
Abstract
Significance:
Alterations in oxidant/antioxidant balance injure pulmonary endothelial cells and are important in the pathogenesis of lung diseases, such as Acute Respiratory Distress Syndrome (ARDS), ischemia/reperfusion injury, pulmonary arterial hypertension (PAH), and emphysema.
Recent Advances:
The endosomal and autophagic pathways regulate cell homeostasis. Both pathways support recycling or degradation of macromolecules or organelles, targeted to endosomes or lysosomes, respectively. Thus, both processes promote cell survival. However, with environmental stress or injury, imbalance in endosomal and autophagic pathways may enhance macromolecular or organelle degradation, diminish biosynthetic processes, and cause cell death.
Critical Issues:
While the role of autophagy in cellular homeostasis in pulmonary disease has been investigated, the role of the endosome in the lung vasculature is less known. Furthermore, autophagy can either decrease or exacerbate endothelial injury, depending upon inciting insult and disease process.
Future Directions:
Diseases affecting the pulmonary endothelium, such as emphysema, ARDS, and PAH, are linked to altered endosomal or autophagic processing, leading to enhanced degradation of macromolecules and potential cell death. Efforts to target this imbalance have yielded limited success as treatments for lung injuries, which may be due to the complexity of both processes. It is possible that endosomal trafficking proteins, such as Rab GTPases and late endosomal/lysosomal adaptor, MAPK and MTOR activator 1, may be novel therapeutic targets. While endocytosis or autophagy have been linked to improved function of the pulmonary endothelium in vitro and in vivo, further studies are needed to identify targets for modulating cellular homeostasis in the lung.
Endosome Transport and Autophagy
The endosomal and autophagic pathways regulate movement of macromolecules within the cell with the primary goal of maintaining cellular homeostasis. Cells are constantly responding to intracellular and extracellular stresses and signals. In settings of increased stress or signaling, the balance of the processes may become tilted, resulting in enhanced macromolecular degradation, diminished biosynthetic processes, and possible cell death. However, the overall purpose of both is to promote cell homeostasis.
Endocytosis is the active process of moving macromolecules and particles from the environment or external membrane into the cell via invagination of the plasma membrane through one of four methods: (i) clatherin-mediated endocytosis (CME), (ii) clatherin-independent endocytosis (CIE), (iii) macropinocytosis, or (iv) phagocytosis (80, 98). CME is dependent on dynamin for membrane scission, whereas CIE can be dynamin dependent (caveolea or Rho-mediated) or independent (cdc42, Arf6, or flotillin) (80, 98). CME is a selective mechanism to internalize surface proteins, whereas CIE is believed to be more of a bulk method of endocytosis.
Macropinocytosis and phagocytosis are endocytic processes driven by an actin ring formed beneath the plasma membrane engulfing either fluid droplets or solid structures, respectively. Macropinocytosis nonselectively takes up fluid, whereas phagocytosis is initiated by the interaction of particles (>0.5 μm) to be engulfed with cell receptors, including Fcγ, C-type lectin, integrins, and scavenger receptors (22). Both processes utilize small GTPases, such as cdc42, Rac, and Rabs, to mediate the actin polymerization and endocytosis, respectively (19). Once endocytosed, the vesicular contents are sorted and can be recycled back to the cell surface, enter into the endoplasmic reticulum (ER) and Golgi complex for post-translational modification of the cargo proteins, or targeted for lysosomal degradation, autophagy, or secreted into the cytoplasm (68) (Fig. 1).
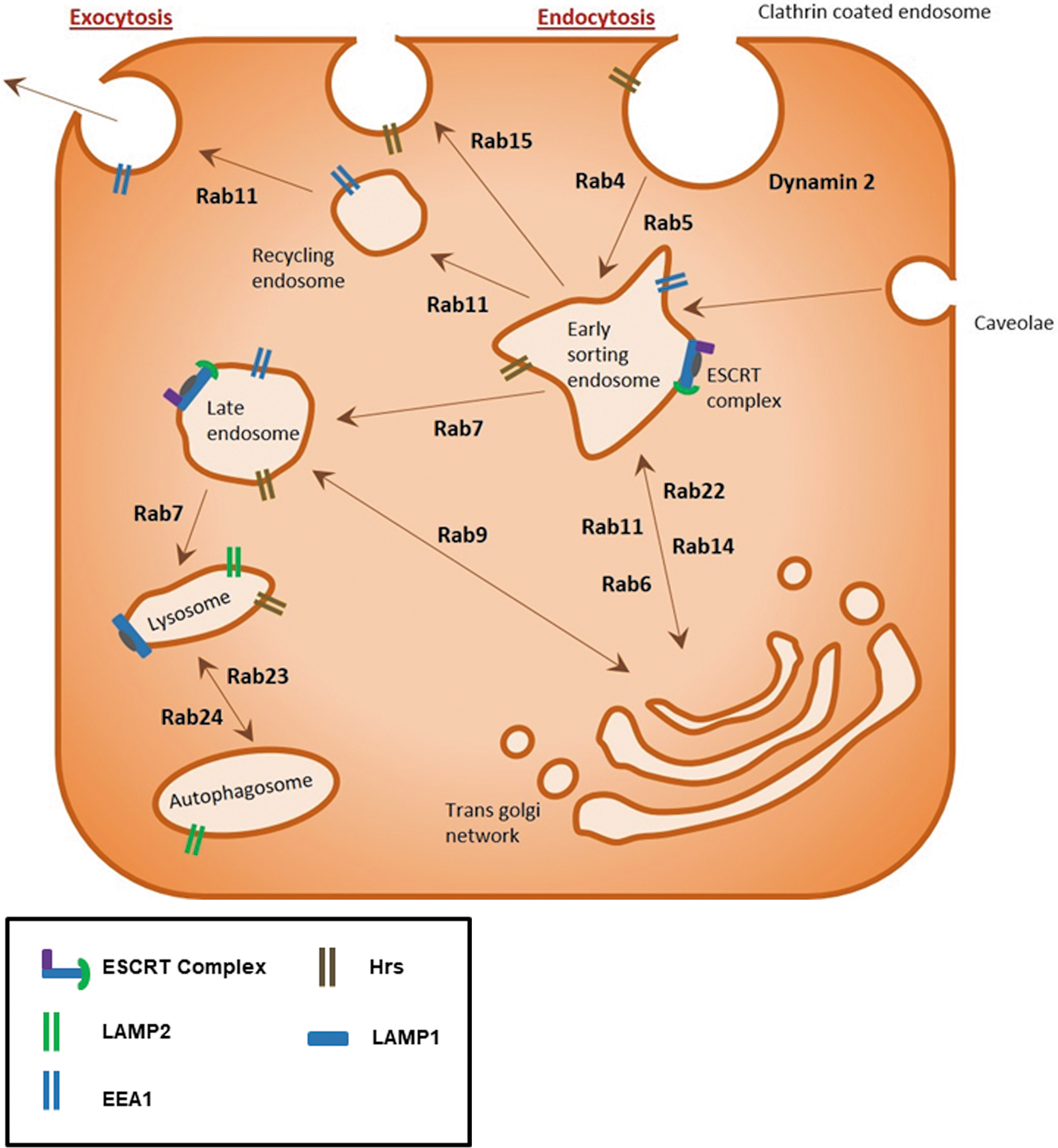
Protein families such as Rab GTPases, adenosine diphosphate (ADP)-ribosylation factor (Arf) G-proteins, endosomal sorting complexes required for transports (ESCRTs), and soluble N-ethylamide-sensitive factor attachment protein receptors (SNAREs) regulate the spatiotemporal control of this dynamic endosomal trafficking system. These families regulate trafficking processes through a variety of highly specialized mechanisms. Similar to the Ras GTPase superfamily, Rab GTPases require guanine exchange factors (GEFs) to promote the guanosine diphosphate (GDP) to guanosine triphosphate (GTP) exchange, allowing the change in conformation needed to bind to the endosomal membrane and mediate trafficking along the microtubule network (114).
