Notch pathway to stimulate self-renewal activity (32). In addition, several recent studies have revealed the role of Nrf2 in the manifestation and maintenance of CSCs. Thus, the total expression and nuclear translocation of Nrf2 increase under the sphere-forming conditions, which enhance self-renewal activity (37, 38). However, it remains largely unresolved how the accumulation of Nrf2 expression contributes to the self-renewal activity. We also observed that the number and the size of mammospheres derived from MCF-7 cells were reduced by silencing Nrf2 as well as pharmacological inhibition of its target protein, GCLC. In addition, ROS levels were found to be decreased in the CD24low/CD44high breast cancer stem-like cells compared with parent cancer cells. In accord with these findings, we noticed a substantially elevated level of intracellular GSH in breast cancer stem-like cells.
Mizuno et al. have shown that aldehyde dehydrogenase (ALDH)-positive clear carcinoma cells retain a lower level of ROS than cells with low ALDH activity, and that increased HO-1 and SOD2 expression is regulated by Nrf2 (28). Notably, Nrf2 can be a therapeutic target for chemoresistant breast cancer cells and CSCs (38, 43). Syu et al. have reported that overexpression of Nrf2 attenuates the apoptosis induced by cisplatin in human breast cancer (MCF-7) cells under hypoxia, and the drug resistance in Nrf2 overexpressing cells was decreased by chemical inhibition of GCLC or its gene silencing (43).
Recently, Ryoo et al. reported that CD44 elevation led to p62-associated Nrf2 activation in doxorubicin-resistant MCF-7 cells (36). Autophagy activation might be an essential cellular process for adaptation and survival of CSCs; however, it is currently not clear how p62 is elevated by autophagic alteration in CSCs (36). It will be important to explore the relevance between autophagy and redox signaling in CSCs. Autophagy can be initiated by increased levels of ROS in various cancer cells, which triggers the activation of Notch1, ALDH, and Nrf2 signaling. The role of autophagy in cancer is still a double-edged sword. Bactericidal antibiotics used in combination with an autophagy blocker decreases tumor growth, whereas the use of an autophagy inhibitor with the anticancer drug leads to increased cell death (1, 34). In addition, many transcription factors including NF-κB, HIF-1α, Nrf2, and Notch are regulated by the cellular redox status. Kipp et al. observed that the activation of Nrf2, HIF, and NF-κB was induced throughout the whole spheroids of colon cancer cells (20). Particularly, HIF and Nrf2 are specifically activated in the core of larger spheroids (20).
An intriguing question is how GSH that accumulates in relatively large amounts in sphere-forming cells can modulate the self-renewal activity of breast cancer stem-like cells. The alteration of nuclear redox conditions influences chromatin conformation and stability. Glutathionylation of histone H3 during the translocation of GSH into the nucleus modulates the chromatin availability to replicative or transcriptional machineries by affecting interactions between H3 and nuclear proteins, such as lamin B receptor and heterochromatin protein 1 (25). In addition, GSH functions as an intracellular metal chelator (11). GSH supplementation improved lipid metabolism through increased expression of AMPK in skeletal muscles (3). Moreover, Dong et al. have revealed that both oxidative stress and ROS inhibition can activate AMPK through different mechanisms in a diabetic rat model. According to this study, AMPK activation by low ROS is due to redox modification, specifically the S-glutathionylation of cysteine residues in its alpha-subunit (10). It has been suggested that FoxO3a phosphorylation by AMPK facilitates the recruitment of the transcriptional complex at the specific target promoter. AMPK activation is associated with an increase in NAD+ levels, leading to activation of SIRT1 and upregulation of its downstream targets including FoxO3a (7). We speculate that the regulation of FoxO3a by AMPK may play a crucial role in fine-tuning of gene expression programs that control redox balance.
It is widely accepted that FoxO3 is a key regulator of self-renewal activity of stem cells depending on cellular redox status (27). Loss of FoxO3 is associated with an increased ROS level as well as decreased expression of SOD2 and catalase (27). The results from our present study corroborate the possible involvement of FoxO3a in reducing the intracellular ROS levels, thereby conferring acquired self-renewal capacity in breast CSCs. Doxorubicin-resistant breast cancer cells show FoxO3a predominantly expressed in the nucleus, which is then available for the phosphorylation of Akt (14). Notably, the Ser-7 residue of FoxO3a is the only phosphorylation site associated with its nuclear enrichment, thus providing a useful molecular marker of nuclear FoxO3a (14).
