
Abstract
Significance:
The pathogenesis and progression of allergic inflammation in the respiratory system are closely linked to oxidative stress. Thioredoxin (TRX) is an essential redox balance regulator in organisms and is induced by various oxidative stress factors, including ultraviolet rays, radiation, oxidation, viral infections, ischemia reperfusion, and anticancer agents.
Recent Advances:
We demonstrated that systemic administration and transgenic overexpression of TRX is useful in a wide variety of in vivo inflammatory respiratory diseases models, such as viral pneumonia, interstitial lung disease, chronic obstructive pulmonary disease, asthma, acute respiratory distress syndrome, and obstructive sleep apnea syndrome, by removing reactive oxygen species, blocking production of inflammatory cytokines, inhibiting migration and activation of neutrophils and eosinophils, and regulating the cellular redox status. In addition, TRX's anti-inflammatory mechanism is different from the mechanisms associated with anti-inflammatory agents, such as glucocorticoids, which regulate the inflammatory reaction in association with suppressing immune responses.
Critical Issues:
Understanding the molecular mechanism of TRX is very helpful for understanding the role of TRX in respiratory diseases. In this review, we show the protective effect of TRX in various respiratory diseases. In addition, we discuss its anti-allergic and anti-inflammatory molecular mechanism in detail.
Future Directions:
The application of TRX may be useful for treating respiratory allergic inflammatory disorders. Antioxid. Redox Signal. 32, 785–801.
Introduction
Inflammation and allergic diseases of the respiratory system are common, and the primary lesions are in the trachea, bronchus, and lungs. Patients with these diseases experience coughing, chest pain, breathing difficulties, hypoxia, and even respiratory failure and death. Due to air pollution, smoking, demographic changes, increased inhalation of mutagens, and other factors, the number of incidents and case fatalities have increased for diseases such as interstitial pneumonia, chronic obstructive pulmonary disease (COPD), bronchial asthma, and influenza (141).
The current primary treatment for respiratory inflammatory and allergic diseases is corticosteroids, β-receptor agonists, and theophylline drugs. These treatments have significant therapeutic effects, but cause some side effects, such as infection and immunosuppression. Therefore, it is vital to develop a new, safe, and effective therapeutic drug.
Thioredoxin (TRX) is a small-molecule secretory protein with antioxidant, redox regulation, anti-inflammatory, and anti-allergic capabilities. TRX has a significant inhibitory effect on respiratory inflammation. It has been used to treat digestive tract inflammation and dermatitis (79). In this review, we will elaborate on the anti-inflammatory and anti-allergic effects, and TRX's mechanisms on respiratory allergic inflammatory diseases.
Molecular Structure and Function of TRX
TRX is a small protein (12 kDa) with highly conserved redox activity in Cys32-Gly-Pro-Cys35. TRX is present in all organisms on earth, ranging from Escherichia coli to human beings (116). TRX's three-dimensional structure is a compact globular protein, and the fold of TRX consists of five β-strands surrounded by four α-helices (77).
Under various stress reactions, such as physical or chemical stimulation as well as microbial invasion, cells produce and release TRX to protect them from damage by regulating the cellular redox status (35). It regulates the redox state of cysteine in glucocorticoid receptor, estrogen receptor, nuclear factor kappa B (NF-κB), mitogen-activated protein kinases (MAPKs), p53, and hypoxia-inducible factor 1 (HIF-1) (79). TRX also regulates a variety of redox-dependent cellular processes, including signal transduction, gene expression, cell growth, and apoptosis (79).
TRX interacts with thioredoxin reductase (TRXR) and nicotinamide adenine dinucleotide phosphoric acid (NADPH) to catalyze protein disulfide bond reduction. This system maintains the cell reduction environment through the reversible thio-disulfide exchange reaction (48, 79), whose primary mechanism of action can be summarized as the transfer of reducing TRX equivalents from the catalytic site cysteine residue (Cys). The reversible oxidation to cystine disulfide (TRX-S2) occurs by the reduction of disulfide substrate X-S2 to X-(SH)2. Then, the oxidized TRX is reduced to Cys from TRX-(SH)2 by NADPH-dependent flavin protein, TRXR (108). Studies have shown that TRX effectively inhibits various types of inflammation without the suppression of immune responses (32). Previous research indicates that TRX plays an essential role in respiratory diseases, such as acute lung injury (ALI), asthma, influenza A (H1N1), and COPD (15, 45, 152).
Therapeutic Effects of TRX on Respiratory Diseases
Influenza A
H1N1 is an acute respiratory infection. Influenza viruses proliferate in the respiratory tract's epithelial tissue after infecting the body, leading to macrophage and neutrophil infiltration, before it creates complications such as ALI and pneumonia (105, 154). In addition, the reactive oxygen species (ROS) produced by the body in the process of resisting the H1N1 virus, such as hydroxyl radicals, superoxides, and nitric oxide, are highly toxic, and their activity is untargeted (98). They also strongly damage lung tissue while fighting pathogenic microorganisms (3, 134, 135), which is also a significant cause of ALI. Acute respiratory distress syndrome (ARDS) is also associated with H1N1 infections (94). These indicate that the pathogenesis of influenza is almost certain to involve not only apoptotic cell death mediated through viral replication in the infected cells but also the injury of noninfected cells by ROS derived from infiltrating neutrophils and macrophages and respiratory tract epithelium (97). These complications have greatly increased the mortality rate of the disease.
Neuraminidase inhibitors are used clinically to inhibit virus reproduction. However, neuraminidase inhibitors do not alleviate the infiltration of macrophages and neutrophils. Glucocorticoids can relieve pulmonary inflammation caused by H1N1, but there are still strong side effects, such as inhibiting T cell-dependent immune responses, which increase the risk of a secondary infection from bacteria and fungi, increasing the patient mortality rate (52, 70, 81, 145). It was also reported that H1N1 viral-infected patients treated with corticosteroid presented more frequently prolonged viral shedding, which was associated with immunosuppression, major morbidity, as well as complication of the influenza (33, 81). It is well known that glucocorticoids regulate the inflammatory reaction in association with the suppression of immune responses. Long-term or high-dose use of glucocorticoids can initiate the mass reproduction of the virus in lung tissues and induce secondary infections, then activate the immune system to produce a large number of cytokines (cytokine storms), and aggravate ALI or ARDS by increasing viral load and predisposing to secondary infection.
TRX plays a significant role in relief from, and repair of, respiratory damage caused by H1N1. Administering TRX to mice ameliorates H1N1-induced ALI through its potent antioxidative and anti-inflammatory actions (151). It has also been shown that human TRX transgenic mice are more resistant to the H1N1 virus, have a higher survival rate, and experience less severe lung histological changes than wild-type (WT) mice (93). Most importantly, the levels of respiratory IgA and serum IgG in human TRX transgenic mice were the same as those in WT mice (93). This indicates that TRX overexpression does not interfere with the immune system. Consequently, TRX may have a significant inhibitory effect on respiratory damage caused by H1N1, which can protect lung tissue and has greater advantages than hormonal drugs.
Chronic obstructive pulmonary disease
COPD is a slow-developing, incurable lung disease characterized by a sustaining limitation in airflow. The pathogenesis of COPD is related to oxidative stress, inflammation, and protease/antiprotease imbalance, and the three are inter-related. Under the stimulation of cigarette smoke or other harmful substances, respiratory tract epithelium secretes inflammatory cytokines and chemokines, including tumor necrosis factor alpha (TNF-α), interleukin (IL)-1β, GM-CSF, and IL-8, inducing the aggregation of macrophages and neutrophils in the lungs (36, 84). Moreover, the oxidative stress of the body is induced under the stimulation of harmful factors. The production of ROS and other substances can obviously increase the inflammatory level of the body and also induce the aggregation of macrophages and neutrophils (88). Macrophages and neutrophils are the main sources of pulmonary elastase. Excessive production of protease causes protease/antiprotease imbalance. Elastase decomposes tissue matrix and degrades and aggravates inflammation. Elastin fragments formed after degradation can attract macrophage precursors to inflammatory sites and aggravate inflammatory reactions (44, 140).
