
Abstract
Significance:
Perinatal brain injury is caused by hypoxia–ischemia (HI) in term neonates, perinatal arterial stroke, and infection/inflammation leading to devastating long-term neurodevelopmental deficits. Therapeutic hypothermia is the only currently available treatment but is not successful in more than 50% of term neonates suffering from hypoxic–ischemic encephalopathy. Thus, there is an urgent unmet need for alternative or adjunct therapies. Reactive oxygen species (ROS) are important for physiological signaling, however, their overproduction/accumulation from mitochondria and endoplasmic reticulum (ER) during HI aggravate cell death.
Recent Advances and Critical Issues:
Mechanisms underlying ER stress-associated ROS production have been primarily elucidated using either non-neuronal cells or adult neurodegenerative experimental models. Findings from mature brain cannot be simply transferred to the immature brain. Therefore, age-specific studies investigating ER stress modulators may help investigate ER stress-associated ROS pathways in the immature brain. New therapeutics such as mitochondrial site-specific ROS inhibitors that selectively inhibit superoxide (O2 •−)/hydrogen peroxide (H2O2) production are currently being developed.
Future Directions:
Because ER stress and oxidative stress accentuate each other, a combinatorial therapy utilizing both antioxidants and ER stress inhibitors may prove to be more protective against perinatal brain injury. Moreover, multiple relevant targets need to be identified for targeting ROS before they are formed. The role of organelle-specific ROS in brain repair needs investigation. Antioxid. Redox Signal. 31, 643–663.
Introduction
The purpose of this review is to summarize reactive oxygen species (ROS)-mediated mechanisms related to mitochondria and endoplasmic reticulum (ER) in the development of hypoxic–ischemic injury in the immature brain. Perinatal brain injury occurs after hypoxia–ischemia (HI) in term neonates (>37 weeks into gestation), perinatal arterial stroke, and states of infection/inflammation (70). Perinatal brain injury leads to devastating long-term disabilities, including cognitive and attention deficits, cerebral palsy, epilepsy, and visual and hearing dysfunctions (70).
Neonatal hypoxic–ischemic encephalopathy (HIE) has been extensively studied in both experimental and clinical settings, although the underlying mechanisms are not yet completely understood. Briefly, a primary phase of energy failure (occurring within minutes after HI injury) involves severe adenosine triphosphate (ATP) depletion due to the failure of mitochondrial respiration, and increased excitotoxicity and associated influx of Ca2+ and Na+. In the secondary (hours to days after injury) and tertiary (days to months) phases of injury, inflammation, protein misfolding and associated ER stress, ROS production, and severe disruption of mitochondrial integrity/function lead to cell death [for review, see Hagberg et al. (68)].
Targeting ROS in the immature brain can be exploited for therapeutic benefits. For instance, adult transgenic mice with enhanced ROS scavenging ability due to genetic overexpression of antioxidant enzymes, such as manganese superoxide dismutase (MnSOD/SOD2) or extracellular SOD, SOD3, show protection against ischemic injury after HI (88). In agreement, genetic deletion of certain superoxide dismutase (SOD) isoforms, for instance, SOD1, results in an increase in ischemic brain damage (123).
In eukaryotic cells, ROS can be generated in multiple organelles, including ER and mitochondria, as a by-product of oxidative protein folding, mitochondrial respiration, and detoxification (55). ROS production affects cell function and homeostasis in both physiological and pathophysiological conditions. The immature brain is particularly vulnerable to ROS-mediated damage due to its high oxygen consumption in relation to its antioxidant capacity, high concentrations of unsaturated fatty acids, poor antioxidative capacity, and increased availability of free iron to catalyze ROS production (72). Increased generation of ROS and an altered redox status are observed in experimental fetal and neonatal models of HI (11), and several agents that reduce ROS are shown to provide neuroprotection (6), for example, allopurinol, deferoxamine, and the lipid peroxidation inhibitor tirilazad reduce brain injury after HI (8, 135). Here, we review the mechanisms of ROS production from mitochondria and ER, targeted antioxidants, and their potential as a therapy for ameliorating perinatal brain injury.
Mitochondrial ROS in Perinatal Brain Injury
The premature brain is particularly susceptible to ROS-induced damage because of inadequate antioxidant stores at birth and impaired upregulation in response to oxidative stress (102). Low levels of ROS production sustain physiological functions, including proliferation, host defense, and signal transduction (55). However, increased levels of intracellular ROS caused by dysfunctional mitochondria serve as a signal to attenuate global protein synthesis (177) acting as a double-edged sword in cellular processes. Mitochondrial energy metabolism is linked to ROS production, and enzymes linked to metabolic pathways can be affected by redox reactions (142).
During HI, oxidative stress induced by ROS “bursts” plays a vital role for mitochondrial dysfunction and subsequent cell death (60, 138). Dysregulated mitochondrial ROS (mtROS) signaling contributes to neuronal loss and neurodegenerative disorders (173). Furthermore, hydrogen peroxide (H2O2) accumulates more in the immature than in adult brain. Due to the limited capacity of H2O2 inactivation (59, 99), ROS defense mechanisms, and relatively high levels of free iron in the immature brain, H2O2 will generate hydroxyl radicals (•OH; via Fenton reaction), which exert toxic effects on the central nervous system (CNS) (60). The analysis of current data supports the hypothesis that in the developing HI brain there is accelerated ROS production in the electron transport chain (ETC) causing oxidative damage. ROS production if left unchecked will indiscriminately attack phospholipids, proteins, and DNA.
Metabolic processes in the mitochondria affecting ROS production
As hubs of cellular metabolism, mitochondria integrate metabolic pathways to contribute to ROS production. For this reason, it is important to analyze the major metabolic pathways that control the redox homeostasis. Mitochondrial oxidative phosphorylation (OXPHOS), which ensures a steady supply of ATP, is also one of the sources of endogenous ROS (113). Rapid movement of electrons through the ETC (the major site of OXPHOS) results in the leakage of electrons that can form superoxide (O2 •−) via univalent reduction of molecular oxygen (O2) (Fig. 1). Mitochondrial complex I (C-I; NADH:ubiquinone oxidoreductase) and complex III (C-III; ubiquinol:cytochrome c oxidoreductase; cytochrome bc1 complex) are the major producers of ROS.
