
Abstract
Aims:
Neuroinflammation and oxidative stress are deemed the prime causes of brain injury after cerebral ischemia/reperfusion (I/R). Since the silent mating-type information regulation 2 homologue 3 (Sirt3) pathway plays an imperative role in protecting against neuroinflammation and oxidative stress, it has been verified as a target to treat ischemia stroke. Therefore, we attempted to seek novel Sirt3 agonist and explore its underlying mechanism for stroke treatment both in vivo and in vitro.
Results:
Trilobatin (TLB) not only dramatically suppressed neuroinflammation and oxidative stress injury after middle cerebral artery occlusion in rats, but also effectively mitigated oxygen and glucose deprivation/reoxygenation injury in primary cultured astrocytes. These beneficial effects, along with the reduced proinflammatory cytokines via suppressing Toll-like receptor 4 (TLR4) signaling pathway, lessened oxidative injury via activating nuclear factor erythroid 2-related factor 2 (Nrf2) signaling pathways, in keeping with the findings in vivo. Intriguingly, the TLB-mediated neuroprotection on cerebral I/R injury was modulated by reciprocity between TLR4-mediated neuroinflammatory responses and Nrf2 antioxidant responses as evidenced by molecular docking and silencing TLR4 and Nrf2, respectively. Most importantly, TLB not only directly bonded to Sirt3 but also increased Sirt3 expression and activity, indicating that Sirt3 might be a promising therapeutic target of TLB.
Innovation:
TLB is a naturally occurring Sirt3 agonist with potent neuroprotective effects via regulation of TLR4/nuclear factor-kappa B and Nrf2/Kelch-like ECH-associated protein 1 (Keap-1) signaling pathways both in vivo and in vitro.
Conclusion:
Our findings indicate that TLB protects against cerebral I/R-induced neuroinflammation and oxidative injury through the regulation of neuroinflammatory and oxidative responses via TLR4, Nrf2, and Sirt3, suggesting that TLB might be a promising Sirt3 agonist against ischemic stroke.
Introduction
Ischemic stroke is a major cause of death and disability worldwide. The outcomes of ischemic stroke damage are far-reaching, generating immense burden to both the family and society (5). To date, ideal drugs or strategies for ischemic stroke are still unavailable. Present strategies for treatment of stroke include recanalization by means of pharmacologic or mechanical thrombolysis and neuroprotective agents (2). So far, recombinant tissue plasminogen activator (rtPA), known as a thrombolytic agent, is the only FDA-approved medical therapy for ischemic stroke (40). However, clinical application of rtPA is limited because of its rigid narrow therapeutic time window, and especially latent ischemia/reperfusion (I/R) injury, which plays a crucial role in aggravating succeeding ischemic brain lesions (35, 42). Therefore, it is of significance to elucidate the mechanisms of cerebral I/R injury and develop more effective strategies or agents to treat cerebral I/R injury.
Emerging evidence suggests that cerebral I/R injury causes a sophisticated cascade of pathophysiologic events, especially neuroinflammation and oxidative stress, which ultimately lead to neuronal injury, even demise (12, 17). Toll-like receptors (TLRs) are known as a transmembrane pattern-recognition receptor family with crucial roles in the mediation of inflammatory responses (49). Up to now, 13 TLRs have been identified in mammals, of which Toll-like receptor 4 (TLR4) is expressed on the cell surface and is identified as the most important pattern recognition receptor involved in cerebral I/R injury. TLR4 is activated in response to cerebral I/R injury, and subsequently directly promotes its pivotal adapter protein myeloid differentiation factor 88 (MyD88) recruitment, resulting in the activation of downstream nuclear factor-kappa B (NF-κB) and thereby releasing proinflammatory cytokines to aggravate cerebral I/R injury in turn (18, 20). Nuclear factor erythroid 2-related factor 2 (Nrf2) is a transcriptional factor involved in antioxidative stress insults (37). Under quiescent condition, Nrf2 localizes to the cytoplasm and is restrained by Kelch-like ECH-associated protein 1 (Keap-1) (27). When cells were subjected to oxidative stress or any other deleterious insults such as cerebral I/R injury, Nrf2 liberates from Keap1 and translocates into the nucleus and then binds to the antioxidant response element (ARE), including hemeoxygenase-1 (HO-1), NAD(P)H-quinone oxidoreductase 1 (NQO1), and glutathione peroxidase (GSH-Px) (1, 33). Intriguingly, accumulating evidence indicates that the Nrf2 signaling pathway is activated during inflammation, which induces the downstream genes of Nrf2 to restrain the inflammatory response, and the Nrf2 signaling pathway cross talks with TLR4 and its downstream genes (e.g., NF-κB) (41); however, whether reciprocity between TLR4 and Nrf2 is involved in the cerebral I/R injury remains still a mystery.
Innovation
The present study, for the first time, discovers that trilobatin (TLB), a novel naturally occurring silent mating-type information regulation 2 homologue 3 agonist from Lithocarpus polystachyus Rehd., mitigates cerebral ischemia/reperfusion injury through regulation of Toll-like receptor 4/nuclear factor-kappa B and nuclear factor erythroid 2-related factor 2/Kelch-like ECH-associated protein 1 signaling. Hence TLB is a novel lead toward the development of a neuroprotective agent.