Currently, the most commonly used drugs are bronchodilators and glucocorticoids. The main types of bronchodilators include β2 receptor agonists and anticholinergic drugs; however, both have side effects. The main side effects of β2 receptor agonists are a rapid heartbeat, muscle tremors, and metabolic disorders (8). And the side effects of anticholinergic drugs include dry mouth, blurred vision, urinary retention, postural hypotension, cognitive problems, and heart rhythm disturbance (136). In addition, long-term inhalation or high-dose use of glucocorticoids may cause an oral candida infection, osteoporosis, and other complications (14).
The most important cause of COPD is oxidative stress, so treatments leading to anti-oxidation may be a good target. Antioxidants inhibit inflammatory gene expression by removing free radicals and oxidants, increasing intracellular thiol levels and controlling NF-κB activation (71).
The concentration of TRX in the serum of smokers is significantly increased (85); this may suggest that the body can resist the damage of cigarette to the body by producing TRX. Some experiments show that TRX plays an essential role in COPD treatment, and the influence of TRX on COPD involves three aspects: scavenging oxygen free radicals, reducing inflammatory reactions, and reducing the damage of elastase to lung tissue. TRX significantly reduces oxidative stress, and this is also well known to all (4). After human TRX transgenic and WT mice were exposed to CS for 6 months, emphysema occurred in WT mice with significant infiltration of neutrophils, but not in human TRX transgenic mice (114). In addition, intraperitoneal TRX administration also effectively inhibited the release of chemokines and cytokines such as IL-6, TNF-α, and RANTES, thus alleviating the smoke-induced lung inflammation in mice (126). Moreover, TRX inhibits elastase-induced emphysema (56). To sum up, we can see that TRX has various intervention effects on COPD.
Asthma
Asthma, as a common chronic airway disorder, is interpreted as the result of airway inflammation and reversible airflow obstruction. Airway inflammation is characterized by increased expression of cytokines, chemokines, and other inflammatory mediators, which together cause remodeling of the airways through resident/infiltrating immune cells such as dendritic cells (DCs) and neutrophils (39, 101). Seasonal mutations, allergic factors, microbial infections, environmental pollution, and overextension are leading causes of asthma.
Clinically, asthma treatment mainly involves corticosteroids, β2 receptor agonists, and aminophylline. Hormone therapy is associated with adverse reactions, correlated mostly to the dose. Hormone therapy is easy to administer in the throat, but it can cause a Candida albicans infection and sore throat (14). The β2 receptor agonist activates the β2 receptor of the heart, activates the sodium-potassium pump, promotes the intracellular transport of K+, and decreases the serum potassium concentration, leading to arrhythmia. Stimulation of skeletal muscle β2 receptors induces tremors (8). Aminophylline use often causes adverse reactions such as palpitations, headache, and vomiting (87).
The inflammatory airway process in allergic asthma is complex, involving predominantly CD4+ T cells, eosinophils (EOS), and mast cells, macrophages, and neutrophils. T memory cells, on molecular presentation from DCs, recognize specific antigens, such as allergens, microbes, and occupational agents. The activated T cells invade into the airway mucosa and induce Th2-mediated inflammatory reaction. A large of research documentations have demonstrated that classical asthma is predominantly a Th2-mediated inflammatory disorder. Allergen-specific IgE production by B cells is driven by IL-4, and IL-5 recruits and maintains a large number of EOS in the allergic tissues (18, 89). IgE cross-linking on the surface of mast cells or basophils leads to degranulation release of immune mediators (74). Further, it activates T cells to subsequently generate and release Th2 cytokines, such as IL-4, IL-5, IL-9, and IL-13. IL-13 can also stimulate airway epithelial smooth muscle cells to secrete eotaxin through activating the extracellular regulated protein kinases (ERK) pathway, thus promoting the migration and activation of EOS (89). A complex inflammatory reaction leads to airway infiltration by T cells, EOS, mast cells, macrophages, and neutrophils. In addition, migration inhibitory factor (MIF), as an important upstream regulator of airway inflammation, promotes eosinophil differentiation, survival, activation, and migration by binding CD74 and CXCR4 on the eosinophil surface (10). Severe asthma will lead to airway remodeling, which is irreversible. IL-13 plays an extremely important role in the development of airway remodeling (65).
The level of TRX in serum of asthmatic patients in attack stage is significantly higher than that in remission stage, and the level of TRX was dramatically amplified with the increasing severity of the asthma attack (155). This suggests that TRX is closely related to asthma pathogenesis.
TRX negatively regulates EOS and inhibits allergic inflammatory reactions (58). Overexpression of human TRX in mice prevents EOS accumulation at the airway inflammation site by inhibiting the production of macrophage MIFs (133).
TRX administration effectively inhibits the production of eotaxin, macrophage inflammatory protein-1α, and IL-13 in mice with asthma, thereby inhibiting airway remodeling and asthma development (47). In addition, goblet cells are the main effector cells for mucus production in respiratory tract, and both endogenous and exogenous TRX have obvious inhibitory effects on goblet cell proliferation (46). TRX inhibits airway hyperresponsiveness (AHR) and eosinophil infiltration in bronchial asthma, providing new ideas for treatment.
Obstructive sleep apnea syndrome
Obstructive sleep apnea syndrome (OSAS) is a condition characterized by recurrent episodes of apnea/hypopnea and subsequent oxygen desaturation during sleep due to upper airway obstruction (109). Some studies have demonstrated that the prevalence of diabetes among OSAS patients is more than 40%, and other studies have reported that OSAS is an independent risk factor for metabolic syndrome, myocardial infarction, and stroke (23, 103, 147).
The pathophysiological process of OSAS is clear. The level of C-reactive protein (CRP) in plasma of OSAS patients increased significantly, suggesting that OSAS is closely related to inflammation (83). The most prominent feature of OSAS is hypoxia, which leads to mitochondrial dysfunction, and it induces ROS production through respiratory chain complex I (68). A large number of studies show that ROS produced by peripheral blood leukocytes in OSAS patients increases (28, 107). ROS not only causes great damage to cells in a nonenzymatic form, resulting in membrane lipid peroxidation and redox damage to proteins, but also breaks DNA (129). More importantly, ROS participates in the MAPK pathway and activates the NF-κB pathway in OSAS (68). With the NF-κB pathway as a pivotal pathway of inflammatory response, the activation of the NF-κB pathway in OSAS patients not only activates neutrophils and endothelial cells but also enhances expression of adhesion molecules such as sE-selectin and sVCAM-1, which increases adhesion of inflammatory cells to vascular endothelium and becomes a risk factor for cardiovascular diseases (43). In addition, TNF-α and IL-6 in plasma of OSAS patients are increased (54), which may also be closely related to the activation of NF-κB and makes OSAS associated with systemic inflammation.
At present, the treatment methods of OSAS include surgical and nonsurgical treatment. Nonsurgical treatment is some improved oral appliances and continuous positive airway pressure (CPAP). Surgical treatment includes palatal implantation, low temperature plasma radiofrequency ablation, and electrical stimulation of upper airway muscles and nerves (73, 123). Common adverse effects to CPAP treatment include mask leakage, mask pressure, dry mouth, stuffed nose, claustrophobia, and difficulties exhaling (11). In addition, the more common complaints on the device pertain to the noise and bulk of the machine, and also side effects of the nasal mask, such as air leaks or ulceration of the bridge of the nose. Nasal congestion, rhinorrhea, and sneezing have also been noted in many cases (38, 66, 95).
