
Abstract
Aims:
Redox homeostasis is tightly controlled and regulates key cellular signaling pathways. The cell's antioxidant response provides a natural defense against oxidative stress, but excessive antioxidant generation leads to reductive stress (RS). This study elucidated how chronic RS, caused by constitutive activation of nuclear erythroid related factor-2 (caNrf2)-dependent antioxidant system, drives pathological myocardial remodeling.
Results:
Upregulation of antioxidant transcripts and proteins in caNrf2-TG hearts (TGL and TGH; transgenic-low and -high) dose dependently increased glutathione (GSH) redox potential and resulted in RS, which over time caused pathological cardiac remodeling identified as hypertrophic cardiomyopathy (HCM) with abnormally increased ejection fraction and diastolic dysfunction in TGH mice at 6 months of age. While the TGH mice exhibited 60% mortality at 18 months of age, the rate of survival in TGL was comparable with nontransgenic (NTG) littermates. Moreover, TGH mice had severe cardiac remodeling at ∼6 months of age, while TGL mice did not develop comparable phenotypes until 15 months, suggesting that even moderate RS may lead to irreversible damages of the heart over time. Pharmacologically blocking GSH biosynthesis using BSO (
Innovation and Conclusion:
Our findings demonstrate that chronic RS is intolerable and adequate to induce heart failure (HF). Antioxidant-based therapeutic approaches for human HF should consider a thorough evaluation of redox state before the treatment.
Introduction
Heart failure (HF) is a frequent cause of human morbidity and mortality worldwide (10, 14). Given the heart's limited regenerative capacity, restoring cardiac function following maladaptive remodeling is a major challenge (61). Although inherited mutations account for most hypertrophic cardiomyopathy (HCM) events (37, 48), the molecular mechanisms underlying noninherited HCMs are poorly understood. Many heart diseases are linked to oxidative stress (20, 49, 63, 64); augmenting antioxidants and modulating redox-sensitive targets have been thought to protect the heart against oxidative stress (16, 50, 51, 55). Since free radical scavenging antioxidants have largely failed to provide cardioprotective benefit (2, 9, 15, 30, 59), activation of endogenous antioxidant systems has been advocated as a possible alternative (1, 12, 34, 57). However, the threshold for antioxidant activation to deliver beneficial effects, and its long-term impact on the heart, is largely unknown. Therefore, investigating the biochemical and physiological responses of the heart under moderate or extreme reductive redox conditions may offer opportunities to identify novel mechanisms with clinical significance.
Innovation
Constitutive activation of nuclear erythroid related factor-2 in the hearts of transgenic mice increased the endogenous antioxidants and caused reductive stress (RS). Specifically, the RS mice (transgenic-high) developed hypertrophic cardiomyopathy with abnormally increased ejection fraction. The transition from compensated hypertrophy to diastolic dysfunction and heart failure, as evidenced in transgenic-low mice with increased age (>15 months), signifies a pathological outcome over time. The characterization of biochemical and functional responses to RS in the heart indicates the potential for damage due to excessive antioxidant signaling. Pharmacologic depletion of glutathione prevents RS-induced cardiac remodeling and dysfunction.
Nuclear factor (erythroid-derived-2)-like 2 (NFE2L2 or Nrf2) is a stress-inducible transcription factor that binds to the antioxidant response element (ARE) and regulates its target expression (38, 40, 41, 62). Under basal conditions, Nrf2 is retained in the cytoplasm by its repressor Keap1 and post-translationally marked for degradation (17, 27, 29). Under stress conditions, the oxidative-sensitive modifications on Keap1 cysteine residues impede the polyubiquitination of Nrf2, allowing the nuclear translocation of Nrf2 to transactivate antioxidant genes (8, 27, 31, 40, 58). Moreover, de novo activation of the Nrf2 gene by mutations was found in patients with multisystem disorders, leading to a sustained upregulation of the Nrf2-dependent antioxidant system (21). Therefore, here we investigated whether a sustained activation of antioxidant systems leads to RS, and how chronic RS affects cardiac structure and function.
In this investigation, using a novel constitutive activation Nrf2 transgenic (caNrf2-TG) mouse model (53), we determined whether chronic RS is sufficient to cause myocardial hypertrophy and pathological cardiac remodeling. We discovered that sustained activity of Nrf2 in the heart perturbs cellular redox systems, resulting in chronic RS. Chronic RS induces functional changes at an early age that progress to irreversible pathological hypertrophy at later ages (6–8 months). This hypertrophy is initially accompanied by an abnormally increased ejection fraction (EF). Subsequently, chronic RS induces diastolic dysfunction and impaired ventricular relaxation leading to HF and reduced survival. In an effort to prevent RS, we used
Results
caNrf2 induces chronic RS in mouse heart
Clinical trials with agents intended to protect against oxidative stress in humans have often resulted in conflicting results (9, 30). The mechanistic underpinnings for these inconsistent effects in heart diseases are largely unknown, in part, due to lack of explicit and suitable in vivo models. To address this gap, we established heart-specific genetic mouse models that provide varied levels of caNrf2, a master regulator of antioxidants and the redox state (53). Using the mouse alpha-myosin heavy chain (α-MHC) promoter to direct the expression of caNrf2 in the heart, we recently established two different transgenic lines that express either low or high levels of caNrf2 at a young age (∼2 months) (53). Cardiac-specific expression of Nrf2 protein was significantly increased in a transgene dose-dependent manner (Fig. 1A), along with augmented Nrf2-ARE (DNA) binding levels in both transgenic-low (TGL) and transgenic-high (TGH) mice (Fig. 1B). In light of the significant and transgene dose-dependent increase in Nrf2-ARE (DNA) binding in TG mice, we analyzed the myocardial redox state in the NTG and TG mice at 6 months of age (23, 25). The levels of the reduced form of GSH (Fig. 1C) and the myocardial GSH/GSSG ratio (Fig. 1C) were significantly increased in the TGH hearts at 6 months of age. Furthermore, the myocardial concentrations of cysteine, a rate-limiting precursor for GSH, were elevated by 150% in TGH hearts (Fig. 1C) as was its GSH-adduct (Cys-SGSH; Fig. 1C), while the levels were unchanged or not significantly different in TGL animals compared with NTG controls.

We then performed RNA sequencing in these experimental groups and results showed transgene dose-dependent expression of various antioxidant genes in TGL and TGH hearts, which were validated by quantitative polymerase chain reaction (qPCR) for randomly selected genes (Fig. 2A, C). At 6–8 months of age, Gclc (2.0-fold vs. 3.5-fold), Gclm (15-fold vs. 52-fold), Gsr (1.5-fold vs. 4-fold), Nqo1 (2-fold vs. 4-fold), Gst-α (3-fold vs. 4-fold), Gst-μ (3-fold vs. 4.2-fold), Gpx1 (1.0-fold vs. 1.5-fold), and catalase (1.0-fold vs. 1.5-fold) were significantly increased in TGL and TGH relative to NTG mouse hearts. These results indicate a dose-dependent response for many antioxidant genes (42, 53).
