
Abstract
Aims:
Acetaminophen (APAP) overdose leads to acute liver injury by inducing hepatic mitochondrial oxidative stress and inflammation. However, the molecular mechanisms involved are still unclear. Farnesoid X receptor (FXR) serves as a therapeutic target for the treatment of liver disorders, whose activation has been proved to protect APAP-induced hepatotoxicity. In this study, we examined whether FXR activation by schaftoside (SS), a naturally occurring flavonoid from Desmodium styracifolium, could protect mice against APAP-induced hepatotoxicity via regulation of oxidative stress and inflammation.
Results:
We first found that SS exhibited potent protective effects against APAP-induced hepatotoxicity in mice. The study reveals that SS is a potential agonist of FXR, which protects mice from hepatotoxicity mostly via regulation of oxidative stress and inflammation. Mechanistically, the hepatoprotective SS is associated with the induction of the genes of phase II detoxifying enzymes (e.g., UGT1A1, GSTα1), phase III drug efflux transporters (e.g., bile salt export pump, organic solvent transporter protein β), and glutathione metabolism-related enzymes (e.g., glutamate-cysteine ligase modifier subunit [Gclm], glutamate-cysteine ligase catalytic subunit [Gclc]). More importantly, SS-mediated FXR activation could fine-tune the pro- and anti-inflammatory eicosanoids generation via altering eicosanoids metabolic pathway, thereby resulting in decrease of hepatic inflammation. In contrast, FXR deficiency can abrogate the above effects.
Innovation and Conclusion:
Our results provided the direct evidence that FXR activation by SS could attenuate APAP-induced hepatotoxicity via inhibition of nuclear factor kappa-B signaling and fine-tuning the generation of proinflammatory mediators' eicosanoids. Our findings indicate that strategies to activate FXR signaling in hepatocytes may provide a promising therapeutic approach to alleviate liver injury induced by APAP overdose.
Introduction
Acetaminophen (APAP)
Innovation
This study demonstrates for the first time that farnesoid X receptor (FXR) could be activated by schaftoside (SS), a newly identified naturally occurring FXR agonist, which can alleviate acetaminophen (APAP) overdose-induced liver injury. The hepatoprotective effect of SS is largely associated with the activation of FXR and fine-tune regulation of eicosanoids levels, consequently resulting in reduced liver inflammatory injury, mitochondrial oxidative damage, and hepatocytes apoptosis. Thus, it can be concluded that activation of FXR signaling is a promising approach to attenuate APAP overdose-induced acute liver injury.
However, despite mounting studies regarding the metabolism and toxicity of APAP, the precise molecular mechanism of cell death in APAP-induced ALI remains to be fully elucidated. Currently, the only available antidote for APAP-induced hepatotoxicity is N-acetyl cysteine (NAC), a ROS scavenger, mainly via restoring the depleted hepatic GSH to increase detoxification of NAPQI (38). However, NAC therapy was found to be effective only at very early stage of acute APAP overdose. Delayed and prolonged NAC administration can cause the elevated liver toxicity and impaired liver regeneration (34, 67). Therefore, it is necessary to develop novel therapeutic agents for late-stage APAP detoxification.
Farnesoid X receptor (FXR, NR1H4), a member of nuclear receptor superfamily of bile acid-activated transcription factor, is highly expressed in liver and intestine (22, 58). Once activated, FXR heterodimerizes with the retinoid X receptor (RXR) and binds to FXR response elements (FXREs) (23, 73), thereby regulating the transcription of genes involved in a variety of biological and pathological processes, including bile acid and cholesterol homeostasis, hepatic inflammation and injury (17, 24, 40). FXR is found to play an essential protective role in various liver injury models, including endogenous bile acid-induced cholestasis and exogenous chemical-induced hepatotoxicity (10, 15, 60). For instance, FXR activation can ameliorate α-naphthylisothiocyanate-induced cholestasis via fine-tune regulation of bile acid homeostasis (10, 15). For chemical/drug-induced liver injury, the role of FXR in APAP-induced hepatotoxicity is especially noteworthy. FXR activation can protect APAP-induced ALI in mice, probably via regulation of APAP metabolism and transport, as well as the regeneration of GSH in the liver (39, 60). However, the precise mechanism by which FXR acts for protection against APAP-induced hepatotoxicity remains unclear. More importantly, the role of FXR in APAP-induced ROS generation and liver inflammation also needs to be addressed.
In this study, we explored the possible relationship between FXR activation, APAP-induced hepatic oxidative stress injury, liver inflammation, and xenobiotic metabolism. The beneficial effect of FXR activation for protecting APAP-induced ALI is associated with the induction of a pattern of phase II detoxifying enzymes and phase III drug transport genes that favor decreased hepatic exposure of APAP or NAPQI via enhancing the detoxification capacity and transport. Furthermore, the hepatoprotective effect of FXR activation is related to the reduced liver oxidative stress and suppressed hepatic nuclear factor kappa-B (NF-κB)-mediated inflammatory signaling, probably by fine-tune regulation of eicosanoids metabolism. Besides, we present a naturally occurring flavonoid compound schaftoside (SS) from Desmodium styracifolium as an FXR agonist for amelioration of APAP-induced hepatotoxicity via FXR-mediated oxidative injury and inflammation. Overall, our results support the importance that activating FXR expression is a promising way to preserve liver function in the context of APAP overdose-induced acute hepatotoxicity.
Results
Pretreatment with SS protects mice against APAP-induced ALI
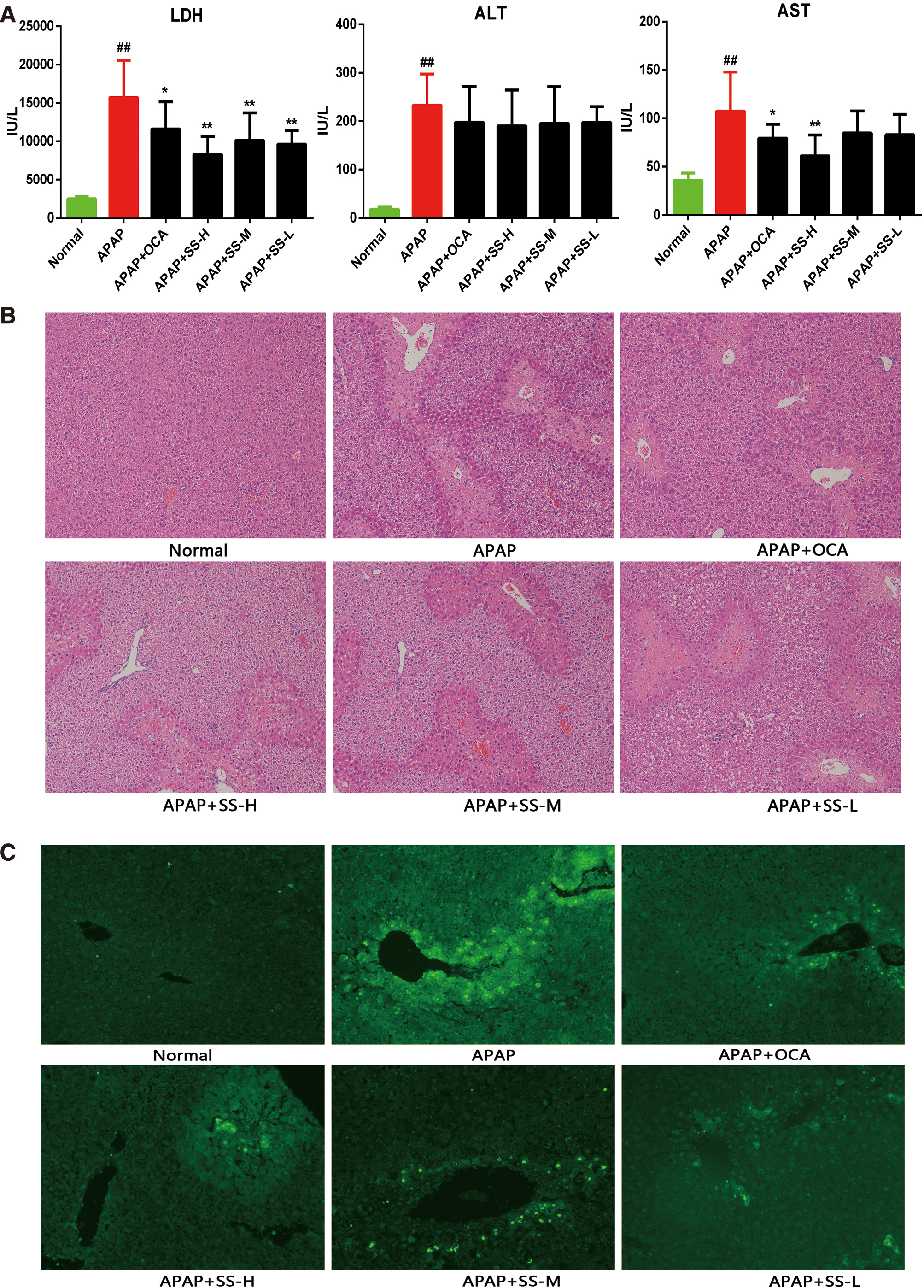
To identify the protective role of SS in APAP-induced ALI, mice were intraperitoneally injected with a lethal dose of APAP (500 mg/kg), and the survival rate was monitored. All APAP-treated mice died after administration of APAP for 24 h (Supplementary Table S1 and Supplementary Fig. S1), while SS pretreatment partially rescued APAP-induced mice mortality (Supplementary Table S1 and Supplementary Fig. S1). Surprisingly, obeticholic acid (OCA), an FXR agonist, exhibited completely reversed APAP-induced death (Supplementary Table S1 and Supplementary Fig. S1). Next, mice were administered with a sublethal dose of APAP (300 mg/kg) to further assess the protective effect of SS. Twelve hours of APAP exposure resulted in notable liver damage to mice, as indicated by the increased level of alanine aminotransferase (ALT), aspartate aminotransferase (AST), and lactate dehydrogenase (LDH) (Fig. 1A). Whereas SS treatment could alleviate APAP-induced mice liver injury, as revealed by decreased serum levels of AST and LDH (Fig. 1A). Meanwhile in SS-treated group, centrilobular hepatic necrosis was also relieved as shown by hematoxylin and eosin (H&E) staining, and the necrotic area was substantially smaller in comparison with the mice treated with only APAP (Fig. 1B). In parallel, DNA fragmentation, a characteristic feature of APAP-induced hepatocyte death (43), was substantially reduced in SS-treated mice compared with the only APAP-treated controls (Fig. 1C). As expected, OCA also exhibited a protective effect against APAP-induced ALI (Fig. 1) (39). Taken together, our results demonstrated that SS treatment could ameliorate APAP-induced hepatotoxicity.