FoxO3a is a transcription factor, which binds to a consensus DNA sequence (G/C/A) (T/C/A) AAA (T/C) called forkhead response element. FoxO3a regulates the transcription of a wide variety of genes, such as Bim and Fas ligand involved in regulating cell survival and death (2). In addition, FoxO3a promotes the transcription of pluripotency factor Nanog gene, and then stimulates enrichment of breast CSCs (22). Interestingly, high expression of FoxO3 was associated with poor prognosis in breast cancer patients who received chemotherapy (22).
Dysregulated expression of Bmi-1 has been frequently observed in several types of cancer (19, 40). Consistent with our results, there has been correlation between Nrf2 accumulation and Bmi-1 expression in cervical and gastric CSCs (17, 49). Our study provides an insight into the mechanism underlying upregulation of Bmi-1 expression by Nrf2 in CSCs (Fig. 8). One of the most salient features of our findings is that nuclear FoxO3a can directly bind to the promoter of the gene encoding Bmi-1.
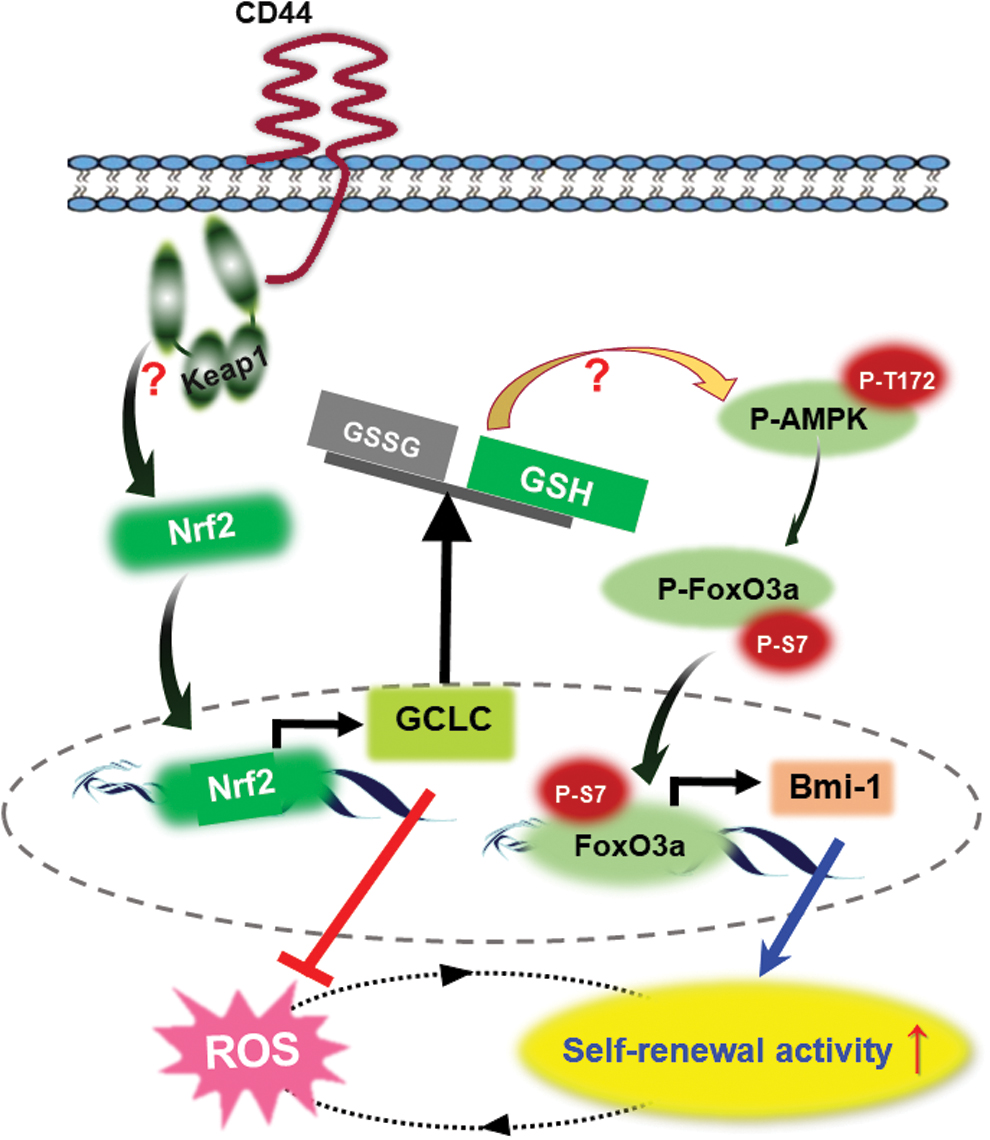
In conclusion, Nrf2-mediated upregulation of GCLC expression and GSH biosynthesis suppress intracellular ROS accumulation. This, in turn, induces the nuclear localization of FoxO3a, which binds to the Bmi-1 promoter, enhancing self-renewal activity and tumorigenic potential of breast CSCs.