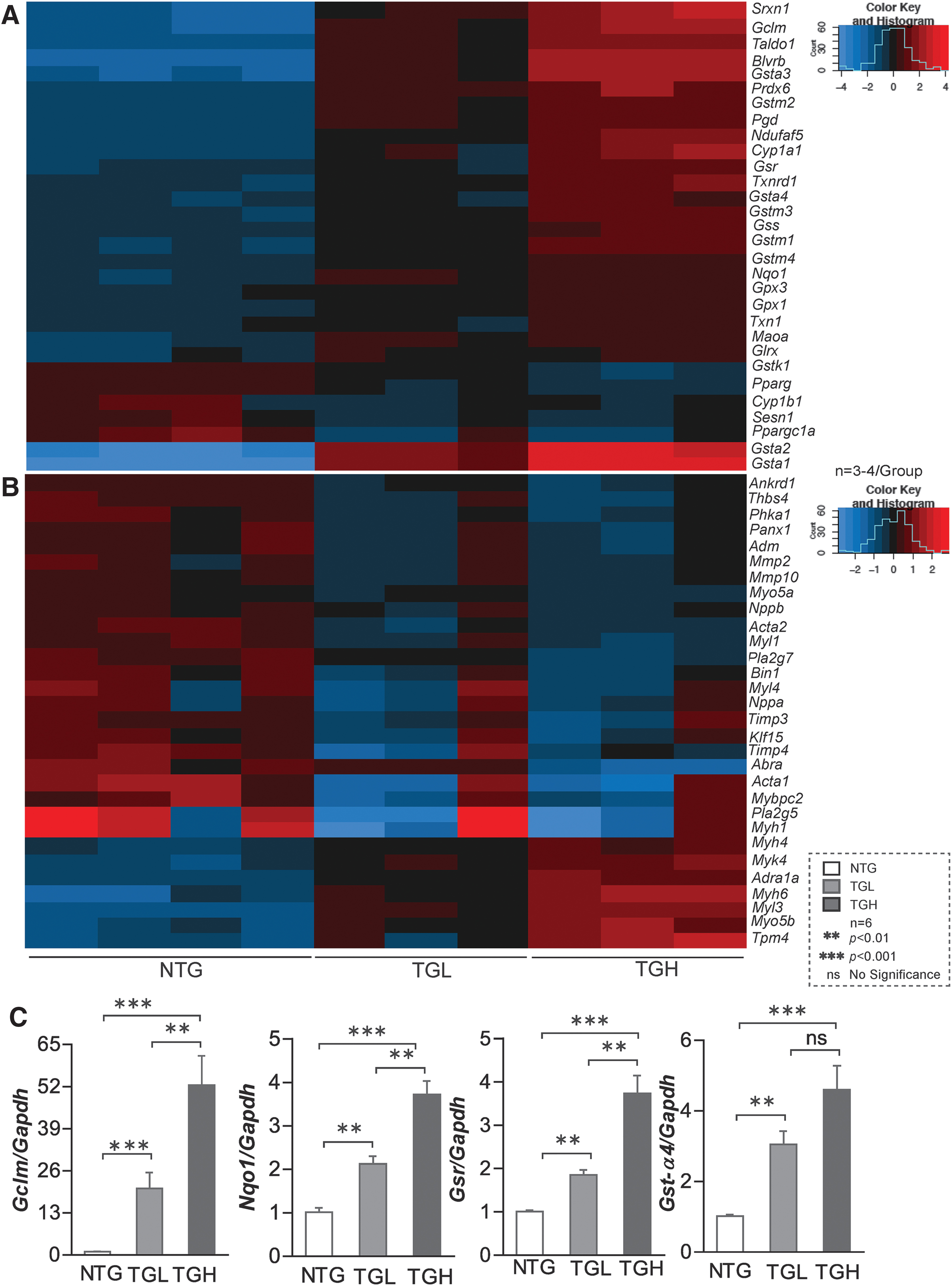
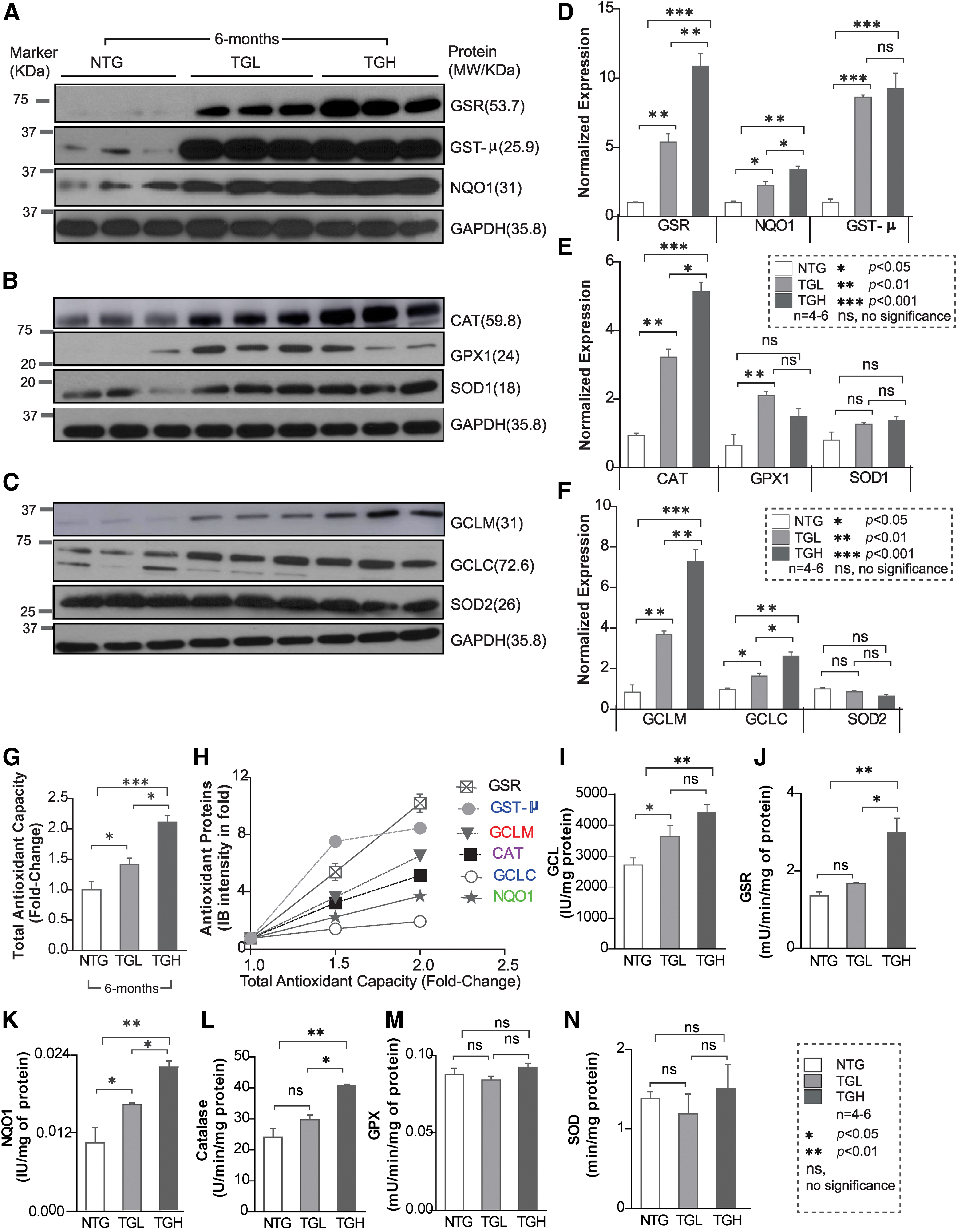
Next, we determined whether the transcriptional changes induced by caNrf2 reflected on protein expression of these antioxidant enzymes. Immunoblot analysis of myocardial cytosol revealed a marked increase in the expression of various antioxidant proteins (Fig. 3A–F). At 6 months of age, glutamate-cysteine ligase catalytic (GCLC) (1.5-fold vs. 2.0-fold), glutamate-cysteine ligase modifier (GCLM) (4-fold vs. 7-fold), glutathione reductase (GSR) (5-fold vs. 10-fold), NAD(P)H:Quinone Oxidoreductase 1 (NQO1) (2-fold vs. 4-fold), glutathione-S-transferase Mu (class) (GST-μ) (8-fold vs. 9-fold), glutathione peroxidase 1 (GPX1) (2-fold vs. 1.5-fold), and catalase (CAT) (3-fold vs. 5-fold) were significantly increased in both TGL and TGH compared with NTG mouse hearts (Fig. 3A–F). Moreover, reactive oxygen species (ROS) levels were significantly decreased in 6-month-old TG mice (Supplementary Fig. S1), indicating diminished ROS signaling and redox imbalance. Concomitantly, significant increases in enzymatic capacities of key antioxidants (GCL: 3.5-fold, NQO1: 4.2-fold, GSR: 2.8-fold, and CAT: 1.6-fold vs. NTG) were also observed in TGH mice (Fig. 3I–N). This strong correlation between the majority of antioxidant enzyme transcripts, protein abundance, enzyme activities, and total antioxidant capacity (TAC) (r 2 = 0.876 – 0.964; p < 0.05; Fig. 3G, H), along with blunted ROS in TG hearts, establishes the reliability of the model in regulating antioxidant pathways and inducing chronic RS.