Pretreatment with SS alleviates APAP-induced oxidative damage in mice liver
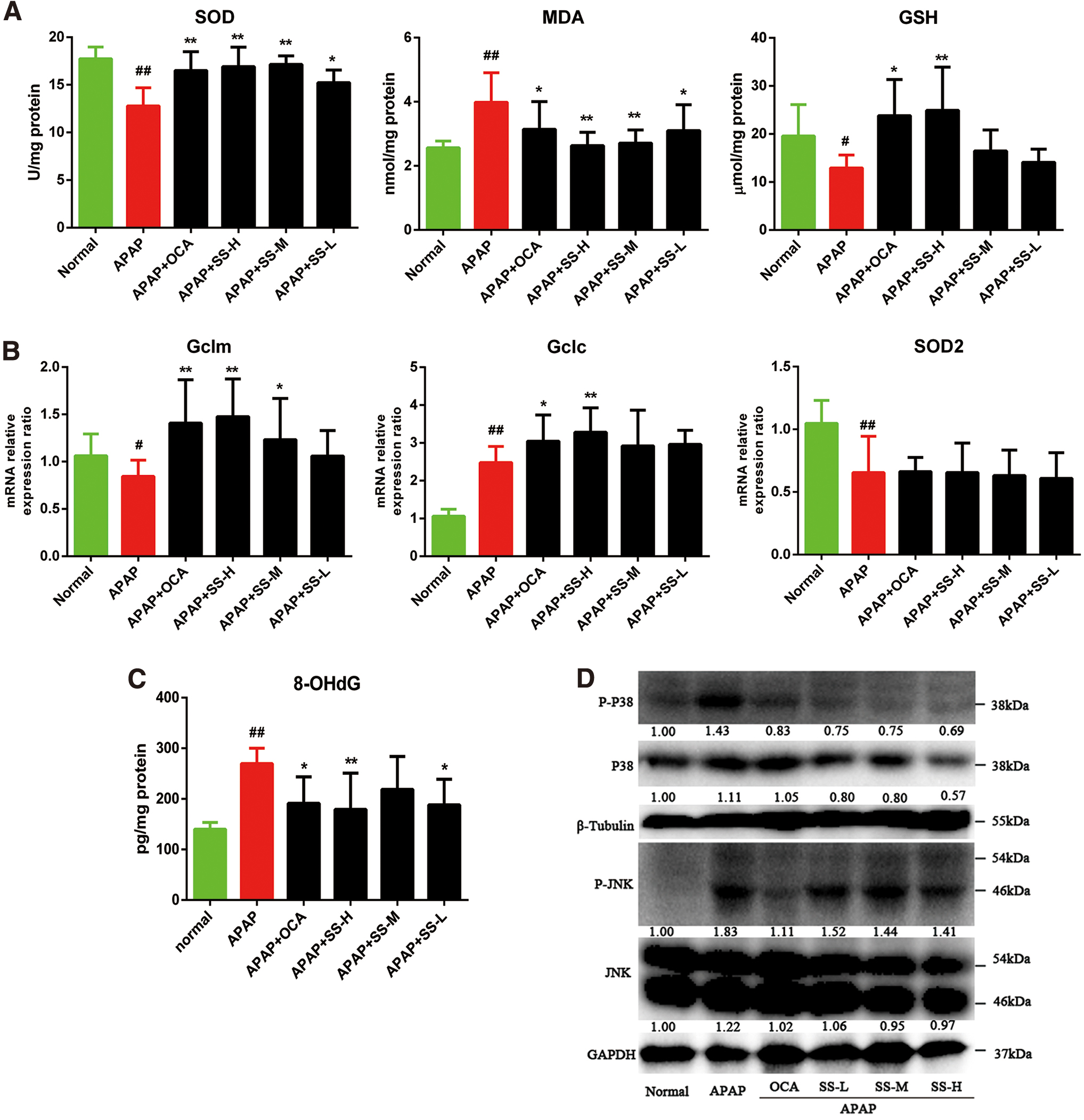
APAP-induced ALI is triggered by hepatic GSH depletion, a hallmark of excessive NAPQI generation during APAP-induced liver toxicity, which further causes oxidative damage (20). Therefore, the protective effect of SS on APAP-mediated oxidative stress was explored. Exposure of mice to APAP for 12 h resulted in a remarkable decrease of hepatic GSH and superoxide dismutase (SOD) levels with an increase of malondialdehyde (MDA) level (Fig. 2A). However, SS pretreatment dramatically increased GSH and SOD levels with reduced MDA levels (Fig. 2A). In agreement with the elevated antioxidative effects in liver, the level of hepatic 8-hydroxydeoxyguanosine (8-OHdG), which is a biomarker of DNA oxidative damage caused by APAP-induced hepatotoxicity, was significantly lowered by SS pretreatment (Fig. 2B). In parallel to the elevated hepatic GSH biosynthesis, gene expression profiles further demonstrated that SS pretreatment could result in enhanced glutamate-cysteine ligase modifier subunit (Gclm) and glutamate-cysteine ligase catalytic subunit (Gclc) gene expression (Fig. 2C), the rate-limiting enzymes in GSH biosynthesis from the modifier subunit of glutamate cysteine ligase (5), but no change in SOD2 was observed (Fig. 2B). It was reported that JNK is activated by sequential protein phosphorylation through a MAPK module, and prolonged JNK activation played an important role in liver cell death. To decipher the molecular basis, phosphorylation level of JNK, an important signaling component of APAP-induced toxicity (53), and the upstream MAPK of JNK (26) were determined (Fig. 2D). Phosphorylation levels of JNK and MAPK were significantly increased after APAP treatment; while SS pretreatment could notably inhibit APAP-induced JNK and MAPK phosphorylation (Fig. 2D, Supplementary Fig. S15). Collectively, our results suggest that the protective effects of SS on APAP-induced liver injury are associated with the enhancement of antioxidative capacity.
Pretreatment with SS attenuates APAP-induced elevation of proinflammatory cytokines generation by suppression of NF-κB signaling pathway
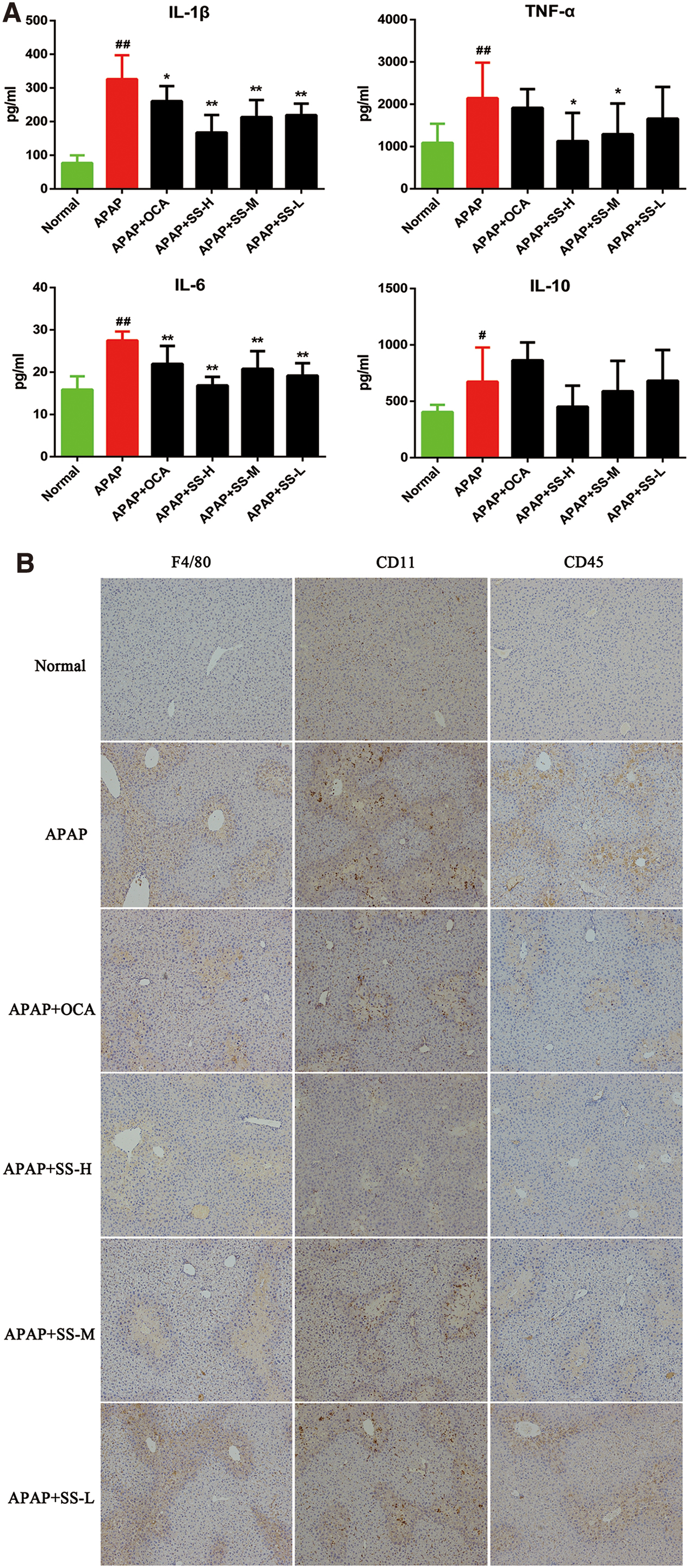
The predominantly accepted concept behind inflammation after APAP overdose is that the process occurs through a sterile inflammatory response (61, 65). Thus, the protective effect of SS in the inflammatory liver injury during APAP exposure was examined. Our results demonstrated that SS could significantly reduce APAP-induced inflammatory liver injury, as shown by suppression of the release of circulating inflammatory cytokines such as interleukin (IL)-1β, IL-6, and tumor necrosis factor (TNF)-α (Fig. 3A). Moreover, IL-6 and CRP, serum acute phase reactants inflammation hallmarks (1, 4), were blunted by SS pretreatment (Supplementary Fig. S2). To further evaluate the anti-inflammatory effect of SS, we examined its influence on inflammatory cell infiltration in liver tissues. Immunohistochemical analysis showed that the expression of F4/80+, CD11b+, and CD45+ in inflammatory cells of APAP-treated liver tissues was significantly increased as compared with that of phosphate-buffered saline (PBS)-treated tissues. However, these proteins expression was attenuated by SS treatment in APAP-treated mice (Fig. 3B). Similarly, the hepatic messenger RNA (mRNA) levels of a master inflammation regulator NF-κB and its downstream proinflammatory genes including IL-1β, IL-6, TNF-α, and inducible nitric oxide synthase (iNOS) were greatly repressed after SS pretreatment. Nevertheless, this treatment did not result in the change of IL-10 and Arg-1 levels (Fig. 3C). Further, SS pretreatment could effectively reduce APAP-induced NF-κB activation, as revealed by reduced NF-κB nuclear translocation (Fig. 3D–E, Supplementary Fig. S15), a major process for NF-κB-mediated inflammation. Together, these data demonstrated that SS can suppress APAP-induced NF-κB activation, and subsequently reduce NF-κB-mediated inflammatory response.