Trilobatin (TLB), a major active constituent of Lithocarpus polystachyus Rehd., is a folk medicine that is used as prophylaxis and treatment of multiple diseases in China, with a long history (46). Of note, previous report demonstrated that TLB exerted attenuation effect on LPS-induced inflammatory response through hindering the NF-κB signaling pathway (6). Moreover, our previous study revealed that TLB also presented apparent potential neuroprotective effect on hydrogen peroxide-induced oxidative injury in a neuron-like PC12 cell via regulating Nrf2/silent mating-type information regulation 2 homologue 3 (Sirt3) signaling pathway (8). Whereas whether TLB can protect against cerebral I/R injury and its underlying mechanisms are associated with reciprocity between the Nrf2 and TLR4 pathways remains still unclear.
Consequently, the focus of this study was designed to investigate whether TLB treatment can elicit neuroprotection against middle cerebral artery occlusion (MCAO)-induced cerebral I/R injury in rats, and oxygen and glucose deprivation/reoxygenation (OGD/R)-induced injury in primary cultured rat astrocytes through mediating the TLR4/Nrf2 signaling pathway.
Results
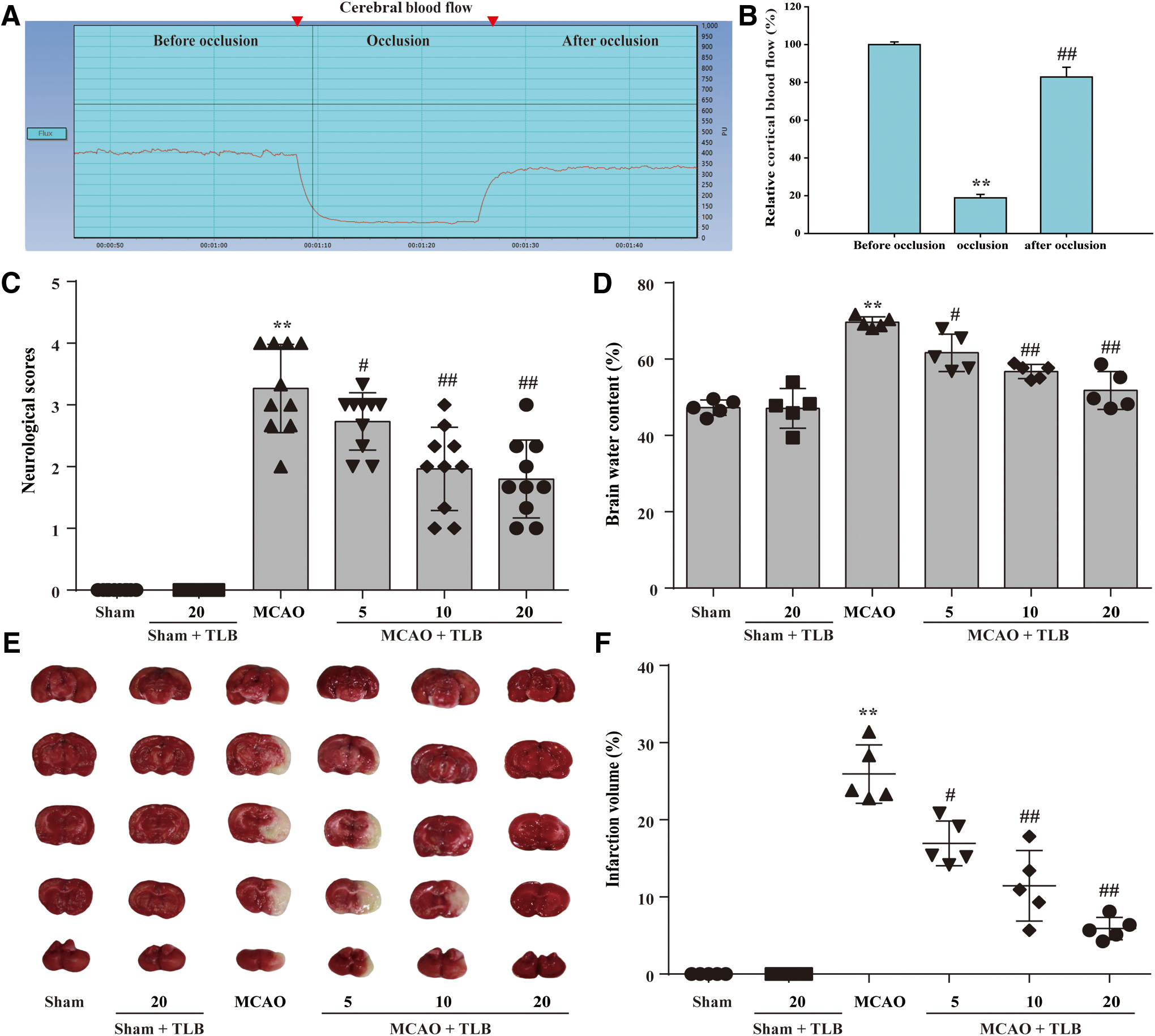