In a clinical setting, OSAS patients have reduced plasma TRX levels after CPAP therapy (124), and TRX-deficient rats are more likely to acquire spatial learning and memory impairment in an intermittent hypoxia environment (150). So there may be a close relationship between TRX and OSAS. There is no direct evidence that TRX has an ameliorative effect on OSAS-induced inflammation, but TRX reduces LPS-induced NF-κB activation by inhibiting p65 and p50 synthesis and promoting I-κb synthesis in macrophages, thus reducing TNF-α and IL-6 production (7). Treating human umbilical vein endothelial cells with IH has improved TRX expression. In contrast, the expression of TRX interacting protein (TBP-2/VDUP1/TXNIP), which is a TRX negative regulator, was suppressed. In addition, after applying estradiol with an antioxidant effect, TRX expression increased further (63). TRX expression was also increased after an induced murine ischemia
In rabbit models of hypoxia
HIF-1 is one of the important regulatory factors in hypoxia. HIF-1 not only increases intracellular glutathione (GSH) level (76) but also promotes the synthesis of pyruvate dehydrogenase kinase, thus inhibiting the synthesis of acetyl-CoA, blocking the circulation of tricarboxylic acid and reducing oxygen consumption. Loss of HIF-1 leads to a decrease in ATP production and an increase in ROS production during hypoxia (118). And TRX plays an important role in HIF-1 stability and activity. TRX can also regulate HIF-1 protein synthesis by promoting p42/p44MAPK phosphorylation and activating p70S6K (159, 160), suggesting that TRX could be used as a safe and effective approach for OSAS.
Interstitial lung disease
Interstitial lung disease (ILD) is a large class of diseases in which inflammation occurs in the pulmonary interstitium, many of which have high morbidity and mortality. The basic pathology is diffuse pulmonary parenchyma, alveolar inflammation, and interstitial fibrosis. Some clinical use of anti-tumor drugs, such as bleomycin and gefitinib, led to ILD formation (156). Inflammatory cells and immune cells continue to accumulate in the alveoli interstitium, damaging the alveolar wall and capillaries, resulting in fibrosis due to improper repair, ultimately leading to irreversible damage to the lung parenchyma. The histologic basis of acute interstitial pneumonia is the infiltration of leukocytes into the pulmonary interstitial space and diffuse alveolar destruction. The majority of chronic ILD are referred to as idiopathic pulmonary fibrosis. Recently, it has become clear that multiple mediators, including ROS, cytokines, chemokines, eicosanoids, prostaglandin, and apoptosis-related genes, may be involved in the establishment of ILD (42). ROS have long been reported to cause oxidative modifications to proteins and to be a major mechanism in pulmonary fibrosis (122).
Due to ILD's complicated etiology, there is currently no unified treatment method in clinics, but anti-inflammatory and anti-fibrosis drugs are widely administered. Both corticosteroids and immunosuppressive agents have an inhibitory effect on the immune system and may induce other lung diseases. The current therapeutic effects are not ideal, and ILD is still not preventable due to its unknown etiology.
In recent years, TRX has been found to act in an anti-inflammatory manner. TRX is weakly expressed in healthy lung tissue (131) but it is strongly expressed in the serum and lung tissue of ILD patients (48). Patients with ILD caused by gefitinib also show significantly increased levels of serum TRX (111). This suggests that TRX may play an essential role in ILD development.
Recent studies in mice have shown that transgenic overexpression of TRX, and systemic administration of recombinant human TRX, can prevent the progression of an acute interstitial inflammatory response induced by IL-18 and IL-2, as well as in the interstitial pneumonia model induced by bleomycin (42). These results indicate that TRX is of great value to ILD research and has excellent prospects in the treatment of the disease.
Acute respiratory distress syndrome
ARDS is a type of respiratory failure characterized by abundant, dysregulated, and self-perpetuating proinflammatory environment with a significant increase in a range of proinflammatory cytokines accompanied by rapid recruitment of neutrophils into the alveolar space, endothelial injury and dysfunction, platelet aggregation and microthrombus formation, interstitial and alveolar edema, alveolar epithelial cell death, and macrophage activation (5, 132).
An essential factor in ARDS pathogenesis is ROS-mediated lung oxidative damage. This leads to cellular damage, including direct DNA damage, lipid peroxidation, protein oxidation, a release of proteases, and inactivation of antioxidant enzymes and antiproteases as well as transcription factor activator protein-1 (AP-1) and NF-κB. The alterations lead to an enhanced expression of proinflammatory genes (90). In addition, the multiple cell types within the inflammatory environment, including macrophages, epithelial and endothelial cells, play direct roles in ARDS pathogenesis. Among these cells, the neutrophil is central to driving this inflammatory state. Increased neutrophil numbers, the presence of neutrophil-derived proteases, and the chemotactic factors that drive neutrophil recruitment are associated with increased ARDS severity and higher mortality rates (115).
TRX reduces lung tissue damage introduced by oxidative stress, and it controls the further development of ARDS. Our previous studies have demonstrated that TRX improves the inflammatory environment in tissues by inhibiting the production and release of cytokines and chemokines such as TNF-α and IL-8. The TRX concentration in bronchoalveolar lavage fluid of ARDS patients is about 3–5 times greater than the TRX concentration in healthy people's plasma (13). The TRX level in the blood and plasma of ARDS patients is generally higher than that of healthy people, indicating that TRX is produced in large quantities by tissue cells and released to the lungs (13). TRX was significantly elevated in alveolar type II epithelial cells and lung macrophages in ARDS patients' lung tissue (13). Moreover, TRX has effects on ARDS induced by seawater inhalation by regulating neutrophil migration and activation, the release of TNF-α and IL-1β, and the redox balance in lung tissue (158). This suggests that TRX provides a new approach to the diagnosis and treatment of respiratory distress syndrome.
Mechanism of TRX Action
Scavenging ROS and antioxidant effects
Oxygen forms ROS with excess electrons in the body, such as superoxide anion radical (O2
The oxidative state results in toxic substances that directly damage alveoli and connective tissue, exacerbating COPD development (139). Excessive ROS formation also activates inflammatory cells, which, in turn, produce more ROS in the lungs and aggravates COPD development. Oxidative stress is also involved in asthma pathophysiology (22) and progression (16). In bronchial asthma, ROS exacerbates airway inflammation by activating transcription factors, such as NF-κB, MAPK, AP-1, and inflammatory mediators.
Oxidative stress is the result of IH, which plays an essential role in OSAS-related cardiovascular and cerebrovascular diseases (29). The process of repeated anoxia/reoxygenation increases ROS production (68). In addition, ROS may act as a signal molecule to regulate some signal transduction pathways, which may lead to pathological changes. ROS targets include MAPK (68), AP-1 (67), sterol regulatory element binding protein (68), GATA4 (68), NF-κB (67, 68), Notch-1 (146), and paraoxonase-1 (146). Tissue damage caused by oxidative stress plays an important role in ALI and ARDS pathogenesis. In response to various inflammatory stimuli, lung endothelial cells, alveolar cells, and airway epithelial cells, as well as alveolar macrophages produce ROS (53, 127).