Pretreatment with SS regulates FXR expression level and genes related to APAP metabolism
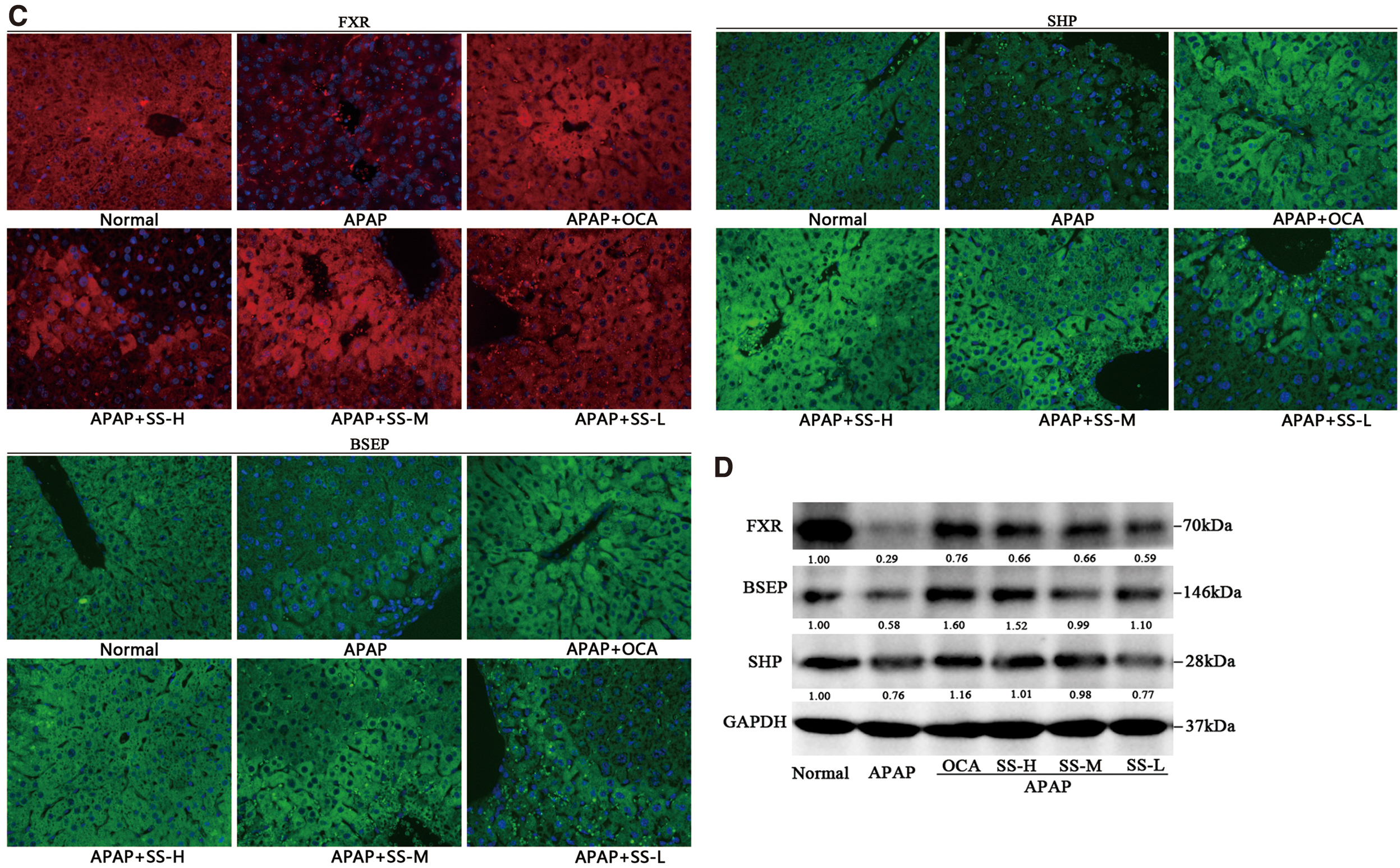
To explore the protective mechanism of SS against APAP overdose, transcriptomics study was used to investigate altered gene expression profiles after APAP overdose. We found that APAP overdose could affect multiple pathways that are involved in the liver damage, such as mitochondrial damage, liver inflammation, hepatocytes apoptosis, and eicosanoids metabolism (Fig. 4A). To further confirm the protective mechanism, metabolizing enzymes and transporters participated in APAP metabolism and elimination were determined. Among phase I enzymes known to facilitate formation of toxic APAP metabolites, expression levels of CYP2E1 and CYP3A11 (human ortholog of CYP3A4) were reduced. However, SS pretreatment resulted in a mild upregulated CYP3A11 gene expression, but not CYP2E1 (Fig. 4B), suggesting a minor role of SS in APAP metabolism by regulation of phase I enzymes. In addition, in terms of phase II enzymes and phase III transporters known to reduce APAP hepatotoxicity by facilitating excretion (12, 33), expression levels of UGT1A11, bile salt export pump (BSEP), organic solvent transporter protein β (OSTβ), NTCP, and OATP1A1 were decreased after APAP administration, while SS pretreatment could reverse this effect (Fig. 4B, D). Intriguingly, APAP treatment could upregulate glutathione S-transferase alpha 1 (GSTA1) gene expression, while SS pretreatment could result in further elevated GSTA1 expression (Fig. 4B). The OCA showed similar regulatory effects (Fig. 4B). It has been found previously that the nuclear receptor transcriptional factor FXR was involved in regulation of APAP metabolism and detoxification (12, 39). We demonstrated that SS treatment could suppress APAP-induced FXR decline (Fig. 4B, C). Protein expression analysis also exhibited that SS could remarkably activate FXR signaling, as indicated by induced expression of FXR and its target genes SHP and BSEP (Fig. 4C, D, Supplementary Fig. S15). Collectively, these results suggest that the protective effects of SS against APAP are largely attributed to altered APAP excretion, which might be associated with the activation of FXR signaling.

SS serves as an FXR agonist to activate FXR signaling in vitro
SS can induce several key genes associated with APAP excretion (e.g., BSEP, OSTβ, NTCP) and detoxification (UGT1A1) in vivo. These genes were mainly regulated by FXR (39), which thereafter prompted us to investigate whether SS can act as FXR agonist to activate FXR signaling, leading to protective effects against APAP-induced liver toxicity. Therefore, the effect of SS in regulation of FXR signaling has been examined in vitro. First, molecular docking study demonstrated that SS formed seven hydrogen bonds with FXR on various residues Arg331, Met265, Gln263, Ser259, and Tyr260 (Fig. 5A and Supplementary Fig. S3). Furthermore, the van der walls interactions were found between SS and FXR mainly through residues Thr270, Ala327, Thr386, Leu298, Asn261, Lys262, Ala291, Met290, Ile335, Leu348, and Ser342, which may help stabilize SS–FXR complex (Fig. 5A and Supplementary Fig. S3). Therefore, the theoretic data pointed out that SS could closely bind with hFXR-ligand-binding domain (LBD) to regulate hFXR signaling. Then, hFXR transactivation study showed that SS could significantly elevate hBSEP luciferase activity in a dose-dependent manner (Fig. 5B). Similar effect was found in the positive assays, where GW4064 (a strong FXR agonist as positive control) and ursodeoxycholic acid (UDCA) (a weak FXR agonist as another positive control) were used separately. SS treatment could significantly increase the gene expression levels of hFXR and BSEP, while inhibiting CYP7A1 gene expression (Fig. 5C). This regulatory effect has been further confirmed by analysis of the corresponding proteins (Fig. 5D, Supplementary Fig. S15). Moreover, SS could promote hFXR nuclear translocation and strengthen hFXR protein expression (Fig. 5E).

Next, to further test whether SS-regulated FXR signaling requires FXR, we examined the expression of FXR downstreaming genes when FXR expression was knocked down by small-interfering RNA (siRNA) technique. The FXR expression was indeed knocked down, indicated by quantitative real-time polymerase chain reaction (qRT-PCR), Western blot, and immunofluorescence assay (Supplementary Fig. S4). As shown in Supplementary Figure S4A–C, the BSEP expression level was elevated, and the CYP7A1 expression level was decreased in the control cells without FXR gene silencing after treatment with GW4064 and SS (Supplementary Fig. S4A–C, Supplementary Fig. S16). In contrast, FXR knockdown greatly abolished GW4064 or SS-regulated BSEP and CYP7A1 expression (Supplementary Fig. S4A–C), indicating that FXR is an essential element for SS-mediated BSEP and CYP7A1 gene expression. Together, these data indicate that SS can serve as an hFXR agonist and activate FXR signaling.
FXR activation by SS protects mice primary hepatocytes against APAP-induced cell death and oxidative stress