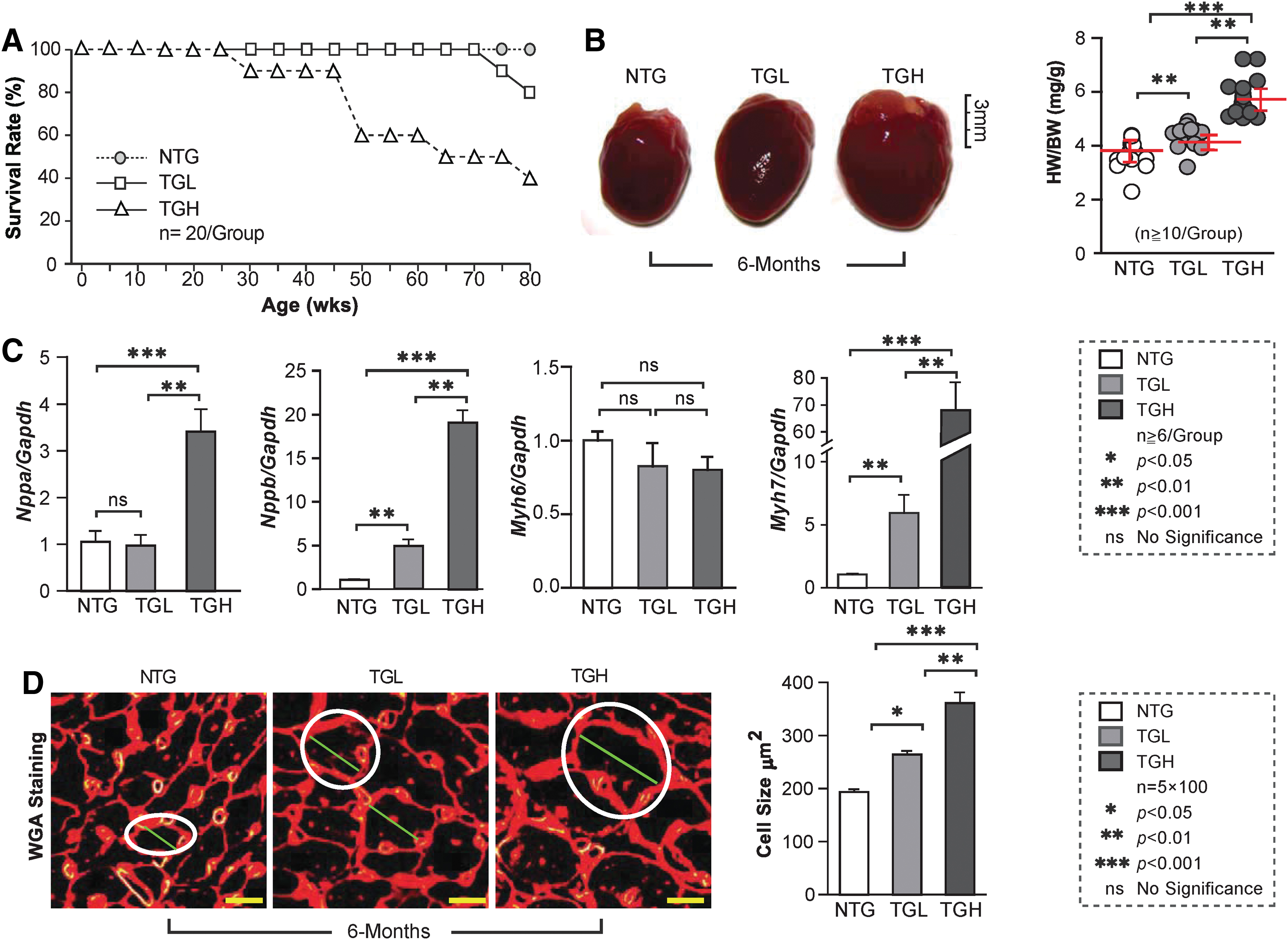
Chronic RS causes maladaptive cardiac remodeling and reduces mouse life span
We next evaluated the impact of RS on survival and morphological and biochemical changes in mouse hearts. Survival of TGL decreased by 20% at 18 months (80 weeks), compared with NTG mice. Most notably, by ∼11 months (50 weeks) of age, ∼40% of TGH animals had died (Fig. 4A). While no effects were observed in 3-month-old mice (Supplementary Fig. S2), progressive increases in cardiac enlargement and heart weight/body weight ratio (HW/BW) were evident in TGH at 6 months of age (Fig. 4B). Transcripts of hypertrophic markers, including ANF (Nppa), BNF (Nppb), and β-MHC (Myh7), were significantly increased in TGL or TGH 6
Chronic RS causes HCM with increased ejection fraction and diastolic dysfunction
To assess the impact of chronic RS on cardiac function, we performed noninvasive measurements of cardiac function and hemodynamics on the NTG and TG mice. Representative echocardiograms (M-mode) obtained in the parasternal short-axis view showed deterioration in cardiac structure and function in TGH mice at ≥6 months of age (Fig. 5A). In TGH, but not TGL hearts, significant concentric LV hypertrophy, decreased chamber volume, and increased cardiac contractility were observed. Compared with NTG mice, only systolic LV posterior wall thickness (LVPWs) increased in TGL mice, whereas both diastolic and systolic LV posterior wall thickness (LVPWd and LVPWs) increased in TGH hearts (Fig. 5C). End-diastolic LV diameter (LVIDd) displayed a significant reduction in TGH, but not TGL mice. Progressive decrease in end-diastolic LV volume (LVVd) was noted in TG mice (Fig. 5B), while the mean end-diastolic interventricular septal thickness (IVSd) increased by ∼50% in TGH mice. Short-axis echocardiography videos also demonstrated hypertrophic with hypercontractility in TGH hearts (Supplementary Video S1).

