
Abstract
Significance:
Increased endothelial permeability and inflammation are two major hallmarks of the life-threatening conditions such as acute respiratory distress syndrome and sepsis. There is a growing consensus in the field that the Rho family of small guanosine triphosphates are critical regulators of endothelial function at both physiological and pathological states. A basal level of reactive oxygen species (ROS) is essential for maintaining metabolic homeostasis, vascular tone, and angiogenesis; however, excessive ROS generation impairs endothelial function and promotes lung inflammation. In this review, we will focus on the role of Rho in control of endothelial function and also briefly discuss a nexus between ROS generation and Rho activation during endothelial dysfunction.
Recent Advances:
Extensive studies in the past decades have established that a wide range of barrier-disruptive and proinflammatory agonists activate the Rho pathway that, ultimately, leads to endothelial dysfunction via disruption of endothelial barrier and further escalation of inflammation. An increasing body of evidence suggests that a bidirectional interplay exists between the Rho pathway and ROS generation during endothelial dysfunction. Rac, a member of the Rho family, is directly involved in ROS production and ROS, in turn, activate RhoA, Rac, and Cdc42.
Critical Issues:
A precise mechanism of interaction between ROS generation and Rho activation and its impact on endothelial function needs to be elucidated.
Future Directions:
By employing advanced molecular techniques, the sequential cascades in the Rho-ROS crosstalk signaling axis need to be explored. The therapeutic potential of the Rho pathway inhibitors in endothelial-dysfunction associated cardiopulmonary disorders needs to be evaluated.
Introduction
Endothelial cell (EC) monolayer covering the luminal space of blood vessels creates a semi-permeable barrier that restricts the passage of fluids, macromolecules, and inflammatory cells between the blood and underlying tissue. The EC barrier is composed of a complex framework of proteins forming adherens junctions (AJ), tight junctions (TJ), and focal adhesions connected with the EC cytoskeleton (45, 107). The endothelium plays a key role in maintaining vascular tone, angiogenesis, and organ differentiation. During various pathological conditions, the disruption of the endothelial barrier results in an influx of plasma protein into extravascular space, leading to tissue edema, which is also accompanied by enhanced inflammatory responses, a common feature of numerous injury syndromes, including acute respiratory distress syndrome (ARDS) (86, 103, 117). Hence, the maintenance and recovery of the endothelial barrier integrity on inflammatory insults and pathologic interventions is regarded as an essential step toward prevention and treatment of a variety of inflammatory disorders, including acute lung injury (ALI).
Although effective pharmacotherapeutics to confront severe pulmonary inflammation and edema are yet to be developed, there has been a substantial advancement in the understanding of molecular and cellular mechanisms involved in the pathogenesis of these disorders.
Small guanosine triphosphates (GTPases), especially Ras-homologous (Rho) family proteins, have been identified as major regulators of endothelial barrier function by controlling endothelial permeability (138). Among these, RhoA is activated downstream of multiple barrier-disruptive agonists such as thrombin, histamine, vascular endothelial growth factor (VEGF), as well as pro-inflammatory molecules such as tumor necrosis factor-α (TNF-α) and gram-negative bacterial endotoxin lipopolysaccharide (LPS) (154, 160). An increase in phosphorylation of myosin light chain (MLC) resulting in the formation of actin stress fibers and activation of actin-myosin contractility has been postulated as a major mechanism of Rho-mediated endothelial hyperpermeability (84, 124).
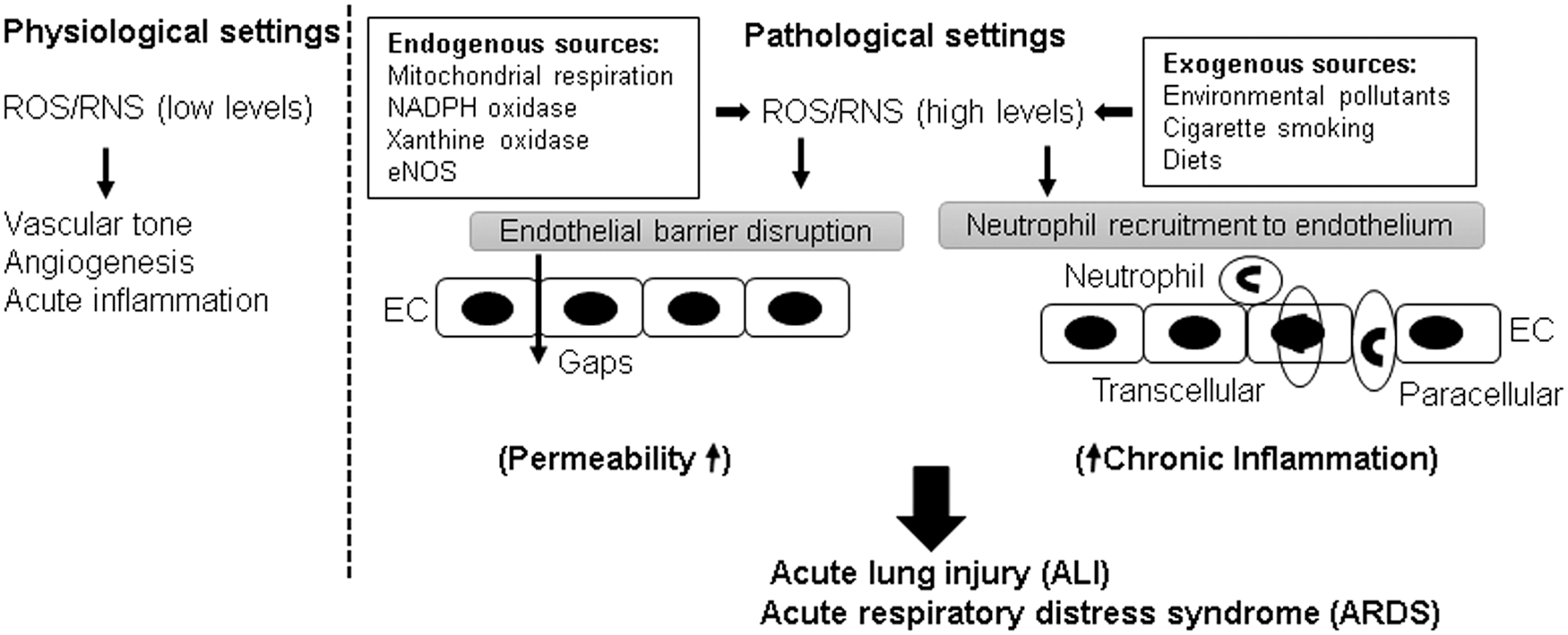
Reactive oxygen species (ROS) play a dual role in the regulation of endothelium (Fig. 1). At physiological levels, ROS-induced signaling axis is necessary for maintaining vascular tone by the endothelium and also facilitates angiogenesis and acute inflammatory responses to counter invading pathogens (34). In turn, excessive ROS generation in pathological settings, that is, hyperoxia, inflammation, environmental pollution has been implicated in endothelial barrier dysfunction (17), although delineation of precise pathologic mechanisms triggered by elevated ROS production awaits further investigation. Earlier studies have provided evidence that ROS increase endothelial permeability both in vitro and in vivo (128, 129). In response to injurious stimuli, EC and neutrophils may directly produce ROS that triggers permeability and inflammatory response. Herein, we will review the roles of Rho and ROS in regulation of endothelial function with the focus on the interconnection between these two signaling pathways in pathological settings of endothelial dysfunction.