We further evaluated the protective effect of SS against APAP-induced hepatotoxicity in mice primary hepatocytes (MPHs) model. It could be found that SS and GW4064 are able to promote nuclear accumulation of FXR (Supplementary Fig. S5), suggesting an activated FXR signaling by SS in MPH. SS pretreatment could effectively ameliorate APAP-induced liver injury as revealed by elevation of cell viability (Fig. 6A) and GSH level (Fig. 6B), and reduction of AST level (Fig. 6C), as well as reduced APAP-induced cell apoptosis (Fig. 6D) in MPHs. Meanwhile, SS pretreatment could also significantly alleviate APAP-induced oxidative stress, as evidenced by suppression of ROS production (Supplementary Fig. S6A) and mitigation of mitochondrial damage (Supplementary Fig. S6B). Next, we investigated the mechanism underlying the protective effect of SS in MPH. JNK signaling has been activated in MPH after exposure to APAP for 12 h, as revealed by the increased protein expression levels of p-JNK and p-MAPKp38 (Fig. 6E). In contrast, SS pretreatment could remarkably suppress the elevated phosphorylation of JNK and MAPK (Fig. 6E). Collectively, these results suggest that SS can relieve APAP-induced hepatotoxicity by reducing cell death and oxidative stress.
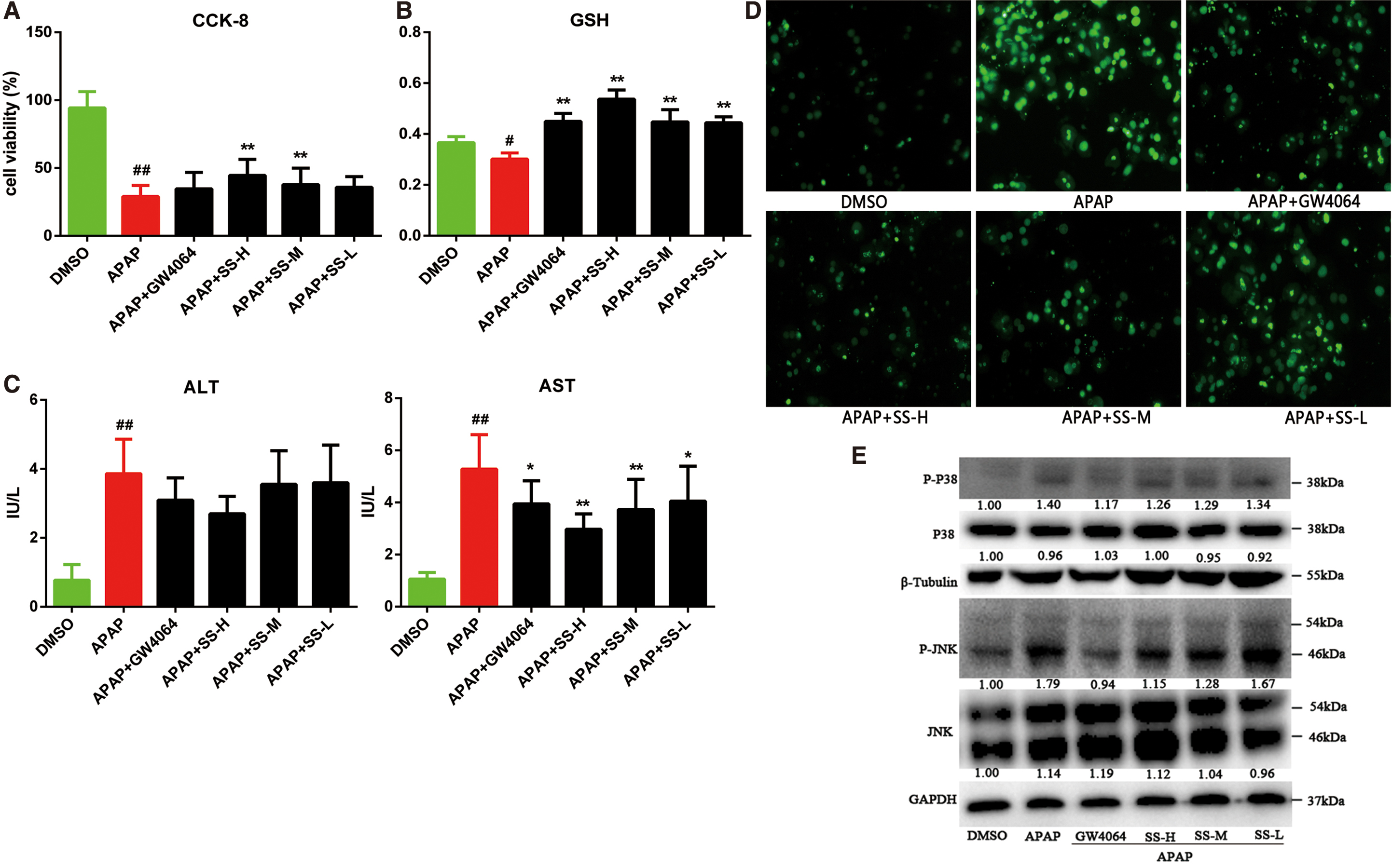
FXR activation by SS alleviates inflammation-mediated cell injury in MPHs
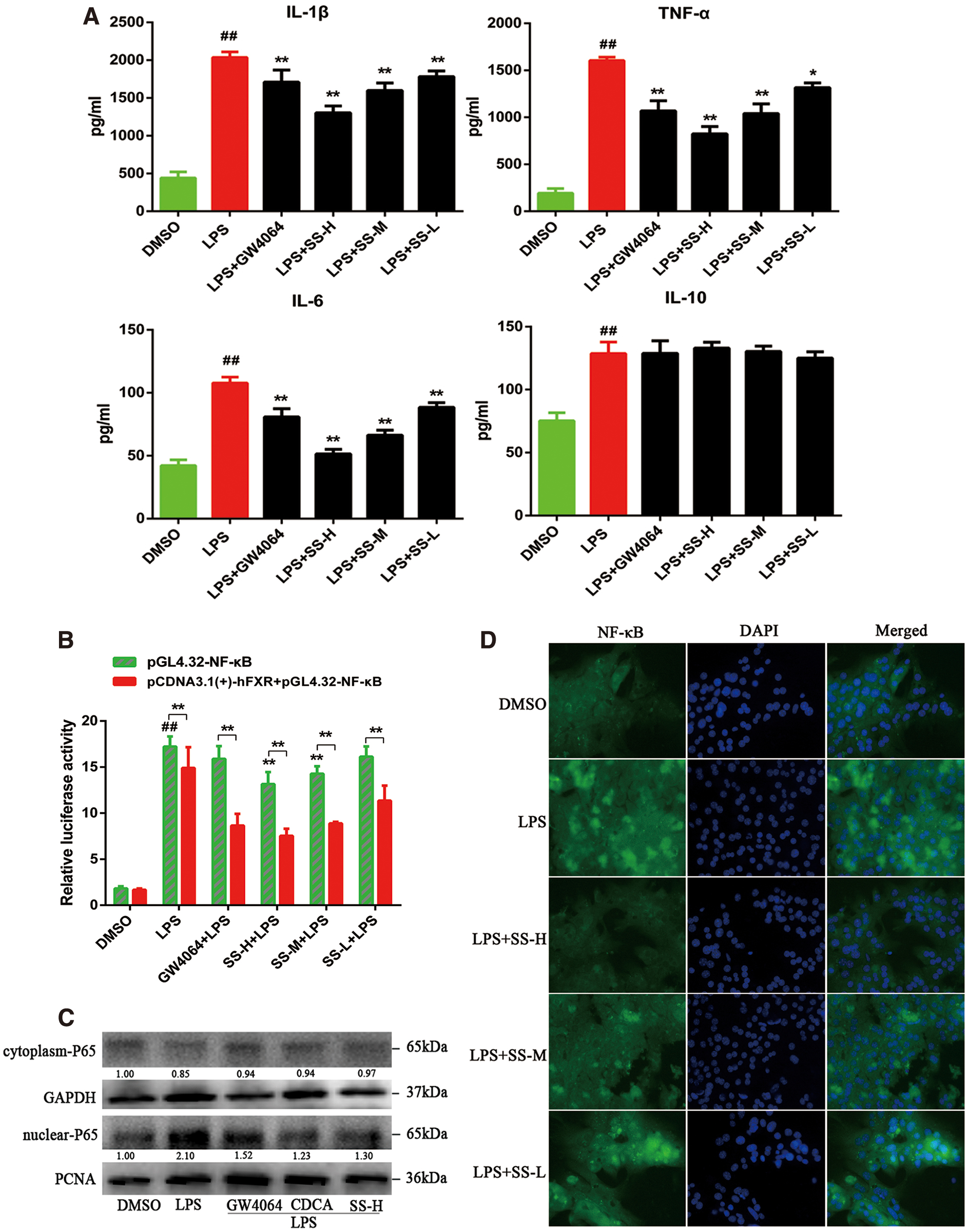
APAP-induced ALI is accompanied by an increase of inflammatory response in the liver (2, 65), and therefore control of inflammation is believed to be a potential therapeutic strategy for alleviation of APAP-induced inflammatory liver injury. We first observed that SS could reduce lipopolysaccharide (LPS)-induced proinflammatory cytokines production in MPHs (such as IL-1β, IL-6, and TNF-α) (Fig. 7A). To demonstrate that the anti-inflammatory property of SS depends on FXR activation in MPHs, NF-κB luciferase activity was evaluated after FXR transfection. SS-mediated repression of inflammation was dependent on the activation of FXR and the inhibition of NF-κB, as demonstrated by the suppressed NF-κB luciferase activity in FXR-transfected cells upon SS or GW4064 treatment (Fig. 7B). SS pretreatment could also notably block LPS-mediated NF-κB p65 nuclear translocation (Fig. 7C, D, Supplementary Fig. S15), and subsequently resulting in repression of proinflammatory genes expression (Supplementary Fig. S7). Collectively, our results demonstrate that activation of FXR by SS can significantly ameliorate LPS-induced inflammatory liver injury.
The hepatoprotective effect of SS against APAP-induced hepatotoxicity is dependent on SS-mediated FXR activation
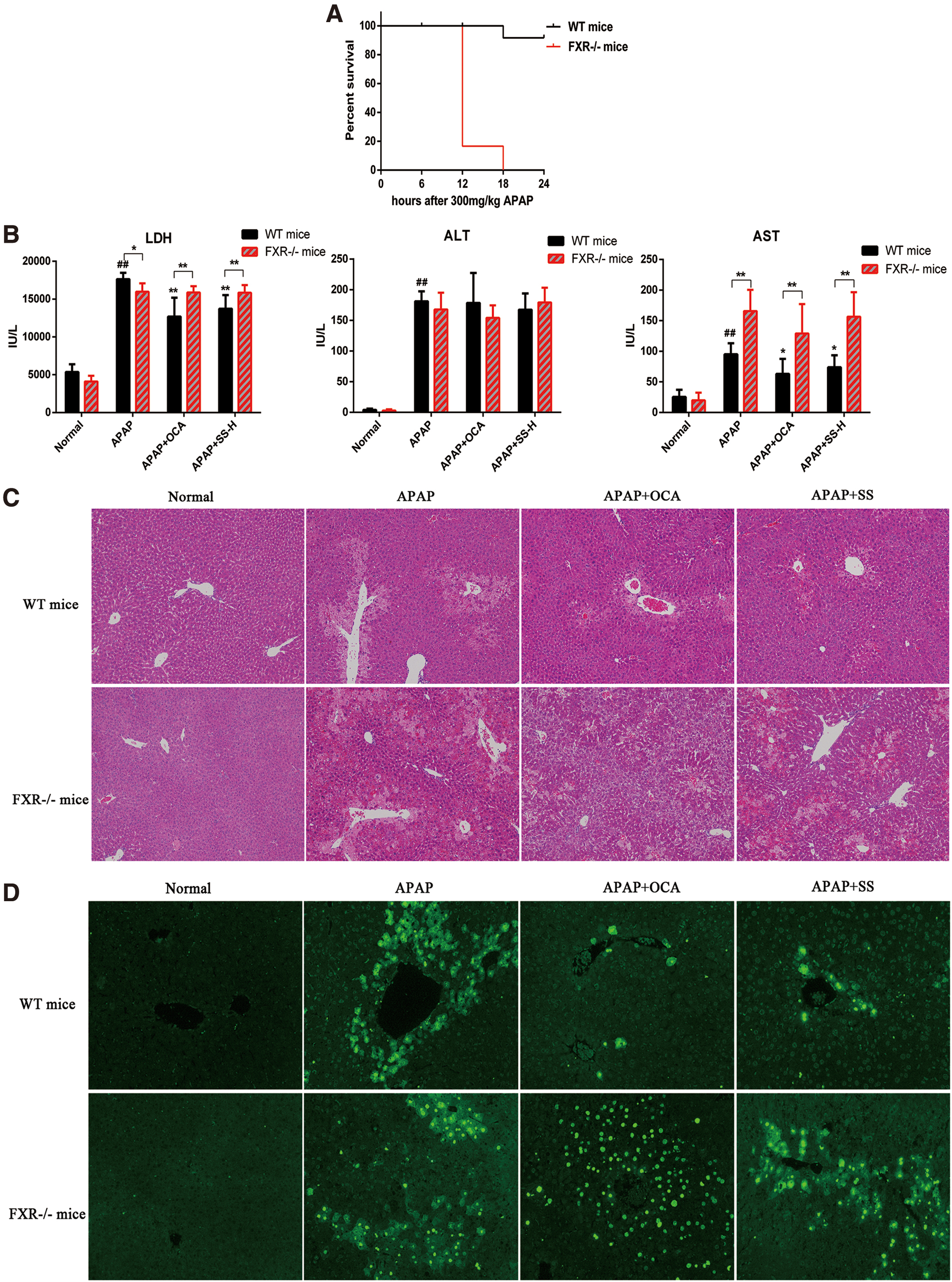
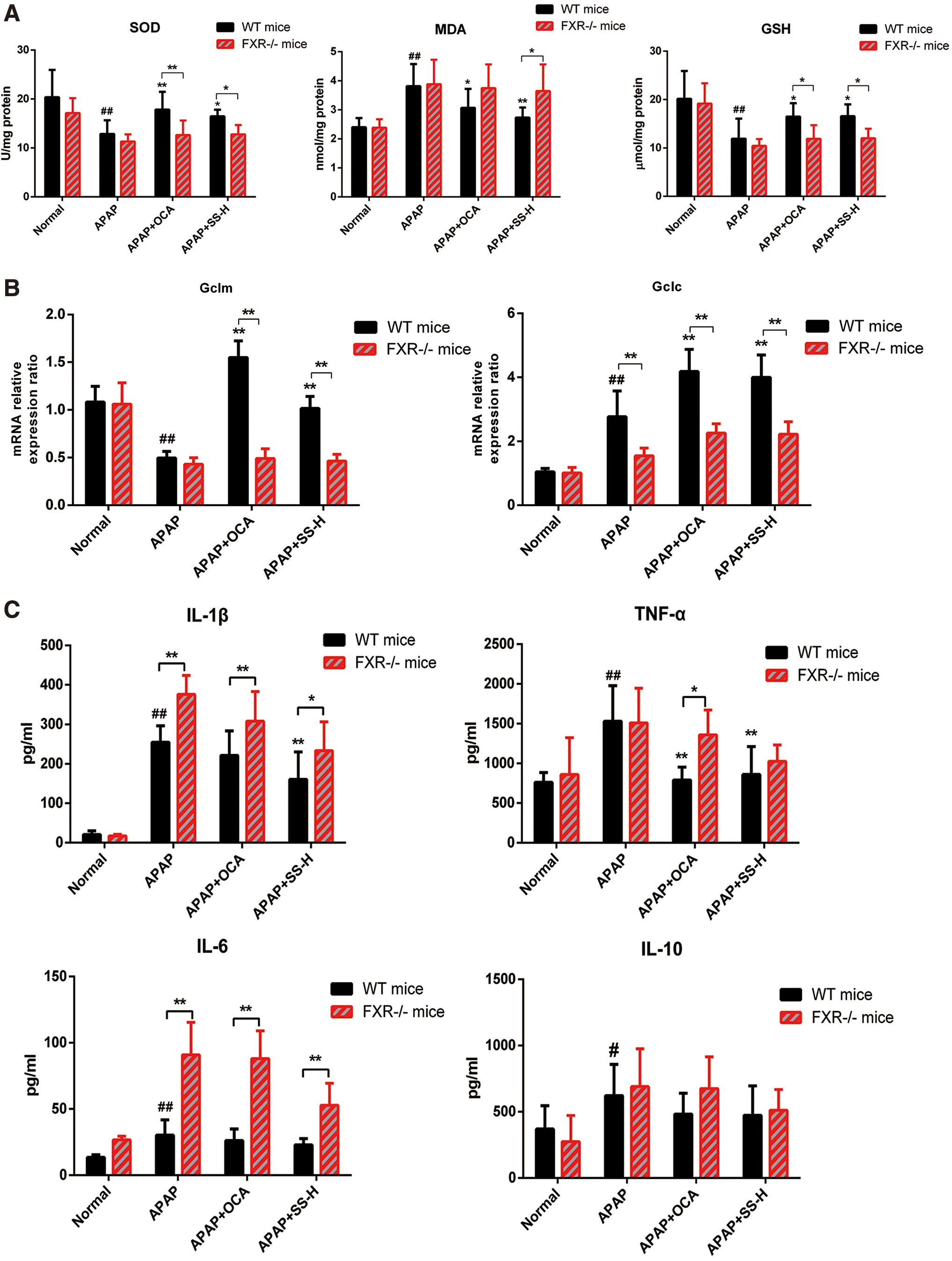
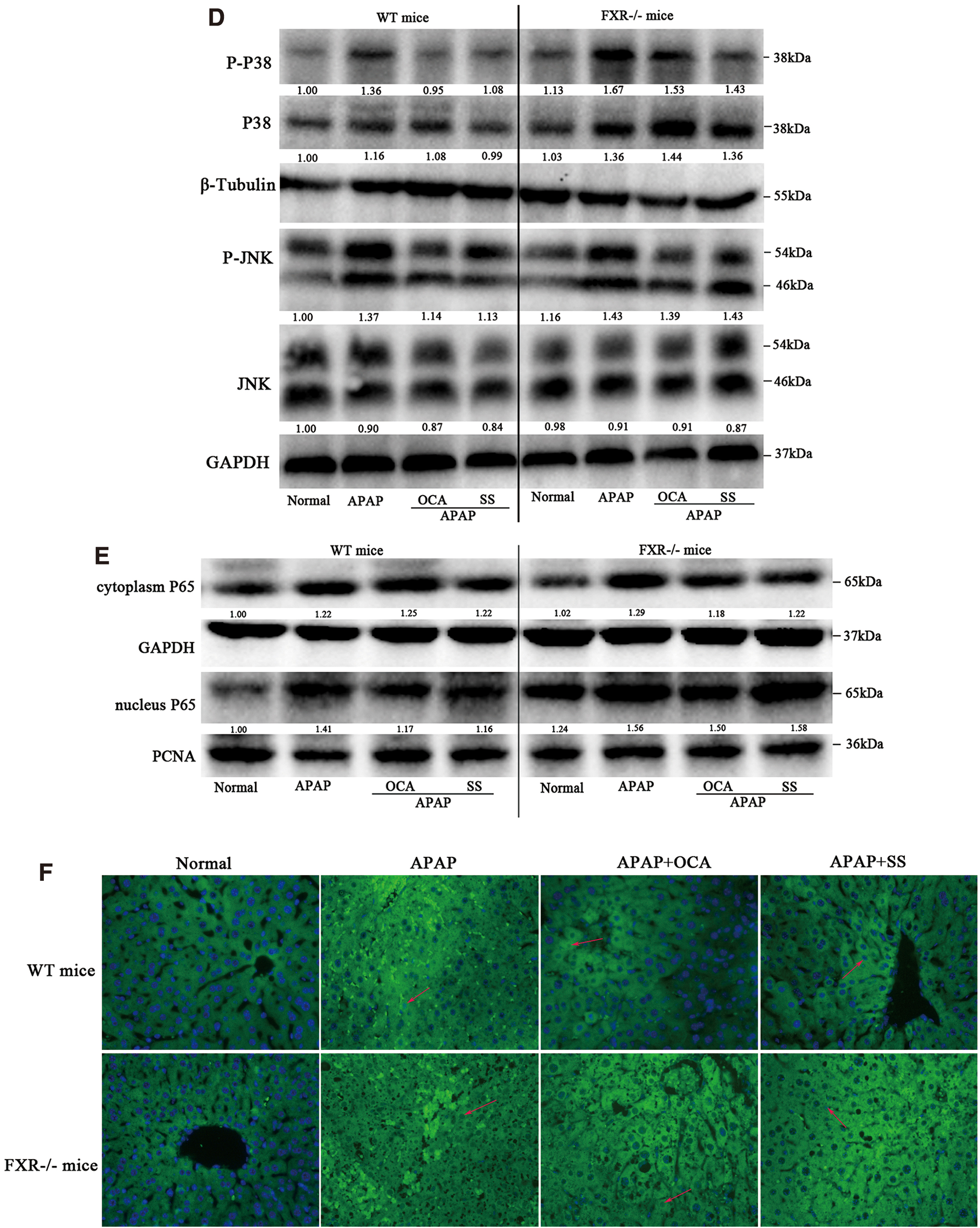
To confirm that the SS-mediated protective effect is dependent on FXR activation, FXR−/− MPHs and FXR−/− mice were conducted for the following study. First, FXR−/− mice exhibited an impaired FXR signaling in mice liver, as indicated by low levels of FXR, SHP, and BSEP (Supplementary Figs. S8, S16). Next, we found that the protective effects of SS against APAP-induced liver toxicity were largely abolished in FXR−/−MPHs, as revealed by loss of protection against APAP-induced cell viability and liver toxicity (Supplementary Fig. S9A). Besides, FXR deficiency could reverse SS-mediated decrease of ROS production (Supplementary Fig. S9B) and oxidative stress (Supplementary Fig. S9C). Similarly, the hepatoprotective effect of SS against APAP-induced ALI in FXR−/− mice was largely abrogated, as shown by lower survival rates (Fig. 8A), higher serum levels of LDH and AST (Fig. 8B), extensive histological liver necrosis (Fig. 8C) compared with wide-type (WT) mice in APAP-treated counterparts (Fig. 8). Meanwhile, the ameliorative effects of SS on oxidative stress and liver inflammation were also nearly abolished in FXR−/− mice, as evidenced by substantial hepatocytes apoptosis (Fig. 8D), higher hepatic MDA levels accompanied with lower levels of GSH and SOD (Fig. 9A) and higher serum proinflammatory cytokine levels (Fig. 9B). However, these effects were dramatically alleviated in WT mice after SS treatment (Figs. 8 and 9), suggesting that FXR deletion resulted in more susceptibility to APAP overdose-induced hepatotoxicity. Mechanistically, FXR−/− mice showed marginal induction of gene expressions related to APAP detoxification and excretion, including UGT1A1, GSTα, OSTβ, and BSEP (Supplementary Figs. S8B and S10) upon SS treatment, and there is no elevation of gene expression for GSH biosynthesis (e.g., Gclm and Gclc), as compared with APAP-treated WT mice (Fig. 9C). Moreover, FXR−/− mice exhibited higher gene expression levels involved in APAP-induced proinflammatory cytokines compared with WT mice in APAP-treated counterparts (Supplementary Fig. S11). Meanwhile, the activation of JNK, MAPK, and NF-κB during APAP overdose was not significantly blocked by SS or OCA treatment in FXR−/− mice (Fig. 9D–F, Supplementary Fig. S15). Reversely, the expression levels of genes involved in APAP detoxification and GSH biosynthesis were increased in WT mice after SS treatment (Fig. 9). Meanwhile, SS could also significantly suppress APAP-induced proinflammatory cytokines gene expression, and reduce phosphorylated JNK and MAPK and NF-κB nuclear translocation in WT mice (Fig. 9 and Supplementary Fig. S11). Furthermore, to look into the potential clinical application of SS for the treatment of liver failure after an APAP overdose, mice were administered with APAP and orally received a single dose of SS 6 h later. After 12 h of SS administration, SS exhibited a good therapeutic effect after 6 h of a toxic APAP overdose, as seen by minimal necrotic areas in livers and decreased serum transaminases level (Supplementary Fig. S12). However, this hepatoprotective effect of SS was largely weakened in FXR knockout (KO) mice (Supplementary Fig. S12). Taken together, these results strongly indicated that protection against APAP-induced hepatotoxicity by SS requires FXR activation.