In parallel to echocardiography, we conducted strain analyses to obtain parasternal long-axis images. At ≤3 months of age, changes in cardiac structure were less pronounced without major functional defects (Supplementary Fig. S3). EF was moderately or markedly increased in TGL or TGH mice, respectively, as were end-diastolic LV mass and longitudinal strain. Collectively, these results indicate a hyperdynamic state in TG mouse hearts (Fig. 5B). Short-axis frozen images illustrate LV hypercontractility and a closer proximity of posterior wall and septal motions during systole in TG mice (Fig. 5D), which may result in a delayed ventricular relaxation leading to progressive diastolic dysfunction.
We also conducted pulse wave/color Doppler analysis on the three mouse groups. LV diastolic function was determined by characterizing mitral valve (MV) motion. While there was a moderate change in the diastolic function at 3 months of age (Supplementary Fig. S3), by 6 months of age, TGH mice had developed overt LV diastolic dysfunction, as indicated by increased mitral inflow velocity (MV E/A ratio) and early diastolic flow velocity (MV E), as well as decreased MV late diastolic flow velocity (MV A) (Fig. 5E, F). This progressive structural remodeling (i.e., concentric hypertrophy) appeared to be coupled with gradual diastolic dysfunction and restrictive filling in TG mouse hearts. Also, at 6 months of age, diastolic dysfunction was much more severe in TGH than TGL mice. The results indicate that chronic RS causes dose-dependent HCM with impaired ventricular diastolic relaxation.
caNrf2-TGL mice with moderate levels of Nrf2 activation increase RS-induced risks of maladaptive cardiac remodeling over time
We next assessed whether sustained proreductive (PR) conditions promote cardiac remodeling over time (or aging) in TGL mice. To pursue this, we conducted echocardiography on TGL mice at >15 months of age (Fig. 6A–C). Compared with NTG controls, EF, LV mass, and IVSd were significantly increased, whereas the LVIDd and LVVd were notably reduced. Profound diastolic dysfunction was apparent by LV hypercontractility and abnormal MV motion, including an enhanced MV E/A ratio (Supplementary Video S2 and Fig. 6D, E). This suggests that even moderate RS may lead to substantial damage to the heart following prolonged exposure. In addition, the redox state (GSH/GSSH ratio) of the 15-month-old TGL mice was greatly increased and was comparable with the 6-month-old TGH mice (Fig. 6F). Furthermore, immunoblotting (IB) showed excessive antioxidant augmentation (GCLC, GCLM, GSR, NQO1) at >15 months of TGL compared with 6 months of TGH mice (Fig. 6G–F), with decreased ROS signaling suggesting that a chronic low dose of antioxidant accumulation also causes pathological cardiac diastolic dysfunction over time.

Hyper-RS accelerates cardiac remodeling and dysfunction in double transgenic mice
Because caNrf2-TGH mice exhibited hypertrophic remodeling and diastolic dysfunction at 6 months of age, but no obvious phenotype at 3 months, we questioned whether robust activation of Nrf2 signaling actually provided the basis for this phenotypic switch. To address this issue, we created an even more severe RS model by crossing the caNrf2-TG with mice expressing a human Nrf2 (hNrf2) transgene (Supplementary Fig. S4A) and tested whether the cardiac remodeling was accelerated earlier than 6 months in the model. We performed qPCR for antioxidant and fetal gene expression, echocardiography for systolic and diastolic function, and immunofluorescence for GSH and 5,5-dimethyl-1-pyrroline N-oxide (DMPO) adducts. Results demonstrated significant increases in antioxidant genes (Supplementary Fig. S4B) with augmentation of GSH-N-Ethylmaleimide (Supplementary Fig. S4C) and decreased DMPO signals (Supplementary Fig. S4D) in double transgenic (DTG) mice, indicating that the hyper-reductive condition reduced the level of ROS.
Furthermore, by 2 months of age, DTG mice developed increased HW/BW ratio (Supplementary Fig. S4E) and increased hypertrophic markers (Supplementary Fig. S4F). In addition, echocardiography showed an increased EF (Supplementary Fig. S4G, H), with an increase in posterior septal wall (LWPD d) and septal wall diastole (IVS d) indicating that structural remodeling was evident in DTG mice by 2 months of age (Supplementary Video S3). Importantly, pulse wave Doppler analysis in the DTG mice (Supplementary Fig. S4I–J) showed increased inflow MV E/A ratio compared with the NTG littermates. Furthermore, DTG mice exhibited a higher amount of antioxidant protein expression compared with caNrf2-TGH mice at ∼2 months of age (Supplementary Figs. S4K, S9), indicating that the hyper-reductive condition led to HCM with restricted diastolic filling by 2 months of age.
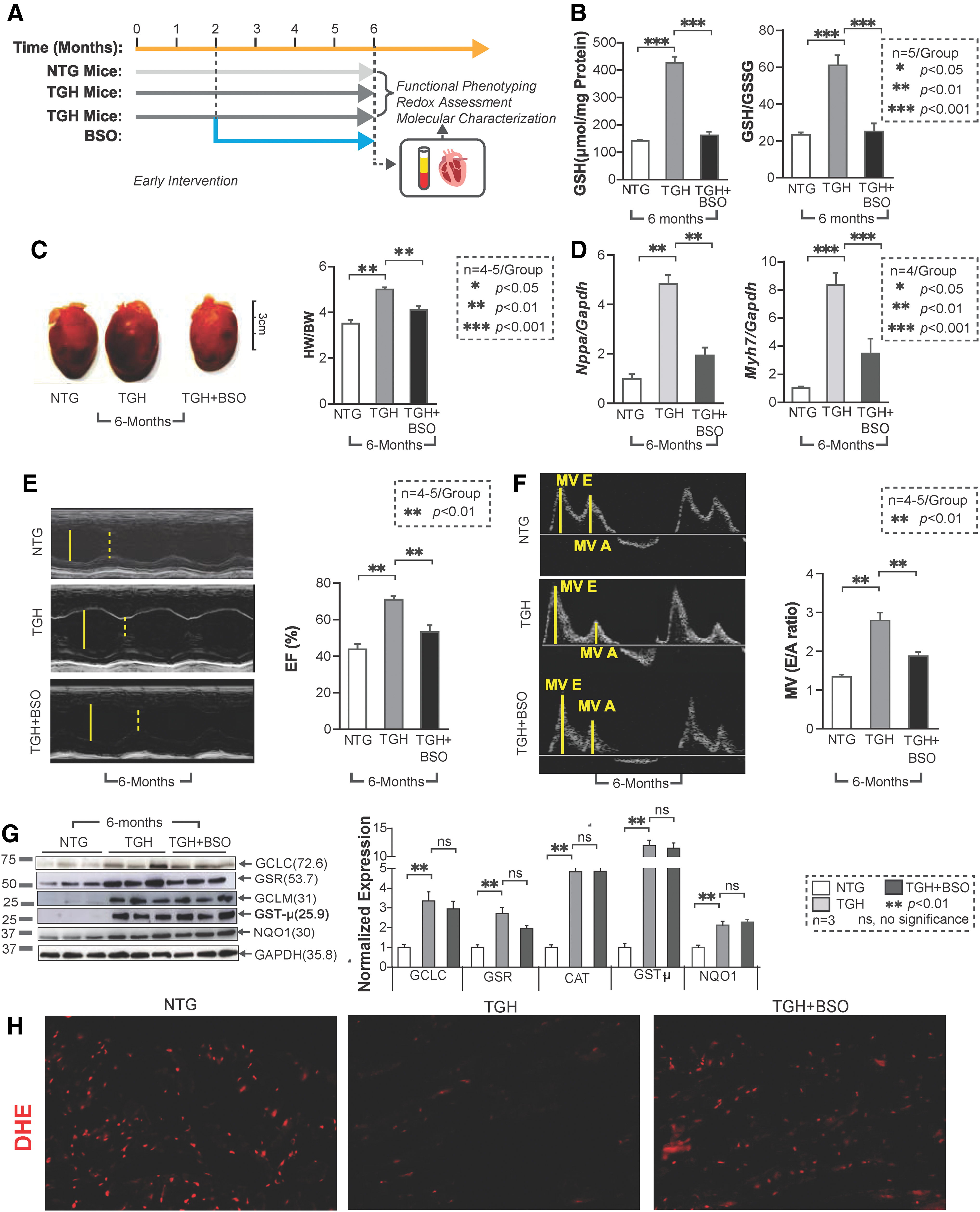
Buthionine sulfoximine-mediated GSH depletion prevents RS and delays onset of cardiac remodeling in TGH mice
As the most abundant nonprotein thiol source, the cellular GSH pool accounts for most of the reducing power accompanying Nrf2 activation. To restore redox balance in RS hearts, we treated TGH mice with BSO in vivo (33, 35) (Fig. 7A). Early BSO intervention (starting at 1.5 months of age and up to 4.5 months) successfully depleted GSH and shifted redox status back to an NTG level (Fig. 7B). Of note, BSO treatment also prevented cardiac hypertrophy, as demonstrated by HW/BW ratio and hypertrophic marker levels (Fig. 7C, D), and significantly improved the diastolic and systolic function of TGH mice (Fig. 7E, F). Furthermore, no significant changes were noted in key antioxidant protein levels (GCLC, GST-μ, and GCLM) (Fig. 7G). These results suggest that pharmacologic depletion of GSH can prevent RS and delay pathological remodeling of the myocardium. During BSO treatment, we also observed that GSR levels were partially restored, suggesting that regulation of some antioxidant enzymes may be sensitive to redox perturbation (GSH levels). It is also possible that their turnover might be regulated by mechanisms independent of Nrf2 levels. Concomitantly, BSO-treated mice showed moderate levels of ROS, confirmed by dihydroethidium (DHE) florescence staining (Fig. 7H), suggesting that early BSO treatment rescues the myocardium from RS and restored the basal ROS signaling.