Rho GTPases: Master Regulators of Endothelial Barrier Function
EC comprise an intact endothelial barrier to control the passage of fluids and solutes between the circulation and the interstitial space. A highly selective permeability of endothelial barrier is essential to maintain tissue fluid homeostasis and to support a normal organ function. Dysregulation in endothelial barrier function often termed as “leaky” endothelium is a prominent feature of many cardiopulmonary disorders (80, 107). In the next sections, we will summarize the mechanisms of endothelial hyperpermeability and the role of Rho in these pathological cascades.
Endothelial permeability
The transport of fluids and macromolecules across the endothelium occurs via two routes: transcellular and paracellular pathways (86). The transcellular pathway is represented by caveolae-mediated vesicular transport of larger macromolecules such as albumin, immunoglobulins (86, 122). Studies have shown that Src kinase-mediated phosphorylation of caveolin-1, a major structural and regulatory component of caveolae, is involved in increased transcellular permeability (43, 110). The paracellular route is regulated by interendothelial junctions composed of AJ and TJ proteins that allow the majority of solutes, cytokines, and other macromolecules trafficking through the EC monolayer (45, 107). Vascular endothelial (VE)-cadherin is a key transmembrane AJ protein forming intercellular junctions in vascular endothelium by providing homophilic adhesion between neighboring EC and via its association with submembrane complex of α/β/γ- and p120-catenin family proteins linked to the actin cytoskeleton (33, 70). Numerous barrier-disruptive agonists increase endothelial permeability by causing phosphorylation-induced internalization and degradation of VE-cadherin, resulting in the weakened AJ assembly with the disruption of VE-cadherin-catenins association (36, 135). An increase in endothelial permeability not only causes an influx of protein-rich fluid into interstitial space but also allows for a rapid migration of neutrophils and uncontrolled flow of inflammatory cytokines, ultimately causing devastating respiratory illnesses that are best exemplified by ARDS.
Role of Rho in endothelial permeability
Vascular endothelium undergoes constant cytoskeletal remodeling in response to various circulating agonists such as thrombin and histamine, bacterial pathogens and endotoxins, and mechanical forces such as cyclic stretch and shear stress. Cytoskeletal reorganization caused by injurious stimuli promotes the formation of paracellular gaps, leading to increased endothelial permeability. Different members of the Rho family small GTPases have contrasting effects on cytoskeletal remodeling and EC permeability (154). Activation of RhoA triggers paracellular gap formation, cell contractility, and EC hyperpermeability response; whereas Rac1 and Cdc42 play a critical role in the maintenance of basal endothelial barrier function and recovery of EC barrier after injury (138). This review will focus on RhoA as a major trigger of EC barrier dysfunction caused by edemagenic agonists, inflammatory mediators, and pathologic mechanical forces. In addition, we will also discuss ROS-mediated regulation of the Rho pathway during endothelial dysfunction.