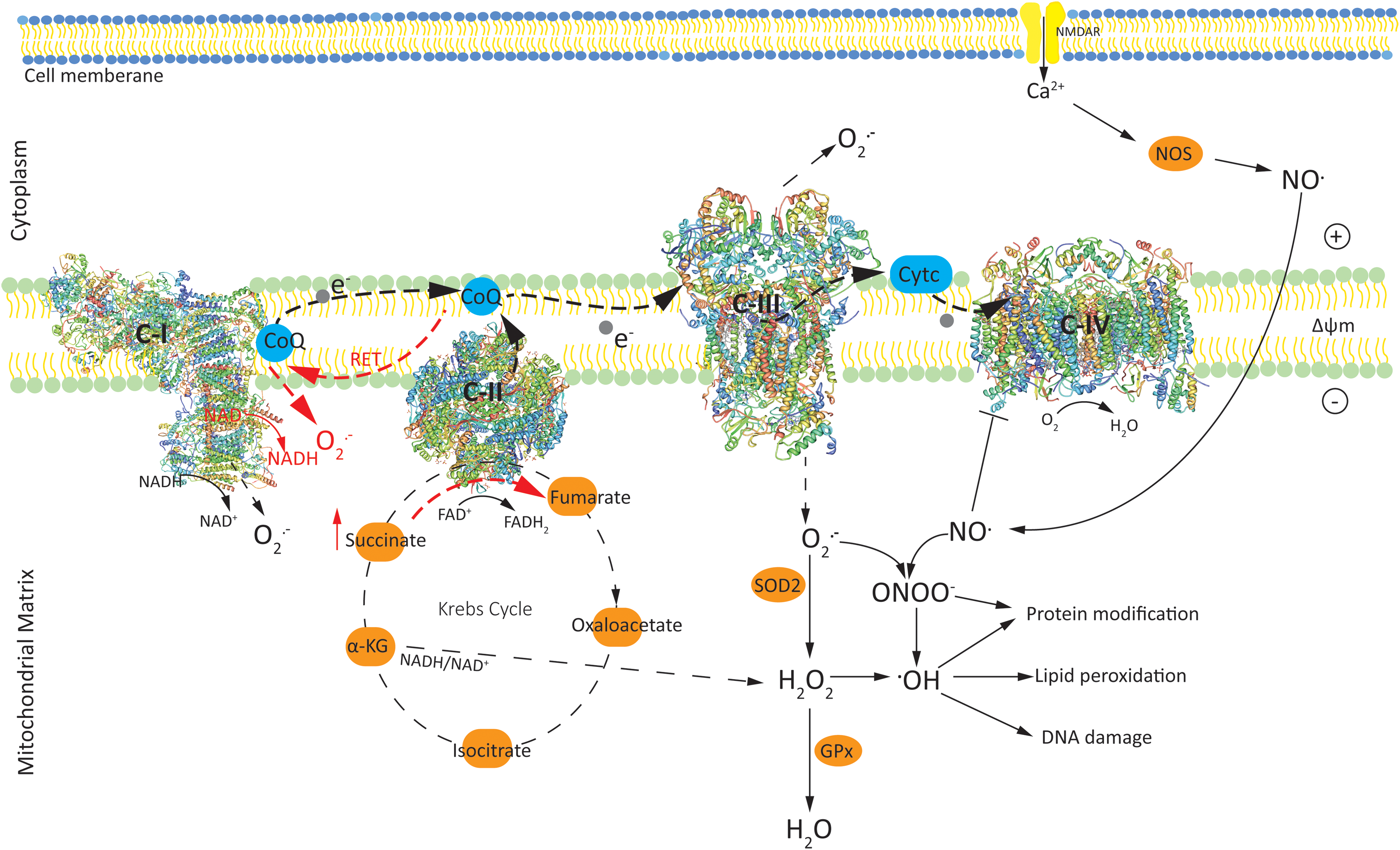
In C-I there are two sites: the flavin in the NADH-oxidizing site (site IF) and the ubiquinone-reducing site (site IQ), producing O2 •−. In C-III, the superoxide is thought to arise from the quinol oxidizing site (site Qo) (18). When supplied with CoQH2 (reduced form of ubiquinone) and when the Qi site is inhibited by antimycin or by pathological events, C-III produces large amounts of O2 •− (180). O2 •− produced from C-I and C-III forms the central ROS in the mitochondrial matrix generated by the core metabolic machinery (124). H2O2 is produced from the dismutation of superoxide (O2 •−) by MnSOD within the mitochondria and functions as a second messenger in a physiologically relevant manner (60), but can also exert toxic effects particularly in immature neurons (118).
It has been shown that generation of O2 •− from C-I during ischemia/reperfusion (I/R) is dependent on electron supply from the mitochondrial citric acid cycle intermediate succinate. Succinate, which accumulates significantly during ischemia and also in the immature brain (96) through the reverse action of complex II (C-II), is rapidly oxidized in the first minutes of reperfusion. This rapid oxidation drives reverse electron transport (RET) at C-I, in which electrons are forced from reduced coenzyme Q (CoQ) in C-II back to C-I, generating large amounts of O2 •− (39). It is shown that when both succinate concentration and mitochondrial membrane potential (Δψm) are high, mitochondria induce RET, which is associated with a high rate of O2 •− production [for a comprehensive review, see Scialò et al. (158)]. Compared with forward electron transport, the rate at which free radicals are generated under RET appears to be of considerable magnitude (159). The generation of mitochondrial superoxide (O2 •−) by RET at C-I causes oxidative damage in pathologies such as I/R injury, but also provides the precursor to H2O2 production (148), which can cross lipid membranes and cause tissue damage (173).
Mitochondrial C-II (succinate-ubiquinone oxidoreductase) or succinate dehydrogenase catalyzes the oxidation of succinate to fumarate, resulting in the donation of electrons to the ETC via the reduction of flavin adenine dinucleotide (FAD) to FADH2. C-II, which is part of both the citric acid cycle and ETC, is a key modulator of mtROS production by other respiratory complexes, particularly C-I (136). Although complex IV (C-IV) does not directly contribute to ROS production, inhibition of C-IV enhances ROS generation during the oxidation of C-I or C-II substrates (33). ROS produced in the ETC depend on many factors, including the concentration and combination of substrates used to feed the respiratory chain (158), and Δψm (170). For example, inhibiting OXPHOS, when C-I or C-II substrates are plentiful, results in hyperpolarized mitochondria as the electron transfer rate along the respiratory chain is diminished, promoting greater O2 •− production (145).
Other sites of ROS generation in the inner mitochondrial membranes functionally connected to the respiratory chain, which are not discussed here in detail, include dihydroorotate dehydrogenase (76) and mitochondrial glycerol-3-phosphate dehydrogenase (122) as not much is known about their role in HI-mediated ROS, but also represent sites of ROS generation. Sites within the matrix can also be important producers of O2 •−/H2O2. In isolated mitochondria under optimal conditions, the mitochondrial 2-oxoacid dehydrogenase complexes have a greater maximum capacity to form O2 •−/H2O2 than the flavin site of C-I (23, 143). Enzymes in the Krebs cycle can generate ROS and significantly contribute to generation of oxidative stress in the mitochondria. Tretter and Adam-Vizi have reported that alpha-ketoglutarate dehydrogenase, an early enzyme in the tricarboxylic acid cycle/Krebs cycle (TCA cycle), significantly contributes to H2O2 production by regulating the NADH/NAD+ ratio (178).
Activation of N-methyl-
The underdeveloped antioxidant system in the immature brain limits the inactivation of H2O2, which makes the developing brain especially sensitive to oxidative stress after perinatal HI during reperfusion and reoxygenation, as discussed below in the next section (59, 99). Oxygen-derived free radicals are formed during reperfusion after ischemia via oxidation of accumulated hypoxanthine by xanthine oxidase and oxidation of arachidonic acid in the presence of lipoxygenase and cyclooxygenases (116, 146). It is shown that allopurinol (a xanthine oxidase inhibitor) and deferoxamine (a chelator of nonprotein-bound iron) preserved cerebral energy metabolism, attenuated development of edema, and improved histologic outcome in the newborn piglets at 24 h after HI on reperfusion and reoxygenation (135).
To summarize, mitochondria integrate metabolic pathways to contribute to ROS production. O2 •− forms the central ROS, and H2O2 is produced from the dismutation of O2 •− by MnSOD. The accumulation of excessive ischemic succinate generates O2 •− by RET at C-I and is considered a critical driver of ROS formation. The underdeveloped antioxidant system in the immature brain limits the inactivation of H2O2, which can cross lipid membranes and cause tissue damage.
Antioxidant systems in the mitochondria
There are a number of reasons that the developing brain may be more sensitive to oxidative stress than the adult brain. The immature brain has lower oxygen requirements (97) than the mature brain. Nevertheless, the antioxidant defence capacity is underdeveloped also in relation to a low respiratory rate, which may cause a risk of ROS overflow in pathological situations such as HI (117). High concentration of lipids in the perinatal brain, primarily due to high polyunsaturated fatty acid content, also leads to an increase in susceptibility to lipid peroxidation (119).