Correlation analysis between different redox levels and its effects on systolic and diastolic function at different ages in Nrf2-transgenic myocardium
Based on the changes in redox conditions associated with structural and functional remodeling in the myocardium of TG (caNrf2) at different ages (3, 6, and 15 months of age), and of DTG (hNrf2:caNrf2; 2 months) compared with NTG mice, we correlated the dose- and time-dependent impact of RS on the severity of myocardial dysfunction. Dose-dependent increases in reductive redox (TGL and TGH) proportionally increased systolic function of the heart at 3 (8% and 41% vs. NTG) and 6 (50% and 72% vs. NTG) months. Of note, TGL mice at 15 months of age showed significantly greater changes in EF (69%) compared with age-matched NTG mice. DTG mice at 2 months of age showed an increased EF (44% vs. NTG), which was comparable with 6-month-old TGL mice (Fig. 8A).

Analysis of diastolic function showed a dose-dependent increase in the MV E/A ratio in TGL and TGH mice (10% and 27% vs. NTG) at 3 months of age, whereas chronic increases (adaptation) in MV E/A ratio of TGH (>100% vs. NTG) mice at 6 months of age resulted in progressive diastolic dysfunction and HF. The TGL mice at 6 months of age showed less change in diastolic function (33% vs. NTG), whereas at 15 months they developed a significant diastolic dysfunction (73% vs. NTG). Surprisingly, DTG mice developed severe diastolic dysfunction (69% vs. NTG) by 2 months of age, which was comparable with the 15-month-old TGL mice. Compared with 3-month-old TGH or 6-month-old TGL mice, the degree of diastolic dysfunction was significantly higher in DTG mice at 2 months (Fig. 8B). We also compared the impact of systolic versus diastolic changes in the context of a reductive environment (Fig. 8C). The analysis demonstrated that increased EF augments MV E/A ratios. However, chronic increase in both leads to an abnormal increase in systolic function with impaired restrictive filling, which results in diastolic dysfunction during chronic RS (Fig. 8A–C).
Discussion
The pathological implications of oxidative stress in various cardiac disorders have been extensively investigated over the past six decades (16, 50, 60). Exogenous supplementation of antioxidants and endogenous activation of antioxidant enzymes are the two major strategies used to curtail this oxidative damage. However, several antioxidant supplement-based interventions have failed to be beneficial (2, 4, 9, 15, 30, 59) and chronic infusion of antioxidants increased ischemia/reperfusion patient mortality (3, 6), leaving the activation of endogenous antioxidant genes a more desirable approach. While it has been assumed that a shift to more reductive redox conditions would be beneficial, it is possible that excessive reductive capacity could actually be harmful.
Single-nucleotide polymorphisms found in human gene disrupt Nrf2 protein expression and have been associated with a broad panel of human diseases (13). Especially, the newly discovered mutations (G31R, E79K, T80K, and G81S) in Neh2 domain (i.e., one of the Keap1-Nrf2 binding domains) led to an elevation of NRF2 level, overexpression in many antioxidant genes, and the manifestation of a multisystem disorder in four pediatric patients (21). Heart defects included thickened bicuspid aortic valve, atrial septal defect, and symptoms of cardiomyopathy. Notably, the level of plasma homocysteine, a redox marker, was reduced among these patients, suggesting that chronic activation of Nrf2 disrupted the redox balance and contributed to disease manifestation. However, the molecular mechanisms by which excessive redox shifts caused maladaptive cardiac remodeling remained largely unknown, partially due to a lack of appropriate animal models.
To investigate whether excessive antioxidant capacity leading to RS causes pathological changes in the heart, we developed a caNrf2 transgenic mouse model (53) with TGL and TGH mice mimicking PR and RS conditions, respectively. The model enables us to study the effects of different levels of RS on myocardial pathophysiology (Fig. 2). In addition, we developed a cardiac-specific DTG mouse expressing human Nrf2 with mouse caNrf2, which induces hyper-RS, accelerated cardiac remodeling, and dysfunction.
A noteworthy finding of the current investigation is that PR conditions in TGL mice appear not to be harmful at a young age, whereas RS in young TGH mice induced pronounced pathological changes (Fig. 4). However, TGL mice did develop significant cardiac remodeling at an older age (>15 months) (Fig. 6). Our data suggest that sustained antioxidant burden (e.g., GSH and antioxidant enzymes), even of relatively low intensity, but over a long period of time led to diastolic dysfunction in mice. To our knowledge, our models are the first transgene dose-dependent (i.e., TGL and TGH) RS models that developed age-dependent, pathological, structural, and functional remodeling of myocardium (Fig. 4).