Materials and Methods
Reagents
Dulbecco's modified Eagle's medium (DMEM), Rosewell Park Memorial Institute (RPMI) 1640 medium, and Dulbecco's modified Eagle's Medium Nutrient Mixture F-12 (DMEM/F-12) were purchased from Gibco BRL (Grand Island, NY). TRIzol®, MitoSOX™, DHE, and DCF-DA were obtained from Invitrogen (Carlsbad, CA). Primary antibodies for Bmi1, CD44, and Foxo3a were purchased from Cell Signaling Technology (Danvers, MA). Primary antibody against GCLC was obtained from Novus Biologicals (Littleton, CO). Primary antibodies against Nrf2 and β-actin were purchased from Santa Cruz (Santa Cruz, CA). Antibodies against CD24 and CD44 were purchased from BD Biosciences (Bedford, MA). Bicinchoninic acid (BCA) protein assay reagent was a product of Pierce Biotechnology (Rockfold, IL). GSH-MEE, BSO, and H2O2 were purchased from Sigma-Aldrich (St. Louis, MO).
Cell culture
Human breast cancer (MCF-7, MDA-MB-231) cell lines were maintained in RPMI 1640 and DMEM medium supplemented with 10% fetal bovine serum (FBS; GenDEPOT, Barker, TX) and 100 ng/mL penicillin/streptomycin/fungizone mixture at 37°C in a humidified atmosphere of 5% CO2/95% air.
Mammosphere formation assay
MCF-7 (5 × 103 cells per well) and MDA-MB-231 (1 × 104 cells per well) cells were cultured in a serum-free DMEM/F12 medium supplemented with B27 (GIBCO), 20 ng/mL epidermal growth factor (Sigma-Aldrich), 20 ng/mL basic fibroblast growth factor (Peprotech, Rocky Hill, NJ), and 4 ng/mL heparin (Sigma-Aldrich) in six-well ultralow attachment surface plates (Corning, NY). After 5 days, primary mammospheres formation was examined using an inverted microscope. To culture secondary mammospheres, primary mammospheres were gently collected and dissociated into a single-cell suspension using 40-μm strainer. Single cells were counted and then seeded again for another 5 days with addition of 1 mL medium every 2–3 days. After 5 days, secondary mammospheres formation was examined using an inverted microscope, and the number of mammospheres formed (>100 μm) was quantified.
Flow cytometric analysis
MCF-7 cells were collected and washed with phosphate-buffered saline (PBS), and dissociated with Accutase solution (Sigma-Aldrich). Cells were then counted and washed with PBS containing 2% FBS and 0.1% Tween-20. Cells were stained with CD24-propidium iodide and CD44-APC from BD Biosciences for 30 min at 4°C. Cells were dissociated into single cells by using 40-μm strainer, and then the population of CD44+/CD24− cells was measured using BD FACSCalibur (Becton Dickinson Biosciences, San Jose). To isolate CSC and non-CSC populations from breast cancer cells, cells were stained with the above antibodies, and the cells were sorted using BD FACSAria™ III cell sorter (Becton Dickinson Biosciences).
Determination of intracellular glutathione levels
Intracellular glutathione levels were determined using the bioluminescent Promega GSH/GSSG-Glo™ assay kit (Promega, Mannheim, Germany) according to manufacturer's protocols. Luminescence was detected using SpectraMax i3 Multi-Mode Microplate Reader (Molecular Devices, Sunnyvale, CA).
Analysis of intracellular ROS accumulation
Intracellular accumulation of ROS was assessed by using redox-sensitive dye DCF-DA. Cells were washed once with Hanks' balanced salt solution and loaded with 10 μM DCF-DA for 30 min at 37°C to assess ROS-mediated oxidation of the fluorescent compound DCF. Fluorescence of oxidized DCF was analyzed by flow cytometry.