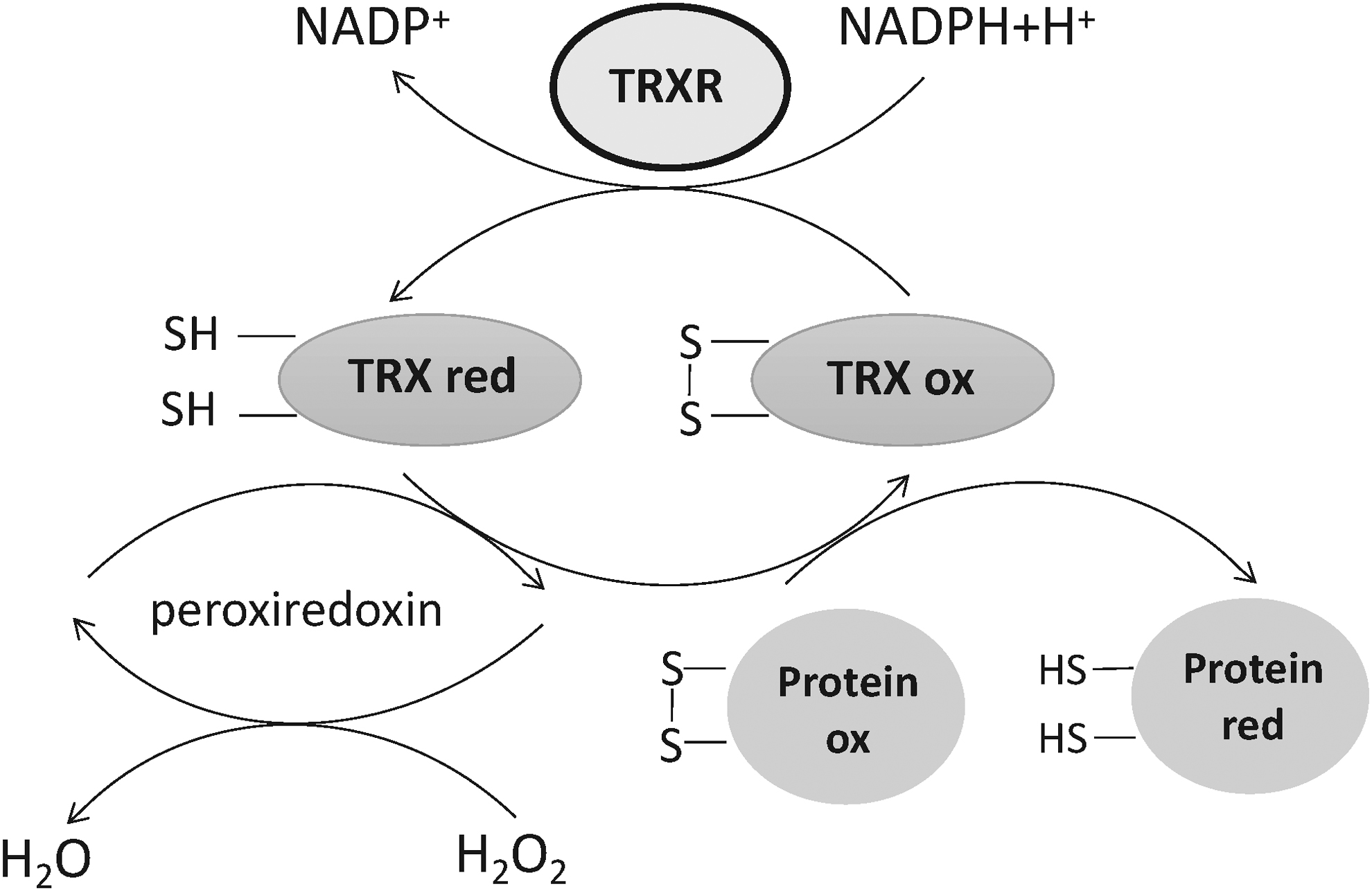
TRX can directly remove ROS (92). Cyanobacteria experiments have shown that TRX plays a role in eliminating H2O2 in photosynthesis (149). In addition, there are also some other redox systems similar to the TRX system, such as the GSH and peroxiredoxin systems (77). The TRX system together with the GSH-glutaredoxin system control the redox environment of mammalian cells, and mammalian TRX system and GSH system can provide the electrons in a cross manner and to serve as a backup system for each other (41, 77). When peroxidase is used to reduce H2O2 in an organism, TRX is required to provide the reduction equivalent (21).
In recent studies, the mitochondrial H2O2 content in the rat brain was measured by polarography in real time to detect the effect of each oxygen system on ROS elimination in mitochondria. The experiment found that when TRXR function was inhibited, H2O2's elimination efficiency was reduced by 80%, which was much higher than the reduction ratio when the peroxidase and GSH systems were inhibited. This suggested that the TRX-TRXR system played the most crucial role in inhibiting and eliminating intracellular ROS (27).
In summary, TRX expression is induced under the condition of excessive ROS production, by eliminating ROS in cells and reducing inflammation caused by increased ROS in the body. At the same time, TRX, TRXR, and NADPH constitute the TRX system, which restored and refolded the oxidatively damaged proteins and regulated the balance of other redox systems, such as the GSH, to maintain the stability of the whole redox balance in cells (Fig. 1).
Inhibition of the production and release of inflammatory cytokines
Infiltration by inflammatory cells, induced by proinflammatory cytokines, is the leading cause of tissue damage during development of a lung condition. The accumulation of activated neutrophils in the lung easily leads to ALI (1, 19). IL-6 activates neutrophils, causing neutrophil infiltration at the inflammatory site, inducing the release of elastase and various oxygen free radicals from neutrophils, and increasing pulmonary vascular permeability and destruction of alveolar surfactants. It also induces neutrophils to release elastase and various oxygen free radicals, resulting in increased pulmonary vascular permeability, destruction of alveolar surfactant, and pulmonary edema (49). In addition, TNF-α, IL-1β, and IL-8 play essential roles in inflammatory cell migration and strengthen oxidative stress during COPD development, further aggravating the condition (2, 69). Endogenous TNF-α plays an important role in pulmonary fibrosis development (31), which can cause secondary ILD. In addition, IL-8 can bind to specific receptors on the surface of neutrophils, inducing the release of leukocytes from proteolytic enzymes and ROS, and aggravating lung tissue damage (100). The presence of these chemokines or other inflammatory response inducers and neutrophils adhere to and penetrate blood vessels, which cause local neutrophil infiltration and stimulate inflammation (30, 78, 128). Neutrophil infiltration in lung tissue is the root cause of ALI induction (34). TRX effectively inhibits the expression of IL-1β, IL-6, IL-8, and TNF-α by blocking the NF-κB pathway or suppressing activation of the inflammatory signals by inhibiting cell surface molecules (130).
Cell experiments have confirmed that TRX is effective on skin epidermal cells and inhibits production and release of cytokines induced by Croton oil (130). Both the in vivo airbag model and in vitro chemotaxis chamber test have shown that TRX inhibited the chemotaxis of macrophages, lymphocytes, and neutrophils (75). In addition, when TRX was added to the medium of human monocyte-derived macrophages, it appeared to inhibit production and release of inflammatory cytokines, such as IL-1β, IL-6, IL-8, and TNF-α induced by LPS.
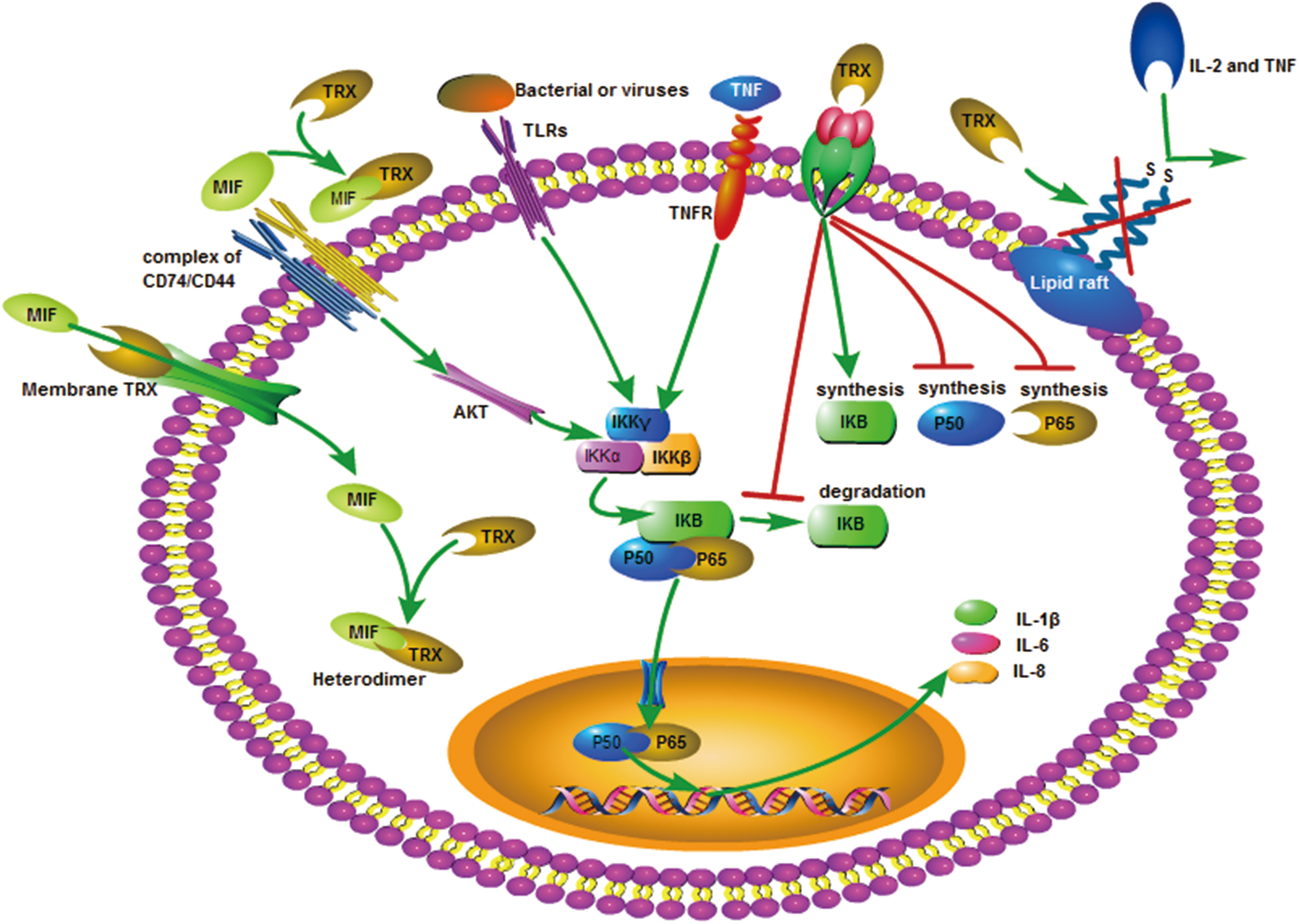
TRX acts on cell membrane receptors to inhibit production of cytokines that inhibit the production of p50 and p65 in cells and promote I-κB synthesis, thereby restricting the activation of NF-κB molecules and inhibiting NF-κB from entering the nucleus, in which the NF-κB signaling pathway is blocked (7) (Fig. 2).