Of note, mice with RS developed a distinctive phenotype, demonstrating HCM with abnormally increased ejection fraction (HCMiEF). Perhaps the model is an extreme example of reductive shift. In the clinical setting, RS and the corresponding phenotype might be less apparent, similar to the recent discoveries of HF phenotype with normal or preserved EF (HFnEF or HFpEF) (5, 7, 65). Based on our finding of an association between redox conditions and disease phenotype, we suggest that HF subjects with systolic dysfunction are more likely to be under oxidative stress and sensitive to treatment with antioxidant supplements or diets enriched in antioxidants. In contrast, HF subjects with chronic hypertension might be under an RS and have increased risks when exposed to/treated with antioxidant drugs (56). Clinically, HFpEF is characterized by diastolic dysfunction (36, 39). Affected individuals tend to be elderly women with diabetes, hypertension, and coronary artery disease (11, 28). The TGH mice exhibited an abnormal increase of EF, developed cardiac hypertrophy, and increased passive myocardial stiffness without evidence of fibrosis. Together, these phenotypic properties suggest that excess antioxidants promote systolic function through progressively increased ventricular stiffness with prolonged relaxation, leading to diastolic dysfunction and HF (Fig. 5).
In the present investigation, we also demonstrated that pharmacological depletion of GSH rescued TG mice from adverse cardiac remodeling. In TGH mice that developed a severe disease phenotype by ∼6 months, GSH depletion with BSO attenuated RS and prevented RS-induced pathological myocardial remodeling (Fig. 7). Our attempts to rescue the TG-mice from adverse cardiac remodeling through pharmacological inhibition of GSH are encouraging. BSO-mediated GSH depletion before developing cardiac hypertrophy (1.5 months of age) prevented RS and rescued the DTG from cardiac dysfunction.
In summary, harnessing the power of cardiac-specific constitutive Nrf2 overexpression in mouse model(s) (PR and RS in TGL and TGH, respectively) can help us understand the influence of antioxidant/redox manipulation on the development of cardiac pathology and disease. Interventions targeting RS at the different stages of disease progression (before and at the onset) may offer new approaches to preventing disease progression.
Conclusions
Short-term Nrf2 activation has demonstrated beneficial effects in different model systems, especially in an oxidative stress environment (32). We report that sustained or chronic Nrf2 activation results in RS, which leads to progressive pathological remodeling and HF through diastolic dysfunction. In the current investigation, we delineated the functional phenotypes and molecular characteristics of genetic models of RS. The results provide new insights by which to evaluate the impact of chronic antioxidant abundance in HF. Under chronic RS, mice developed HCMiEF, pointing to a potential direction for further investigation in HF subjects. Furthermore, our results show that, in addition to upregulation of genes involved in GSH metabolism, protein folding response also significantly disrupted under RS. Hence, we speculate that enhanced GSH accumulation might impair the protein folding mechanisms. However, it requires proteome-wide characterization for RS-induced changes in protein features (e.g., expression, turnover, and posttranslational modifications) to better comprehend the molecular interplays bridging excessive reductive shift and pathological remodeling of heart.
Detailed Methods
Reagents
RNeasy kit (74106), QuantiTect SYBR Green PCR, and QuantiTect reverse transcription kit (205313; Qiagen) were purchased from Qiagen, Inc. (Valencia, CA). Secondary antibodies for immunofluorescence conjugated with Alexa Fluor 488/594 anti-rabbit (A11008/A11012) and anti-mouse (A21042/A11005) were obtained from Life Technologies Corporation (Carlsbad, CA). The anti-rabbit (PI-1000) or anti-mouse (PI-2000) secondary antibodies for immunoblots (horseradish peroxidase-conjugated with IgG) were purchased from Vector Laboratories (Burlingame, CA). Primers for qPCR were designed using the Harvard Medical School PrimerBank website and purchased from Integrated DNA Technologies (IDT) (Coralville, IA). Protein Assay reagent (#500-0006) was procured from Bio-Rad (Hercules, CA). The Trans-AM Nrf2 promoter binding assay kit (50296) was from Active Motif (Carlsbad, CA). All other chemicals, including bovine serum albumin (BSA), RNAlater, reduced and oxidized GSH as standards, were purchased from Sigma Aldrich unless otherwise stated.
Animal models
All animal experimental procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Utah and University of Alabama at Birmingham. Two distinct transgenic mouse lines were used. The heart-specific caNrf2 transgenic model (TGL and TGH) has been described in our recent publication (53). The DTG mice were generated by crossing the caNrf2-TGH mice with hNrf2-TG mice. Briefly, to overexpress human Nrf2 in the myocardium, the Nrf2 construct was cloned in a mouse α-MHC promoter. The α-MHC-hNrf2 fragment was injected through pronuclear injection, and founders (C57BL/6) were identified by genotyping and qPCR for transgene expression levels. Based on genotyping, the TG-founders (hNrf2, both male/female) were confirmed, and breeding colonies were maintained separately under controlled conditions in the Animal Research Facility at the University of Utah and University of Alabama at Birmingham. caNrf2-TGH mice at the age of ∼1.5 months were treated with buthionine sulfoximine, an inhibitor of gamma-glutamylcysteine synthetase (GCLC), which is involved in GSH synthesis.
Necropsy and tissue collection
Mice (2–18 months of age, n = 4–6/group/gender) were anesthetized with isoflurane and euthanized by cervical dislocation. Hearts were immediately excised, perfused with ice-cold phosphate-buffered saline (PBS), and cut along the coronal plane. Two to three cross sections were obtained and stored in 10% zinc formalin and/or optimal cutting temperature (OCT) medium for immunohistochemistry/immunofluorescence imaging. The apex tissue biopsy (∼20 mg/each) was collected for RNA studies and stored in 300 μL of RNAlater solution. Before paraffin embedding and sectioning, tissue samples were kept in 10% zinc formalin for 24 h.
RNA isolation, next-generation RNA sequencing, and real-time qPCR
Heart tissue from NTG, caNrf2 TGL, and TGH (n = 6) at 6 months of age was homogenized using the QIAshredder kit, and RNA was subsequently extracted with RNeasy mini kits according to the manufacturer's instructions. Concentrations of freshly isolated RNA were obtained using a NanoDrop ONEc (Thermo Scientific, Waltham, MA). Two micrograms of RNA from each sample (n = 3–4/group) was used to perform next-generation RNA sequencing and analyzed as previously described (42), and 1.25 μg of RNA was used to synthesize cDNA with a QuantiTect reverse transcription kit. Twenty-five to 50 ng of cDNA template was used with 1 pmol primer in a 10 μL SYBR green reaction mix (204056; Qiagen) and was amplified in a Roche LightCycler 480 (Roche, Basel, Switzerland). Primers were designed with NCBI primer tools (Table 1 for sequences) and procured from IDT. Real-time qPCR was performed for randomly selected genes represented in heatmaps to valid the RNA sequencing, relative expression was quantified using Ct values, and expression fold-change was calculated by normalization to the Ct of housekeeping genes Gapdh or Arbp1 using the 2−ΔΔCt method (45, 47, 53).