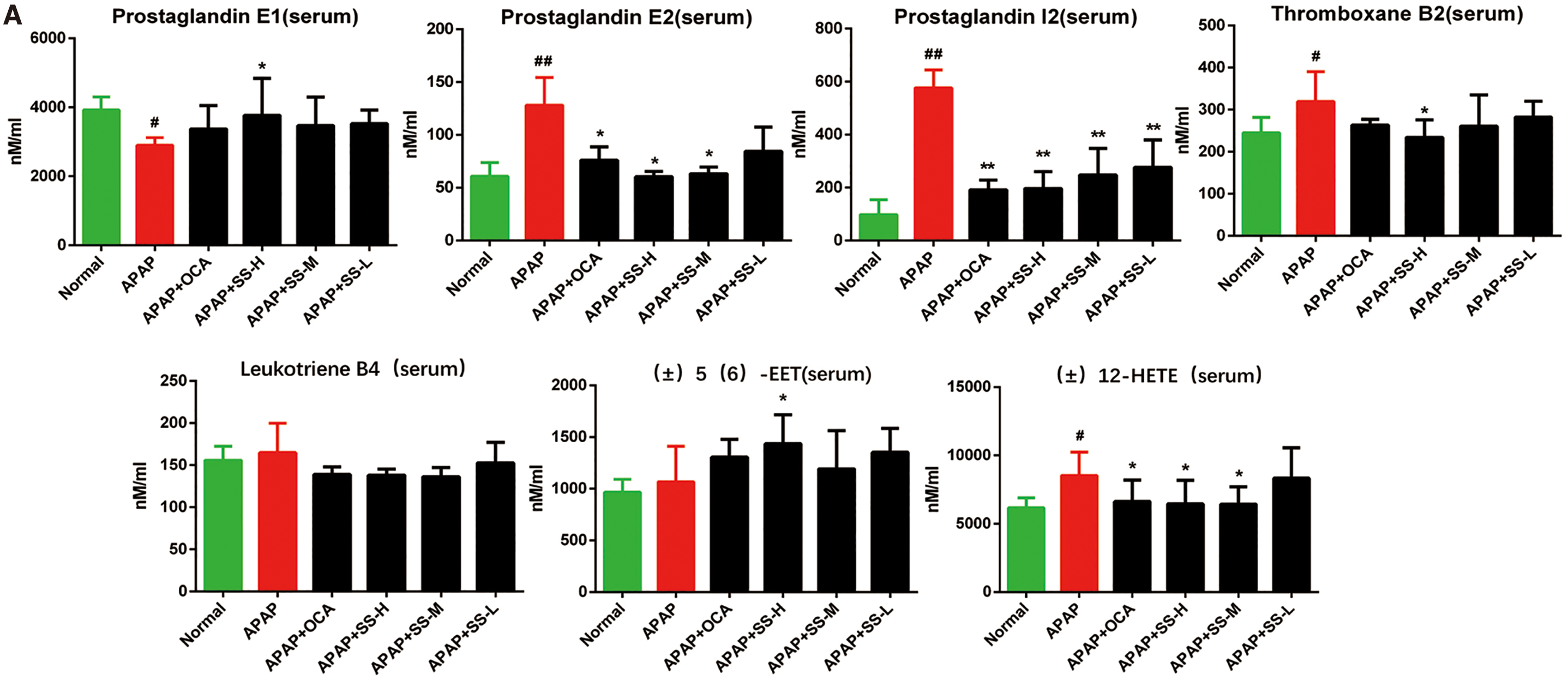
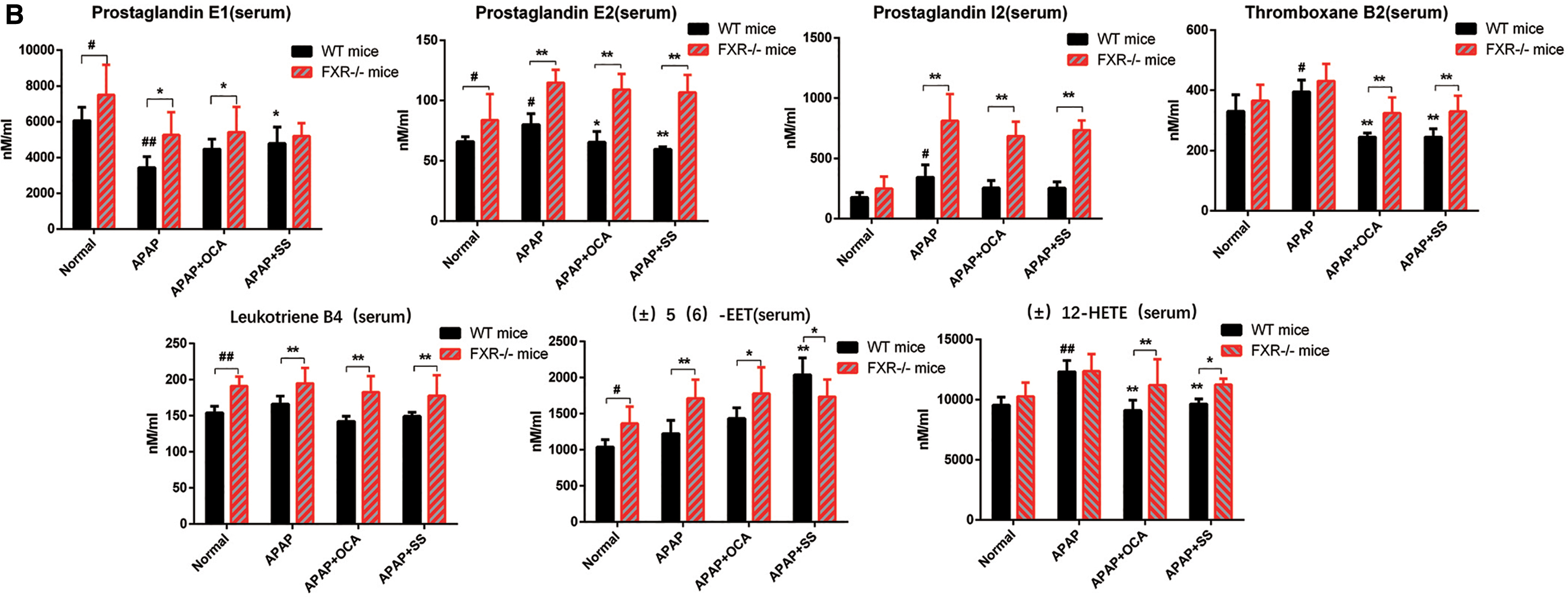
Activation of FXR by SS relieve abnormal eicosanoids metabolism induced by APAP overdose
Figure 4A exhibits that APAP overdose could alter the expression level of genes involved in the metabolic pathways of eicosanoids, a category of lipid regulators derived from metabolites of arachidonic acid (AA), which are involved in inflammatory process (9, 18, 72). Serum and hepatic levels of eicosanoids were then determined to assess the SS-mediated anti-inflammatory liver injury. The result indicated that SS or OCA treatment could fine-tune eicosanoids generation by suppressing the increased proinflammatory eicosanoids levels (such as prostaglandin I2 [PGI2], prostaglandin E2 [PGE2], thromboxane B2 [TXB2], and (±)12-HETE) and elevating the decreased anti-inflammatory eicosanoids levels (such as prostaglandin E1 [PGE1] and (±)5(6)-EET) (Fig. 10A). In contrast, FXR-deficient mice showed a basal elevation of eicosanoids level (Fig. 10B). FXR deficiency could result in a sharp elevation of serum proinflammatory levels of PGI2, PGE2, TXB2, and (±)12-HETE during APAP overdose as compared with that of WT mice, and this effect could not be reversed by SS or OCA treatment (Fig. 10B). Besides this, the elevated anti-inflammatory lipids PGE1 and (±)5(6)-EET were largely blocked in FXR−/− mice compared with WT mice (Fig. 10B). In addition, hepatic eicosanoids levels exhibited a similar pattern after SS or OCA treatment (Supplementary Fig. S13). In parallel, hepatic gene expression profiles showed that SS or OCA could notably repress the expression levels of Cox-2 (Ptgs2), Ephx1, and Alox12, which largely converts AA into proinflammatory lipid (PGE2, TXA2, TXB2, etc.), leukotrienes (LTA4, leukotriene B4 [LTB4], LTC4, etc.), as well as 5-, 8-, 12-, and 15-HETEs. The treatment can also induce the expression levels of Cyp2j6, Cyp2c29, Cyp2c50, and Cyp2c54, which are enzymes that catalyze AA to anti-inflammatory lipids EETs (Fig. 10C). To further probe the effect of SS/OCA in eicosanoids-induced hepatotoxicity, MPH was then treated with AA. Terminal-deoxynucleotidyl transferase mediated nick end labeling (TUNEL) study showed that AA could induce MPH apoptosis, while cotreatment with SS/OCA could attenuate this effect (Supplementary Fig. S14A). Mechanistically, exposure to AA led to inhibition of FXR and BSEP expression and elevated proinflammatory signaling in MPH. However, this effect was reserved by SS/OCA treatment (Supplementary Fig. S14B). Consistently, SS/OCA treatment could obviously alter the expression levels of genes in eicosanoids metabolic pathways, by regulation of anti/proinflammatory genes (Supplementary Fig. S14C). In contrast, this protective effect of SS/OCA was largely abolished in FXR KO MPH compared with that of FXR WT MPH (Supplementary Fig. S14D, E), suggesting an essential role of FXR in attenuation of AA-induced liver injury. Overall, our results exhibit that activation of FXR by SS or OCA can attenuate APAP-induced inflammatory liver injury via regulation of the pro- and anti-inflammatory eicosanoids homeostasis.

Discussion
This study highlights the critical role of FXR in regulating APAP-induced hepatotoxicity through reduction of hepatic oxidative stress, hepatocyte death, and hepatic inflammatory response. Our study also demonstrates that a novel FXR agonist SS exerts protective effects against APAP-induced ALI in mice. The protection effect is primarily via hepatic FXR signaling activation, thus resulting in decreased hepatic damage and liver inflammation.
APAP-induced ALI is considered to be the combination of two processes (71): the first is the enhanced oxidative stress caused by NAPQI, an intracellular toxic metabolite of APAP (48); while the second is the result of the acute necrotic inflammatory response (48, 65). In the first process, excessive oxidative stress induced by NAPQI represents the initial step of liver injury, and the initial GSH depletion reflects uncontrolled ROS generation in the liver (48). The excessive mitochondrial ROS generation in turn induces JNK activation and promotes JNK translocation to mitochondria, forming a vicious ROS-JNK feed-forward loop (27, 62 –64), which further results in excessive oxidative stress, and eventually causes hepatocyte death (62 –64). However, persistent JNK activation during cell death circumstances depends on the binding of p-JNK to the phosphorylated Sab, an outer membrane p-JNK “receptor” and substrate (27, 62 –64). The tight binding between these two proteins results in interruption of electron transport in mitochondrial signaling pathway, and consequently causes ROS release, which continues to activate MAPK cascade (62 –64). In this context, JNK inhibition or deficiency protects mice against APAP-induced ALI (14, 27, 37), suggesting that JNK is a key oxidative stress and apoptotic factor in APAP-induced hepatotoxicity (27). In agreement with the previous findings, we observed that APAP administration could result in a remarkable elevation of ROS and activation of p-JNK with reduction of GSH in mice liver and in hepatocytes. However, FXR activation by SS or OCA could decrease APAP-induced hepatotoxicity, by the suppression of sustained JNK phosphorylation and excessive ROS generation, primarily due to the elevated GSH level (Fig. 2). This is in agreement with several previous studies, which had shown that SS could exert antistress and antioxidant effects by regulation of JNK signaling pathway (35, 66). Our results indicated that SS as a novel JNK inhibitor could suppress APAP-induced JNK activation and protect against APAP-induced hepatotoxicity via reduction of oxidative stress.