Western blot analysis
Breast cancer cells were lysed in radioimmunoprecipitation assay lysis buffer (150 mM NaCl, 0.5% Triton × 100, 50 mM Tri-HCl (pH 7.4), 25 mM NaF, 20 mM EGTA, 1 mM dithiothreitol, 1 mM Na3VO4, 0.1 mM PMSF, protease inhibitor cocktail tablets) for 15 min on ice followed by centrifugation at 13,000 g for 20 min. The protein concentration of the supernatant was measured by using BCA reagents (ThermoScientific, Rockford, IL). Protein (30 μg) was separated by running through 8%–12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis gel and transferred to the polyvinylidene fluoride membrane (Gelman Laboratory, Ann Arbor, MI). The blots were blocked with 5% nonfat dry milk/Tris-buffered saline buffer containing 0.1% Tween-20 (TBST) for 1 h at room temperature. The membranes were incubated overnight at 4°C with indicated primary antibodies. The blots were rinsed three times with TBST buffer for 10 min each. Washed blots were incubated with 1:5000 dilutions of horseradish peroxidase-conjugated secondary antibody (Thermoscientific) for 1 h and washed again three times with TBST buffer. The transferred proteins were visualized with an enhanced chemiluminescence detection kit (Amersham Pharmacia Biotech, Buckinghamshire, United Kingdom).
Preparation of cytosolic and nuclear proteins
Cells were gently washed twice with ice-cold PBS, scraped in 1 mL PBS and centrifuged at 12,000 g for 30 s at 4°C. Pellets were suspended in 200 μL of hypotonic buffer A (10 mM HEPES, pH 7.9; 10 mM KCl; 2 mM MgCl2; 1 mM DTT; 0.1 mM EDTA; 0.1 mM PMSF) for 15 min on ice, and 12.5 μL of 10% Nondiet P-40 solution was added for 5 min. The mixture was then centrifuged for 6 min at 12,000 g, washed once with 400 μL of PBS, and suspended in 70 μL of buffer C (50 mM HEPES, pH 7.9; 50 mM KCl; 300 mM NaCl; 0.1 mM EDTA; 1 mM DTT; 0.1 mM PMSF; and 10% glycerol) for 20 min on ice and centrifuged for 6 min at 14,000 g. The supernatant containing nuclear proteins was collected and stored at −70°C after determination of the protein concentration.
Immunofluorescence microscopy
To determine the localization of indicated proteins, we adopted the immunocytochemical method that utilized a monoclonal antibody recognizing Nrf2 and FoxO3a. Cells (1 × 105 cells/600 μL in four-well chamber slides) were fixed in 95% methanol/5% acetic acid solution for 10 min at −20°C. After a rinse with PBS, cells were blocked for 1h at room temperature in fresh blocking buffer (0.5% Tween 20 in PBS, pH 7.4, containing 10% normal goat serum). Dilutions (1:100) of primary antibodies were made in PBS with 1% bovine serum albumin, and cells were incubated overnight at 4°C. After three washes with PBS containing 0.1% Tween 20 (PBST), the cells were incubated with FITC-conjugated secondary antibody in PBST with 3% bovine serum albumin for 1h at room temperature. Cells were rinsed with PBS, and stained cells were analyzed under a fluorescence microscope and photographed.
Reverse transcription-polymerase chain reaction
Total RNA was isolated from each cell by using TRIzol reagent (Invitrogen) according to the manufacturer's protocol. One microgram of total RNA was reverse transcribed with murine leukemia virus reverse transcriptase (Promega, Madison, WI). Polymerase chain reaction (PCR) was carried out in a thermos-cycler using specific primers for Nrf2, GCLC, Bmi-1, and GAPDH. The primers employed were (forward and reverse, respectively): Nrf2, 5′-ACTGGTTGGGGTCTTCTGTG-3′ and 5′-CGGTATGCAACAGGACATTG-3′, 263 bp; GCLC, 5′-TGAAGGGACACCAGGACAGCC-3′ and 5′-GCAGTGTGAACCCAGGACAGC-3′, 167 bp; Bmi-1, 5′-CCAGGGCTTTTCAAAAATGA-3′ and 5′-GCATCACAGTCATTGCTGCT-3′, 271 bp; GAPDH, 5′-GCATGGCCTTCCGTGTCCCC-3′ and 5′-CAATGCCAGCCCCAGCGTCA-3′, 216 bp. Amplification products were resolved by 1.5%–2% agarose gel electrophoresis, stained with ethidium bromide and photographed under ultraviolet light.