Chronic ROS exposure can inactivate the iron/sulfur centers of C-I, II, III, and aconitase (an enzyme that catalyzes the stereospecific isomerization of citrate to isocitrate in the tricarboxylic cycle), resulting in cessation of mitochondrial energy production (35). ROS exposure can also result in oxidative damage to mitochondrial and cellular proteins, lipids, and nucleic acids (119). Several antioxidant enzyme systems operate to combat deleterious ROS production in mitochondria. O2 •− produced by the ETC is converted to H2O2 by mitochondrial SOD2 (Fig. 1), and H2O2 is further converted to H2O by glutathione peroxidase (GPx) in the presence of glutathione (38). Conversion of H2O2 to •OH in the presence of free iron leads to increased neurotoxicity in the immature nervous system (59).
Adenine nucleotide translocator (ANT) an abundant mitochondrial inner membrane protein plays a major role in detoxification of ROS (20). The content of the ANT protein increases more than twofold in the first 3 postnatal weeks and the expression of ANT in rat brain is required for the development of OXPHOS (157). The prime function of ANT is to function as an uncoupler and facilitate exchange of ATP and adenosine diphosphate (ADP) across the inner mitochondrial membrane, which is important for both ATP production and maintenance of normal Δψm. These functions of ANT protect the mitochondria from increased ROS generation associated with increased Δψm (92), suggesting that the postnatal enrichment of the ANT protein in rat brain mitochondria is an essential factor for the development of OXPHOS capacity in the early postnatal period (157). ANT also largely reduces the mitochondrial and nuclear signs of apoptosis induced by NO• and ONOO− in intact cells (18).
Experimental evidence presented leaves little doubt that changes encompassing all the major mitochondrial antioxidant systems are involved in neonatal brain injury, and therefore, modulation of the mitochondrial antioxidant systems may in the future become a target for therapy.
Mitophagy and ROS production
Mitochondrial mitophagy is defined as the selective degradation of damaged mitochondria through the autophagosomal/lysosomal pathway (149). Mitochondria undergo continuous fission and fusion through highly regulated processes in response to metabolic demands (71) (Fig. 2). In neonatal rats, it is shown that HI brain damage can increase the degree of mitophagy, and the inhibition of mitophagy can aggravate the condition (106). Induction of mitophagy and neuronal cell death has been suggested to be sex dependent, and deficient elimination of damaged mitochondria contributes to male vulnerability to neuronal death and long-term neurobehavioral deficits in neonatal rat following HI (52).

Mitophagy is triggered by mild oxidative stress in a mitochondrial fission-dependent manner (61). Damaged mitochondria, which are not degraded spontaneously, age and produce surplus ROS. Excessive amount of ROS is not only toxic to mtDNA but can also promote lipid peroxidation (2), impair cellular function, and induce apoptosis (124). Mitochondrial dynamics and morphology are increasingly shown to be regulated by ROS and reactive nitrogen species (RNS) (71). High concentrations of exogenous H2O2 can induce dose- and time-dependent mitochondrial fragmentation in C2C12 myocytes and human umbilical vein endothelial cells, leading to increased expression of fission and fusion genes (84, 156). It is shown that the mechanism by which exogenous H2O2 induces mitochondrial fragmentation in C2C12 myocytes involves increased dynamin-related protein 1 (DRP1) activity (82) (Fig. 2).
MitoNEET, a dimeric mitochondrial outer membrane protein, is a key regulator of mitochondrial function and lipid homeostasis (189). MitoNEET is known to modulate the oxidative capacity of cardiac mitochondria, but its function during reperfusion injury is unknown (67). Mdivi-1, a quinazolinone derivative originally identified as a DRP1 inhibitor (29), inhibits fission and is shown to decrease ROS formation in response to nutrient overload stress (85), suggesting that mitochondrial fusion decreases the tendency for mtROS generation or release (179). Mitophagy and biogenesis need to go hand in hand to ensure efficient ATP production and degradation of damaged mitochondria (175). To conclude, mitophagy is a key process playing an essential role in reducing production of ROS and mutation of mitochondrial DNA by maintaining mitochondrial quantity and quality.
Effect of inflammation on ROS production
Bacterial infection is an important cause of death and long-term morbidity in neonates, especially in infants born preterm (70). Pre-exposures to infectious agents (such as TOLL-like receptor agonists) have also been shown to enhance brain injury after HI (57, 121, 168). Immune cells in the newborn brain such as microglia contribute to the generation of a chronic or mild inflammatory environment by regulating ROS production through mitochondria-mediated mechanisms (100). Microglias achieve immune regulation depending on the mitochondrial dynamics and metabolic state allowing them to respond with an appropriate cytokine response to each situation, which is crucial for the correct establishment of immune responses (120).
Lipopolysaccharide (LPS) exposure in microglia cells results in excessive mitochondrial fragmentation and a metabolic shift from OXPHOS to glycolysis, which goes hand in hand. Although not applicable to all immune cells, in microglia, fragmented mitochondria produce excess ROS, increased Δψm, and succinate accumulation (126). Therefore, in microglia, mitochondrial fragmentation is an important mechanism to promote ROS production thereby enhancing the proinflammatory response in the CNS (126). Thus, targeting mitochondrial fragmentation by means of metabolic blockers presents new opportunities to counteract inflammatory diseases.
mtROS and sirtuins
Sirtuins are NAD+-dependent histone deacetylases, with several histone and nonhistone targets [for review, see Houtkooper et al. (78)]. First discovered in yeast, seven mammalian homologues have been identified (Sirt1–7), which can be found in different locations in the cell. Sirt1 (105) and Sirt6 (202) regulate antioxidant pathways mainly in the nucleus and cytosol, whereas Sirt3 plays an important antioxidant role in the mitochondria (9). For instance, Sirt3 suppresses ROS production by deacetylating NADH dehydrogenase (ubiquinone) 1 alpha subcomplex 9 of C-I of the ETC (4), and isocitrate dehydrogenase of the TCA cycle (199). Furthermore, Sirt3 activates SOD2 directly via deacetylation and via increased transcription through deacetylation and nuclear translocation of transcription factor forkhead box O 3a (FOXO3a) (37). Sirt3 physically interacts with FOXO3a and promotes its binding to the SOD2 promoter region (83).
Sirt1 also deacetylates FOXO3a, however, Sirt1-mediated antioxidant effects involve deacetylation of both FOXO3a and peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC1α) because knocking down either of these proteins prevents Sirt1-mediated induction of antioxidant genes, at least in bovine aortic endothelial cells (133). Unlike Sirt1, Sirt3 is not known to deacetylate PGC1α (130). In fact, one study has shown that Sirt3 is a downstream target gene of PGC1α in murine muscle and liver cells, and its expression is mandatory for the PGC1α-dependent activation of antioxidant enzymes, including SOD2 and GPx, in C2C12 murine myocytes (95).
Most of the current information on Sirt3 and its associated regulatory pathways comes from non-neuronal cells, and the role of Sirt3 in neurodegenerative conditions remains relatively unexplored (162). Nevertheless, studies using in vitro oxygen/glucose deprivation (OGD) models of cerebral ischemia or oxidative/excitotoxic models of stress have consistently shown that overexpression of Sirt3 is neuroprotective via inhibition of oxidative stress in both primary neurons and neuronal cell lines (see Table 1 for summary) (49, 50, 93, 161, 187).