TLB suppressed MCAO-induced injury through decreasing neurologic deficits, infarct volume, and cerebral edema
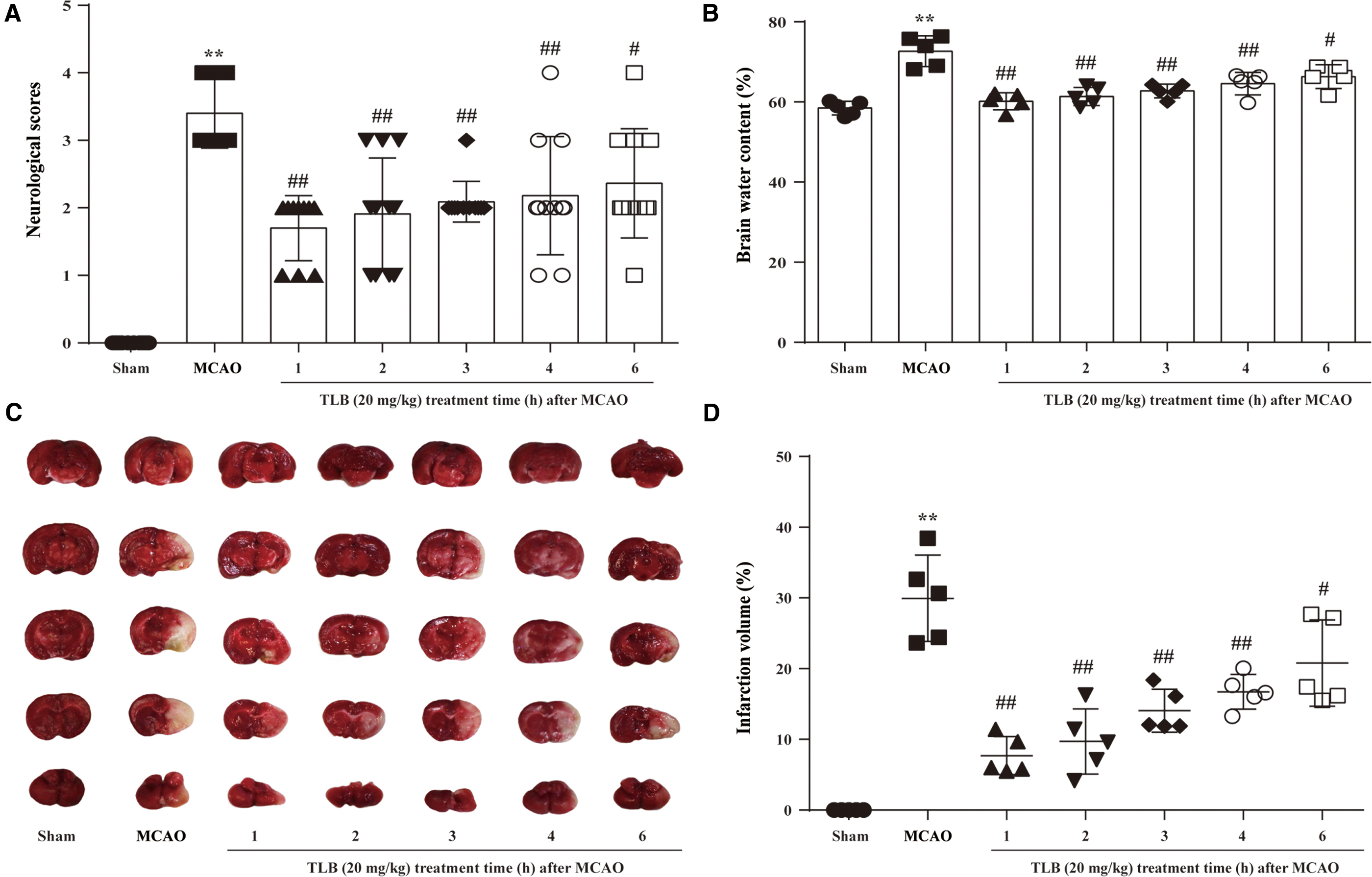
The inhibitory effects of TLB on cerebral I/R-induced outcome 3 days after 2-h MCAO were measured by neurologic deficits, cerebral edema, and infarct volume in rats. First, regional cerebral blood flow (rCBF) was reduced to below 20% and recovered to more than 80% of baseline, indicating that a successful MCAO model was accepted (Fig. 1A, B). The results showed that TLB ameliorated cerebral I/R-induced neurological deficits (Fig. 1C) and cerebral edema (Fig. 1D), and reduced infarct volume (Fig. 1E, F), respectively. These findings suggested that TLB dose dependently inhibited cerebral I/R-induced injury. Furthermore, the time window for TLB treatment after MCAO was explored, due to an appropriate time window for treating cerebral I/R injury being crucial for the therapeutic effects of TLB in the clinic. TLB was administered at 1, 2, 3, 4, and 6 h after MCAO and neurological deficits, cerebral edema, and infarct volume were also measured in rats, respectively. The results showed that TLB (20 mg/kg) significantly reduced the neurologic deficits, cerebral edema, and infarct volume after MCAO within 4 h, while these effects were decreased at 6 h after MCAO (Fig. 2).