Transient transfection of siRNA and plasmid
MCF-7 and MDA-MB-231 cells were plated at a confluence of 60% in 60-mm dish and grown in complete growth media. Nrf2, CD44 and FoxO3a siRNA (25 nM) and Nrf2 plasmid were transfected into MCF-7 cells with lipofectamine RNAiMAX or lipofectamine 2000 (Invitrogen) reagents. The siRNA sequences were (forward and reverse, respectively) as follows: Nrf2, 5′-AAGAGUAUGAGUGGAAAAACTT-3′ and 5′-GUUUUUCCAGCUCAUACUCUUTT-3′; CD44, 5′-UAUUCCACGUGGAGAAAATT-3′ and 5′-UUUUUCUCCACGUGGAAUACA-3′; FoxO3a, 5′-CAGUUCUAACUUCACUGUU-3′ and 5′-AACAGUGAAGUUAGAACUG-3′. Plasmid of pcDNA3.1-EGFP-C4-human Nrf2 was obtained from Adggene (plasmid #21549).
ChIP assay
After formaldehyde crosslinking, cells were harvested, and the ChIP assay was performed using EZ ChIP kit according to the manufacturer's protocol (Millipore). Chromatin was immunoprecipitated with antibody against the FoxO3a or IgG as a negative control. Input and bound DNA were purified using the Genomic DNA Extraction Kit (SolGent, Daejeon, Korea). PCR was performed against FoxO3a binding region of the Bmi-1 promoter. The primer sequences were 5′-TTGTTTGGATCTGAGTTCGTGTG3′ (forward) and 5′-ATCGCATCGTTTCCTCCGTG-3′. Reaction conditions were as follows: initial denaturation (5 min at 94°C) was followed by 40 cycles for 1 min at 94°C, 1 min at 55°C, and 1 min at 72°C. PCR was completed by 10 min at 72°C, and products were separated on 2.5% agarose gel.
Knockdown of Nrf2 in breast cancer cells
A set of two human (SH-003755) specific SMART vector 2.0 lentiviral shRNA particles (108 TU/mL) for NFE2L2 were purchased from Thermo Fisher Scientific Dharmacon (Lafayette, CO). The vector had an human cytomegalovirus promoter, a TurboGFP reporter gene, and a puromycin selection gene. SMART vector 2.0 nontargeting shRNA control particles (108 TU/mL) were designed as a negative control. MDA-MB-231 cells were cultured in DMEM with 10% FBS in the presence of antibiotics, and viral particles (NFE2L2 and nontargeting control) were added with a multiplicity of infection of 3.0 for cells. TurboGFP-expression was analyzed using microscopy and GFP-positive cells selected using puromycin (5 μg/mL). The selected cells were maintained in culture media containing puromycin at 0.5 μg/mL until further use. Nrf2 silencing was verified by Western blot and mRNA quantification.
Mouse studies
Animal experiments were conducted in accordance with Guide for the Care and Use of Laboratory Animals, and approved by the Institutional Animal Care and Use Committee of Seoul National University (SNU protocol #160622-10). For a xenograft tumor model, MDA-MB-231 (2 × 106 cells per mouse in 50 μL containing Matrigel) was injected into 6-week-old female BALB/c nu/nu mice. After 10 days, tumor volume was determined by measuring the diameters of tumors with a caliper every 2 or 3 days. Tumor volumes were calculated according to the following formula: (length) × (width) × (height) × 0.52. For an orthotopic tumor model using mammosphere cells, MDA-MB-231 mammosphere cells (1 × 105 cells per mouse in 50 μL containing Matrigel) were injected into 6-week-old female nude mice at mammary fat pad. Bioluminescence imaging was obtained by the IVIS spectrum micro CT and Living Image (ver. 4.2) software (PerkinElmer).
Public data resources
The clinical data of breast cancer were obtained from both TCGA and GSE2034. The Cancer Genome Atlas (TCGA) clinical data for breast cancer patients (n = 1108) were retrieved from cBioPortal, including RNA-seq gene expression profiles. Dataset for recurrent breast cancer patients was obtained from Gene Expression Omnibus. Gene expression profiles of breast cancer from GSE2034 (nonrelapse [n = 179] and relapse [n = 107]) and GSE1456 (nonrelapse [n = 119] and relapse [n = 40]) were used for analysis.
Statistical analysis
When necessary, data were represented as means±SD of at least three independent experiments, and statistical analysis between groups was performed using Student's t-test.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study was supported by the Global Core Research Center (GCRC) grant (No. 2011-0030001) and Basic Science Research Program grant (No. 2013R1A1A2012429 and 2016R1A6A3A11935082) from the National Research Foundation (NRF) of Republic of Korea.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Figure S7
Supplementary Figure S8
Supplementary Figure S9
Supplementary Figure S10
Supplementary Figure S11
Supplementary Figure S12
Supplementary Figure S13
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.