The majority of the 70 plus Rab GTPases are involved in endocytic processes. Rab5, and effector protein early endosome antigen 1 (EEA1), associate with clathrin-coated endosomes, internalized at the plasma membrane, to mediate trafficking to the early endosome compartment (108). Rab4 and Rab11, in turn, mediate the short-loop and long-loop recycling of endosomes back to the plasma membrane, respectively (97). Rab7 and Rab9, however, promote the formation of late endosomes of cargo sorted in the trans-Golgi network (41). Early endosomes at the cell periphery undergo fusion and fission with other endosomes to create larger endosomes within the cell to allow sorting of the macromolecular contents during trafficking.
Arf G-proteins also interact with GEFs and guanine activating proteins to regulate GDP/GTP exchange and GTP hydrolysis, respectively. Arf proteins localize throughout the cell at membrane surfaces, including the plasma, endosomal, lysosomal, and secretory membranes (17). Six Arf proteins have been identified whose functions include recruitment of proteins that coat the membrane during endocytosis to promote sorting, enzymes that modify membrane lipid content, and proteins involved in cytoskeletal tethering, scaffolding, and folding (17).
Unlike Ras, Rho, and Rab GTPases, Arf G-proteins are myristoylated, which promotes their localization to membranes. Arf1 and Arf3–5 are primarily in the ER-Golgi complexes regulating retrograde transportation from the Golgi to ER and at the trans-Golgi network. Arf6 is localized at the plasma membrane and is involved in cortical actin distribution, endosomal trafficking, and rapid endosomal recycling via interactions with the microtubule motor adaptor protein, c-Jun N-terminal kinase-interacting protein-4 (17). Arf1 and Arf6 regulate intercellular junctions, adherens, and tight junctions, through the regulation of cadherin molecules at the cell surface (51, 111).
In contrast to Rab proteins, ESCRT proteins play a significant role in the delivery of cargo to the lysosome via the generation of multivesicular bodies (MVB) (67). The ESCRT protein complex is formed at endosomes during MVB sorting by sequential recruitment of ESCRT-0, -I, and -II and the ubiquitinated cargo. The ESCRT proteins form a subdomain on the endosome, which then invaginates incorporating the ubiquinated cargo. ESCRT-III functions to bud off the intraluminal vesicles within the MVB. Once MVB fuse with lysosomes, the content of the intraluminal vesicles is degraded (9). Perturbations in any of these highly regulated pathways can result in disordered trafficking of key molecules. The ESCRT complex has been implicated as having a role in the maturation process of the autophagosome (54, 73).
SNAREs are a superfamily of proteins involved in the membrane fusion of intracellular organelles, with the exception of the mitochondria (89). For membrane fusion to occur, SNARE proteins form a four-helix bundle on the adjacent membranes complex with one R-SNARE (or vesicle (v)-SNARE) on one membrane and 3 Q-SNARE (or target cell (t)-SNARE) proteins on the other membrane (89). SNARE proteins mediate merging of the membranes in close proximity and provide energy to promote the fusion of the membranes. Adenosine triphosphate (ATP)-dependent SNARE folding drives this membrane fusion. Once fused, the SNARE proteins are unfolded via the ATPase N-ethylmaleimide-sensitive factor and synaptosomal nerve-associated protein (89). Interestingly, the membrane fusion function of SNAREs is also crucial for exocytosis, autophagosome-lysosome fusion, and autophagosome biogenesis (20, 61).