FXR plays an important role not only in maintaining bile acid homeostasis (25) but also in the modulation of redox balance (45). Accumulating evidence showed that the antioxidant activity of FXR is correlated with its function of liver regeneration and cell proliferation in oxidative stress-induced liver injury (39, 45, 57). Fxr-null mice have shown spontaneously enhanced hepatic oxidative stress level (45), while FXR activation could protect liver against APAP-induced oxidative stress through elevation of hepatic GSH level (39). Mechanically, activation of FXR by GW4064 could significantly increase hepatic GSH level and induce the expression of Gclm and Gpx1 gene (39), both of which were rate-limiting enzymes in GSH biosynthesis and have been found to harbor several FXREs in their proximal promoter region. In contrast, this effect was not observed in Fxr-null mice (39), indicating the essential role of FXR in control of GSH biosynthesis. Furthermore, FXR antagonized JNK signaling pathway by activating SOD3 to suppress ROS production (57), while JNK inactivation could attenuate CCl4-induced ALI in Fxr-null mice (53), demonstrating a direct regulation pattern between FXR and JNK signaling in ALI. Recently, flavonoid C-glycosides such as SS could reduce glucose-induced oxidative damage via reduced p-JNK signaling (66), hinting a potential ameliorative role of SS against APAP-induced ALI. Interestingly, we have currently reported that SS could enhance hepatic FXR and ileal liver X receptor α mRNA expression and prevent lithogenic diet-induced cholesterol gallstone disease (41), supporting a vital evidence linked to FXR signaling. To elucidate the effect of SS on hepatoprotection associated with FXR activation, in this study, SS was characterized as an FXR agonist for the first time according to the following evidence: first, SS could activate hFXR transactivation, as indicative of induction of hBSPE-luc activities; second, molecular docking study showed that SS could bind to FXR-LBD pocket; third, SS could regulate transcriptional expression levels related to several FXR-dependent downstream target genes, such as BSEP and CYP7A1 in vitro and in vivo; fourth, more importantly, FXR knockdown or knockout could abrogate SS-mediated expression of BSEP and CYP7A1 in vitro and in vivo. Consistent with the alleviative oxidative stress (66), we observed that SS-mediated FXR activation could ameliorate APAP-induced hepatotoxicity by induction of hepatic Gclm and Gclc gene expression in a FXR-dependent mechanism, resulting in elevation of hepatic GSH level and decrease of APAP-induced ROS level (Fig. 2). In contrast with WT mice, FXR-null mice exhibited little inductive effect of hepatic Gclm and Gclc after SS treatment, consequently demonstrating a relatively low GSH level and sustained excessive ROS level in liver and hepatocytes (Fig. 9). To further uncover the mechanisms underlying the protective effect of SS via FXR activation, we first investigated the influence of FXR activation on APAP metabolism and elimination since it was regarded as a major contributing factor to the pathogenesis of APAP-induced hepatotoxicity. The results indicated that FXR activation by SS or OCA had marginal inductive effect on the phase I metabolizing enzymes such as CYP2E1 and CYP3A11, both involved in the generation of toxic metabolite NAPQI. However, SS/OCA-mediated FXR activation strongly induced phase II detoxifying enzymes (e.g., UGT1A1 and GSTA1) as well as drug efflux transporters (e.g., BSEP and OSTβ) (Fig. 4), suggesting that FXR activation could promote the conversion from APAP to pharmacologically inactive glucuronide and sulfate conjugates, which were further excreted into the urine or bile, therefore decreasing NAPQI-mediated oxidative stress. In contrast, FXR−/− mice exhibited low levels of UGT1A1, GSTA1, OSTβ, and BSEP, SS or OCA treatment showed limited elevation effects on these genes expression (Supplementary Figs. S8B and S10). Next, we evaluated the influence of FXR activation on APAP-induced sustained JNK activation, which is a key downstream event of APAP metabolism but upstream of APAP-induced necrosis. Consistently, knockout of FXR could cause the enhanced JNK phosphorylation in FXR−/− mice liver and FXR−/− MPHs (Fig. 9). Conversely, FXR activation by SS or OCA could significantly suppress JNK phosphorylation (Fig. 2), indicating that FXR could counteract APAP-mediated toxic JNK activation (59). Together, our findings provide a mechanistic support for the protective role of FXR activation by SS or OCA in APAP-induced liver injury.