TRX inhibits the migration and activation of neutrophils
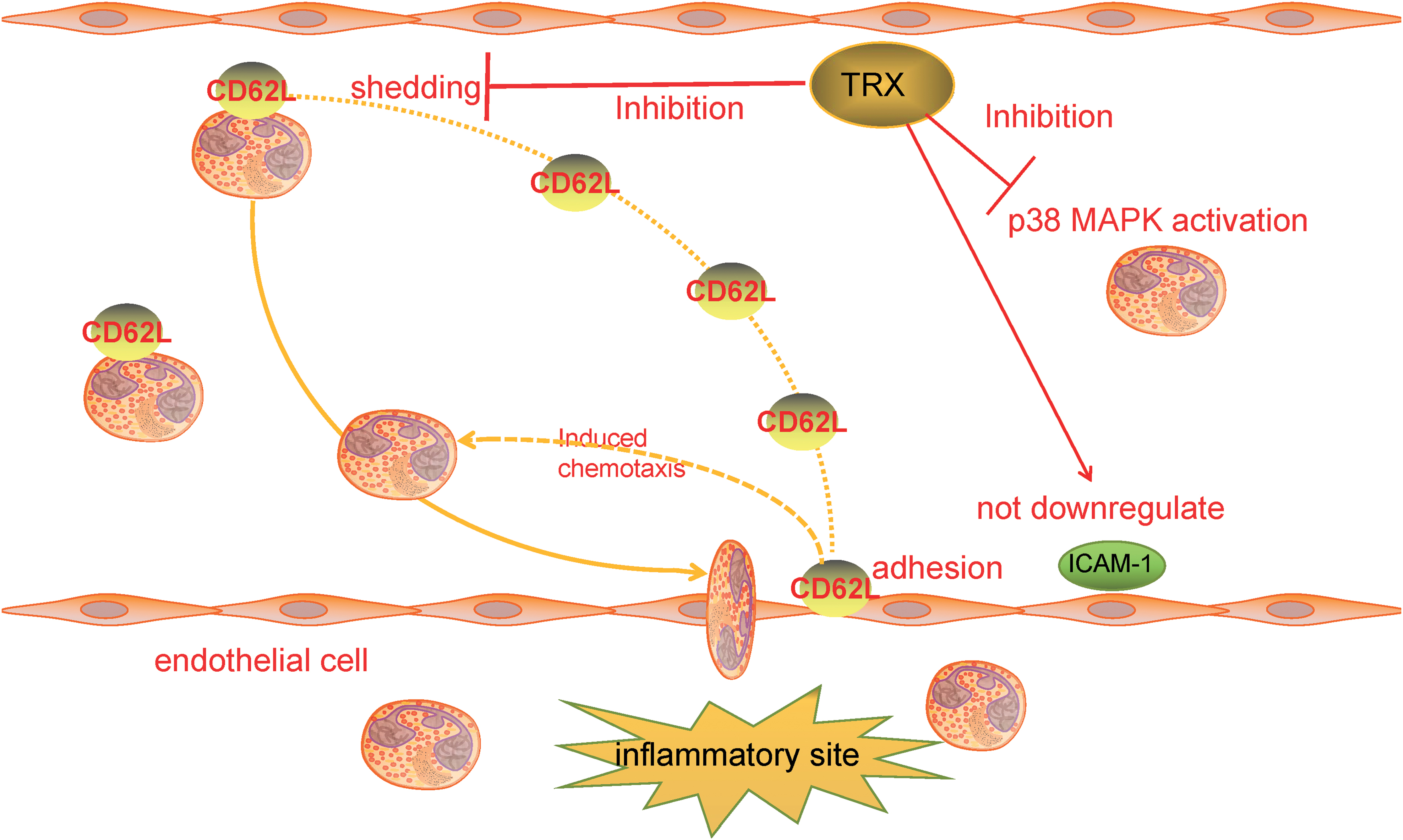
TRX has a significant inhibitory effect on the connection between neutrophils and vascular endothelial cells. Nakamura et al. demonstrated that TRX blocks the LPS-stimulated activation of neutrophil p38 MAPK and the adhesion of neutrophils to endothelial cells through a mouse airbag model.
CD62L is a shedding molecule on neutrophils that plays a vital role in guiding neutrophils to adhere to the vascular endothelium and penetrate blood vessels. It has been demonstrated that TRX inhibited LPS-induced downregulation of CD62L exfoliation on neutrophils and reduced CD62L attachment to endothelial cells, whereas the mutant TRXC32S/C35S does not inhibit neutrophils' adhesion to endothelial cells (91).
In an LPS-induced inflammation rat model, systemic TRX administration significantly reduced neutrophil infiltration in bronchial and lung tissues but did not directly reduce the increase of LPS-induced intercellular adhesion molecule-1 (ICAM-1) on endothelial cells (137). In addition, TRX did not inhibit neutrophil infiltration by reducing ICAM-1 levels. In conclusion, TRX can effectively inhibit and relieve tissue damage by preventing the migration and activation of neutrophils (Fig. 3).
TRX inhibits the migration and activation of EOS
The recruitment and activation of EOS play an essential role in the progression of some allergic diseases, such as asthma. Activated EOS secretes eosinophilic cationic proteins, which mediate the destruction of the airway epithelium and induce mast cells to release histamine, causing airway inflammation (119). EOS are also closely associated with AHR, as well as airway epithelial injury and shedding, and they are the primary inflammatory cells that cause chronic asthma (104).