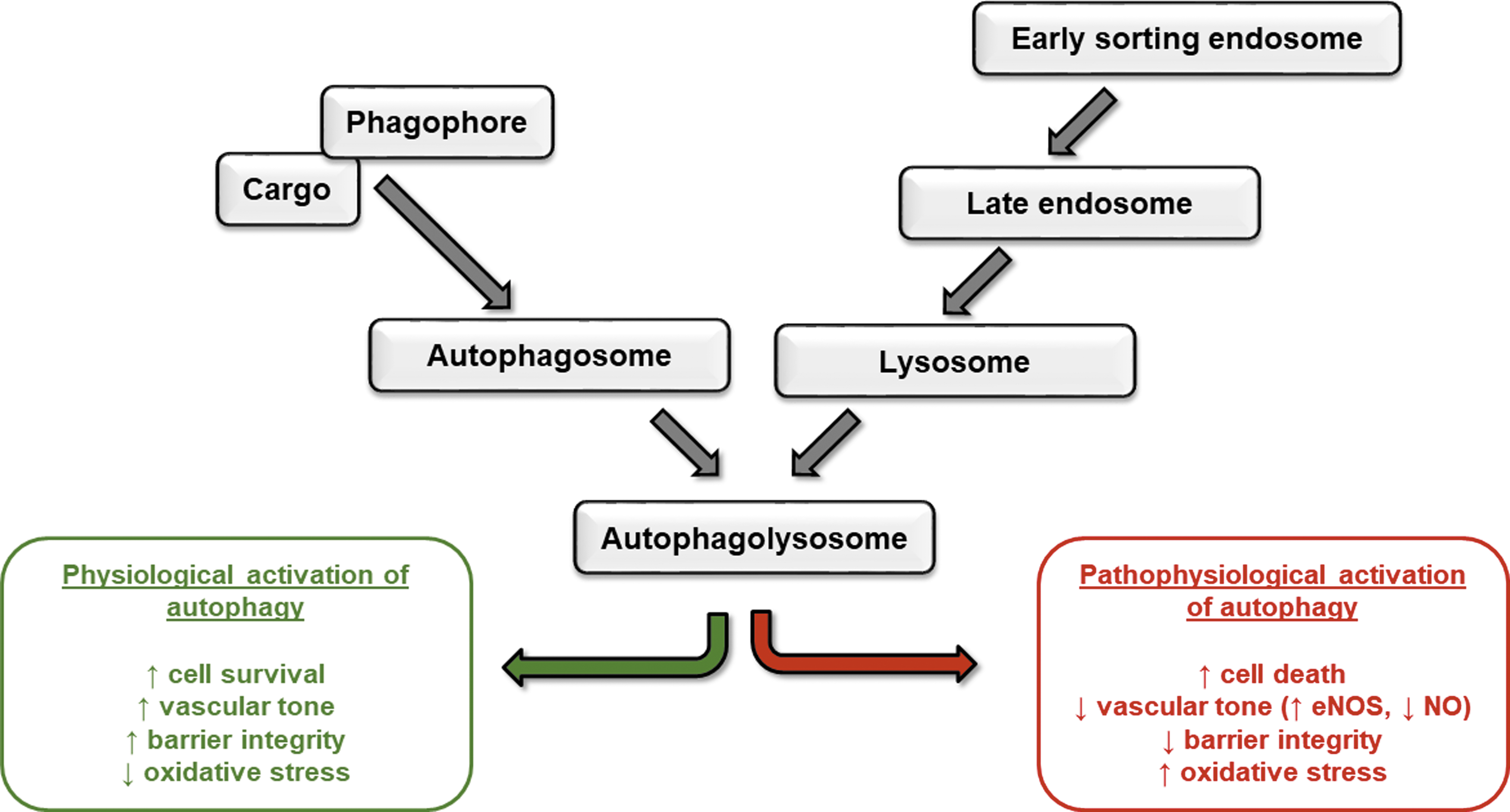
Similar to the endocytic pathway, autophagy is both a recycling and a degradative system, whereby intracellular molecules are targeted to endosomes or lysosomes, respectively. Autophagy occurs in settings of cell stress and serves to promote cell survival. In settings of excessive autophagy, cell death ensues. As described by Mizushima and Komatsu (60), there are three main types of autophagy, chaperone-mediated autophagy (CMA), microautophagy, and macroautophagy, with each resulting in the degradation of the vesicle contents (Fig. 2).

CMA is a precisely targeted event, whereby the protein cargo is recognized via a pentapeptide motif (lysine-phenylalanine-glutamic acid-arginine-glutamine) (KEFRQ)-like motif, a chaperone protein, heat shock cognate 71 kDa protein (HSC70), and its co-chaperone proteins, and subsequently internalized by a lysosomal surface receptor in conjunction with lysosomal-associated membrane protein (LAMP)2A and degraded (37). During microautophagy, subcellular small molecules enter in bulk directly into late endosomes via engulfment and, subsequently, fuse with MVB for degradation (74). Inhibition of the ESCRT complex I blocked microautophagic degradation in MVB, suggesting a role for these proteins in this process (74) and crosstalk between the endosomal and autophagic pathways.
Although initially believed to be a nonselective process, there is evidence in yeast of parallel chaperone-mediated events using HSC70 and KFERQ-like motif proteins in microautophagy and subsequent binding to phosphatidylserine on the lipid bilayer (94). HSC70 was also noted to serve as a chaperone in eukaryotic cells, which selectively targets cytosolic proteins to late endosomes that then undergo microautophagy (74). Additional chaperone proteins are believed to complex cargo proteins for microautophagy that targets select organelles, such as micromitophagy, microlipophagy, and micropexophagy, in yeast (94). The CMA process is upregulated in settings of oxidative stress due, in part, to increased LAMP2A transcription (38). Finally, macroautophagy, often just referred to as “autophagy,” is described as the most commonly occurring form of this process.
Autophagy proceeds via five main steps: initiation, nucleation, elongation, lysosome fusion, and cargo degradation. Initially, an isolation membrane or phagophore is formed from the ER, Golgi, mitochondrial, plasma, or recycling endosomal membrane. Formation of the phagophore requires altered activation and/or recruitment of a number of signaling molecules, including mammalian target of rapamycin (mTOR). In settings of cellular homeostasis, the mTOR complex 1 (mTORC1) binds to and inactivates Unc-51 like autophagy activating kinase (ULK1) or autophagy-related genes (Atg) 1 (39). On cell starvation, mTORC1 is inactivated, releasing and activating ULK1, which, in turn, binds the membranes to initiate the phagophore formation via recruitment of other proteins, including FIP200, Atg13, and Atg101 (101).
In addition to mTOR, adenosine monophosphate kinase (AMPK) has been shown to activate ULK1 directly via phosphorylation (39), as well as indirectly via Raptor-mediated inhibition of mTOR (3, 101). With the activation of ULK1, additional proteins are recruited that are necessary for the phagophore elongation. Beclin 1, originally identified as having antiapoptotic properties through its binding to B cell lymphoma 2, is a necessary component in autophagy via the formation of complex with class III-type phosphatidylinositol 3 kinase complex (29, 52).