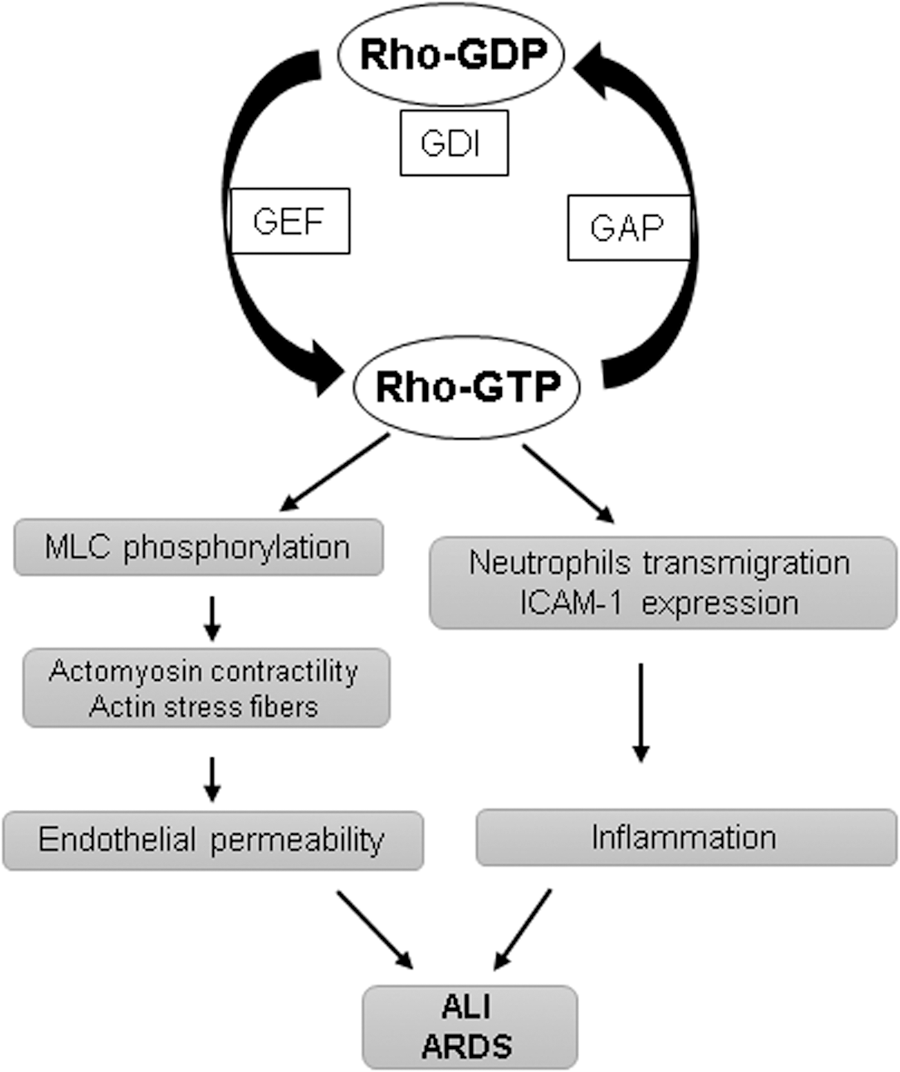
Small GTPases act as molecular switches for numerous signaling pathways of cell migration, adhesion, proliferation, and differentiation by cycling between GTP-bound active and GDP-bound inactive states (Fig. 2). The switch between Rho-GTPase-active and -inactive form is regulated by three different classes of regulators (9, 134). In resting cells, Rho is maintained in the inactive GDP-bound state by its interaction with GDP dissociation inhibitors (GDIs) present in the cytosol. On stimulation, Rho gets dissociated from GDI and translocates to the cell membrane where Rho-specific guanine nucleotide exchange factors (GEFs) such as GEF-H1, p115RhoGEF become activated and convert Rho to the GTP-bound active state. Once activated, Rho exerts its functions through its effectors, and GTPase activating proteins (GAPs) such as p190RhoGAP induce GTP hydrolysis and revert Rho to the cytosol in its GDP-bound inactive form.
A negative role of Rho in endothelial barrier function has long been appreciated, with the observations that inhibition of RhoA and its effector Rho-associated kinase (ROCK) reduces baseline endothelial permeability both in vitro and in vivo (1, 8). However, this established dogma is challenged by a study reporting that basal Rho kinase activity is required for the maintenance of endothelial barrier (149). These opposing effects of RhoA have to be interpreted with caution since there is a wide variation in the signaling pathways activated by the Rho family GTPases depending on the cellular context and specific agonists. Moreover, a crosstalk between various Rho family members provides checks and balances for precise regulation of EC barrier function. Contrasting effects of Rho GTPases have been further illustrated by findings that Rac1, which is typically involved in the enhancement of endothelial barrier, may increase endothelial permeability in a p21-activated kinase-dependent manner in certain conditions (140, 141). Nevertheless, the role of increased RhoA activity in endothelial barrier dysfunction is more clearly understood and will be briefly overviewed next.
The increased actin-myosin contractility with the formation of actin stress fibers has been postulated as a major mechanism of RhoA-induced endothelial hyperpermeability (124). The actomyosin contractility is mediated by the phosphorylation of regulatory MLC, and Rho is directly involved in the regulation of MLC phosphorylation. RhoA, through its effector ROCK, can induce MLC phosphorylation at Ser19 and Thr18 or it can also increase MLC phosphorylation by inhibiting myosin phosphatase activity with the phosphorylation of its regulatory subunit MYPT1 at Thr686 and Thr850 residues (3, 84). Among various barrier-disruptive stimuli, the initial studies showed that thrombin induces endothelial permeability by the RhoA-MLC signaling axis (13, 52). In addition to RhoA activation, thrombin can also phosphorylate MLC in calcium/calmodulin-dependent activation of MLC kinase (37, 147). Further, Src-mediated Tyr phosphorylation in EC activates MLC kinase and thrombin utilizes this pathway to induce endothelial permeability (51, 131). The extensive studies have established thrombin-induced RhoA activation as a model of acute endothelial barrier disruption. The indispensable role of RhoA in endothelial dysfunction is further bolstered by the findings that during agonist-assisted recovery of endothelial barrier dysfunction, RhoA activity returns to basal levels, whereas barrier-protective Ras family small GTPase Rap1 becomes activated (11, 14). Accordingly, pharmacological inhibitors of the RhoA pathway have been shown to offer protection against endothelial barrier disruption in various models of endothelial dysfunction.