Complete List of Real-Time Quantitative Polymerase Chain Reaction Primer Sequences
Protein isolation and IB
Heart tissues from NTG, caNrf2 TGL, and TGH mice (n = 4–8) at 3–18 months of age were homogenized in a cytosolic extraction buffer (10 mM HEPES, 10 mM KCl, 0.1 mM EDTA, 0.5 mM MgCl2, with freshly prepared 0.1 mM phenyl methylsulfonyl fluoride (PMSF), 1 mM dithiothreitol, and 1% Triton X-100, pH 7.9) and centrifuged at 5000 rpm for 5–6 min. The pellet was processed to obtain nuclear fractions using nuclear extraction buffer (20 mM HEPES, 420 mM NaCl, 0.1 mM EDTA, 1.5 mM MgCl2, 25% glycerol with 0.5 mM PMSF, and 1 mM dithiothreitol, pH 7.9) and centrifuged at 8200 rpm for 10 min as previously described (26, 45, 47, 53). Protein concentrations were determined with the Bradford reagent, and equal amounts of protein were resolved on 10%–12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis. Proteins were then transferred to polyvinylidene difluoride membranes (EMD Millipore Corp., Billerica, MA) and blocked in tris-buffered saline-Tween 20 (TBST) containing 5%–10% nonfat dry milk or BSA. Membranes were then incubated overnight at 4°C or 2 h at room temperature with their respective primary antibodies, including antibodies against GCLM, GCLC, NQO1, GSR, GSTμ, SOD1, SOD2, GPX1, NRF2, and CAT proteins (see Table 2 for details). The primary antibodies were diluted with 1%–2% BSA in TBST for incubation. Following 2 × 10-min washes with TBST, horseradish peroxidase IgG (Vector Laboratories)-conjugated secondary antibody (anti-rabbit and anti-mouse) incubation followed by electrochemiluminescence-based detection (Pierce, Rockford, IL) of antigen/antibody complex was performed, and the signals were captured using autoradiography films or images were obtained with an Amersham Imager 600 (GE Healthcare Life Sciences, Chicago, IL). Bio-Rad dual-color protein standards (#1610374) were compared to determine the size of the proteins. Protein loading was monitored by stripping the membrane with a mild stripping buffer (1.5% glycine, 0.1% SDS, and 1% Tween 20) and reprobing for glyceraldehyde 3-phosphate dehydrogenase (GAPDH)/LAMIN-B1 expression. The signal densitometry was quantified using ImageJ software and then normalized to the intensity of NTG sample (housekeeping protein GAPDH as internal standard). Mean arbitrary units normalized with GAPDH were used to calculate protein fold-change in TGL and TGH mice relative to NTG (n = 4–6/group).
Complete List of Primary Antibodies and Sources
Nrf2 binding activity assay
Efficiency of Nrf2 DNA-binding activity in NTG and TG mice hearts (n = 6/group) was evaluated at the age of 6–8 months using a commercially available Trans AM Nrf2 kit (Active Motif) as previously described (53). Briefly, the nuclear protein extracts (10 μg) were incubated with immobilized wild-type or mutated competitor oligonucleotides bearing the ARE consensus sequence. The bound Nrf2 was detected using an anti-Nrf2 primary antibody (100 μL of a 1:1000 dilution) reaction followed by washing and incubation with the horseradish peroxidase-conjugated secondary antibody (100 μL of a 1:1000 dilution) before chromogenic reaction with the tetramethylbenzidine substrate. The color forming reaction was terminated by addition of stop solution and the optical density was read at 450 nm. Incubation with normal rabbit polyclonal IgG was also performed separately to determine the specificity of the Nrf2 antibody.
Characterization of redox status markers
GSH redox measurements
Reduced GSH and oxidized GSH (GSSG) levels of NTG and caNrf2-TG hearts were measured using a GSH assay kit (Cat. No. 703002; Cayman Chemical, Ann Arbor, MI). Myocardial 2-(N-morpholino) ethanesulfonic acid (MES) lysates were prepared in MES buffer (acidic; pH 6.0) and centrifuged for 5 min at 5000 rpm in 4°C. An aliquot of the supernatant was used for protein determination and equal volume of 10% meta-phosphoric acid (MPA; Cat. No. 239275; Sigma) was added to the remaining samples to precipitate the proteins. The supernatants were equally mixed with 10% MPA to precipitate proteins. The MPA extracts were then treated with 4 M triethanolamine (TEAM) reagent. The TEAM-treated extracts were aliquoted for GSH and GSSG quantification. The extracts for GSSG measurements were further processed with 1 M 2-vinylpyridine, and the enzymatic recycling assay was executed according to the manufacturer's instructions. The GSH and GSSG standards were prepared and treated in the same way with the heart samples and the redox state was determined (26, 38, 43, 47, 53).
High-performance liquid chromatography-based assay of thiol/disulfide redox in the cardiac tissue
Upon tissue collection, ∼50 mg of cardiac tissue from NTG and caNrf2-TG mice (n = 6/group) was added to 0.5 mL of 5% (w/v) perchloric acid containing 0.2 M boric acid and 10 μM γ-Glu-Glu (internal standard) solution. The sample was briefly sonicated on ice and centrifuged for 5 min at 5000 rpm in 4°C. The supernatant was aliquoted (300 μL/tube) and stored at −80°C until derivatization, which was conducted at the Emory Clinical Biomarkers laboratory. The protein pellet was further resuspended in 200 μL 1 N NaOH and subjected to protein assay (23 –25).
Derivatization
A 60 μL aliquot of 50 mM iodoacetic acid solution was then added to the 300 μL supernatant aliquot. The samples were vortexed, and pH adjusted to 9.0 ± 0.2 with 1 N KOH/tetraborate solution. The tubes were incubated at room temperature for 20 min for complete precipitation of potassium perchlorate. Following incubation, 300 μL of 75 mM dansyl chloride solution (prepared in acetone) was added to the tubes, and samples were vortexed well and stored in the dark at room temperature for 20 h. Following derivatization, chloroform (500 μL) was added to each tube, vortexed, centrifuged, and samples stored at 4°C in the dark until HPLC analysis.
HPLC analysis
Samples were centrifuged for 2 min and an aliquot of the upper aqueous layer was transferred to an autosampler vial. The instrumentation used is a gradient HPLC module 2695 (Waters) with Empower software. The Waters 2475 multifluorescence detector was used with excitation at 335 nm and emission detected at 518 nm. Separations were obtained on a 25 cm by 4.6 mm, 5 μm silica, LC-NH2 Supelco column from Sigma Aldrich (Cat No. 58338). Flow rate for HPLC was maintained constant at 1.0 mL/min. The solvent gradient used was as follows: initial solvent conditions were 80% A [80% (v/v) methanol/water] and 20% B [acetate-buffered (pH 4.6) methanol solution] run at 1 mL/min for 10 min. A linear gradient to 20% A and 80% B was run over the period from 10 to 30 min. From 30 to 46 min, the conditions were maintained at 20% A and 80% B and returned to 80% A and 20% B from 46 to 48 min. Equilibration time for the next run was 12 min [Solvent A: 80% (v/v) methanol/water and Solvent B: acetate-buffered (pH 4.6) methanol solution prepared by mixing 640 mL of methanol, 200 mL of acetate stock, 125 mL of glacial acetic acid, and 50 mL of water].
Calculations
Integrated peaks provided by the HPLC software corresponding to GSH, GSSG, Cys, CySS, and mixed disulfide (CySSG) were used and concentrations were calculated relative to the integrated peak of the internal standard (γ-Glu-Glu) and normalized to the protein concentration. The individual concentrations, expressed in molar values, were used with the Nernst equation to calculate redox potentials. At pH 7.4, an Eo value of −264 mV was used for the GSH/GSSG couple, and an Eo value of −250 mV was used for the Cys/CySS couple.