Unlike other vesicles, phagophore double membranes are formed de novo through the synthesis of phosphatidylinositol-3-phosphate and the recruitment of microtubule-associated protein-1 light chain-3 (LC3) or Atg 8 and the Atg12-Atg5-Atg16 complex. Through a series of post-translational modifications, Atg8 is covalently attached to phosphatidylethanolamine (PE) of the lipid bilayer. Atg8-PE conjugation on the outer layer of the autophagosome is believed to regulate the size of the compartment, whereas that on the inner surface serves in the selection of cargo that is internalized (29, 32). Once the phagophore matures into the autophagosome, the outer membrane fuses with lysosomes with the release of the inner-membrane bound cargo, termed autophagic body, which is subsequently degraded, and the primary cell building blocks, such as amino acids and fatty acids, are recycled back to the cytosol (32).
In settings of excessive autophagy, cells may undergo autophagic or apoptotic cell death. Autophagy-dependent cell death occurs in settings of autophagic degradation of (i) ferritin (ferroptosis) (33) or (ii) protein tyrosine phosphatase, non-receptor type 13 (FAS-driven extrinsic apoptosis) (24); (iii) via necroptosis by utilizing the necrosome scaffolding complexes in autophagy or by cellular inhibitor of apoptosis protein (cIAP)-1 and cIAP-2 degradation (23); and (iv) by autosis, which is mediated via Na+/K+ ATPase (48). Apoptosis or programmed cell death is a well-studied regulated cell death (RCD) process that may occur by signals that are initiated intrinsically or extrinsically and can occur in a caspase-independent manner. More in-depth reviews on apoptosis are available (27, 85).
Regulated endosomal trafficking is key for efficient autophagy; indeed, the autophagosome membrane has been demonstrated to originate from the endosomal membrane (57). Maturation of the autophagosome, by fusion with the lysosome, forms the autophagolysosome where autophagy can occur. This fusion process allows the delivery of key lysosomal components such as hydrolases to degrade autophagocytosed products, the H+-vacuolar ATPase to acidify the vesicle, and transporters and permeases needed to allow efficient recycling of autophagic degradation products (82).
Endosomal proteins form the molecular machinery required for formation of the autophagosome. For example, several Rab GTPases (Rab11, Rab33b, Rab5, and Rab7) as well as the EEA1 have all been demonstrated to impact autophagy by using in vitro studies (8, 31, 87, 100). Several of these trafficking molecules regulate dynamics of the recycling endosome to regulate fusion between MVB and the autophagosome (Rab11), push the recycling endosome into the late endosome/lysosome pathway (Rab7), or promote trafficking of endosomes from the Golgi to the maturing autophagosome (Rab33b) (49, 88, 90). These proteins sort endosomes early in the trafficking pathway, promoting the formation of the lysosome and subsequent maturation into the autophagolysosome, which enables autophagic degradation of targeted cargo.
In addition to LC3/Atg5/Atg7-mediated autophagic flux, studies also indicate the presence of an independently mediated autophagosome formation pathway (65). This LC3/Atg5/Atg7-independent autophagy is linked to the fusion of Rab9-positive endosomes with the autophagosome. Interestingly, both Rab7 and Rab9 GTPase are also involved in autophagosomal membrane formation, which occurs during mitophagy (30, 103). Recent papers suggest that Rab5 mediates the closure of the autophagosome before fusing with lysosomes in yeast cells (10, 115). More recently, data demonstrate that Rab5 recruits ESCRT to complete the autophagosomal scission (115). It is clear that there is crosstalk between the endocytic and autophagic processes and therefore likely that Rab GTPases mediate these two key pathways, which control cellular homeostasis.
The master regulator of the lysosome, mTORC1, also plays a key role in autophagy. In nutrient-rich settings, mTORC1 reduces autophagy by inhibiting catabolic pathways, in close coordination with AMPK, and promotes biosynthesis pathways to increase cell growth (78). mTORC1 responds to several upstream stress signals, such as amino acids and growth factors, via H+-vacuolar ATPase and Rheb GTPase localized to the lysosome surface (77, 84). Amino acid signals from the lumen of the lysosome to the Ragulator complex result in the recruitment of mTORC1 to the lysosome (70). The Ragulator complex has, therefore, been the focal point of much autophagy research, with studies indicating that the H+-vacuolar ATPase-Ragulator interaction activates the GTP-loading of Rag GTPases, leading to mTORC1 recruitment, its activation by Rheb, and the subsequent inhibition of autophagy (77).
There are a range of proteins that comprise the Ragulator complex, including GTPases RagA-RagD and scaffold proteins late endosomal/lysosomal adaptor, MAPK and MTOR activator 1–5 (62, 77). LAMTOR1 provides the scaffold for Rag GTPases to recruit mTORC1, and the LAMTOR2/3 and LAMTOR4/5 heterodimers anchor the complex to the lysosome membrane (63). As such, inhibition of LAMTOR1 in mouse embryonic fibroblasts has been demonstrated to promote autophagy, through decreased mTOR phosphorylation and increased LC3II formation, in settings of nutrient deprivation (107). Interestingly, the Ragulator complex also regulates mitogen-activated protein kinase kinase signaling from the late endosome, associated with lysosome biogenesis, membrane dynamics, and organelle transport along the cytoskeleton (63). Members of the Ragulator complex, such as LAMTORs, therefore represent interesting molecular targets to modulate autophagy in settings of disease.
Function of Endocytosis and Autophagy in Healthy Endothelium
The flow of macromolecules in the endothelial cells through endosomal and autophagic trafficking is important in regulating functional vasculature, angiogenesis, and responding to aging and changes in the environment. Inhibition of the basal levels of either process leads to endothelial cell apoptosis, permeability, migration, and vessel regression. Conversely, excessive activation of endocytosis or autophagy can exacerbate vascular injury, leading to pathological progression of diseases (Fig. 3).
Endocytosis is important in the trafficking of many cell surface molecules, including adherens and tight junction proteins, receptor tyrosine kinases (RTK), as well as proteins localized near the plasma membrane, such as Rho and Rac GTPases. We have shown upregulation of recycling endosomal proteins, p18 or Rab4, to increase vascular endothelial (VE)-cadherin surface expression in pulmonary endothelial cells; protect against barrier dysfunction (11, 13); and promote migration, proliferation, and angiogenesis (11, 12, 93). The long-loop recycling Rab GTPase, Rab11a, provides the platform for LC3-positive autophagosomes to mature and has also been shown to protect the pulmonary endothelium against leak, through improved adherens junction formation (104). Other studies have similarly shown tight junction proteins, including zonula occludens, junctional adhesion molecule-A, claudins, and occludin, to be regulated by endosomal trafficking affecting endothelial barrier function [reviewed in (86)].