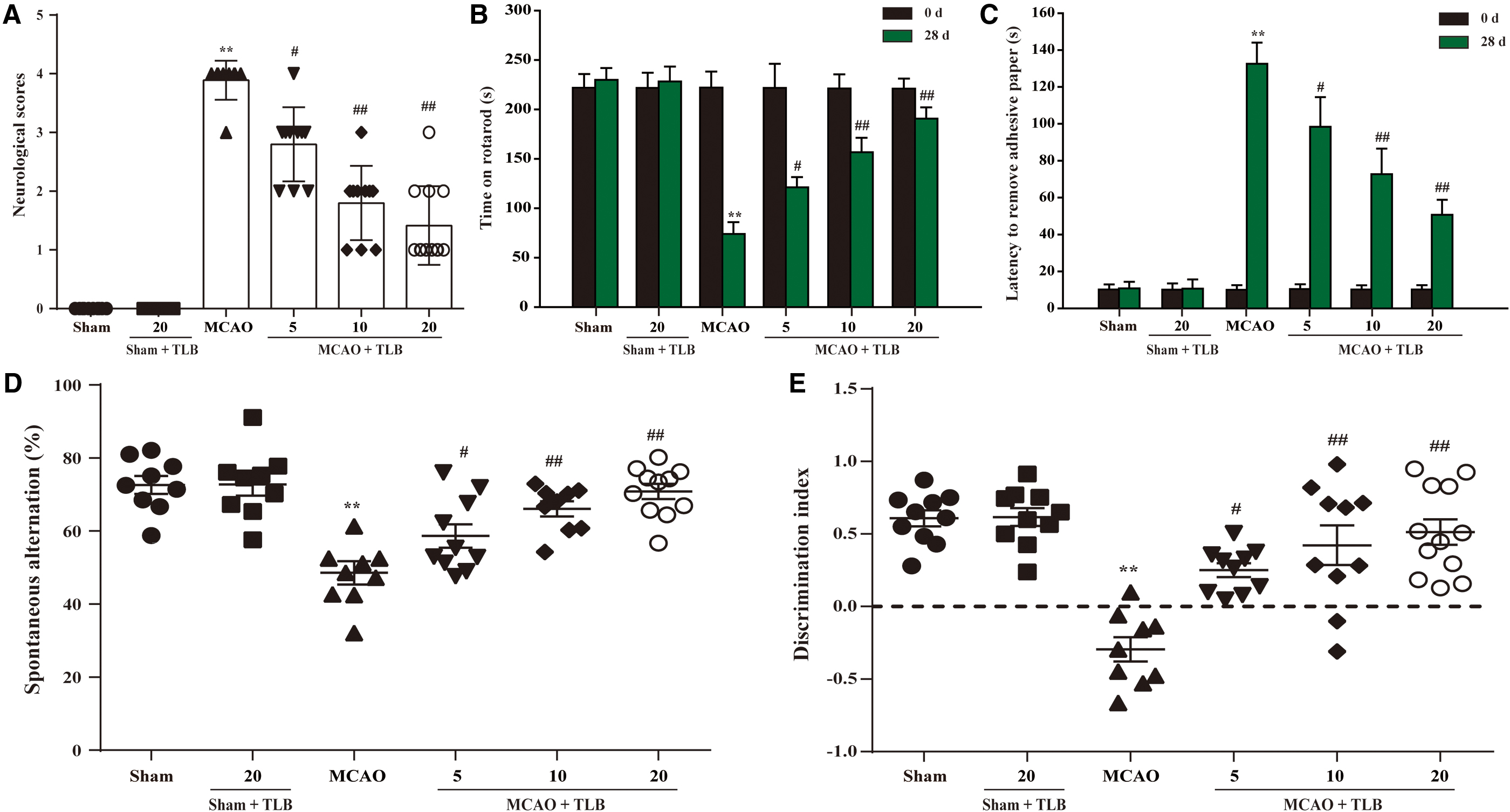
TLB restored long-term neurological functions at 28 days after MCAO in rats
To further explore the effect of TLB on long-term (28 days after MCAO) neurological function recovery, a battery of sensorimotor and cognitive tests, including neurological scores, rotarod, adhesive tape removal, Y-maze and NOR tests, were performed at 28 days after MCAO in rats. The results showed that the neurological scores of rats treated with TLB were markedly lower than those of rats after MCAO at day 28 (Fig. 3A). TLB obviously improved the performance in rotarod and adhesive tape removal from 28 days after MCAO in rats (Fig. 3B, C). In addition, TLB conspicuously increased the percentage of correct spontaneous alternations in Y-maze test (Fig. 3D) and the discrimination index (DI) of novel from familiar objects in the NOR test (Fig. 3E). Collectively, these findings indicated that TLB effectively restored long-term neurological functions after MCAO in rats.