Assessment of total antioxidant activity
Myocardial TAC was determined in homogenates of NTG, caNrf2 TGL, and TGH mice using the TAC assay kit (OxiSelect: STA-360; Cell Biolabs, Inc.) according to the manufacturer's instructions. The conversion of copper (II) ion (Cu2+) into copper (I) ion (Cu+) by the antioxidants was measured as total reducing capacity of the nonenzymatic antioxidants present in the myocardial extracts. In brief, heart extracts were prepared immediately upon tissue harvesting (n = 6–8/group). Equal amounts of heart tissues (20–25 mg) were homogenized with 200 μL of ice-cold PBS, centrifuged, and 20 μL of supernatant was mixed with 180 μL of reaction buffer; initial absorbance was measured at 490 nm. Copper ion reagent (50 μL) was then added to the mixture to initiate the reaction and incubated on an orbital shaker for 5 min and 50 μL of 1 × stop solution was added to stop the reaction. The color development was measured at 490 nm. Antioxidant capacity was represented as fold change in relation to NTG (53).
Immunofluorescent microscopy
General protocol
For immunofluorescent staining (n = 3–6/group), heart tissue was embedded in O.C.T. freezing medium and 10 μm cryostat sections were obtained. In preparation for staining, sections were incubated with 4% paraformaldehyde for 15 min and washed thrice with PBS followed by permeabilization with 0.25% Triton X-100. Following three washes with PBS, tissue sections were blocked with 5% normal goat serum in PBS for 1 h to prevent nonspecific binding of antibodies. Sections were then incubated for 1 h at room temperature or overnight at 4°C with primary antibodies. Following primary antibody incubation, all sections were washed with PBS thrice and incubated with one of the following secondary antibodies at 1:1000 for 1 h at room temperature: Alexa Fluor 488 goat anti-mouse IgM (μ-chain) (A21042), anti-rabbit IgG (H+L) (A11008), or Alexa Fluor 647 chicken anti-mouse IgG2a (y2a) (A21241), anti-rabbit IgG (H+L) (A21443) (Life Technologies, Carlsbad, CA). Finally, the sections were washed thoroughly with PBS (3 × 10 min each) and mounted with Fluoroshield/DAPI (ab104139; Abcam, Cambridge, MA). Images were acquired with an Olympus BX43 fluorescent microscope or with a Nikon A1 confocal microscope using a 60 × objective. At least three to four micrographs were taken for each section and used to calculate the appropriate intensity for each target (18, 52 –54).
WGA staining
Cryostat sections used for WGA staining were labeled with 5 μg/mL WGA in PBS (Alexa Fluor 594 Conjugate, W11262; Molecular Probes, Eugene, OR) in a light-protected chamber maintained at 37°C for 30 min. After washing and fixation with mounting medium, cardiac cells were imaged with a confocal microscope set to 60 × . Cardiomyocyte cell size was measured using NIH ImageJ software (52).
DMPO staining
For the evaluation of myocardial DMPO protein adducts, mice were injected intraperitoneally with two 1 g/kg doses of DMPO (D048-10; Dojindo Molecular Technologies, Inc., Kumamoto, Japan) at 2 and 1 h before sacrificing. DMPO is a free-radical spin trap used to detect the free radical in the biological samples. DMPO staining in tissues/cells using anti-DMPO antibodies detects DMPO-protein adducts, an indirect estimation of oxidant levels (ROS) (52). The procured hearts were fixed in 10% neutral buffered formalin and soaked in 30% sucrose for 24 h. Then, the tissues were fixed in cryostat OCT solutions and stored at −20°C before sectioning. Cryostat sections (5 μM) were prepared and blocked with 1% casein PBS for 1 h at room temperature. Slides were then incubated overnight at 4°C with 5 μg/mL mouse anti-DMPO primary antibody (kindly provided by Dr. Mason). Sections were washed, conjugated with secondary antibodies, and mounted with DAPI. Images were obtained with a confocal microscope at 60 × magnification.
DHE staining for the determination of tissue level of ROS superoxide
The DHE, a lipophilic, cell-permeable, red fluorogenic dye, was used to measure the level of ROS (superoxide only). Briefly, the frozen sections (10 μm) of the myocardium were incubated with 5 μg/mL of DHE in PBS in a light-protected chamber maintained at 37°C for 30 min. Then tissue sections were washed with PBS for two to three times to remove the excess DHE stain and then fixed with the Fluoroshield mounting medium. The images were captured by randomly selecting 3–5 fields/section using an Olympus BX43 fluorescent microscope (18, 53, 54).
Noninvasive echocardiography analysis of cardiac function
NTG (at 1.5, ∼3, ∼6, and >15 months of age), caNrf2-TG (at ∼6 and >15 months of age), and DTG (hNrf2xmcaNrf2 at ∼2 months of age) mice were anesthetized with 1%–2% isoflurane, supplemented with 100% oxygen, and chest area was shaved in preparation for echocardiography analyses (n = 5–10) using the Vevo2100 Imaging System (Fujifilm VisualSonics, Inc., Ontario, Canada). A 38 MHz probe was used to capture images at maximum (50 μM) resolution. Long-axis B-mode was used for strain analysis to calculate EF, end diastole/systole left ventricular mass. The parasternal short-axis M-mode was utilized in the determination of fractional shortening, wall thickness, and chamber dimension during systole and diastole. Pulse Wave/Color Doppler analysis was used to measure early (E) filling and late atrial (A) filling velocity, as well as MV tissue motion (E′) in systole and diastole. Three consecutive cardiac cycles from B- and M-mode images were used for measuring for each variable (43, 52).
Statistical analysis
All data are represented as mean ± standard error of the mean. When three or more groups were analyzed, a one-way ANOVA with post hoc Tukey multiple comparison test was performed. Student's t-test was used to calculate statistical significance. All analyses were performed using GraphPad Prism 7. Significance: *p < 0.05; **p < 0.01; ***p < 0.001; and ns, no significance.
Footnotes
Acknowledgments
The authors thank Dr. James Fang, MD, and Dr. John Michael, MD, for providing resources at the University of Utah. They acknowledge Dr. Ivor Benjamin and Dr. Jeffrey Robbins for providing the mutant-CryAB mice, CryAB-antibodies, α-MHC promoter constructs. The authors also thank Dr. Sethu Palaniappan for providing the fluorescence microscope facility. They are grateful to Dr. Susan Tamowski, Director, Transgene—Targeting Mouse Facility, for her excellent technical support to generate the caNrf2-TG mice. They also thank Dr. Michael Crowley (Heflin Center for Genomic Sciences at UAB) for providing access to the genomic core facilities. The authors acknowledge the support from Mr. Wayne E. Bradley for assisting with analyses/interpretation of echocardiography. They thank Dr. Madhusudhanan Narasimhan for editorial assistance.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study was supported by funding from NHLBI (2HL118067 and HL118067), NIA (AG042860), the AHA (BGIA 0865015F), the University of Utah Center for Aging Pilot grant (2009), the Division of Cardiovascular Medicine/Department of Medicine, University of Utah, and the start-up funds (for N.S.R.) by the Department of Pathology and School of Medicine, the University of Alabama at Birmingham, AL, and UABAMC21 grant by the University of Alabama at Birmingham, AL.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Figure S7
Supplementary Figure S8
Supplementary Figure S9
Supplementary Video S1
Supplementary Video S2
Supplementary Video S3
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.