RTK were believed to function at the plasma membrane to regulate endothelial cell function; however, it is now recognized that several RTK instead signal from the cytosol via endosomes. For example, vascular endothelial growth factor receptor 2 (VEGFR2) and fibroblast growth factor receptor 1 are localized to endosomes. Once bound by vascular endothelial growth factor-A or -C, VEGFR2 internalization occurs via the clathrin/dynamin-mediated pathway (7), which then leads to Rab5-mediated trafficking to early endosomes and recycling via Rab11 (5) or via macropinocytosis (6). In addition, unbound VEGFR2 is noted to undergo constitutive endocytosis and recycling to the plasma membrane via Rab4 (40), as well as shear stress-induced caveolin-dependent endocytosis of VEGFR2.
Interendothelial cell junctions are transiently disrupted by some edemagenic agents, such as thrombin, via actions of Rho A and Rac 1. In settings of low levels of Rho B or deficiency, Rac 1 is recycled via endosomes to the plasma membrane promoting barrier function. It has been shown that endosomal Rho B diminishes Rac 1 plasma localization by targeting it to late endosomes for degradation, causing delayed barrier recovery (56). As Rac 1 is involved in other endothelial cell functions, such as migration, it is likely that this molecule is also impacted by endosomal trafficking.
Autophagy has been shown to regulate the levels of nitric oxide (NO) in settings of increased shear stress on static endothelial cell monolayers and ex vivo perfusion of the carotid artery (25) and in aging (44). The changes in shear stress correlated with increased levels of p62, beclin and LC3, autophagy markers; increased endothelial nitric oxide synthase (eNOS); showed a reduction in endothelin-1 (25); and increased Rab4 expression (106). Beclin and p62 were also reduced in the brachial artery endothelial cells of older relative to younger human subjects and mice, with a corresponding decrease in endothelial-dependent dilation and NO bioavailability with an increase in oxidative stress and inflammation (44). Further, in settings of high shear stress, autophagy was required to promote endothelial cell alignment with the flow, protect against endothelial cell apoptosis or senescence, and plaque formation (99).
Similarly, autophagy attenuates oxidant injury to endothelial cells. Ha and colleagues showed that curcumin induced autophagy in human umbilical vein endothelial cells exposed to hydrogen peroxide via increased LC3II and number of autophagosomes to promote cell viability (26). After cigarette smoke exposure, elevated autophagy and senescence have been observed in endothelial cells and lungs, respectively (Fig. 4).
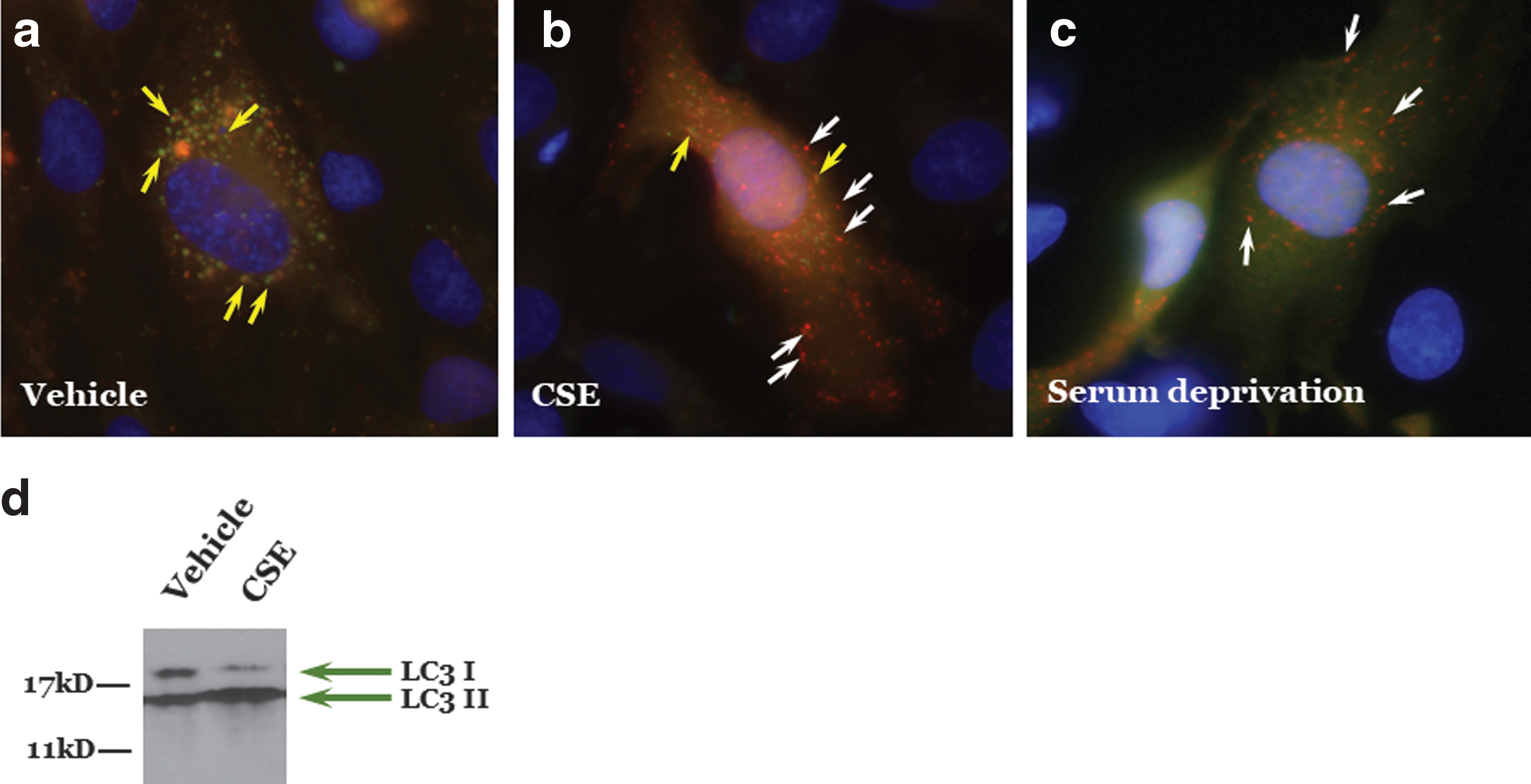
Other studies have shown that induction of autophagy prevented against stroke by diminishing reactive oxygen species (ROS) and improving vascular relaxation of the carotid (43), improved NO signaling in endothelial cells isolated from patients with type 2 diabetes (21), and improved endothelial-dependent dilation in aged animals (44). Rab26, a pro-autophagic GTPase that targets activated Src to LC3-positive autophagosomes, protects the pulmonary endothelial barrier by stabilizing adherens junctions (18, 46). Rather confusingly, Rab5a and Rab9, implicated as pro-autophagic GTPases, negatively regulate barrier function in the vasculature with overexpression of the dominant-negative, GDP-locked Rab9 mutant in the pulmonary endothelium attenuating bacteria- and endotoxin-induced barrier disruption both in vitro and in vivo (11, 105). These and other studies suggest that autophagy is necessary for proper vascular response and disruption of autophagy can lead to a dysfunctional endothelium.