Besides thrombin, a plethora of studies has shown that activation of RhoA serves as a central pathway in mediating the barrier-disruptive effects of a wide range of other stimuli. For example, TNF-α-induced stress fiber formation and apoptosis in EC is mediated by RhoA activation (120, 121). However, later studies suggested a more prominent role of Rac1 inactivation due to a decrease in cyclic adenosine monophosphate (cAMP) levels rather than RhoA activation in the TNF-α-induced breakdown of endothelial junctions (127). Similar cAMP-dependent mechanisms as well as RhoA have been implicated in the endothelial barrier disruption caused by LPS (16, 46, 64, 77). VEGF, histamine, and lysophosphatidic acid also increase EC permeability via the RhoA signaling pathway (109, 142, 150). Transforming growth factor-β (TGF-β)–induced endothelial barrier disruption also involves the RhoA pathway (12, 31). Certain bacterial pathogens and their virulent cell wall components are also known to evoke endothelial permeability via RhoA activation (22, 159). Finally, activation of the RhoA pathway has been observed in EC exposed to pathologic mechanical forces, which led to endothelial barrier disruption (10, 132).
Rho in inflammation
Robust inflammatory pulmonary host response to various chemical, mechanical, and pathogenic challenges is a typical feature of ALI and ARDS (102). EC play a critical role in the initiation of inflammation. Inflammatory agents rapidly stimulate the expression of intercellular adhesion molecule (ICAM)-1, vascular cell adhesion molecule (VCAM)-1 by vascular EC, which facilitate the recruitment of neutrophils to the endothelium (90, 156). Rho GTPases mediate integrin-dependent signaling and, thus, regulate cell adhesion and migration (91). The RhoA pathway has been implicated in the trans-endothelial migration of neutrophils (30, 126). Consistently, fasudil, a Rho-kinase inhibitor, has been shown to inhibit leukocyte adhesion in large blood vessels (137). In line with the role of RhoA in exacerbating inflammation, various studies have demonstrated the protective effects of fasudil on different models of lung inflammation (42, 96, 163). Further, RhoA is activated in EC during inflammation (112), and RhoA activation is directly involved in the increased expression of ICAM-1 in EC exposed to thrombin and lysophosphatidic acid (4, 133). It is important to note that inflammation and endothelial permeability are inter-related pathological events. A number of inflammatory agonists such as TNF-α and LPS cause profound EC hyper-permeability (54, 162). It has been demonstrated that chemoattractant-mediated activation of neutrophils also induces a rapid increase in permeability (53). On the other hand, weakening of EC AJ by cell incubation with VE-cadherin blocking antibody simultaneously increased endothelial permeability and neutrophil transmigration (55). In turn, neutrophil extravasation further boosts endothelial inflammation and the generation of RhoA-activating pro-inflammatory cytokines (97, 119). Taken together, these results suggest that increased EC permeability, inflammation, and neutrophil transmigration form a vicious circle of vascular dysfunction, which is mediated by pathologic RhoA signaling.
Differential regulation of Rho GTPases in various EC types
EC originated from different vascular beds possess different characteristics. The best example is higher levels of basal transendothelial electrical resistance in microvascular EC compared with EC from large vessels (80). The studies have also shown the differential regulation of endothelial barrier function by Rho GTPases in various EC types. Baumer et al. demonstrated that cytotoxic necrotizing factor (CNF)-mediated simultaneous activation of RhoA, Rac, and Cdc42 increases permeability in macrovascular EC but stabilizes barrier function in microvascular EC (8). The inhibition of the Rho pathway with ROCK inhibitor Y-27632 prevents CNF-induced barrier disruption in macrovascular EC, suggesting that Rac-mediated neutralization of Rho effects is weak in these EC. Further, a cell-type-specific crosstalk between p38MAPK and Rho has been shown to exist during Staphylococcus aureus-derived lipoteichoic acid and peptidoglycan-induced endothelial dysfunction in micro-and macrovascular EC (157). The inhibition of p38MAPK with its pharmacological inhibitor SB203580 represses Rho activation in microvascular EC but has no effect on macrovascular EC. The molecular/cellular factors governing EC heterogeneity have now been actively investigated by various groups, including our own, and future studies will unravel the mechanisms defining cell type-specific regulation of Rho GTPases and their downstream effects in these processes.
Rho inhibitors as therapeutic targets for lung injury
Considering a major role for Rho as a trigger of endothelial permeability, inhibition of Rho signaling axis may serve as an attractive therapeutic target to treat lung syndromes with profound endothelial hyperpermeability. Indeed, the inhibitors of ROCK, the downstream effector or RhoA, have been shown to protect against lung injury and inflammation in various in vivo models. For example, ROCK inhibitor Y-27632 and fasudil prevented LPS-induced ALI, pulmonary inflammation, and coagulation in mice (41, 96). In other studies, fasudil inhibited LPS-induced vascular leak in guinea pigs (144), blunted systemic inflammation and endotoxin-induced ALI (42), attenuated sepsis-induced ALI (153), and alleviated various parameters of endothelial dysfunction in paraquat-induced ALI in rats (163). Importantly, these ROCK inhibitors have already been clinically approved for cerebral vasospasm and glaucoma in Japan and China. Ongoing clinical trials in Japan are evaluating therapeutic effects of fasudil, Y27632, and other Rho/ROCK inhibitors in various diseases such as idiopathic pulmonary fibrosis, renal failures, erectile dysfunction, and corneal endothelial disorders (47).
Notably, fasudil has shown promising therapeutic potential for another endothelial dysfunction-associated disease pulmonary arterial hypertension (PAH) with its effect to suppress hypertension in animals and humans as well as to decrease pulmonary artery pressure and improve vascular remodeling in rats [reviewed in Hensley et al. (61)]. The clinical trials have already provided evidence that fasudil therapy improves pulmonary artery pressure and pulmonary vascular resistance in PAH patients (49). Consistently, more recent clinical trials showed similar beneficial outcomes of fasudil in patients with congenital heart disease and severe PAH (158). Thus, its proven role in mitigating endothelial injury-related pathologies and encouraging findings from preclinical animal studies suggest the therapeutic potential of ROCK inhibitors against ARDS, a devastating respiratory illness with 40% mortality and currently without efficient pharmacological treatment.