Microarray data analyses
Differentially expressed genes (DEGs) were determined in the condition of both p-value <0.05 and fold change (FC) >1.5. As shown in Figure 4A, 1014 DEGs were identified in MCAO versus sham groups, while 363 DEGs were identified in TLB versus MCAO groups. Furthermore, 52 out of the 415 DEGs responding to TLB treatment were associated with DEGs elicited by cerebral I/R injury, as evidenced by Venn diagram. Moreover, hierarchical clustering analysis showed that the expression profiles of the 415 DEGs in sham and TLB groups were significantly different to that of the MCAO group (Fig. 4B). Moreover, the KEGG pathway and enrichment of Gene Ontology (GO) terms for the selected 415 DEGs involved were depicted. We revealed that the top three pathways enriched from the KEGG database were positive regulation of MAPK cascade, negative regulation of endopeptidase activity, and negative regulation of inflammatory response (Fig. 4C). As shown in Figure 4D–G, the top 10 biological processes, cellular components, and molecular functions for upregulated or downregulated DEGs were listed in bubble plots, which showed that the beneficial effects such as positive regulation of MAPK cascade, positive regulation of angiogenesis, and negative regulation of apoptotic process were included. As presented in Figure 4H, protein/protein interactions (PPIs) in the 415 DEGs identified after TLB treatment were depicted. There were 742 interaction pairs among these DEG-encoded proteins, the size of circles represents the degree of protein connection to others. Notably, TLR4 was involved in 19 interactions, suggesting that TLR4 might be the dominant relevant signaling molecule. Furthermore, we also found that TLR4 interacted with Nrf2, and Nrf2 interacted with Sirt3 using ClusterONE analysis. Of note, to validate the microarray results, the expressions of TLR4, Nrf2, and Sirt3 were determined by quantitative real-time polymerase chain reaction (qRT-PCR). The results showed that TLB downregulated TLR4 and upregulated Nrf2 and Sirt3 than those of the MCAO group, in accordance with the array data (Fig. 4I–K).

TLB reduced MCAO-induced astrocyte and microglial activation
The effect of TLB on glial activation was measured by immunohistochemical (IHC) staining using the antiglial fibrillary acidic protein (GFAP) antibody to mark astrocyte, and the anti-Iba-1 antibody to mark microglia. The results showed that the number of GFAP-positive cells and Iba-1-positive cells was markedly elevated after MCAO than those of the sham group; however, the increased number of activated astrocyte and microglia was attenuated by TLB (Fig. 5).

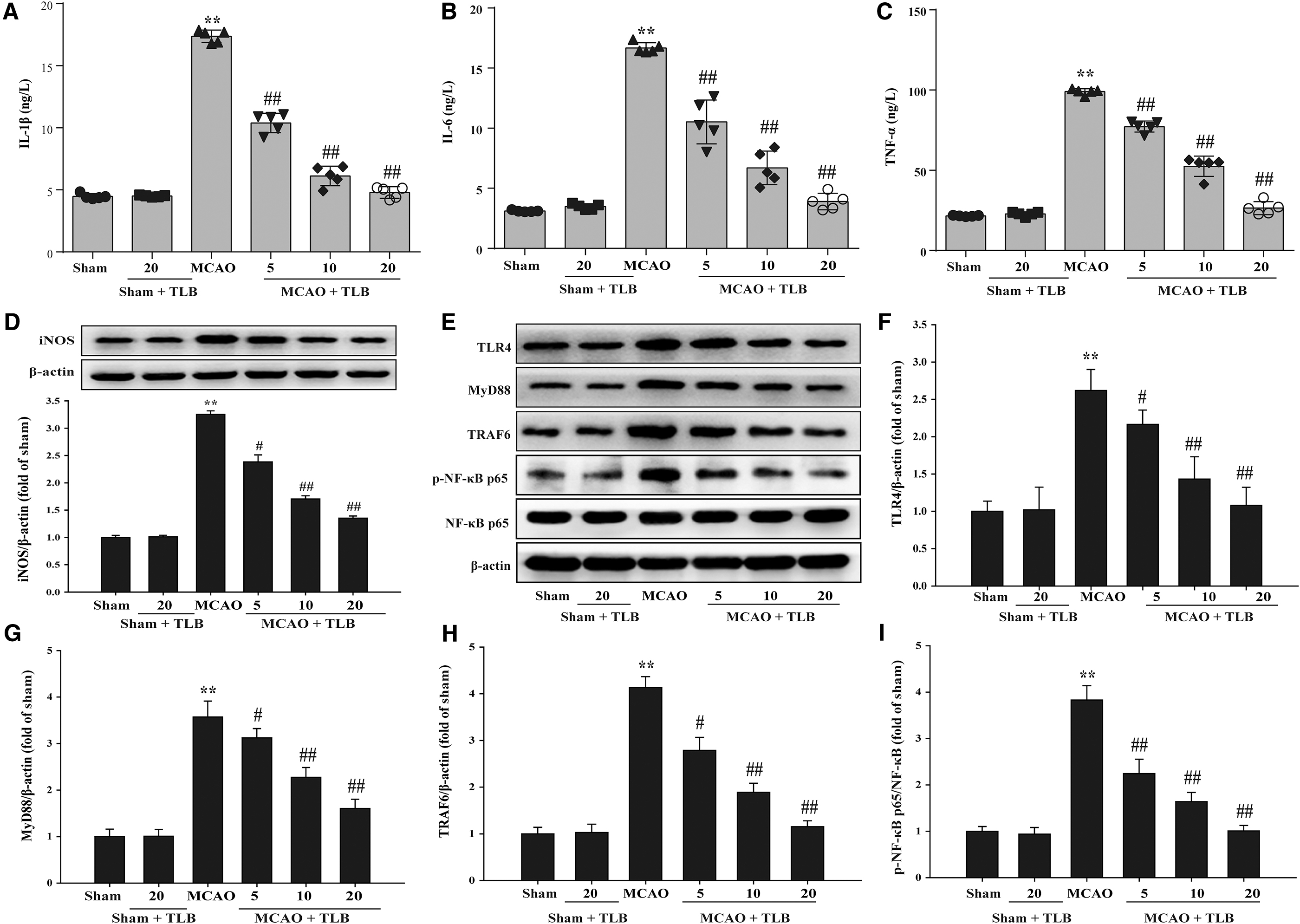
TLB inhibited inflammatory cytokine and TLR4 signaling pathway after MCAO
Inflammatory cytokine levels and inducible nitric oxide synthase (iNOS), TLR4, MyD88, TRAF6, and NF-κBp65 in the brain tissues of rats after MCAO were detected using according inflammatory cytokine enzyme-linked immunosorbent assay (ELISA) kits and Western blot, respectively. The results indicated that the levels of interleukin-1β (IL-1β), interleukin-6 (IL-6), tumor necrosis factor α (TNF-α), and iNOS expression were markedly augmented in the MCAO group than those of the sham group, as well as expressions of TLR4, MyD88, and TRAF6, and phosphorylation level of NF-κBp65. Whereas TLB reduced IL-1β, IL-6, TNF-α, and iNOS expression (Fig. 6A–D), as well as expressions of TLR4, MyD88, and TRAF6 and phosphorylation level of NF-κBp65 (Fig. 6E–I).
TLB attenuated oxidative injury and activated Nrf2/Sirt3 signaling pathway after MCAO