Excessive inflammatory mediators (e.g., TNF-a, IL-6, and IL-1β) and elevated neutrophil activation in both mice and humans contribute to early phase APAP-induced hepatotoxicity via NF-κB signaling pathway (3, 16, 28, 65), and anti-inflammatory therapies can attenuate these detrimental inflammatory effects from APAP overdose (19, 69), which suggests that APAP-induced ALI is, at least in part, mediated by inflammation (65). In APAP-induced liver injury, the released DAMP molecules such as nuclear DNA fragments, mitochondrial DNA, and some mitochondrial and nuclear proteins from damaged hepatocytes can trigger an irreversible secondary inflammatory injury through abnormal activation of the innate immune system (65, 68, 71), although controversial data regarding the role of inflammatory response in APAP-induced ALI have been generated in different preclinical models (61, 65). Therefore, suppression of inflammation plays a potential therapeutic strategy for liver protection against APAP-induced injury, especially in the early phase of APAP overdose. FXR knockout results in strong hepatic inflammation (59). FXR activation could suppress NF-κB signaling and ameliorate thioacetamide-induced hepatic inflammation (56), which indicates a negative crosstalk between the FXR and NF-κB signaling pathways. However, despite mounting evidence in favor of FXR role for protection against chemical-induced liver injury, the protective role of FXR in APAP-induced hepatic inflammatory injury remains largely unknown. In this study, we exhibited that APAP exposure could induce hepatic inflammatory injury, which is consistent with the previous reports. Conversely, FXR activation by SS or OCA could significantly attenuate APAP/LPS-induced liver tissue inflammatory injury or hepatocytes inflammation. Similar to our results, a recent study implied that FXR activation by hedragonic acid decreased hepatic inflammatory responses and protected mice from liver injury induced by APAP overdose via FXR activation (42). However, the underlining mechanism for hedragonic-acid-mediated FXR activation and inhibition of NF-κBp65 signaling pathway remains unclear thus far. In this study, we indicated that SS- or OCA-mediated suppression of NF-κB p65 nuclear translocation was largely abolished in FXR−/− mice or FXR−/− MPHs (Fig. 9E, F and Supplementary Fig. S11), implying an essential role of FXR in repression of NF-κB signaling. Taken together, our results showed that FXR activation by SS or OCA could ameliorate APAP-induced liver inflammatory injury primarily accounted for the suppression of NF-κB signaling.
Accumulating evidence points out that eicosanoids metabolism is dysregulated during APAP overdose, which is associated with the inflammation process (30, 51). Inflammatory homeostasis is tightly regulated by pro- and anti-inflammatory eicosanoids (9, 18, 72), and the regulation of eicosanoids synthesis plays a vital role in the inflammatory processes. Three major enzymatic pathways consisting of cyclooxygenase (COX), lipoxygenase (LOX), and CYP450 enzymatically convert AA into different eicosanoids lipid metabolites (18). Namely, the COX enzymes convert AA into prostaglandin H2 through a cyclic pathway of eicosanoids biosynthesis, and further metabolize into prostaglandins (such as PGE2, PGD2, and PGI2) and thromboxane (TXA2, TXB2) (18, 49, 47); many of them exhibit proinflammatory properties (18, 47, 49). While LOX enzymes catalyze AA to leukotrienes (such as LTA4, LTB4, and LTC4) as well as 5-, 8-, 12-, and 15-HETEs (6, 18, 36, 70), and CYP450 enzymes convert AA to HETEs and EETs (6, 18, 36, 70). EETs were reported to have anti-inflammatory effect by suppression of various proinflammatory cytokines (54, 55), whereas leukotrienes and HETEs were well-known proinflammatory lipid mediators (11, 21, 50), which significantly promoted TNF-α and IL-6 production in macrophages (50). The synthesis of eicosanoids could alter the intracellular redox balance, and contribute to ROS generation and inflammation induction (13). Intriguingly, 5-LOX deficiency or inhibition protected mice against APAP-induced liver toxicity via decreasing hepatic oxidative stress and liver inflammation (31, 46). Therefore, these relationships between inflammation homeostasis and lipid metabolism prompt us to explore whether APAP overdose regulates the metabolic balance between pro- and anti-inflammatory eicosanoids. Consistent with the elevated eicosanoids levels in APAP overdose (51), the proinflammatory eicosanoids levels (e.g., PGI2, PCE2, TXB2, and (±)12-HETE) were significantly elevated, which highly correlated with the enhancement of hepatic injury and hepatic inflammation, while the anti-inflammatory eicosanoids levels were remarkably reduced after APAP exposure (Fig. 10A). However, SS/OCA could significantly reverse anti-inflammatory and proinflammatory eicosanoids generation (Fig. 10A). This reversal effect of SS/OCA is largely dependent on the regulation of eicosanoids metabolism, namely by inducing anti-inflammatory eicosanoids genes, such as Cyp2j6, Cyp2c29, Cyp2c50, and Cyp2c54, and downregulating proinflammatory eicosanoids genes, such as Ptgs2 and Ephx1, and Alox12. Contrarily, FXR deficiency could notably abolish SS/OCA-mediated reversal effect (Fig. 10B, C), suggesting a key role of FXR in regulation of eicosanoids generation. This suggestion is supported by recent studies showing that FXR activation by OCA could reprogram high-fat diet-induced AA metabolism dysregulation via suppression of proinflammatory leukotrienes level and elevation of anti-inflammatory EET levels (23), subsequently leading to inhibition of NF-κB signaling and hepatic inflammation (23). To further evaluate the FXR's role in the regulation of eicosanoids levels, MPHs were treated with AA directly. The result indicated that AA could indeed suppress FXR signaling and elevate inflammatory signaling by inducing proinflammatory eicosanoids genes (Supplementary Fig. S14A), leading to enhanced MPH apoptosis (Supplementary Fig. S14). However, this effect was largely attenuated by SS/OCA in WT MPH rather than FXR KO MPH (Supplementary Fig. S14), indicating that the protective effect of SS/OCA is FXR dependent. Collectively, our present data suggest that anti-inflammatory effects of SS upon APAP overdose might be correlated with FXR-mediated generation of pro- and anti-inflammatory eicosanoids.
In conclusion, for the first time, we have demonstrated that SS exhibits protective effects against APAP overdose-induced hepatotoxicity via FXR-mediated suppression of oxidative stress and inflammation. Our findings highlight the importance of FXR in APAP-induced ALI by regulation of oxidative stress and inflammation. More importantly, naturally occurring SS has been proven to be an effective FXR agonist, thus offering a potential therapeutic agent against APAP-induced ALI.
Materials and Methods
Chemicals and reagents
SS (C26H28O14; molecular weight [Mw] = 564.49) was purchased from Chengdu Herbpurify Co., Ltd. (Sichuan, China); OCA (C26H44O4; Mw = 420.63) was purchased from Dongguan Walixi Chemical Co. Ltd. (Guangdong, China); and GW4064 (C28H22Cl3NO4, Mw = 542.84) was purchased from Sigma Chemical Co. (St. Louis, MO). FXR (ab129089), BSEP (ab71793), CYP7A1 (ab65596), CD11b (ab133357), CRP (ab229110), IL-6 (ab7737), CD45 (ab10558), and GADPH (ab8245) primary antibodies were purchased from Abcam (Cambridge, United Kingdom). NF-κB p65 (#8242), JNK (#9252), P-JNK (#4668), P38 (#9212), P-P38 (#4631s), β-Tubulin (#2128), and proliferating cell nuclear antigen (PCNA, #13110) primary antibodies were purchased from Cell Signaling Technology (Denvers, MA). SHP (sc-30169) and F4/80 (sc-377009) primary antibodies were purchased from Santa Cruz Biotechnology (CA). PGE1, PGI2, (±)5(6)-EET, PGE2, LTB4, TXB2, and (±)12-HETE were purchased from Avanti Polar Lipids, Inc. Human FXR siRNA (M-003414-01) and nontargeting control siRNA (D-001210-01-20) were obtained from Dharmacon (Lafayette, CO). pGL3-CMV Renilla luciferase plasmids were provided by Dr. Ming Huang (College of Pharmacy, Sun Yet-Sen University). The NF-κB reporter vector pGL4.32 [luc2P/NF-κB-RE/Hygro] and the Dual-Glo luciferase reporter assay system were purchased from Promega (Madison, WI).
Animal models
Male C57BL/6 mice were purchased from the Animal Laboratory Centre of Guangdong Province (Guangzhou, China). Nr1h4 knockout (FXR−/−) mice (007214-B6.129X1(FVB)-Nr1h4tm1Gonz/J) were purchased from The Jackson Laboratory (Bar Harbor, ME). They were kept in a specific pathogen-free animal facility at a constant temperature of 22°C ± 2°C and a relative humidity of 50%–70%, with a 12 h light/dark cycle and free access to standard laboratory chow and water. The mice were acclimatized to laboratory conditions for 1 week before experimentation. All animal care and experimental studies were approved by and in accordance with the guidelines of the Animal Ethics Committee of Guangzhou University of Chinese Medicine.
Thirty-six male C57BL/6 mice (8 weeks old) were randomly divided into six groups (n = 6): normal, APAP, APAP+OCA (20 mg·kg−1), APAP+SS-H (160 mg·kg−1), APAP+SS-M (80 mg·kg−1), and APAP+SS-L (40 mg·kg−1). For another set of experiments, both FXR knockout (FXR−/−) mice (8 weeks old) and male WT mice were grouped as normal, APAP, APAP+OCA, APAP+SS-H (n = 6). Mice received a daily gavage of SS or OCA in 0.5% carboxymethyl cellulose for 7 days. In the seventh day of dosing SS or OCA, Overnight fasted mice received 300mg/kg body weight of APAP (dissolved in 55°C ultrapure water) by intraperitoneal (i.p.) injection. Mice were sacrificed 12 h after APAP injection, and then blood and livers were collected. The survival rate of mice was recorded within 24 h of APAP exposure. Part of the liver sample was fixed in 10% formalin for histology, and the rest of the tissue was stored at −80°C for further study.
Liver histology, immunohistochemistry, and immunofluorescence
The liver specimens were prepared and fixed in 10% formalin, embedded in paraffin, cut into liver sections (4-μm thick), processed for routine H&E staining, and examined under a light microscope (Olympus). Liver histology was blindly assessed by an experienced hepatopathologist.
Immunohistochemistry and immunofluorescence were performed according to standard procedures. Liver tissue sections were dewaxed and rehydrated in ethanol at different concentration gradients, then the sections were incubated with 3% H2O2 in distilled water for 10 min, followed by a blocking solution of 10% goat serum in PBS for 20 min. For immunohistochemistry, liver sections were incubated with the primary antibodies F4/80 (dilution 1:200; Santa Cruz), CD11b (dilution 1:500; Abcam), and CD45 (dilution 1:500; Abcam) at 4°C overnight. After being washed in PBS, the sections were incubated with antirabbit secondary antibody in PBS for 30 min at 37°C, then incubated with avidin-biotin peroxidase solution (SABC kit; Boster, China) and colorized with a 3,3′-diaminobenzidine (DAB) kit (Boster). Finally, the sections were counterstained with hematoxylin and observed under a microscope (Olympus, Tokyo, Japan). For immunofluorescence, liver sections or cells were incubated with the primary antibodies FXR, BSEP, CRP, IL-6 (Abcam), NF-κB (Cell Signaling Technology, Denvers, MA), and SHP (Santa Cruz Biotechnology, CA) at 4°C overnight. After being washed in PBS, the sections were incubated with FITC- or Alexa 594-conjugated IgG secondary antibodies at room temperature for 1 h. Nuclei were counterstained by incubation in 1 μg/mL 4′,6-diamidino-2-phenylindole (DAPI; Solarbio, China) for 15 min followed by exhaustive washing in PBS. Coverslips were mounted in Fluorescence decay resistant medium (Boster), and the sections were visualized with a fluorescence microscope (Olympus).