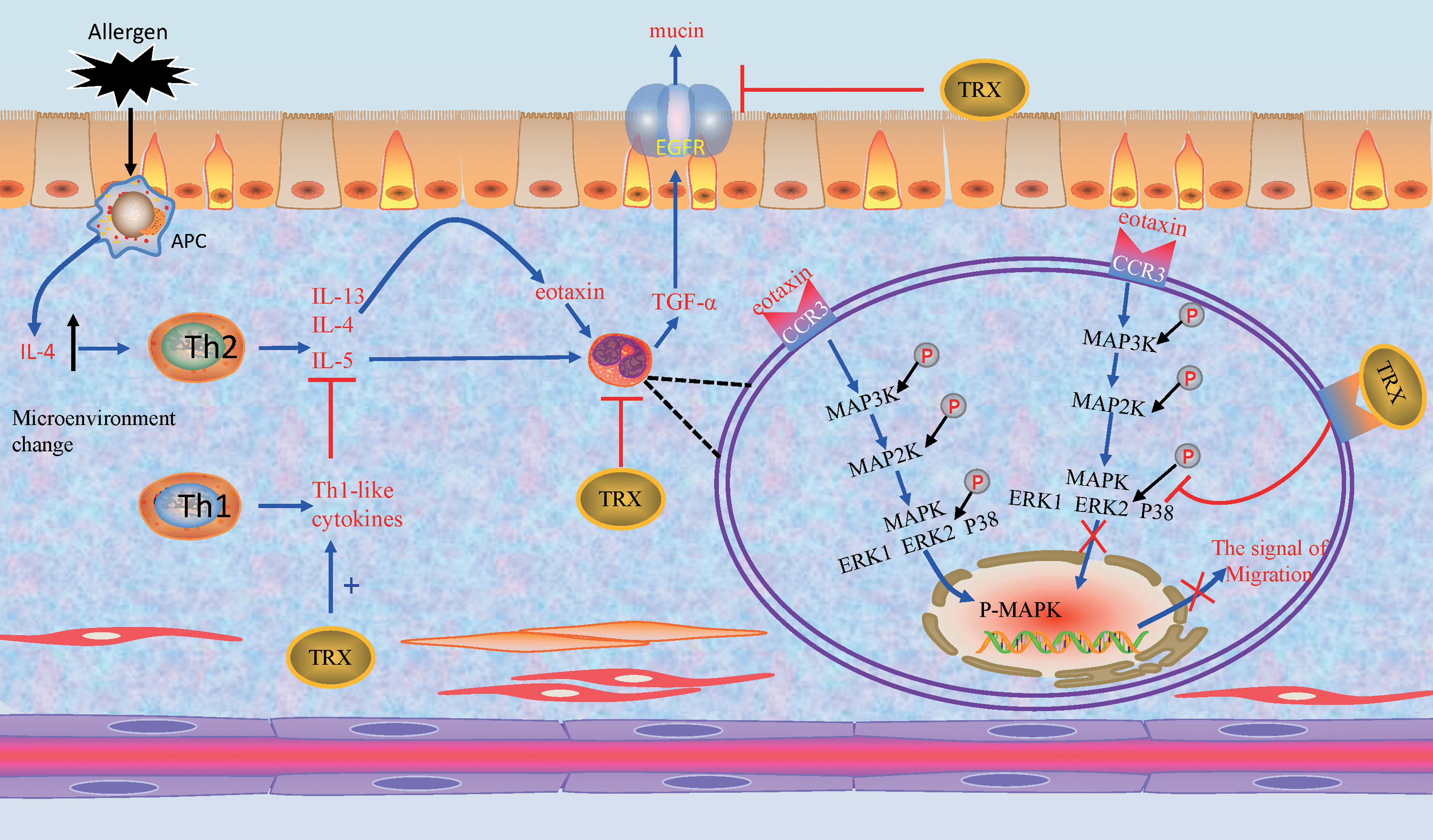
TRX acts as a regulator to minimize allergic inflammation by inhibiting eosinophil infiltration and activity (148). Eosinophil recruitment and activation are induced by cytokines and chemoattractants, such as eotaxin and IL-13. Eotaxin is produced by inflammatory cells and the airway epithelium and induces degranulation and chemotaxis of EOS through activation of ERK2 and p38 MAPK (9, 50).
TRX inhibits the migration and activation of EOS by regulating the cellular signal pathway, the extracellular Th1/Th2 balance and the molecule interacted with EOS-produced cytokine. After EOS were treated with TRX, TRX inhibited eotaxin-induced phosphorylation of extracellular signal-regulated kinase 1/2 and p38 by reducing the activation of ERK 1/2 and p38 MAPKs (58). Exogenous TRX inhibits airway remodeling by downregulating the production of chemokines MIP-1α and eotaxin in the lung, and TRX also modulates Th2 cytokines, such as IL-13, which induce antigen-presenting cells to produce eotaxin (47). When Th2 cytokine responses are increased, TRX induces Th1-like cytokines such as IL-1α, IL-1β, IL-1Ra, and IL-18 expression, thereby inhibiting Th2-like cytokine expression (45). TRX does not directly affect the proliferation and differentiation of Th1/Th2, but it inhibits inflammation by regulating the production and release of Th1/Th2 cytokines, because lymphocytes isolated from TRX-Tg mice are similar to WT mice in their ability to produce Th2 cytokines such as IL-4, IL-5, and IL-13 once they leave the high TRX environment in vivo (133). Moreover, the expression of MIF was significantly reduced in the lung tissue of TRX-Tg mice (45). In addition, activated EOS produce transforming growth factor-α, which acts on epidermal growth factor receptor (EGFR) of airway epithelial cells, inducing epithelial cells to synthesis mucin, which is closely related to tracheal inflammation (12). And overexpressing TRX in cell inhibit the synthesis and activation of EGFR (17, 37). All in all, TRX exerts anti-allergic effects by regulating eosinophil activation and migration (Fig. 4).
Specific effects with lipid rafts
Lipid rafts are specialized domains in the cell membrane, which are rich in cholesterol and glycosphingolipids (106). Recently, researchers have made the latest annotations regarding the formation of lipid rafts. They inserted gangliosides with fluorescent markers into the cell membrane and discovered that substances such as cholesterol and the CD59 receptor protein in the ganglioside and plasma membrane act rapidly to form lipid rafts. These molecules can quickly leave, and other molecules form new lipid rafts to play other roles (55).
There is currently no consensus about whether exogenous TRX can enter cells and play a specific role. However, there is a close relationship between TRX and lipid rafts. Researchers have shown that exogenous TRX can enter cells relatively slowly, and its internalization may be associated with lipid rafts (59). Other experiments have shown that the active site variant, rTRX-c35s, can be rapidly internalized into Jurkat T cells, suggesting that the active redox site of TRX is significant for internalizing TRX into cells (60).
Lipid rafts are platforms for cell surface receptor complex assembly. TRX is bound to have complex regulatory effects on lipid rafts, and its mechanism is inseparable from the interaction between disulfide bonds because activation of many cytokine receptors is achieved through disulfide bonds mediated disulfide. For example, erythropoietin receptors are activated by the homodimerization of disulfide bonds (143), and the reduction of disulfide bonds of CD132 leads to reduced binding to IL-2, thereby affecting signal transduction (20). TRX reduces the disulfide bond in CD30, a member of the TNF receptor family, thereby altering cytokine binding (138). The D2 domain of the CD4 receptor contains disulfide bonds. TRX, with redox activity, interacts with disulfide bonds in D2 and downregulates CD4 receptors on the cell surface (82).
All the experiments described earlier show that TRX alters receptor activity by interacting with disulfide bonds in cell surface receptor complexes, suggesting that TRX has a broad regulatory effect on lipid rafts. At present, toll-like receptors (TLR) on the cell surface are closely related to ROS and inflammation. In addition to TLR4-dependent signaling that leads to MyD88-dependent activation of NF-κB, and production of downstream inflammatory factors (40, 144), TLR are also involved in IL-1β processing and secretion (26). However, it is unclear whether TRX's anti-inflammatory effect is also directly related to TLR. Experiments have shown that TXNIP deficiency ameliorated high fat diet-induced nonalcoholic steatohepatitis through modulating TLR2-NOD-like receptor protein 3 inflammasome axis (86) (Fig. 2).
Inhibition of macrophage MIF
As a member of the TRX superfamily that shares a redox-active motif, -Cys-Xxx-Xxx-Cys-, MIF is a class of pleiotropic immunoregulators with unique structures, which function similar to chemokines and have proinflammatory responses, cell-directed migration, and release other cellular inflammatory factors (64). Under the stimulation of proinflammatory factors, MIF is secreted by bacterial metabolites, mononuclear/macrophages, B/T lymphocytes, endothelial cells, epithelial cells, endocrine cells, and smooth muscle cells (110).