Reactive oxygen species (ROS) and malondialdehyde (MDA) levels, and superoxide dismutase (SOD) and GSH-Px activities were detected using the according kits, and Nrf2, Keap1, HO-1, and NQO1 expressions were detected by Western blot. The results indicated that ROS and MDA levels were elevated and SOD and GSH-Px activities were reduced after MCAO; however, these change were suppressed by TLB (Fig. 7A–D). Furthermore, nuclear Nrf2 level was increased and cytosol Nrf2 level was decreased after MCAO than those of the sham group; However, TLB significantly upregulated Nrf2 expression of the nucleus and decreased it in the cytoplasm than those of MCAO group (Fig. 7E–G). Notably, TLB not only downregulated the Keap-1 expression and upregulated NQO-1 and HO-1 expressions than those of MCAO group (Fig. 7E, H–J), but also increased Sirt3 expression (Fig. 5K).

Prediction of drug targets of TLB against MCAO-induced injury
In addition, the results further exhibited the strong binding affinity between TLB and Sirt3, with binding energy of −5.44 kcal/mol. To investigate the activity and structure relationship of TLB, we synthesized an analogue of TLB, which was named as Tr1. TLB and Tr1 have a similar structure, except for the glucose of TLB. Tr1 and Sirt3 with binding energy of −4.09 kcal/mol indicated that Tr1 failed to bind with Sirt3. Thereafter, the presumptive binding modes and the pocket of amino acid were tested by a molecular docking, including LYS205, SER152, ARG365, PRO355, HIS354, LEU 173, and ASP172, which further confirmed that TLB bound to the hydrophobic pocket of Sirt3, consistent with the results both in vivo and in vitro (Fig. 8). These findings indicated that TLB might directly bind to Sirt3 to exert its pharmacological activities.

TLB protected against OGD/R-induced injury in astrocytes via inhibiting inflammatory cytokine and TLR4 signaling pathway
To further explore the role of TLB during cerebral I/R, the OGD/R model in primary cultured rat astrocytes was applied to mimic the cerebral I/R. We first detected the cytotoxicity of TLB and Tr1, an analogue of TLB (Fig. 9A), in astrocytes to determine suitable in vitro treatment concentrations by a 3-(4,5-dimethythiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. TLB or Tr1 exerted no effect on astrocytes below 50 μM within 48 h (Fig. 9B, C). Therefore, 50 μM or less of TLB and Tr1 was used in the following experiments. Next, the cell viability and cytotoxicity were determined using MTT assay and lactate dehydrogenase (LDH) assay, respectively. The results showed that TLB protected against OGD/R-induced astrocyte injury in a concentration-dependent manner, however, 50 μM Tr1 did not improve the cell viability (Fig. 9D). Moreover, TLB also reduced the amount of LDH release in a concentration-dependent manner. However, 50 μM Tr1 did not alter the LDH level (Fig. 9E). In addition, OGD/R resulted in astrocyte shrink and depletion in cell numbers, even death. Whereas TLB reversed these changes after OGD/R, as evidenced by observation of light converted microscopy. However, 50 μM Tr1 did not alter these changes (Fig. 9F). Furthermore, inflammatory cytokine levels and iNOS, TLR4, MyD88, TRAF6, and NF-κBp65 in the astrocytes after OGD/R were detected using according inflammatory cytokine ELISA kits and Western blot, respectively. The results demonstrated that the levels of IL-1β, IL-6, TNF-α, and iNOS expressions were markedly enhanced in the OGD/R group than those of control group, as well as expressions of TLR4, MyD88, and TRAF6 and phosphorylation level of NF-κBp65. Whereas TLB mitigated IL-1β, IL-6, TNF-α, and iNOS expressions (Fig. 9G–J), as well as expressions of TLR4, MyD88, and TRAF6, and phosphorylation level of NF-κBp65 (Fig. 9K–M).