ROS in Endothelial Regulation
ROS are generated during normal cellular metabolism from incomplete reduction of molecular oxygen. Superoxide anions (O2 −), hydroxyl radicals (OH−), hydrogen peroxide (H2O2), and hypochlorous acid (HOCl) represent the most common ROS with high oxidizing potencies (146). At low physiological levels, ROS act as important signaling molecules in various cellular functions; initial ROS-mediated cellular responses protect cells against oxidative stress and play an important role in maintaining redox homeostasis (44). But an excessive ROS production during pathological conditions causes tissue injury and organ failure that has been ascribed to pathogenesis of ALI and ARDS (23, 81). Elevated ROS levels may directly induce EC dysfunction and, thus, lead to the development of various cardiopulmonary disorders (7, 101). Also, nitric oxide (NO) functions as an important signaling molecule in maintaining vascular tone and promoting angiogenesis. However, some of the NO metabolites known as reactive nitrogen species (RNS), such as peroxynitrite (ONOO−), formed after the reaction between NO and superoxide anion trigger endothelial dysfunction (105, 106). Deleterious cellular effects of both ROS and RNS have been attributed to their role in the oxidation of proteins, lipids, and oxidant-induced DNA damage.
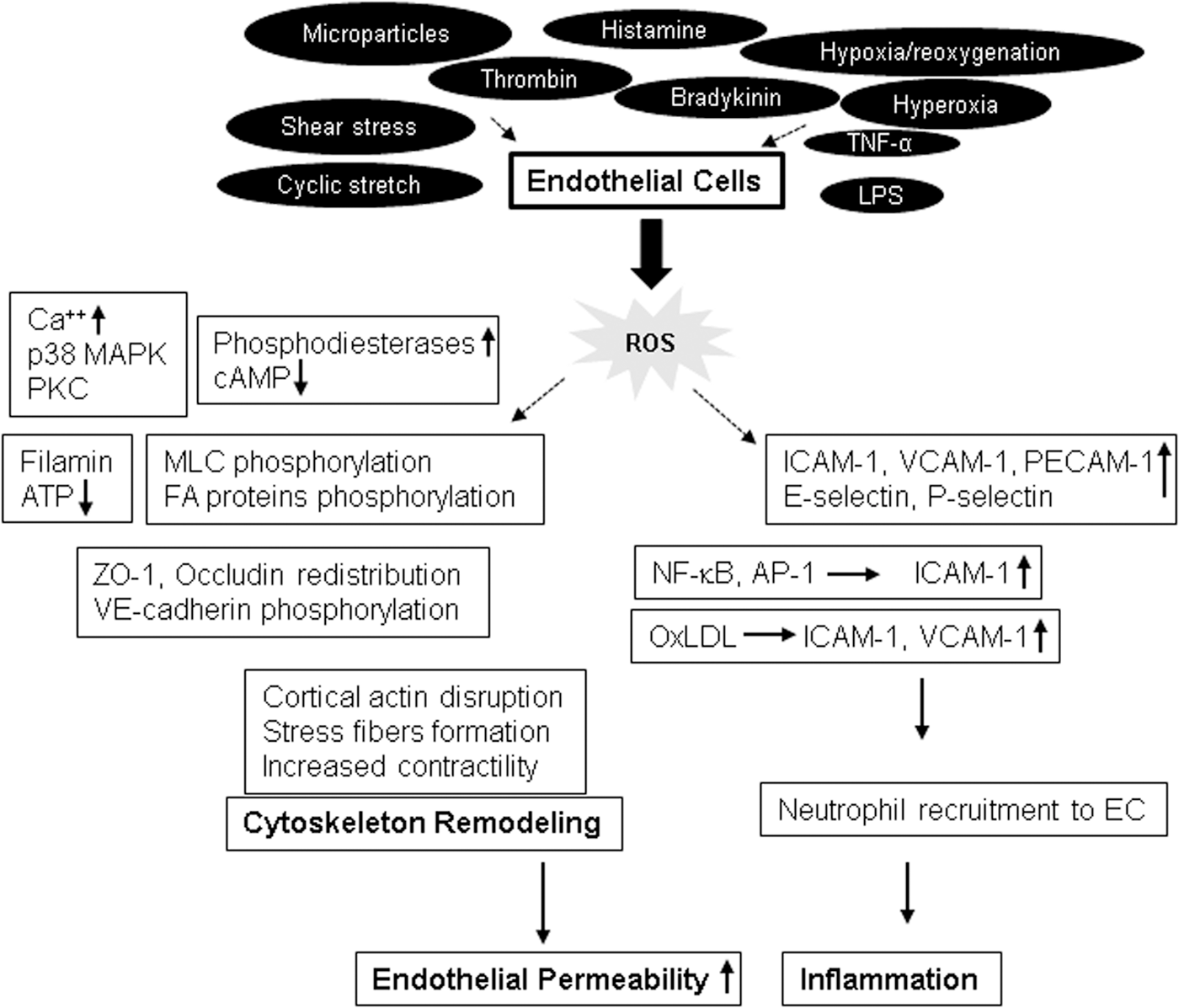
Major exogenous sources of ROS/RNS for pulmonary endothelium include environmental pollutants, cigarette smoking, and diet (17). During infection and inflammation, the endothelium becomes exposed to a wide variety of ROS and inflammatory cytokines, a typical phenomenon associated with the development of ALI and its progression to ARDS. In these settings, neutrophils serve as a principal source of ROS for the endothelium, as activated neutrophils adhere to the EC surface. The induction of respiratory burst by neutrophils is facilitated by NADPH oxidase 2 (NOX2) present in these cells (87). Numerous studies have demonstrated an ample ROS generation by activated neutrophils in response to phorbol esters, TNF-α, and chemoattractants (100, 113).