MIF promotes macrophage activation. Activated macrophages that re-secrete MIF increase the amount of vascular cell adhesion molecule-1 (VCMA-1) and ICAM-1 to promote leukocyte and vascular endothelial adhesion, bind to the CXCR2/4 receptor, and promote the recruitment of leukocytes at the inflammatory site. In addition, MIF also mediates monocyte aggregation by increasing the expression of monocyte chemoattractant protein-1 (6). Moreover, NF-κB is the acting site of MIF. MIF is mediated by the CD74/CD44 receptor, which activates NF-κB via the AKT pathway and induces the expression of inflammatory factors, such as IL-1β and IL-8 (112). In addition, MIF has at least two catalytic activities, tautomerase and redox. The redox activity of MIF is closely related to the TRX family (6, 64, 110, 112).
Various experiments have shown that TRX can act directly on MIF and inhibit inflammation induced by MIF. TRX overexpression was shown to significantly suppress the level of MIF in a dextran sulfate, sodium-induced, colitis mice model. In addition, TRX significantly inhibited MIF release by human monocytes (125).
Further, in a smoke-induced mouse lung inflammation model, the mRNA expression of MIF in the spleen of TRX transgenic mice was lower than in the control group, indicating that TRX overexpression inhibited MIF production in the spleen (113). When the human TRX cDNA gene was transfected into Jurkat cells, TRX overexpression inhibited MIF expression (61). MIF has a specific affinity for TRX. Extracellular MIF can only internalize into cells by binding to TRX on the cell membrane. Exogenous and intracellular TRX combine with MIF to form a compound, thereby affecting the MIF-induced inflammatory response (120). In summary, TRX reduces the damage response of tissues and organs by directly affecting the internalization of MIF and the MIF-mediated, inflammation-inducing signaling pathway (Fig. 2).
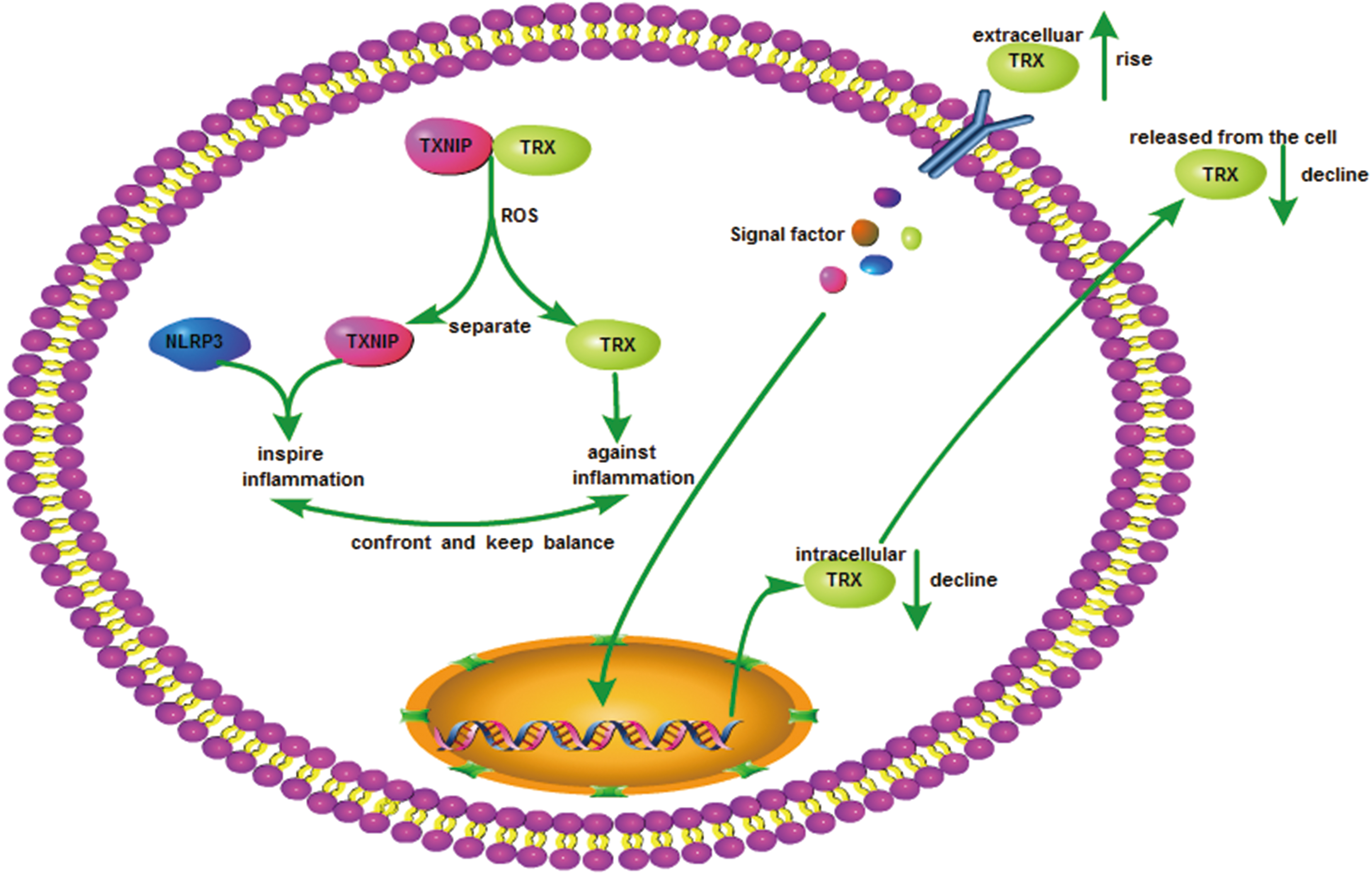
TRX/TXNIP, redoxisome: a tool to balance redox
TXNIP, a type of TRX-binding protein, mediates oxidative stress, inhibits cell proliferation, and induces apoptosis by inhibiting the TRX system function (79). TRX and TXNIP constitute a redox-like protein compound (redoxisome), which regulates a variety of redox-sensitive signals and is of great significance for maintaining intracellular and extracellular redox balance and monitoring the process of inflammatory responses (142).
TXNIP has a significantly adverse regulatory effect on TRX expression and reducibility. TRX expression is significantly inhibited in cells transfected with TXNIP vectors. In addition, TXNIP only binds to reduced TRX at healthy active sites, which weakens TRX's reductive activity, but does not affect TRX in the oxidized state (96).
In one study, endogenous TRX expression was significantly reduced in LPS-activated macrophages after adding exogenous TRX to the culture medium (59). Extracellular TRX maintains the dynamic balance of redox in the cells by inhibiting intracellular TRX expression, thereby affecting redoxisome-dependent inflammatory signaling pathways. The mechanism could involve negative feedback inhibition, and there may be a receptor on the cell membrane that senses the extracellular TRX concentration and regulates the production of intracellular TRX by controlling specific signaling pathways.
Intracellular oxidative stress induces the aggregation or isolation of intracellular redoxisome components, including TRX and TXNIP. Without oxidative stress in the cell, TXNIP is in a binding state with TRX. When the intracellular ROS content increases, TRX and TXNIP are dissociated. The mechanism of this process could involve the competitive binding of ROS to TRX. The free TXNIP obtained after dissociation binds to and activates the NLRP3 inflammasome, which induces IL-1β expression in the manner of NLRP3-ASC-caspase-1 dependency, thus inducing inflammatory reactions (161). The NLRP3-dependent inflammatory response was induced by injecting monosodium urate crystal to TXNIP–/– and WT mice. As a result, the contents of IL-1β and neutrophils in the peritoneal lavage fluid of TXNIP–/– mice were significantly lower than in WT mice (117).
In summary, there is a relatively complex relationship among extracellular TRX, intracellular TRX, and TXNIP. With the change of redox state, the organism regulates TRX and TXNIP levels to maintain the balance of redox. Therefore, it could be seen that redoxisome regulates the inflammation signal pathway (Fig. 5).