In Vitro Studies Showing Neuroprotection by Sirt3 Against Different Types of Stresses, Including Ischemic, Oxidative, and Excitotoxic Stress
AMPK, 5′-adenosine monophosphate-activated protein kinase; ATP, adenosine triphosphate; ETC, electron transport chain; H2O2, hydrogen peroxide; MPTP, mitochondrial permeability transition pore; mTOR, mammalian target of rapamycin; NMDA, N-methyl-
Pharmacological activation of Sirt3-FOXO3a-SOD2 axis is neuroprotective in adult mouse models of stroke (192, 196), however, intriguingly, knocking out Sirt3 is also found to be neuroprotective in these models. Accumulation of sphingolipid ceramide in mitochondria after cerebral I/R injury suppresses C-III activity, resulting in increased ROS accumulation (65). Novgorodov et al. (132) showed that Sirt3 directly activates ceramide synthases 1, 2, and 6 via deacetylation, and neuroprotection in Sirt3 KO mice against middle cerebral artery occlusion (MCAO) is likely due to reduced accumulation of ceramide in mitochondria. Recently, Verma et al. (188) addressed this anomaly more elegantly by showing that neuroprotection in Sirt3 KO mice against MCAO is due to a compensatory upregulation of Sirt1 because pharmacological inhibition of Sirt3 using AGK7 increased ischemic damage, which was associated with downregulation of both Sirt1 and 3.
Notably, Sirt1, 3, and 6 share a number of substrates (78), and any in vivo intervention must carefully explore the interplay between these three sirtuins. This is particularly important in relation to cerebral I/R injury because both Sirt1 and 6 are shown to provide neuroprotection against adult cerebral I/R injury (184, 202). For instance, Sirt6-mediated neuroprotection against adult cerebral I/R injury involves attenuation of oxidative stress, which is mediated via nuclear factor erythroid 2-related factor 2 (NRF2) as evident from the loss of neuroprotection in NRF2 KO mice (202).
There is a scarcity of research on the role of sirtuins in perinatal brain injury. While the roles of Sirt3 and 6 remain uninvestigated, a few studies show the involvement of Sirt1 in perinatal brain injury. For instance, Carloni et al. (27, 28) showed that melatonin-mediated protection against neonatal HI is associated with the restoration of Sirt1 activity. In another study, melatonin protected neurons in the dentate gyrus of neonatal rats challenged with LPS (160). These protective effects were mediated through upregulation of the Sirt1/NRF2 pathway, which resulted in the attenuation of inflammation and oxidative stress.
ER-Associated ROS in Perinatal Brain Injury
Basal ROS production in ER and ER stress
ER is the major site for folding and maturation of secretory and membranous proteins due to its highly oxidative environment. Being the largest reservoir of Ca2+ in the cell, ER also maintains Ca2+-homeostasis in the cell, and influences ROS production from mitochondria under certain physiological and several pathological conditions (114). Oxidative protein folding in ER is one of the major contributors of ROS in the cell under physiological conditions. In eukaryotic cells, disulfide bond formation in the nascent proteins is achieved through exchange of electrons between cysteine residues of the substrate protein and the ER oxidoreductase, protein disulfide isomerases (PDI) (25) (Fig. 3). Electrons are then transferred to an acceptor molecule, for instance, ER oxidoreductase 1 (ERO1) or peroxiredoxin IV, to continue the process of protein folding, and are finally transferred to molecular O2, resulting in the production of H2O2 (204). Therefore, the main ROS produced in ER is H2O2, although there are reports of production of O2 •− as well (155).
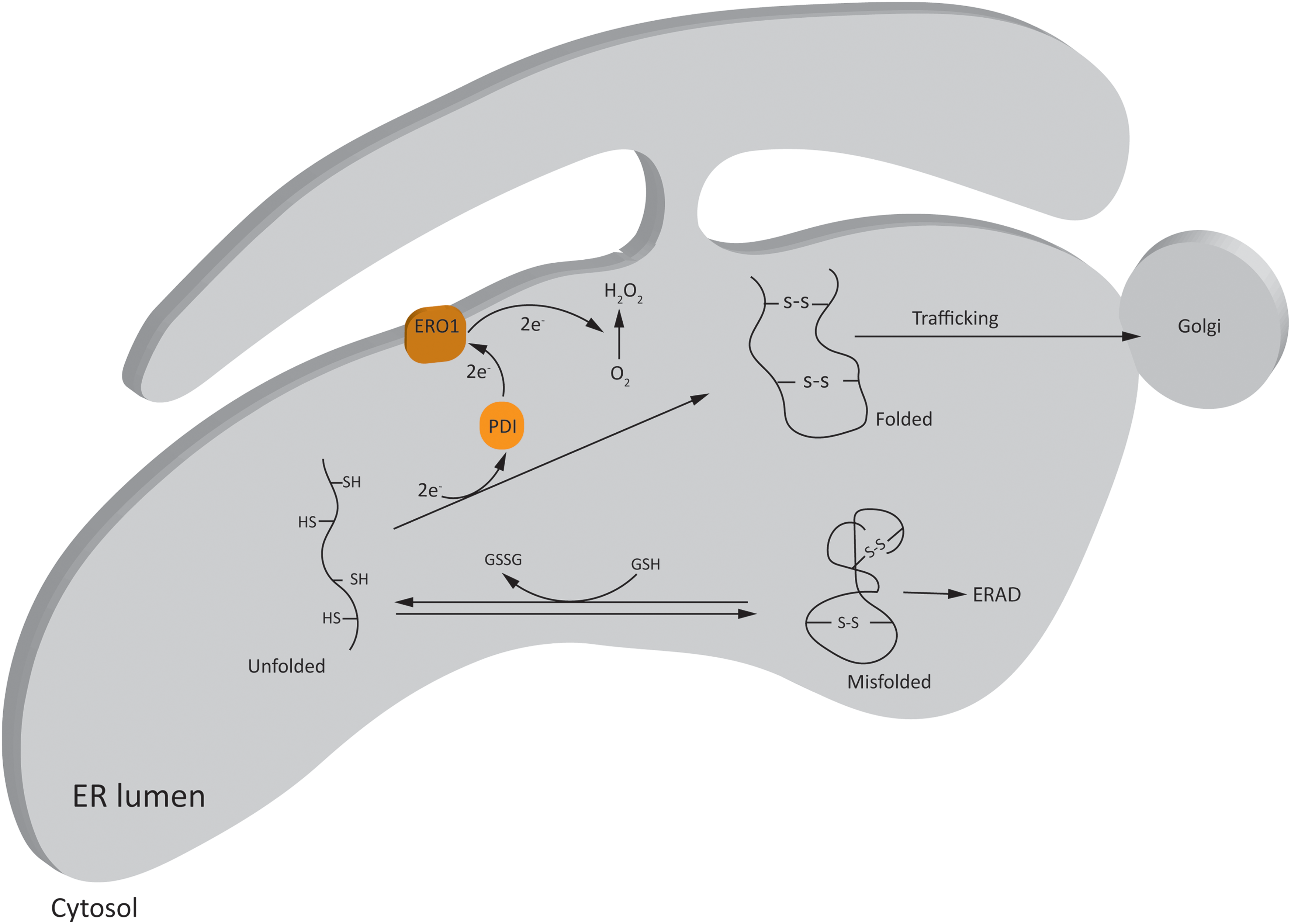
The optimal redox requirements for protein folding in ER are facilitated by a low ratio of reduced glutathione (GSH) to oxidized glutathione (GSSG; 3 to 1:1 compared with the cytosolic ratio of 30 to 100:1) (80). Unlike mitochondria, low levels of GSH and other antioxidant defense systems under basal conditions (111) render ER particularly susceptible to oxidative stress in the case of folding overload.
Proper protein folding in ER is dependent on several intracellular factors. These include energy homeostasis, glycosylation (for trafficking of proteins to Golgi apparatus), and the maintenance of intra-ER oxidative environment and high Ca2+ concentration (155). The latter is required for the proper functioning of redox-dependent enzymes PDI and ERO1, and Ca2+-binding chaperons such as calnexin and calreticulin. Perturbations of these factors, for instance, by extracellular stressors including HI and infection, cause accumulation of mis/unfolded proteins and trigger a process called ER stress (25). To restore protein homeostasis (proteostasis), cells activate the unfolded protein response (UPR).