EC may also become a prominent source of ROS on challenge with injurious stimuli (Fig. 3). For example, an earlier study showed that EC produce superoxide ions after the treatment with interleukin (IL)-1 and interferon-γ (104). Consequently, other studies have demonstrated an increased ROS production by EC in response to various agonists, including shear stress (28), cyclic stretch (25), hypoxia/reoxygenation (99, 165), hyperoxia (19), microparticles (18, 108), TNF-α (27), LPS (118, 136), histamine (72), bradykinin (67), and thrombin (66). ROS production in EC is mediated by the mitochondrial electron transport chain, NOXs, especially NOX2 and NOX4 isoforms, uncoupled endothelial nitric oxide synthase, and xanthine oxidase (20, 74).
ROS-mediated endothelial permeability
Published studies provide substantial evidence that ROS have profound negative effects on endothelial barrier function (115, 148). Although the exact mechanism behind ROS-induced increase in endothelial permeability remains to be fully elucidated, elevation of intracellular Ca++ levels and activation of p38MAPK have been suggested as potential ROS targets. The role of ROS in disrupting endothelial barrier has been further supported by in vivo findings of pulmonary edema formation caused by ROS challenge (6, 7, 76, 128). Some of these studies showed a simultaneous increase in capillary hydrostatic pressure in conjunction with edema formation (76), whereas others showed that the development of edema without capillary pressure increases (6, 128). An involvement of ROS-induced endothelial dysfunction has also been observed in animal models of ischemia/reperfusion injury (5, 82). Briefly, Armstead et al. showed an increase in blood
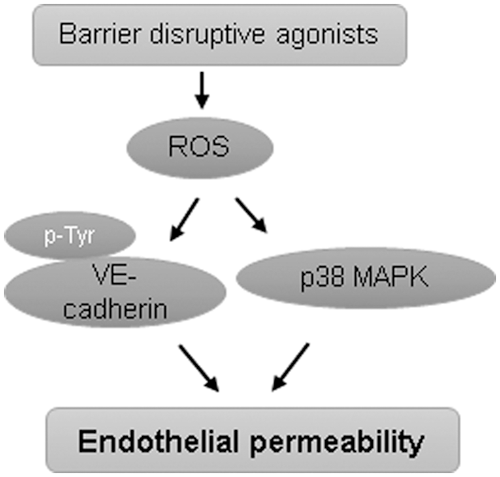
Endothelial barrier integrity is maintained by a dynamic equilibrium between barrier-protective intercellular and cell-matrix adhesive forces and barrier-disruptive actomyosin contractile forces (45). Both sets of these forces are generated by the actin-cytoskeleton network, and the shift in the balance toward contractile forces increases endothelial permeability (15, 130). Analysis of potential mechanisms of ROS-induced endothelial barrier disruption has shown that ROS induce EC cytoskeletal remodeling and enhance the expression of surface adhesion molecules in affected endothelium (101). Exposure of EC to H2O2 or xanthine oxidase caused an abrupt cell shape change along with the formation of intercellular gaps (63, 129). Alterations in calcium homeostasis and activation of protein kinase C (PKC), p38MAPK, and phosphodiesterases have been attributed to ROS-induced increase in endothelial permeability in these and other studies (129, 143, 148). A role of ROS-mediated calcium signaling pathways in endothelial barrier regulation has been summarized in detail in an excellent recent review (38) and is not the scope of this review. Interestingly, ROS challenge caused a decrease in intracellular cAMP that was linked to increased EC permeability (60, 143). The studies from our group and others have established an important role of cAMP in upregulating EC barrier function by either PKA-dependent activation of Rac or PKA-independent Epac-Rap1 signaling cascade. Thus, ROS-mediated decrease in cAMP levels may induce endothelial permeability by regulating the activity of these Rho GTPases. Taken together with findings of decreased barrier-protective Rac1 activity and upregulation of RhoA activity as described earlier during endothelial dysfunction, these results highlight the functional crosstalk between ROS signaling and Rho GTPases in control of EC permeability.
ROS-induced cytoskeletal remodeling involves the formation of actin stress fibers and the disruption of cortical actin, a feature associated with increased cell contractility (99, 164). A role of oxidative stress in the reorganization of actin cytoskeleton was further addressed in the study by Crawford et al., where overexpression of superoxide dismutase inhibited reoxygenation-induced actin remodeling (35). ROS are known to cause changes in actin organization by targeting some actin-binding proteins such as filamin, which is involved in actin filament crosslinking and connecting actin microfilaments to membrane glycoproteins (59, 60). They reported that human umbilical vein endothelial cell (HUVEC) exposed to H2O2 show the rapid translocation of filamin from membrane cytoskeleton to cytosol, which was accompanied by a decrease in cAMP levels, resulting in the formation of interendothelial gaps. A drop in ATP levels after prolonged oxidative stress has also been reported to induce the disruption of actin microfilaments and depolymerization of microtubules (MT), leading to an increase in endothelial permeability (63, 68).
EC exposure to H2O2 increased MLC phosphorylation (164), suggesting ROS-induced activation of MLC phosphorylation-dependent signaling axis of endothelial dysfunction. EC exposure to ROS increased protein tyrosine phosphorylation at focal adhesions, which was also linked to ROS-induced increase in permeability and neutrophil adherence on endothelium (21, 56). The tyrosine phosphorylation of focal adhesion kinase, paxillin, and p130cas in response to ROS exposure induced the formation of actin stress fibers and increased neutrophils adherence on HUVEC. ROS have also been implicated in disruption of TJ proteins. H2O2 caused redistribution of occludin on the cell surface, limiting its association with another TJ protein zonula occludens-1 (ZO-1) and leading to an increase in endothelial permeability (83). Likewise, H2O2-caused increase in brain EC permeability was mediated by altered expression and localization of actin, occludin, and ZO-1 (92). Rac overexpression-mediated ROS production has been shown to increase endothelial permeability due to ROS-mediated increase in tyrosine phosphorylation of VE-cadherin, leading to its disappearance from cell junctions and internalization (151). In agreement with the role of ROS-mediated VE-cadherin phosphorylation in inducing endothelial permeability, it has been shown that TNF-α causes endothelial dysfunction via NOX-mediated VE-cadherin phosphorylation (116). The increase in endothelial permeability due to loss of VE-cadherin from cell junctions via ROS-mediated phosphorylation represents a noncontractile mechanism of endothelial barrier disintegration where Rho GTPases are not involved. Further, redox-dependent activation of p38MAPK has also been shown to directly induce endothelial permeability (148). These evidences suggest that ROS can directly cause endothelial barrier disruption independent of Rho activation (Fig. 4).