The Role of TRX in the Immune System
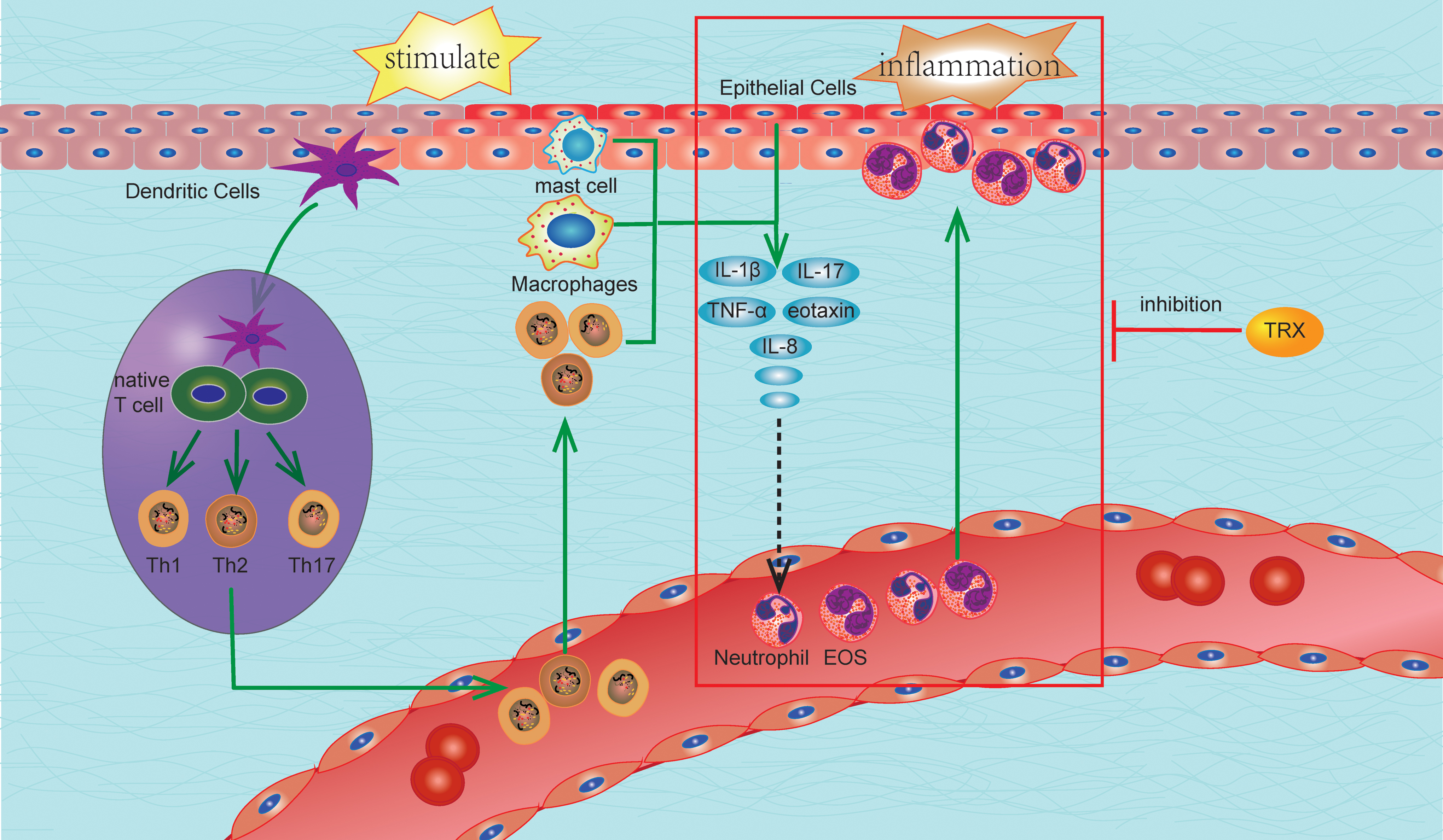
TRX, as an effective anti-inflammatory protein, has been shown to play a role in various inflammatory responses and has substantial advantages over other anti-inflammatory drugs. At present, the anti-inflammatory effects of corticosteroids commonly used in clinical practice interfere with the division and proliferation of lymphoid tissues due to antigens affecting lymphocytes' metabolism and inducing apoptosis, which have strong immunosuppressive effects.
A series of studies have shown no significant differences in the population and differentiation of immune cells, such as mast cells, DCs, and lymphocytes, between TRX overexpression and WT animals (121). In one study, TRX transgenic and WT mice were sensitized by applying the 2,4-dinitrofluorobenzene (DNFB) solution. In the sensitization stage, there was no significant difference between TRX overexpression and WT mice in the quantity and distribution of the activation of cutaneous DCs and hapten-specific cells in the lymph nodes. This suggests that the overexpression of TRX did not affect the original immune response during the sensitization period of allergic contact dermatitis. However, during the activation period, the infiltration of neutrophil and IL-17 mRNA expression in the skin of TRX transgenic mice was significantly lower than in WT mice (32). Therefore, the anti-inflammatory and anti-allergic mechanisms of TRX may be different from corticosteroids' inhibition of the immune system. In addition, its anti-inflammatory effects mainly depend on the inhibition of chemotaxis and infiltration of inflammatory cells, as well as the inhibition of inflammatory cytokine expression (Fig. 6).
Although TRX has a very good anti-inflammatory effect, TRX plays an anti-inflammatory role mainly by eliminating ROS and inhibiting cytokines. The production of ROS is the main way for neutrophils to play an antibacterial role. Cytokines such as TNF-α are also important inflammatory mediators against bacteria. These substances are important defense lines of the body. Excessive removal of them may increase the possibility of infection (24, 57). However, once excessive amount is produced, it will cause damage to the body, and appropriate anti-inflammatory substances are needed to remove it at this time. However, as an anti-inflammatory substance, the applicable index of TRX application is not clear. Second, the receptor of exogensis TRX on the surface of the cellular membrane is not well known. These problems are also the targets of our next exploration.
Conclusions and Perspectives
A large amount of evidence has demonstrated that TRX has excellent anti-inflammatory effects on a wide range of inflammatory disorders. In this review, we focus on TRX's therapeutic effects and mechanisms on allergic inflammatory diseases of the respiratory system. TRX exerts its protective functions in many respiratory diseases, such as ILD, asthma, H1N1, COPD, OSAS, and ARDS. In addition, TRX's most distinctive feature is that it has no interference suppression to the immune system, and, therefore, it differs from other existing anti-inflammatory and allergic agents.
We summarized seven aspects of TRX's anti-inflammatory and anti-allergic mechanisms: (i) antioxidant effect, (ii) inhibition of production and release of inflammatory cytokines, (iii) anti-leukocyte chemotaxis and activation, (iv) suppressing migration and activation of EOS, (v) regulating lipid raft, (vi) inhibition of the MIF, and (vii) redox-dependent interactions between TRX and TXNIP. The mechanism of TRX explains its protective effects under inflammatory and allergic conditions.
TRX is effective not only for respiratory inflammation but also for digestive tract inflammation and dermatitis (79). Altogether, TRX is expected to completely or partially replace traditional drugs as a new type of safe anti-inflammatory and anti-allergic drugs. Recent studies have also demonstrated that TRX is a new drug target to treat Parkinson's syndrome and Alzheimer's disease (72, 157). In addition, TRX has a significant effect on the treatment and improvement of hypertension in the elderly (25). All these facts suggest that the modulation of cellular redox regulation has emerged as a potential clinical approach to a significant number of diseases, such as allergies, inflammation, neurodegeneration, and vascular system diseases.
Further, increased TRX levels have been associated with disease conditions, such as acute kidney injury (51), Schering's syndrome (62), AIDS, acute coronary syndrome, acute pancreatitis, and other diseases (152). It suggests that TRX could also be used as a new clinical test for clinical diagnosis of the diseases suggested earlier.
Footnotes
Acknowledgments
The authors deeply appreciate Pro.Takashi Inamoto for pointed advice and discussion for writing this article.
Funding Information
No funding was received for this article.