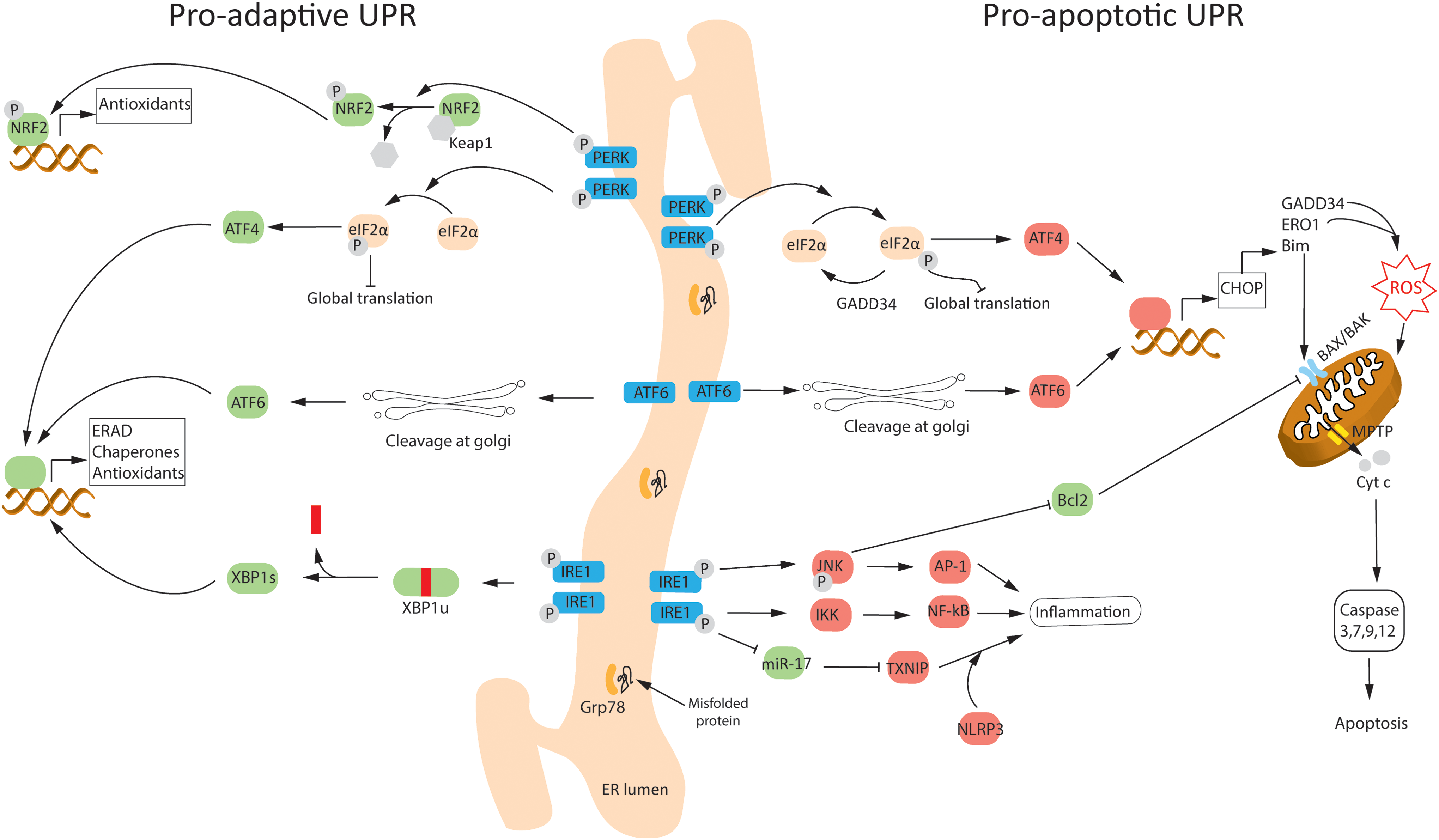
UPR triggers three ER stress-sensor pathways, namely inositol-requiring protein (IRE1), activating transcription factor-6 (ATF6), and RNA-activated protein kinase-like endoplasmic reticulum kinase (PERK) (Fig. 4). These pathways reduce messenger RNA (mRNA) translation globally (to decrease protein folding load in ER), increase expression of protein folding chaperons (to enhance the folding capacity of ER), and activate components of the ER-associated protein degradation (ERAD) pathway and/or autophagy (to get rid of terminally mis/unfolded proteins). The aim of UPR is to promote cell survival; however, severe/chronic ER stress, or pharmacological/genetic inhibition of UPR components, leads to UPR-mediated cell apoptosis (25, 155). Importantly, both the survival and apoptotic signals are simultaneously activated during UPR (155).
UPR is activated in almost all neurodegenerative disorders, including aging-related neurodegenerative conditions and acute CNS injuries (201), for instance, adult stroke (129) and neonatal HIE (26, 31). The immature brain is more sensitive to ER stress post-HI than the mature brain due to the poorer induction of chaperone protein expression, for instance, the 78 kDa glucose-regulated protein or binding immunoglobulin protein (Grp78/BiP) (169).
Under normal physiological conditions, the three transmembrane ER stress sensors are inactivated via binding with the most abundant protein chaperone in the ER lumen, Grp78 (155). Accumulated mis/unfolded proteins competitively bind to Grp78, causing the activation of ER stress sensors (Fig. 4). IRE1 possesses a kinase and an endoribonuclease domain in its cytosolic portion. On dissociation from Grp78, IRE1 undergoes dimerization and transautophosphorylation. This activates the prosurvival arm of IRE1, promoting the splicing of a 26 kDa intron from the mRNA of X-box binding protein 1 (XBP-1), leading to its successful translation (24). The transcription factor XBP-1 in turn activates the expression of chaperone proteins Grp78, Grp94, and calreticulin (to enhance ER protein folding capacity), antioxidant enzymes catalase and SOD1 (to reduce ROS accumulation), and ERAD components, including ER degradation-enhancer, mannosidase alpha-like protein 2 (EDEM2; to degrade terminally mis/unfolded proteins) (36, 101). IRE1, together with PERK, can also induce autophagy, which can inhibit the UPR in a negative feedback loop as noted after neonatal HI (26).
On the contrary, severe/chronic ER stress activates the proapoptotic arm of IRE1, where kinase activity of IRE1 recruits tumor necrosis factor receptor-associated factor 2 (TRAF2) to ER membrane. TRAF2 recruits apoptosis signal-regulating kinase 1 (ASK1), which leads to phosphorylation/activation of c-Jun N-terminal kinases (JNK) and downstream inhibition of antiapoptotic B cell lymphoma 2 (Bcl-2) protein, leading to apoptosis (36). IRE1 may also lead to apoptosis via cleavage of ER-localized procaspase-12, either via clustering and activation of procaspase-12 with TRAF2 (198), or via activation of calcium-dependent proteases, calpains, as shown in the OGD model of cerebral ischemia (7). TRAF2-mediated activation of JNK/activator protein-1 (AP-1) and nuclear factor κ-B (NF-κB) signaling also leads to activation of proinflammatory cytokines, thus linking ER stress with the inflammatory pathways (164) (Fig. 4).
Transient phosphorylation of IRE1 and XBP-1 splicing has been shown to occur immediately after HI in term-equivalent neonatal rodents, which is followed by upregulation of Grp78/94 and EDEM2 (7, 26, 31). Moreover, activation of proapoptotic and proinflammatory pathways during development of neonatal HI injury, including activation of caspase-3, 9, and 12 (176), and JNK and NF-κB, is well documented (69). Finally, inhibition of endoribonuclease domain of IRE1α using a specific inhibitor, STF-083010, is shown to protect against neonatal HI via suppression of nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing-3 (NLRP3)-mediated inflammasome formation (32).