Homogenization and preparation of tissue extracts
Frozen livers were homogenized in ice-cold saline (0.9% sodium chloride solution) using ultrasonic processor Uibra Cell VCX800 (Sonics & Materials, Newtown, CT). The whole extraction process has been done on ice all the time. Liver extracts were centrifuged at 10,000 g for 20 min at 4°C, and the supernatant was aliquoted and stored at −80°C.
Analysis of serum or liver homogenates biochemical indices
ALT, AST, LDH in serum, and MDA, SOD, and GSH in liver homogenates were measured using reagent kit from Nanjing Jiancheng Bioengineering Institute (Nanjing, China).
Enzyme-linked immunosorbent assay
Inflammatory cytokines including TNF-α, IL-1β, IL-6, and IL-10 and 8-OHdG level in serum samples or cell culture media were determined using enzyme-linked immunosorbent assay (ELISA) kit from Cusabio (Wuhan, China) according to the manufacturer's instruction.
Cell culture
HEK293T and Huh-7 cells were purchased from the cell bank of the Shanghai Institute (Shanghai, China). The cells were cultured by Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 0.1 mg/mL streptomycin at 37°C in humidified 5% CO2 incubator. All culture reagents including FBS and DMEM high-glucose culture medium were purchased from Gibco.
Isolation of MPHs
MPHs were isolated from WT and FXR−/− male mice as previously described (8). The viability of isolated hepatocytes was exceeded 88% assessed by trypan blue dye exclusion. Hepatocytes were cultured in DMEM containing 10% FBS, 100 U/mL penicillin, and 0.1 mg/mL streptomycin. After incubating for 4 h at 37°C in humidified 5% CO2 in an air incubator, adherent MPH cells were cultured overnight, and then treated with SS or GW4064 followed by APAP or LPS for the study.
TUNEL analysis
Paraffin-embedded liver biopsy sections or MPHs were stained using the one-step TUNEL Apoptosis assay kit (Beyotime, Beijing, China) according to the manufacturer's instruction.
Determination of mitochondrial membrane potential
Mitochondrial membrane potential was measured using a JC-1 staining kit (Beyotime) according to the manufacturer's instructions. In brief, DMEM-washed cells were stained with JC-1 dye for 20 min. Then, the cells were observed under a fluorescence microscope (Olympus).
Cell viability
MPHs were seeded into 96-well plates. Cells were treated with SS or GW4064 for 24 h, and then followed by exposure to APAP for 12 h. The cell viability was measured by cell counting kit-8 (CCK-8; Dojindo, Kyushu, Japan). In brief, 10 μL of CCK-8 reagent was added to each well, and incubated at 37°C for 2 h, and subsequently measured the absorbance using Enspire Multimode Plate Reader (PerkinElmer, Waltham, MA) at 450 nm.
RNA interference assay
Huh-7 cells were seeded on six-well plates. After reaching ∼80% confluence, nontargeting siRNA, 200 nM negative control siRNA or siRNA targeting human FXR (siGENOME SMARTpool, Dharmacon) were transiently transfected to Huh-7 cells using lipofectamine 2000 (Invitrogen, Carlsbad, CA). After 24 h transfection, cells were treated with dimethyl sulfoxide (DMSO), GW4064, and SS for 48 h, and then harvested for real-time quantitative PCR and Western blot assay.
Dual-luciferase reporter assay
hFXR expression plasmid was constructed by cloning genes encoding FXR into pCDNA3.1(+)-3Flag-C vector. hBSEP promoter reporter was constructed by cloning a genomic DNA fragment upstream of the transcription start site (−1448 to +83) contained an FXRE into pGL3-Basic vector. For transactivation assay, HEK293T cells in 96-wells culture plate were transiently transfected with pCDNA3.1(+)-entry vector, pCDNA3.1(+)-hFXR (100 ng per well), pGL3-basic-hBSEP (200 ng per well) or pGL4.32-NF-κB (200 ng per well) and pGL3-CMV Renilla luciferase plasmid (10 ng per well) as an internal control using Lipofectamine 2000™ (Invitrogen). Twenty-four hours after transfection, transfected cells were treated with 0.1 mL of fresh supplemented culture medium containing DMSO (0.1% v/v; vehicle control), GW4064 (10 μM), UDCA (50 μM), SS (12.5–50 μM) for 24 h, respectively, followed by incubation with LPS (2000 ng/mL) for 24 h. At the end of incubation period, firefly and Renilla luciferase activities were measured on Enspire Multimode Plate Reader (PerkinElmer, CA) using the Dual-Luciferase Report Assay system (Promega, Madison, WI) according to manufacturer's instruction. Firefly luciferase activities were normalized to the Renilla luciferase controls.
Determination of Intracellular ROS Levels
The intracellular ROS levels of cells were measured using an intracellular ROS assay kit (Beyotime) according to the manufacturer's instructions. In brief, the cells were inoculated into a 24-well microtitration plate, then 500 μL of ROS detection reagent mixture was added, and the plate was incubated for 30 min at 37°C in a 5% CO2 humidified incubator. Then, the cells were observed under a fluorescence microscope (Olympus).
Quantitative real-time PCR analysis
Total RNA was extracted using TRIzol reagent. The quality and quantity of total RNA were determined using the NanoDrop 2000 Spectrophotometer (Thermo Fisher Scientific, Waltham, MA). Reverse transcription was performed using the SuperScript™ VILO™ cDNA Synthesis Kit, and qPCR was carried out using Power SYBR® Green PCR Master Mix (Applied Biosystems, Carlsbad, CA) in an ABI StepOnePlus system (Applied Biosystems) according to the manufacturer's protocol. The primer sequences used for amplification of FXR, BSEP, CYP3A11, CYP2E1, NTCP, OATP1A1, OSTβ, UGT1A1, GSTA1, SOD2, Gclm, Gclc, NF-κB, IL-1β, TNF-α, IL-6, IL-10, Arg1, iNOS, Ephx1, Cyp2j5, Cyp2j6, Cyp2c29, Cyp2c50, cyp2c54, Ptgs1, Ptgs2, Alox5, Alox12, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) are listed in Table 1. For standardization and quantification, GAPDH mRNA was amplified simultaneously. The comparative Ct method was used to estimate the relative quantification for gene expression according to the following formulae: ΔCt = Ct (test gene) − Ct (GAPDH); ΔΔCt (test gene) = ΔCt (test gene in treatment group) − ΔCt (test gene in vehicle control group); mRNA expression fold change = 2−ΔΔCt, which indicates the relative mRNA level of the corresponding transcript compared with control samples.
Primer Information for Gene Amplification
BSEP, bile salt export pump; FXR, farnesoid X receptor; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; Gclc, glutamate-cysteine ligase catalytic subunit; Gclm, glutamate-cysteine ligase modifier subunit; GSTA1, glutathione S-transferase alpha 1; IL, interleukin; iNOS, inducible nitric oxide synthase; NF-κB, nuclear factor kappa-B; OSTβ, organic solvent transporter protein β; TNF, tumor necrosis factor; UGT1A1, UDP glucuronosyltransferase family 1 member A1.
Western blot
Samples from cells and liver tissues (100 mg) were rinsed in cold PBS (pH 7.4), lysed and homogenized in RIPA lysis buffer containing a cocktail of protease inhibitors on ice and centrifuged at 12,000 g for 15 min at 4°C. The supernatant was collected, and the protein concentration was measured using bicinchoninic acid protein assay kit (Beyotime Biotechnology, Beijing, China). In addition, nuclear and cytoplasmic proteins were extracted by Nuclear and Cytoplasmic Protein Extraction kit according to manufacturer's instructions (Beyotime Biotechnology, Beijing, China). Equal concentrations of protein were separated using a 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE; Bio-Rad Laboratories, Hercules, CA) and transferred to Immuno-Blot PVDF membranes (0.22 μm; Bio-Rad Laboratories). The membranes were then blocked with 5% bovine serum albumin in Tris-buffered saline containing 0.1% Tween 20, and incubated overnight at 4°C with primary antibodies targeting FXR (1:4000), BSEP (1:500), CYP7A1 (1:1000), NF-κB p65 (1:1000), JNK (1:1000), P-JNK (1:1000), P38 (1:1000), P-P38 (1:1000), PCNA (1:2000), β-Tubulin (1:2000), and GADPH (1:3000). The blots were then incubated with HRP-conjugated secondary antibodies and detected by Immobilon Western Chemiluminescent HRP Substrate (Millipore, Billerica, MA) using Molecular Imager ChemiDOC™ XBS imaging systems (Bio-Rad Laboratories).
Molecular docking
The structure of SS was obtained from the PubChem database (Pubchem CID 442658). The three-dimensional structure of hFXR cocrystalized with chenodeoxycholic acid was obtained from the RCSB Protein Data Bank (PDB code: 1OSH). The hFXR ligand chenodeoxycholic acid served as template molecule to evaluate the ligand affinity for hFXR. The cocrystalized structure was prepared using SYBYL2.1.1 to correct structural errors, such as broken bonds and missing loops. Then, chenodeoxycholic acid was removed, and hydrogen atoms were added to prepare the structure for docking experiments, with all water molecules and ligands from the protein structures removed. The prepared structure was submitted to FlexX for molecular docking. The residues around chenodeoxycholic acid (template ligand) were selected as the binding pock for docking SS to hFXR-LBD. The docking complex was carried out with the program Sybyl version 2.1.1. The docking results were analyzed, and the figures were created in Discovery Studio Visualizer.
Detection of eicosanoids by ultra performance liquid chromatography-tandem mass spectrometry
The determination of eicosanoids was performed on a triple-stage quadrupole ultra performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS) (TSQ Quantum; Thermo Fisher, San Jose, CA) with an Accela MS pump and an Accela autosampler (Thermo Fisher).
One hundred microliters of liver tissue homogenates or serum and 200 μL acetonitrile with PGE2 containing four deuterium atoms at the 3,3′,4,4′ positions (PGE2-d4, 50 ng/mL) as internal standard were added, the mixture was then vigorously vortexed for 10 min and centrifuged at 4°C, 13,000 g for 10 min, then 10 μL supernatant was analyzed by UHPLC-MS/MS.
Chromatographic separation of eicosanoids was carried out using Poroshell 120 SB-Aq (2.1 × 100 mm, 2.7 μm; Agilent). The flow rate was 0.7 mL·min−1 with the following gradient elution: (0–1.5min: 5%–5%A; 1.5–2min: 5%–100%A; 2–5 min 100%–100%A; 5–5.5min 100%–5%A; 5.5–7min 5%–5%A), acetonitrile (A) 0.1% formic acid (B) as mobile phase. MS/MS quantification was performed on multiple reaction monitoring positive/negative mode using a TSQ Quantum triple quadrupole mass spectrometer with electrospray ionization. The MS/MS parameters of analytes are summarized in Table 2. The other parameters were set as follows: the ion source spray voltage was set at 3500 V and the capillary temperature at 300°C. Nitrogen was used as sheath and auxiliary gas, and the pressures were set at 30 and 10 arbitrary units, respectively.
The Ultra Performance Liquid Chromatography-Tandem Mass Spectrometry Parameters of Eicosanoids
ESI, electrospray ionization.
Data analysis
All results in the figures and text are expressed as the mean ± SEM. The data were evaluated using GraphPad Prism Version 6.0 (GraphPad Software, La Jolla, CA). The statistical significance of multiple comparisons was analyzed using one-way analysis of variance followed by least significant difference or Dunnett's. A value of P < 0.05 was considered statistically significant for all tests.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by the Natural Science Foundation of China (81773969, 81102883, 81573566, 81800738, 81673872, and 81873091), Guangdong Science and Technology Collaborative Innovation Center for Sport Science (2019B110210004), the high-level talents project of Guangdong Province (YUE [2014] 104), and the Science Program for Overseas Scholar of Chinese Medicine (XH20160103).
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Figure S7
Supplementary Figure S8
Supplementary Figure S9
Supplementary Figure S10
Supplementary Figure S11
Supplementary Figure S12
Supplementary Figure S13
Supplementary Figure S14
Supplementary Figure S15
Supplementary Figure S16
Supplementary Table S1
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.