ROS in endothelial inflammation
EC play a major role in the immune and inflammatory responses in both the vasculature and surrounding tissues. Elevated levels of ROS increase the expression of EC adhesion molecules ICAM-1, VCAM-1 and platelet-endothelial cell adhesion molecule-1 (PECAM-1) that facilitate an enhanced adhesion and extravasation of neutrophils in the endothelium. Several studies have demonstrated that increased oxidative stress drives neutrophil recruitment and trafficking in the endothelium. Both in vitro and in vivo ROS exposure to EC resulted in an upregulated expression of ICAM-1, PECAM-1, and P-selectin and a subsequent increase of neutrophil adhesion to EC, which was abolished by EC incubation with antibodies against these EC adhesion proteins (50, 98). Conversely, antioxidants such as vitamin E and α-tocopherol have been shown to inhibit leukocyte adhesion to EC (75, 161). Along with the EC surface adhesion molecules, platelet-activating factor (PAF) is also suggested to play a role in neutrophil adhesion. This notion is supported by the experimental evidence that inhibition of PAF or its receptors blocked the ROS-induced leukocyte adhesion to EC (50, 71).
Another pathological consequence of ROS-induced neutrophil-EC interaction is the production of proinflammatory cytokines that also regulate the expression of EC adhesion molecules. The study by Griffin et al. demonstrated that TNF-α in conjunction with IL-17 activated EC by increasing the E-selectin and P-selectin expression, leading to enhanced leukocyte recruitment to EC (57). Likewise, LPS stimulation upregulated the expression of ICAM-1 in epithelial cells and VCAM-1 in renal mesangial cells in an ROS-dependent manner (29, 93). Another pro-inflammatory molecule, oxidized low-density lipoprotein (OxLDL), also induced ICAM-1 and VCAM-1 expression in vascular EC that was mediated by oxidative stress (32).
Some redox-dependent transcription factors such as nuclear factor-kappa B (NF-κB) and activator protein-1 (AP-1) control gene transcription of EC adhesion molecules and regulate ROS-dependent neutrophil adhesion in the endothelium. In this line, H2O2 rapidly increased ICAM-1 mRNA transcripts and protein expression at the EC surface, causing enhanced neutrophil adhesion (98). Further, a definitive role of ROS in transcriptional regulation of EC adhesion molecules was demonstrated in studies with anti-oxidants N-acetyl cysteine and NF-κB inhibitor pyrrolidine dithiocarbamate, which suppressed ICAM-1 expression (48, 73). In addition, changes in glutathione levels in EC showed both transcription-dependent and transcription-independent expression of adhesion molecules dictating neutrophil-endothelial adhesion (85). A study has also reported that OxLDL induces monocyte adhesion to EC via the ROS-p38MAPK-NF-κB signaling pathway (26).
Crosstalk Between Rho and ROS in Endothelial Regulation
The fact that both RhoA and ROS trigger endothelial dysfunction by inducing MLC phosphorylation suggests that a crosstalk may exist between these two signaling pathways (Fig. 5). An increasing body of evidence suggests that ROS regulate RhoA activity, and RhoA, in turn, controls cellular redox balance by modulating the enzymes involved in the generation of ROS (65, 111). It has been demonstrated that Rho GTPases RhoA, Rac1, and Cdc42 contain a conserved redox-sensitive motif, and ROS target this motif to induce GTP nucleotide exchange, leading to Rho GTPases activation (62). The reversible oxidation of cysteine on this motif causes the nucleotide displacement favoring the GTP exchange and activation of Rho GTPases. These findings were further validated by another study that showed that two cysteine residues located at the redox-sensitive motif of RhoA are critical for ROS-induced activation of RhoA and subsequent cytoskeletal reorganization (2). The presence of one extra redox-sensitive cysteine residue in RhoA that is absent in Rac1 and Cdc42 is suggested to regulate the functional differences between these GTPases (62). Oxidative stress also activates RhoA by PKC-dependent phosphorylation and activation of p115RhoGEF (24). This signaling axis activates endothelial arginase, which competes with NOS for

Studies have suggested that nitrosative stress also regulates RhoA activity, thereby mediating endothelial dysfunction. For instance, LPS-induced endothelial barrier disruption was mediated by nitration of Tyr34 in RhoA (123). On the other hand, Rac-mediated NOX-dependent ROS generation activates p190RhoGAP, which inhibits RhoA activity (114). The opposing redox-dependent role of RhoA and Rac during hypoxia/reoxygenation-induced endothelial permeability has been described (155). Hypoxia inhibits Rac, leading to the activation of Rho, which causes stress fibers formation, breakdown of AJ, and increased endothelial permeability. In contrast, reoxygenation robustly activates Rac, inducing the enhancement of cortical actin cytoskeleton and strengthening AJ by inhibiting Rho activity. These findings suggest toward the existence of a redox-dependent, self-regulatory crosstalk between barrier-protective and barrier-disruptive members Rho GTPase family. It is likely that Rac-mediated ROS production inhibits Rho activity through NOX. This notion is supported by a report showing that activation of NOX1 inhibits Rho, whereas inhibition of NOX1 hyperactivates Rho (125).