On dissociation from Grp78, PERK also dimerizes and gets phosphorylated. During prosurvival, UPR-activated PERK phosphorylates eukaryotic translation initiation factor 2 subunit α (eIF2α), leading to transient downregulation of global protein synthesis, while maintaining translation of a specific PERK target, activating transcription factor 4 (ATF4) (112) (Fig. 4). ATF4 promotes cell survival by inducing the expression of chaperones, and genes involved in amino acid metabolism, redox homeostasis, stress response, and antioxidant defense (e.g., heme oxygenase-1 via NRF2). The antioxidant defense is also induced directly via PERK-mediated phosphorylation of NRF2 (47), which is discussed in more detail in the ER stress-associated ROS and NRF2 section.
However, chronic activation of ATF4 during prolonged or severe ER stress causes downstream activation of C/EBP homologous protein (CHOP, aka GADD153) and DNA damage inducible gene 34 (GADD34). GADD34 restores protein synthesis by dephosphorylating p-eIF2α via interaction with protein phosphatase 1 (PP1) (112). Inhibition of GADD34/PP1complex using salubrinal is shown to be neuroprotective in rodent models of excitotoxicity (165) and focal ischemia (128), suggesting that the PERK-eIF2α axis plays a crucial cell survival role in the acute phase of injury. CHOP is the master regulator of the proapoptotic branch of UPR because it inhibits the expression of prosurvival/antiapoptotic genes, Bcl-2 and Bcl-2-associated X protein (BAX), and upregulates the expression of proapoptotic genes, Bcl-2 interacting mediator of cell death (Bim) and p53 upregulated modulator of apoptosis (PUMA) (193). Immediate although transient phosphorylation of PERK and eIF2α occurs after neonatal HI in rodents, whereas the expression of CHOP and GADD34 increases in the subacute phase (24 h post-HI) as injury develops (7, 26, 31, 110).
On dissociation from Grp78, ATF6 translocates to the Golgi apparatus, where it is cleaved by site-1 and site-2 proteases to produce a cytosolic fragment that translocates to the nucleus and initiates transcription of chaperone genes, Grp78/94, and ERAD components, to promote ER folding capacity (75) (Fig. 4). ATF6 can also induce the expression of Bim and activate CHOP (via unknown mechanisms) to induce apoptosis (140). Cortical neurons exposed to OGD showed decreased expression of full-length ATF6 (7). The active cleaved form of ATF6 was not detectable; however, the expression of partially glycosylated form of ATF6, which shows increased migration to Golgi apparatus, was increased (7). ATF6 protein levels increased following HI in neonatal rats, although the authors did not discuss which isoform of ATF6 was measured (110).
In summary, chronic ER stress induces proapoptotic UPR, which contributes to cell death during neonatal HI. Not surprisingly, neuroprotection against neonatal HI and in vitro OGD stress via different paradigms is shown to involve attenuation of chronic ER stress [for recent review, see Thornton et al. (174)].
ER stress-associated ROS production
ER stress and oxidative stress accentuate each other via a positive feedback mechanism and collectively regulate the fate of the cell (25). ER stress-associated ROS production has been primarily elucidated using non-neuronal cells or adult neurodegenerative experimental models (25, 129). Therefore, the mechanisms are largely unknown in the immature brain. Nevertheless, amelioration of ER stress by neuroprotective antioxidants, for instance, melatonin (27) and basic fibroblast growth factor (110), exemplifies the cross talk between ER stress and oxidative stress during neonatal HI.
ROS are both a trigger and a consequence of ER stress. Exogenous oxidants, including peroxides, ROS generators, metal ions, and lipid oxidation products, can induce ER stress by disturbing protein folding (155). However, the strength of resulting ER stress is dependent on the type of cell and stimulus. For instance, 7-ketocholesterol induces full UPR in macrophages, an effect that is reversible via the antioxidant N-acetyl cysteine (NAC) (104). Exogenous ROS, for instance, H2O2, do not induce UPR in renal tubular cells (197), induce a partial UPR (upregulation of Grp78 but not CHOP) in primary murine cortical neurons (17), and a full UPR (upregulation of CHOP and downstream Bax-Bim) in neural stem cell line, C17.2 (34), and neuroblastoma cell line, SH-SY5Y (195). These effects can be reversed by 4-phenylbutyrate, a synthetic chaperone. SOD1 overexpression in adult mice and rats suppressed MCAO-induced activation of ATF4 and CHOP, resulting in neuroprotection (73). This was associated with reduced lipid peroxidation in ER, confirming that oxidative stress damages ER and triggers ER stress during adult brain ischemia. Although SOD1 overexpression was not neuroprotective against neonatal HI (53), the rapid induction of oxidative stress and ER stress following neonatal HI (11, 31) suggests that both these stresses likely accentuate each other and contribute to neuronal death and subsequent brain injury in the immature brain (174).
ER oxidoreductases and ROS
ER stress triggers ROS production through different pathways, including the overactivation of ER oxidoreductases, Ca2+-mediated stimulation of mtROS production, and activation of NADPH oxidase (NOX) (Fig. 5).

GSH is oxidized to allow repair of mispaired disulfide bonds in the substrate proteins, via the PDI-ERO1-H2O2 cycle. However, prolonged ER stress causes futile repetition of this cycle due to an increase in un/misfolded proteins, leading to accumulation of H2O2 and depletion of antioxidant GSH (74). As UPR progresses, activation of ERO1α and GADD34 by CHOP further increases the accumulation of H2O2 (172). ERO1α also opens the ER Ca2+-release channel, inositol-1,4,5-triphosphate receptor (IP3R), which activates Ca2+/calmodulin-dependent protein kinase (CaMKII) in the cytoplasm (103). Both Ca2+ (via activation of NOS) and CaMKII (via activation of NOX2) increase ROS production, which causes more ER stress due to activation of CHOP (via double-stranded RNA [dsRNA]-activated protein kinase) and opening of another ER Ca2+-release channel, ryanodine receptors (RyRs; due to oxidation of their thiol groups) (41, 104). Ca2+ released from ER enters mitochondria and increases mtROS production (discussed in the ER-mitochondrial cross talk and ROS section), which can lead to cell death via opening of the mitochondrial permeability transition pore (MPTP) (15). Indeed, Ca2+ released from IP3R/RyRs and the subsequent mitochondrial dysfunction are implicated in neuronal and oligodendroglial death following excitotoxic injury (151, 152).
Taken together, ER stress-induced ROS increase total cellular ROS production (termed ROS-mediated ROS release), which further aggravates ER stress (155). This vicious cycle of ER and oxidative stress severely hampers cellular homeostasis, leading to cell death (25). Because UPR-mediated ROS production from mitochondria is an important event in ROS-induced cell death, this is discussed in more detail in the following section.
ER-mitochondrial cross talk and ROS
ER and mitochondria are in close physical and functional apposition to each other via structures known as mitochondria-associated ER membranes (MAMs). MAMs cover about 20% of outer mitochondrial membrane in mammalian cells (147). The interorganelle distance between ER and mitochondria at MAMs is between 10 nm (smooth ER) and 50 nm (rough ER) (63, 185). MAMs are considered to provide the communication hub that facilitates gathering and cross talk between ER and mitochondrial components during health and disease. For instance, MAMs may facilitate ER to mitochondrial Ca2+ transfer because of their enrichment in IP3R/RyRs and mitochondrial voltage-dependent anion channels (VDACs) (171) (Fig. 5). Similarly, the ER-mitochondrial cross talk during different stages of UPR may also be facilitated via MAMs because several ER cofactors and chaperons are located at MAMs, including ERO1α and PERK (183).