Endosomal Signaling in the Pulmonary Endothelium in Lung Diseases
Acute respiratory distress syndrome
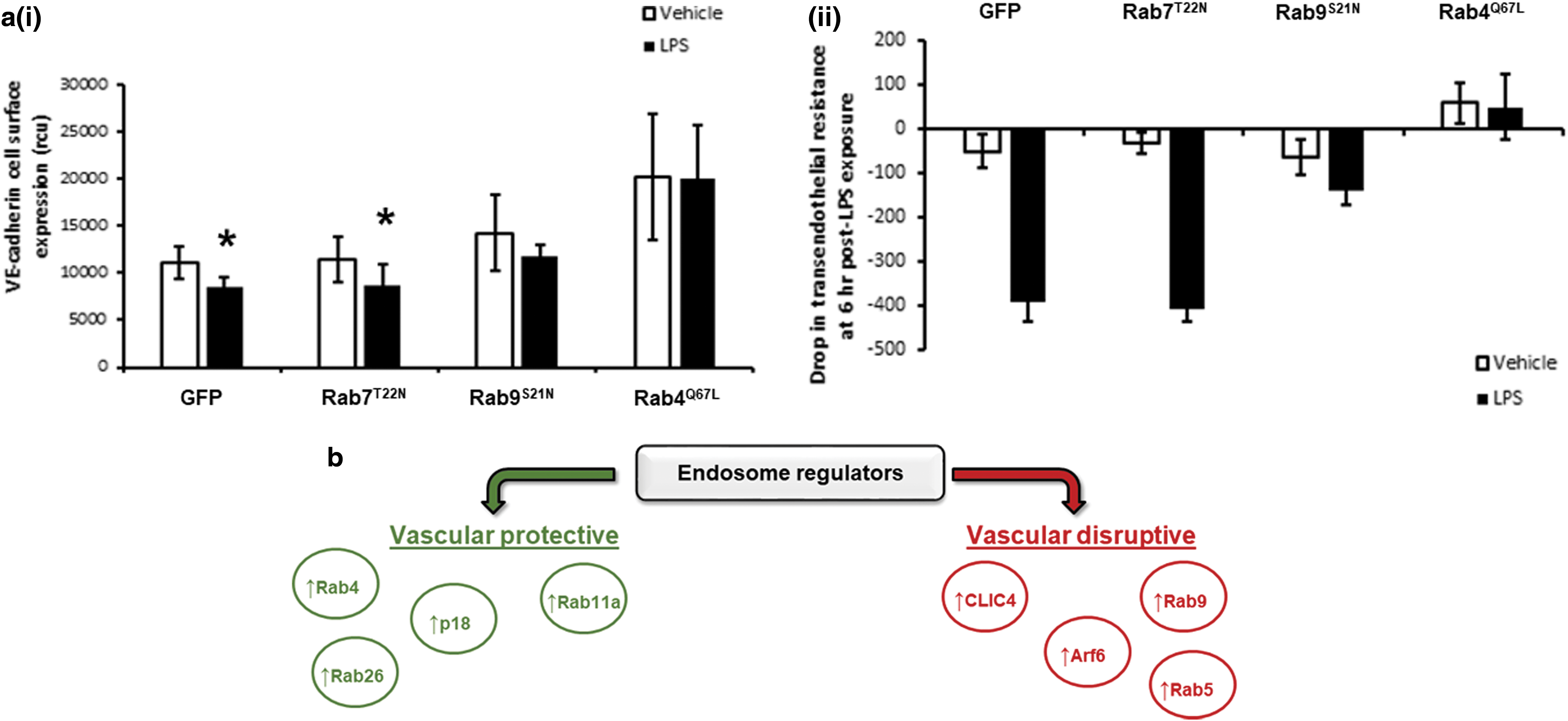
Although studies demonstrate a role of the endosome in regulating the endothelium, less is known about the role in the lung. We and others have demonstrated that members of the endosomal cycling pathways, Rab GTPase family, Rab4, Rab5, Rab9, and Rab11, play a key role in regulating microvascular permeability and edema formation in both in vitro and in vivo models of acute respiratory distress syndrome (ARDS) (11, 104, 105). Activation of the pro-recycling GTPases, Rab4, and Rab11, and inhibition of pro-degradative GTPase, Rab9, elevates trafficking of the junctional protein, VE-cadherin, to the adherens junction, thereby improving barrier integrity and reducing lipopolysaccharide (LPS)- and thrombin-induced vascular leak (Fig. 5) (11, 104). Conversely, inhibition of GTP-bound Rab5, the GTPase associated with endosome internalization, attenuated LPS-induced barrier disruption (105), indicating the importance of this small GTPase family in regulating the pulmonary microvasculature.
Pulmonary hypertension
Recent studies have shown expression of the chloride intracellular channel 4 (CLIC4) to be upregulated in the lungs of pulmonary arterial hypertension (PAH) patients and rodents (102) and in endothelial colony-forming units from PAH patients (1). Likewise, Arf6 was also increased in endothelial colony-forming units from PAH patients (1). CLIC4 localizes to endocytic vesicles, where it interacts with Arf G-proteins triggering the acidification of the vesicles to lysosome and targeting bone morphogenetic protein receptor II (BMPRII) for degradation (1). As such, CLIC4 was identified to regulate pulmonary endothelial cell function (81, 96), and targeted disruption of CLIC4 or Arf6 expression was identified to attenuate PAH (1).