The direct involvement of Rho GTPases in ROS generation is best exemplified by Rac-mediated, NOX-induced ROS production, where Rac plays the role of an essential component of an activated NOX complex at the cell membrane that, ultimately, produces ROS (69). Rac also directly interacts with NOS to regulate NO production (39) and interacts with SOD in a redox-dependent manner during the conversion of superoxide ion to hydrogen peroxide (58). The interaction of Rac1 with SOD has pathological implications as observed in amyotrophic lateral sclerosis. Some SOD mutants linked to the amyotrophic lateral sclerosis firmly are associated with Rac1, causing the sustained activation of Rac with excessive generation of ROS (58). Interestingly, Cdc42, an Rho GTPase closely related to Rac, is not involved in ROS generation by the NOX mechanism, but it directly binds to flavocytochrome b558 (40). This competitive binding of Cdc42 and Rac to flavocytochrome antagonizes the ROS-producing capabilities of Rac. Nonetheless, ROS-induced activation of Cdc42 by p47phox-mediated inhibition of Cdc42 negative regulator, Cdc42GAP, has been reported in smooth muscle cells (94).
Recent studies have suggested that Rho GTPases mediate a positive redox feedback loop. In this regard, actin binding protein profilin-1-mediated endothelial dysfunction is shown to be dependent on its positive feedback loop activation by ROS-activated RhoA/Rho-kinase (95). Briefly, advanced glycation end products-induced endothelial permeability is mediated by co-ordination of increased expression of profilin-1 and elevated production of ROS that leads to RhoA activation. Herein, a positive feedback loop consisting of prolifin-1-mediated ROS production and ROS-Rho-mediated expression of prolifin-1 regulates oxidative stress and endothelial dysfunction. An earlier study has demonstrated that neutrophil migration is controlled by a Rac-regulated and redox-mediated feedback loop (89). The authors proposed that Rac-mediated NOX activation inhibits phosphatase and tensin homologue (PTEN). Such inhibition of PTEN results in accumulation of PtdIns (3,4,5) P3. This signaling cascade creates a positive feedback loop to activate Rac and promote cell migration or activation of cortical cytoskeleton motility that is essential for recovery of intercellular gaps and reannealing of cell
An active interplay between ROS production and RhoA activation in mediating EC dysfunction was established by a study from our group. The results showed that LPS-induced endothelial permeability and lung inflammation is mediated by oxidative stress-caused destabilization of MT and ensuing RhoA activation (88). LPS challenge increased ROS levels in EC that caused the disassembly of the MT network, leading to the release of the MT-bound RhoA activator, GEF-H1. ROS-mediated and MT-dependent activation of RhoA was responsible for enhanced endothelial permeability and inflammation both in vitro and in vivo. In agreement with these findings, recent data suggest that ROS-mediated MT destabilization plays a pivotal role in S. aureus-induced endothelial permeability and inflammation (78). The exposure of EC to bacteria increased ROS levels, which activated histone deacetylase 6 (HDAC6), causing MT disintegration. The destabilization of MT increases endothelial permeability and inflammation either via the breakdown of EC junction assembly or via release of GEF-H1 and activation of the Rho pathway. This pathway also mediated lung endothelial permeability and inflammation caused by air pollution factor, particulate matter (79). These findings strongly suggest that the ROS-HDAC6-MT-Rho signaling pathway of endothelial dysfunction appears to be a universal mechanism triggered by many barrier-disruptive and proinflammatory agonists (Fig. 6).

Actin cytoskeletal reorganization represents another major impact of ROS-Rho GTPase crosstalk in endothelial function. Various Rho GTPases regulate endothelial barrier integrity by constant cytoskeletal remodeling based on the cell needs. The actin reorganization is essential for cell adhesion and migration, and ROS are known to influence these processes. Actin itself is prone to redox modification and oxidation of cysteine-374 is shown to alter its structural and functional organization on oxidation, resulting in decreased actin polymerization (145). ROS-mediated glutathionylation of actin is another mechanism that inhibits actin polymerization (152). Moreover, published studies demonstrate that Rac1 and other NOX components directly bind to actin and actin activates NOX (139). Taken together, these findings clearly suggest the existence of tri-directional interplay between Rho GTPases, ROS, and actin remodeling. It can be postulated that Rho GTPases, especially Rac1, are involved in the ROS production, and these GTPases regulate actin cytoskeleton organization either directly or via ROS-mediated redox signaling. In turn, ROS can directly activate Rho GTPases.
In summary, the bidirectional regulatory interaction between RhoA and ROS signaling appears to play a crucial role in inducing endothelial dysfunction and pathogenesis of several cardiopulmonary disorders, including ALI and ARDS. Basal physiological levels of ROS are essential for the maintenance of vascular homeostasis, whereas uncontrolled generation of excessive ROS impairs normal endothelial function by increasing permeability and inducing inflammation. RhoA activation is widely regarded as a central mechanism of endothelial permeability and inflammation in response to a broad range of barrier-disruptive and inflammatory agonists. A growing body of evidence suggests that ROS also activate RhoA to induce endothelial dysfunction, and Rho mediates a positive feedback loop of ROS generation. The existence of such a vicious cycle of ROS-RhoA signaling will have detrimental effects on endothelial barrier integrity, which may augment tissue injury and organ failure. A better understanding of ROS-induced regulation of Rho and interplay between the two in triggering endothelial dysfunction will be key to the development of new therapeutics against a broad range of disorders associated with dysregulated redox balance and hyperactivated RhoA signaling.
Footnotes
Acknowledgments
The studies in the authors laboratory are supported by the grants HL076259, HL087823 (K.G.B.) from the National Heart, Lung, and Blood Institute and GM122940 (K.G.B.) from the National Institute of General Medical Sciences.