The number of MAM sites increase during proadaptive UPR, promoting cell survival via Ca2+ uptake and maintenance of mitochondrial bioenergetics (21). However, functional alterations in these connections activate the proapoptotic arm of UPR (22). Due to methodological constraints (owing to the nanoscale size of MAMs), it is not yet known if localized ROS signaling occurs at MAMs (46). Nevertheless, using synthetic mito-ER linkers, Booth et al. (15) showed that Ca2+ microdomains, created by ER Ca2+-release channels at the MAMs, stimulate the release of H2O2 from mitochondrial cristae. This creates localized H2O2 nanodomains at the MAMs, stimulating ER Ca2+-release channels via positive feedback to sustain the efflux of Ca2+ from ER (25, 155).
In the initial stages of neonatal HI, neurons show enlargement of ER and increase in chaperone protein calreticulin (31). Dilated ER accumulates Ca2+ deposits (139). As a protective mechanism, this enlargement of ER is suggested to sequester Ca2+ away from mitochondria (31), and possibly decrease the concentration of misfolded proteins in the ER lumen (10). However, as the injury progresses, this protective mechanism is lost, resulting in the loss of calreticulin and release of Ca2+ (31). Mitochondrial uptake of Ca2+, possibly through MAMs, causes opening of MPTP. This releases cytochrome c (cyt c), leading to an increase in ROS via inhibition of C-III (18). Furthermore, cyt c binds to IP3R, stimulating a sustained efflux of Ca2+ from ER in a feed forward loop (14). Accumulation of Ca2+ in mitochondria further increases ROS production by stimulating TCA cycle dehydrogenases, which results in increased oxygen consumption, and by activating NOS, which generates NO, leading to inhibition of C-IV activity (18, 191). In addition, the futile cycle of disulfide bond formation and reduction in ER consumes energy and GSH, causing an increase in mtROS production via enhanced mitochondrial respiration and reduction in ROS detoxification (129).
Attenuation of ER-mitochondrial Ca2+ cross talk, perhaps at MAMs, is neuroprotective against neonatal HI. For instance, pretreatment with dantrolene, an RyR antagonist that prevents release of Ca2+ from ER, showed a mild reduction in infarct size after neonatal HI (66). Inhibition of glycogen synthase kinase-3β (GSK-3β), which is associated with IP3R in MAMs of mouse heart and isolated cardiomyocytes, prevented activation of IP3R, thus reducing Ca2+ transport across MAMs and subsequent cell death (64). GSK-3β is activated after neonatal HI, and pharmacological inhibition of GSK-3β reduces brain injury (79). Whether this neuroprotection involves functional attenuation of MAMs deserves investigation.
NOX, cytochrome P450, and ROS
The NOX enzyme complex contributes significantly to ROS production in the cell (Fig. 5). For instance, NOX catalyzes the production of O2 •− in activated microglia, such as during NMDA-mediated excitotoxicity (54). NOX catalyzes the transfer of an electron from NADPH to molecular O2 via FAD and heme, resulting in the formation of O2 •−. The NOX family consists of 7 members, including NOX1–5 and dual oxidase (Duox) 1–2. The NOX complex consists of cytosolic subunits (p47phox, p67phox, p40phox, and Rac), which bind to the membrane-associated catalytic subunits (gp91phox and p22phox) to form the active NOX complex. NOX is localized in the ER, plasma membrane, focal adhesions, nucleus, mitochondria, and cytoskeleton (77, 155). Neurons primarily express the NOX2 isoform, which is also expressed in microglia, and is responsible for ROS production on microglial activation (81, 154). NOX4 is also expressed in neurons and its expression increases following adult stroke (182). Both NOX2 and 4 are implicated in ER stress-induced ROS production and cell death, effects that are ablated after knocking down these NOX isoforms (104, 134). Recently, NOX4 was shown to form a complex specifically with p22phox subunit in the ER membrane of primary human and mouse fibroblasts (200), and the orientation of NOX4-p22phox complex in the ER membrane suggested release of ROS into ER lumen (200).
Expression of NOX2 subunits, gp91phox and p22phox, increases in ischemic brain following neonatal HI (54, 94), with an earlier and more dramatic increase in the expression of p22phox (54). However, the role of NOX2 in the development of perinatal brain injury is not yet clear. While genetic inhibition of NMDA receptor activation suppresses HI-induced expression of NOX2 and the associated O2 •− production/brain injury (94), both the genetic and pharmacological inhibitions of NOX2 either aggravate or have no effect on HI and excitotoxic brain injury (54). On the contrary, genetic inhibition of NOX2 causes 40% reduction in brain injury following adult ischemic stroke (181). These findings emphasize once again the difference between the mature and the immature rodent brain with regard to injury mechanisms. While NOX2 appears to be the most important NOX member in the development of adult cerebral ischemic injury (87), it does not appear to be critical during perinatal brain injury. The dramatic increase in p22phox assembly subunit of NOX4 following neonatal HI (54), however, suggests that this NOX isoform may play a more important role in perinatal brain injury, but this needs to be investigated.
Cytochrome P450 (CYP) is a monooxygenase that catalyzes the oxidation of its substrate by transferring electrons from NADH/NADPH to O2 via CYP reductase [for an extensive review, see Cederbaum (30)]. The majority of CYP is present in ER (especially from the liver, and also the brain and other organs), with low amounts detected in mitochondria and plasma/nuclear membrane. In the CYP monooxygenase cyclic reaction, the oxygen atoms from O2 are utilized to produce a monooxygenated substrate and an H2O molecule via several CYP-oxygen intermediates. The ROS are produced when the transfer of electrons from NADPH/NADH are not tightly regulated to the transfer of oxygen atom to the substrate, resulting in the reduction of CYP-oxygen intermediates (109). The major ROS produced via the CYP catalytic cycle include O2 •− and H2O2. Notably, ROS production can also occur in the absence of substrate (termed “electron leakage”) (109).
CYP substrates include endogenous molecules, such as fatty acids and hormones, and exogenous molecules, including xenobiotics. Arachidonic acid is released after cerebral ischemia due to the NMDA-mediated influx of Ca2+ into the cell, which stimulates phospholipase A2 (90). Several CYP enzymes catalyze the ω-hydroxylation of arachidonic acid to produce a number of epoxyeicosatrienoic acids (EETs), hydroxyeicosatetraenoic acids (HETEs), and di-HETEs (150). Overexpression of CYP 2J2 is shown to provide neuroprotection against adult cerebral ischemia in mice via increase in the production of EETs (107). Conversely, pharmacological inhibition of CYP 4A-mediated 20-HETE production using N-hydroxy-N′-(4-n-butyl-2-methylphenyl)formamidine (HET-0016) is found to be neuroprotective against HI injury in neonatal piglets (194). HET-0016 is also shown to enhance the neuroprotective efficacy of therapeutic hypothermia against neonatal HIE (203).
ER stress-associated ROS and NRF2
During proadaptive UPR, PERK recruits the master regulator of antioxidant defense, NRF2. Cells that are deficient in NRF2 are more sensitive to an ER stress inducer and undergo apoptosis in less stressful situations than normal (48). Under physiological conditions, the NRF2 protein is kept low by constitutive breakdown by the proteasome. Stress conditions and phosphorylation can route NRF2 from degradation to transport to the nucleus. NRF2 binds to specific DNA sites termed “anti-oxidant response elements”/“electrophile response elements” to initiate transcription of an array of cytoprotective genes. Activated PERK phosphorylates NRF2, which perhaps in combination with other stimuli releases NRF2 from its sequester protein kelch-like ECH-associated protein 1 (KEAP1) (Fig. 4) (48). This can initiate NRF2-dependent transcription of genes coding for antioxidant proteins such as GSH synthesizing enzymes.
It is important to note that NRF2 has multiple phosphorylation sites, some of which lead to activation and some of which cause inactivation/degradation of NRF2. Activating effects have been shown by protein kinase C (12), PI3K (127), JNK, extracellular regulated kinase (190), and PERK (48). Inhibitory effects have been reported for the mitogen-activated protein kinase (MAPK), p38, and GSK-3β (40, 91). The level of NRF2 is also regulated by protein p62, an important factor in autophagocytosis, several microRNAs (miRNAs), and epigenetic modifications such as histone acetylation/methylation and DNA methylation (153).