Other studies have shown that the vasomotor tone of the pulmonary vasculature is regulated via endocytosis of eNOS in vivo. Caveolin-1 binding of eNOS in the caveolae serves to inhibit its function (58). On endocytosis, caveolin-1 is phosphorylated, releasing eNOS from an inhibitory state promoting the synthesis of NO (58). The vasodilatory response pulmonary vasculature in caveolin null mice is attenuated (53). Heritable mutations of caveolin-1 predispose individuals to developing PAH (4). A recent study identified a frameshift mutation in the carboxyl terminus of caveolin-1; it disrupts its retention in the ER, resulting in its degradation and diminished caveolae formation (15). Thus, it is likely that the development of pulmonary hypertension (PH) is due, in part, to disrupted endocytosis of surface proteins, such as BMPRII, eNOS, or other proteins that are important in regulating the pulmonary vasculature.
Autophagic Signaling in the Pulmonary Endothelium in Lung Diseases
Recent reviews have summarized evidence for RCD and autophagy in lung diseases (2, 59, 79). In many lung diseases, oxidative stress alters protein folding, revealing amino acid motifs that are subsequently recognized by CMA machinery and targeted for autophagy, thereby restoring homeostasis. In addition, mitochondrial injury may result in mitophagy and removal of damaged organelles. However, unchecked autophagy can also have deleterious effects, including excessive apoptosis and impaired angiogenesis.
Table 1 summarizes the effects of various lung injuries on autophagy in lungs and endothelial cells and the effects of autophagy on function. In general, autophagic processes are identified in lungs and cultured cells by increased expression of autophagy markers, such as increased ratio of microtubule-associated protein 1 LC3-II to LC3-I and characteristic autophagosome morphology detected by transmission electron microscopy. Effects of autophagy on cell or organ function are generally inferred by the functional effects of autophagy inhibitors (e.g., 3-methyladenine [3-MA]), activators (e.g., rapamycin), or by altering the expression of autophagy regulators (e.g., early growth response-1 [Egr-1]).
Autophagy in Lung Diseases
Summary of literature demonstrating autophagy, including endothelial cell autophagy, has been demonstrated in lungs after various insults.
ARDS, acute respiratory distress syndrome; ND, not done; PA, pulmonary arterial; PAH, pulmonary arterial hypertension; PH, pulmonary hypertension.
Pulmonary hypertension
PH is characterized by vascular remodeling with abnormal proliferation of vascular cells, including endothelial cells, with increased intimal thickness and formation of plexiform lesions in PAH. Studies in animal models have demonstrated initial endothelial apoptosis, followed by proliferation of apoptosis-resistant endothelial cells (75). Autophagy has been demonstrated in lungs of humans with PAH and may serve a protective role against endothelial injury that initiates vascular remodeling in PH [reviewed in (109)].
Lee et al. (45) demonstrated increased expression of the autophagy marker, LC3B-II, in the endothelium of lungs from humans with PAH, with increased LC3B-II protein levels in lung tissue from PAH (IPAH) versus non-PH controls, and in hypoxia-induced PH in mice. Genetic downregulation of LC3B-II or Egr-1 (transcriptional regulator of LC3B-II) resulted in exacerbation of PH in mice and increased hypoxia-induced proliferation of endothelial and smooth muscle cells, suggesting that autophagy keeps vascular cell growth in check and may protect against PH development. Similarly, using the Sugen/hypoxia model of severe PH in rats, Kato et al. (36) demonstrated increased expression of autophagy markers LC3 and beclin-1 in parallel with worsening PH and endothelial cell proliferation.
Rapamycin, an activator of autophagy, decreased PH and endothelial cell proliferation, with a concomitant increase in endothelial cell autophagy and apoptosis, suggesting that endothelial cell autophagy suppressed the progression of endothelial cell proliferation, resulting in diminished PH (36). Indeed, this is the case in methamphetamine-associated PAH, which occurs in individuals with a single nucelotide polymorphism that reduces expression of the methamphetamine metabolizing enzyme, carboxylesterase 1 (66). Pulmonary microvascular endothelial cells that are deficient in carboxylesterase 1 demonstrated reduced autophagic flux and increased apoptosis in response to methamphetamine. These results suggest that autophagy is protective against methamphetamine-induced PAH.
However, others have demonstrated deleterious effects of autophagy in PH. Persistent PH of the newborn is characterized by decreased lung blood vessel density. Teng et al. (95) reported increased expression of autophagy markers in pulmonary artery endothelial cells isolated from lungs of lambs with persistent PH of the newborn. Inhibition of autophagy increased in vitro angiogenesis, suggesting that autophagy impairs angiogenesis in persistent PH of the newborn.
In humans with HIV infection and in a simian model of HIV-associated PAH exposed to morphine, Dalvi et al. (16) observed hyperproliferative and apoptosis-resistant endothelium that expressed autophagy markers. Since opioid use increases the risk of developing PH in HIV-infected individuals, the authors assessed the effects of HIV-tat and morphine on cultured pulmonary endothelial cells. They noted increased autophagy flux and apoptosis in human pulmonary microvascular endothelial cells when treated with HIV-tat protein and morphine in an ROS-dependent manner. Autophagy inhibitors increased and activators diminished apoptosis. In addition, HIV-tat plus morphine-induced increases in ROS were regulated by autophagy and autophagy inhibition increased ROS, suggesting that autophagy is regulating ROS. They concluded that autophagy of pulmonary endothelial cells may increase the extent of angioproliferative remodeling and thus the severity of HIV-associated PH.
Similarly, Haslip et al. (28) studied a mouse model of PH caused by intermittent hypoxia. They found that mice deficient in mitochondrial uncoupling protein 2 (UCP2) had more severe PH after 5 weeks of intermittent hypoxia. Mouse lung endothelial cells deficient in UCP2 demonstrated increased mitophagy and apoptosis, similar to human pulmonary artery endothelial cells from humans with PAH. They also concluded that endothelial cell autophagy was deleterious in PH. Mao et al. (55) demonstrated that hypoxia-induced pulmonary vascular angiogenesis is dependent on increased autophagy.