From this, it follows that the activation of NRF2 after ER stress is dependent on the molecular context at that particular time. For example, depending on the severity and length in time of inflammation, NRF2 can be both up- and downregulated by activation of different kinases. For instance, in a situation where NRF2 is upregulated as a result of acute inflammation (42 –45), the oxidative stress and brain injury by free radicals generated by ER stress and mitochondria are reduced. On the contrary, chronic inflammation can decrease NRF2 protein levels and reduces NRF2-mediated transcription via p38, GSK-3β, and/or decreased acetylation of histone 3/4 (42, 44, 45). For example, the decrease in NRF2 parallels the sensitization of neonatal HI brain injury caused by peripheral inflammation, when animals are injected with LPS 72 h before HI (43, 186). The sensitization by LPS was attenuated by NAC, which normalized the reduced GSH levels and decreased apoptosis (186).
The NRF2 system also induces enzymes and other proteins that regulate the detrimental effects of labile free iron, that is, activation of NRF2 can limit the increase in the Fenton reaction and in elevated lipid peroxidation (1). A decreased antioxidant NRF2 system due to inflammation, and followed by stress such as HI, will likely increase the proportion of iron-mediated ferroptosis with increased lipid peroxidation and •OH-mediated mitochondrial damage compared with a situation where the NRF2-system is intact [for recent reviews, see Abdalkader et al. (1) and Stockwell et al. (167)]. Analysis of inflammatory markers in the cerebrospinal fluid of preterm infants show a proinflammatory profile compared with term infants, and the cord blood of preterm infants has higher levels of free iron (13, 137). Speculatively, the elevated level of inflammation will decrease the NRF2-mediated antioxidant capacity, which in combination with high free iron will elevate the risk of oxidative stress that may be accompanied by the later cognitive dysfunction observed in preterm infants with elevated concentrations of circulating inflammation-associated proteins (98).
In summary, activation of NRF2-mediated transcription to increase the antioxidant capacity is an important protective function of the proadaptive UPR. Prolonged inflammation can reduce this ability of a cell to activate NRF2 (153) and one consequence is lower levels of glutathione. This may lead to pronounced H2O2 production in ER-stress and mitochondria after HI because of the decreased capacity of the glutathione-dependent GPx reaction to convert H2O2 to water. Thus, in a situation with HI combined with inflammation, therapeutic strategies that can elevate glutathione, such as NAC (186), have a stronger neuroprotective potential compared with conventional antioxidants.
Therapeutic Implications
ROS or RNS species are produced in excess during inflammation and I/R, and have been implicated as major mediators of perinatal brain injury (59). NAC, an antioxidant and a GSH precursor, provides neuroprotection (up to 78% reduction of brain injury) in neonatal rats (186). This is associated with improvement in the redox state and inhibition of apoptosis due to increased glutathione levels. As a major site of ROS production, targeting mtROS species is now being considered a therapy for inflammatory and neurodegenerative diseases (108). Many mitochondria-targeted antioxidants have been developed by conjugation to the lipophilic triphenylphosphonium (TPP) cation such as SKQ1, MitoTEMPO, and CoQ10 (86).
Mitochondria-targeted ubiquinone (MitoQ) has shown protective effects in the ischemic brain (3), and against oxidative damage in mitochondria (58). MitoQ comprises coenzyme Q10 and lipophilic TPP tail, which can accumulate within the mitochondria preventing mitochondrial oxidative damage (Fig. 6). MitoQ accumulates in the inner mitochondrial membrane and into the mitochondrial matrix 100- to 500-fold depending on the membrane potential (89), where it is recycled into the active ubiquinol form (125). This reduction enables MitoQ to function as a highly effective antioxidant by reacting with ROS and inhibiting ONOO− formation (5). Similarly, administration of SkQ1, a cardiolipin peroxidation inhibitor, mediates mitochondrial protection from excessive ROS accumulation by breaking the chain reaction of lipid destruction. SkQ1 decreases the mitochondrial fragmentation after cerebral ischemia and prevents progression of the apoptotic cascade (163).

MitoTEMPO, a specific scavenger of mitochondrial O2 •−, can easily pass through the lipid bilayers and accumulate in the mitochondria (131). It must be noted that SkQ1 and MitoTEMPO failed to exert long-term beneficial effects in a model of murine polymicrobial sepsis (144). S1QELs and S3QELs are site-specific suppressors of mitochondrial O2 •−/H2O2 generation that act on site IQ of C-I and have no effect on OXPHOS (18). The molecule Mdivi-1, which was originally identified as a specific inhibitor of the fission mediator DRP1, also acts as a reversible mitochondrial C-I inhibitor and regulator of ROS production (16) (summarized in Fig. 6). Most of the abovementioned agents have not been tested in animal models of perinatal brain injury but could serve as important tools in the future to dissect and understand ROS-related pathways and maybe even as potential therapies.
Only partial neuroprotection was observed against neonatal HI by two studies that used specific ER stress inhibitors, STF-083010 (IRE1α RNase-inhibitor) (32) and dantrolene (an RyR antagonist) (66). Because ER stress and oxidative stress accentuate each other, and occur in both acute and subacute phases of neonatal HI (preceding the delayed phase of neuronal death via programmed necrosis) (31), a combinatorial therapy utilizing both antioxidants and ER stress inhibitors may prove to be more protective. Furthermore, since the immature brain is particularly vulnerable to ER stress following HI because of poor expression of chaperone proteins (169), a therapeutic approach combining antioxidants with synthetic chaperons, for instance, 4-phenylbutyrate, which is shown to be neuroprotective against adult HI injury (141), may offer a promising avenue for testing.
Conclusions and Future Directions
Compared with the mitochondria-associated oxidative stress, ER stress-mediated oxidative stress has not been well studied in the context of perinatal brain injury (174). Considering that the findings from mature brain cannot be simply transferred to the immature brain (54, 62), therapeutics that specifically target ER stress may help delineate the ER stress-associated ROS pathways involved in the development of perinatal brain injury.
Antioxidants targeting ROS production have shown improved outcomes experimentally, but these compounds show poor clinical translation. Although antioxidant therapy still holds promise, scavenging ROS is probably not the most effective way of limiting their impact on surrounding tissue. The majority of antioxidants target ROS only after they are formed (115), giving adequate time to ROS to cause damage before antioxidants can act. Targeting the source of ROS production rather than ROS themselves may offer better neuroprotection than classical antioxidants. Speculatively, drugs modulating specific ROS producing sites in individual complexes such as C-I or C-III or strengthening the endogenous antioxidant systems may be more effective and serve better as neuroprotectants. Understanding the protective versus the damaging effects of ROS could be the key to developing a safe and effective therapy for perinatal brain injury. The role of ROS in brain repair in the immature brain is largely unexplored.
Footnotes
Acknowledgments
We gratefully acknowledge the support from ERA-net (EU; VR 529-2014-7551), Wellcome Trust (WT094823), Swedish Medical Research Council (VR 2015-02493, H.H.; VR-2017-01409, C.M.), National Institute of Health (GM044842, M.S., C.M.), Brain Foundation (H.H., C.M.), Ahlen Foundation (H.H., C.M., S.N.), Tore Nilsons Foundation (S.N.), grants from the Swedish state under the agreement between the Swedish government and the county councils, the ALF-agreement (426401, H.H.; 722491, C.M.), and the Leducq Foundation (DSRRP34404), Torsten Söderberg (M98/15, C.M.), Frimurare Barnhusdirektionen (S.N.) to enable this study to be completed.
Author Disclosure Statement
No competing financial interests exist.