Thus, whether autophagy is protective or deleterious in PH depends on whether it limits or increases abnormal endothelial proliferation or whether it prevents normal angiogenesis. More work is needed to determine the impact of autophagy on the pulmonary endothelial function in the setting of increased ROS and PH.
Acute respiratory distress syndrome
ARDS is a syndrome of pulmonary edema, hypoxemia, and low lung compliance, frequently associated with inflammation and increased oxidant stress. Edema in ARDS is caused by increased pulmonary microvascular permeability, resulting in water and protein flux out of the vasculature and into the lung interstitium and alveolar gas space. Injury to the lung endothelium and epithelium are critical to increased permeability edema in ARDS.
Autophagy can be protective in ARDS caused by bacterial pneumonia by facilitating removal of pathogens by macrophages (xenophagy) (72). Similarly, autophagy can protect against sepsis-induced mitochondrial damage and downstream inflammasome activation and cytokine release, demonstrating a role for autophagy in regulating innate immune responses (64).
Hyperoxia (95% oxygen for 48–72 h) is a mouse model of severe oxidant lung injury characterized by inflammation, increased vascular permeability, and endothelial cell apoptosis. Silencing of lung endothelial hemoxygenase-1 (HO-1) increased inflammation, worsened survival, and increased endothelial apoptosis after hyperoxic exposure (113). Hyperoxia decreased expression of autophagy markers in both mouse lungs and cultured lung endothelial cells, an effect that was exacerbated by HO-1 suppression. These results indicate that hyperoxia decreases endothelial autophagy and suggest that HO-1 modulates this protective mechanism.
On the other hand, the lungs of humans and mice with H5N1 influenza infection demonstrated accumulation of autophagosomes and increased the ratio of LC3B-II:LC3B-I autophagy marker expression (91). Studies of alveolar epithelial cells showed that H5N1 virus, but not H1N1, induced autophagy and decreased viability. Inhibition of autophagy by silencing Atg5 or by 3-MA prolonged survival and decreased edema resulting from H5N1 pulmonary infection. Thus, in the case of lung injury caused by H5N1 viral pneumonia, autophagy in epithelial cells appears to be deleterious.
Similarly, recent studies indicate that autophagy enhanced LPS-induced endothelial barrier dysfunction. Zhang et al. (110) reported that a chemical inhibitor of autophagy, chloroquine, and silencing of Atg7 blunted LPS-induced autophagosome formation in human pulmonary microvascular endothelial cells, decreased cell viability, and exacerbated LPS-induced monolayer permeability. Similarly, Slavin et al. (83) reported that the autophagy inhibitor, 3-MA, reduced inhaled LPS-induced lung edema both when given prophylactically and as a treatment with minimal effects on inflammation and proinflammatory cytokine expression. In isolated endothelial cells, 3-MA or silencing of Atg5 blunted thrombin-induced barrier dysfunction and reversed LPS-induced endothelial barrier dysfunction. Thus, autophagy appears to play a key role in promoting the breakdown of the endothelial barrier in response to edemagenic agents.
The role(s) of autophagy in acute lung injuries resulting in ARDS are, therefore, complex and dependent on cell type studied and cause of lung injury. It has been proposed that therapeutic approaches to ARDS should not focus on autophagy alone, but on autophagy plus protection against oxidative stress, such as that conferred by Nrf2, a transcription factor that promotes genes that regulate oxidative stress (71).
Ischemia/reperfusion lung injury
Ischemia/reperfusion lung injury can occur with pulmonary thromboembolectomy and lung transplantation and is characterized by lung inflammation and edema. Autophagy markers were increased in rodent lung models of ischemia/reperfusion injury (47, 112) Inhibition of autophagy by 3-MA alleviated and rapamycin activation of autophagy aggravated lung injury (47, 112). Pulmonary microvascular endothelial cells exposed to hypoxia, followed by reoxygenation, displayed increased mitophagy and apoptosis (47). These results suggest a role for autophagy in endothelial injury caused by ischemia/reperfusion.
Emphysema
Emphysema is characterized by loss of alveolar/capillary septum and increased apoptosis of epithelial and endothelial cells (34, 35). Damaged organelles, such as mitochondria (59) and cilia (42), can be disposed off through autophagy (14) in chronic obstructive pulmonary disease. Mitophagy and ciliophagy have been demonstrated in bronchial epithelial cells. It is not clear to what extent mitophagy occurs in lung microvascular endothelial cells in emphysema.
With regard to the lung endothelium, we have reported that acute exposure to cigarette smoke increased lung vascular permeability and endothelial monolayer permeability through an oxidant-mediated effect (50). We also found that cigarette smoke extract increased cultured lung endothelial cell apoptosis and expression of markers of the unfolded protein response (eIF2a phosphorylation) and autophagy (the increased ratio of LC3B-II:LC3B-I) (76).
Using a mouse model of emphysema caused by intratracheal instillation of cadmium, Surolia et al. (92) demonstrated worsening of emphysema in HO-1 knockout mice. Exposure of lung endothelial cells to cadmium increased both apoptosis and expression of autophagy markers. Cadmium exposure of endothelial cells from HO-1 knockout mice increased apoptosis and decreased autophagy; whereas overexpression of HO-1 or treatment with the autophagy activator, rapamycin, decreased apoptosis and increased autophagy. These results indicate that HO-1 induces protective autophagy in lung endothelial cells and is protective against cadmium-induced emphysema.
Table 1 demonstrates that autophagy, including endothelial cell autophagy, has been demonstrated in lungs after various insults. However, the effects of autophagy on lung or endothelial cell function—usually assessed by the effects of autophagy inhibitors or activators or by suppression of key autophagy effectors—vary among injuries. Thus, there is a need for additional study of the role of autophagy in lung diseases.
Footnotes
Acknowledgments
This work was supported by Diabetes United Kingdom grant 15/0005284 (H.C.), RO1 HL123965 (E.O.H.), VA Merit Review (S.R.), U54 GM115677 (S.R.), and P20 GM103652 (E.O.H. and S.R.). Some of the research reported in this article was supported with the use of facilities at the Providence VA Medical Center. The views expressed in this article are those of the authors and do not necessarily reflect the position or policy of the Department of Veterans Affairs.