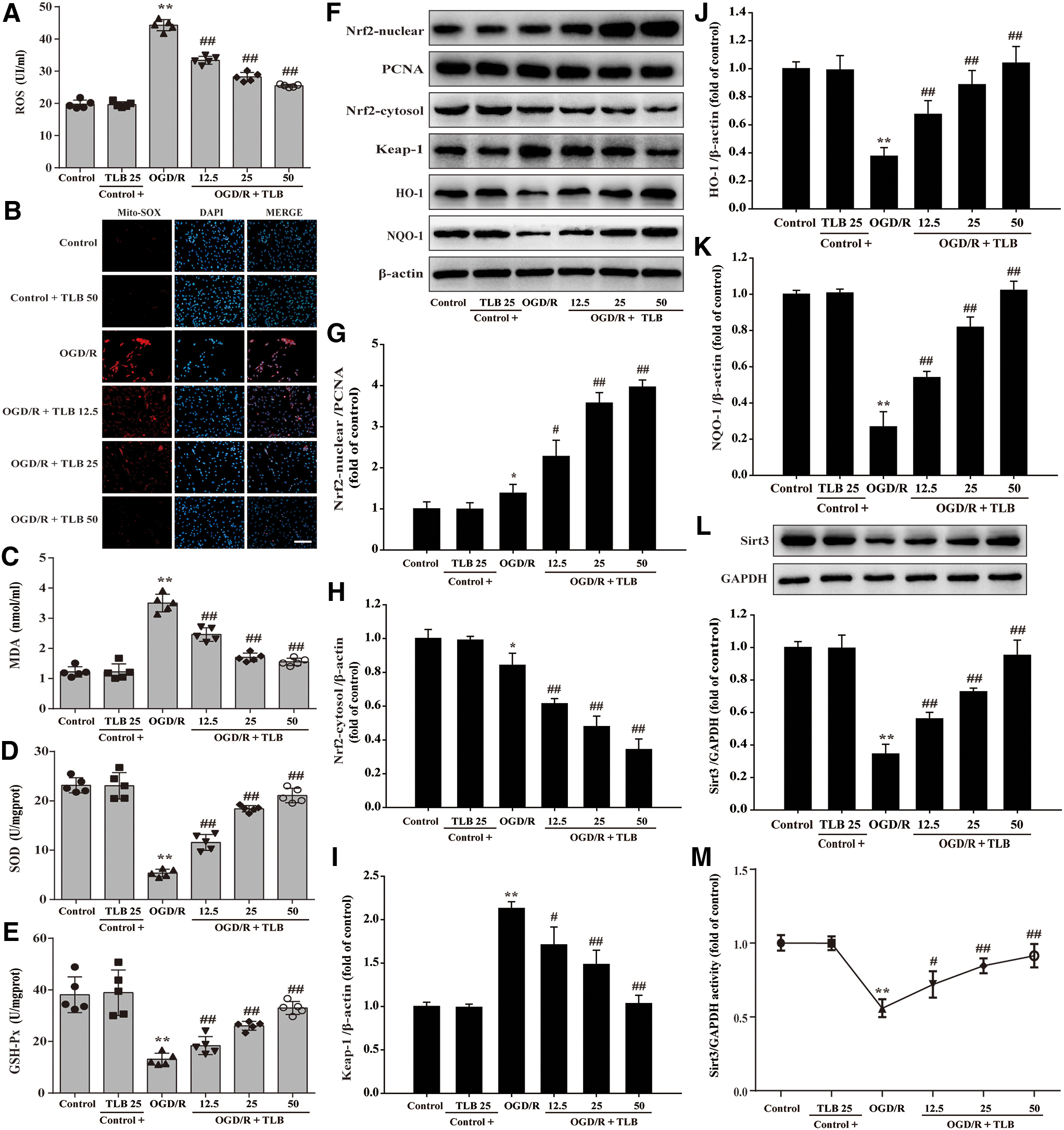
TLB attenuated oxidative injury and activated Nrf2/Sirt3 signaling pathway after OGD/R insult in astrocytes
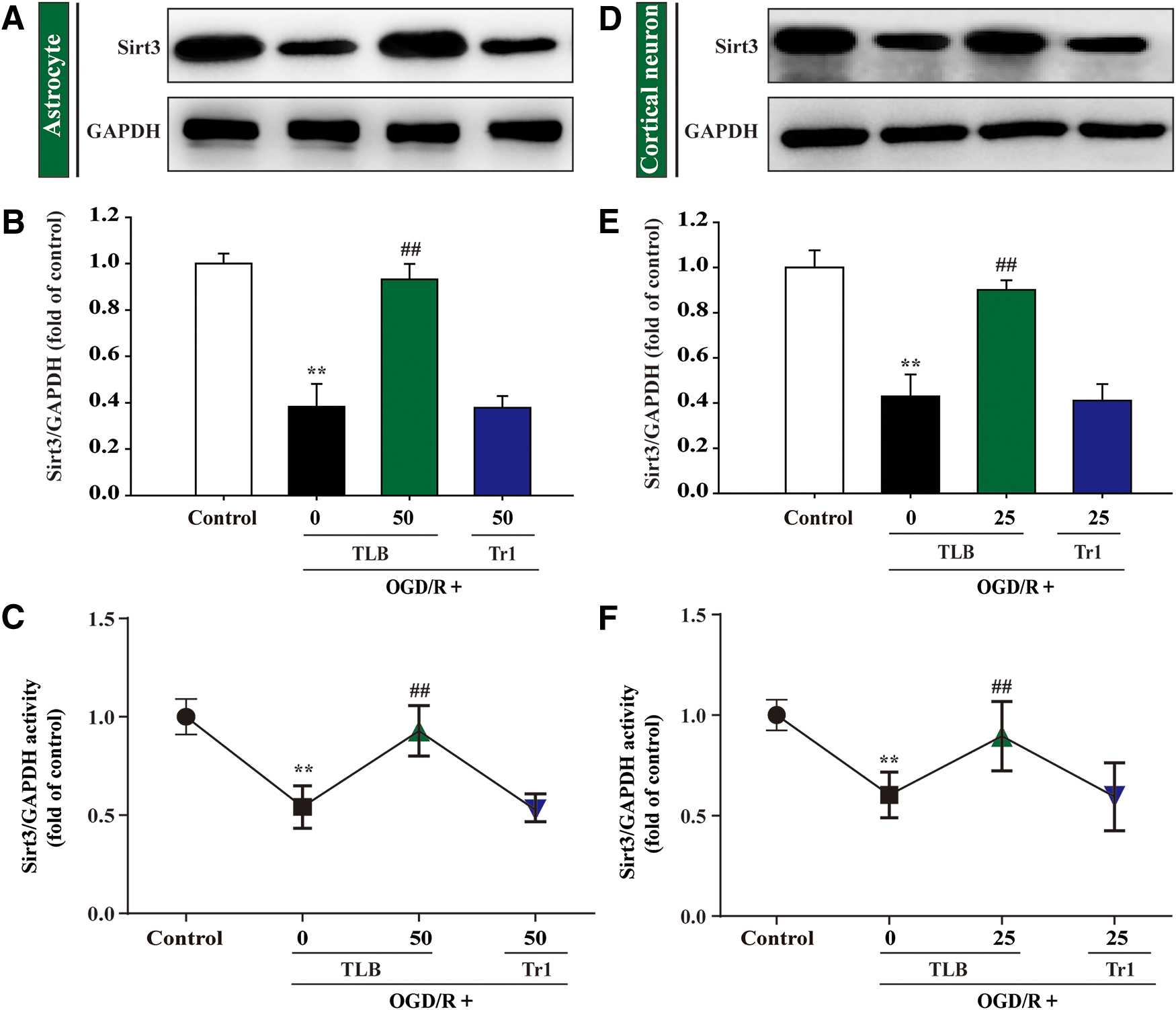
MDA level, SOD, GSH-Px activities, intracellular ROS, and mitochondrial superoxide anion (O2 •−) generation were detected using according kits and MitoSOX staining, respectively, and NRF2, Keap1, HO-1, and NQO1 expressions were evaluated by Western blot. The results indicated that the red fluorescence was enhanced after OGD/R than that of control group, which suggested that OGD/R accelerated O2 •− generation in mitochondria. Moreover, the generation of intracellular ROS and MDA was also elevated and SOD and GSH-Px activities were reduced after OGD/R; However, these change were restrained by TLB (Fig. 10A–E). Furthermore, nuclear Nrf2 level was increased and cytosol Nrf2 level was decreased after OGD/R than that of control group; However, TLB upregulated Nrf2 expression of the nucleus and downregulated it of the cytoplasm (Fig. 10F–H). Notably, TLB not only downregulated the Keap-1 expression and upregulated NQO-1 and HO-1 expressions than those of OGD/R group (Fig. 10I–K), but also increased Sirt3 expression and its activity (Fig. 10L, M). Whereas 50 μM Tr1 did not alter Sirt3 expression and its activity (Fig. 11A–C).
TLB protected against OGD/R-induced injury in primary cortical neurons
The effect of TLB on OGD/R-induced injury in primary cortical neurons was also investigated. TLB or Tr1, an analogue of TLB, exerted no effect on neurons below 25 μM within 24 h (Fig. 12A, B). Therefore, 25 μM or less of TLB and Tr1 was used in the following experiments. Next, cell viability and cytotoxicity were determined using MTT assay and LDH assay, respectively. The results showed that TLB concentration dependently protected against OGD/R-induced neuronal injury, however, 25 μM Tr1 did not improve the cell viability (Fig. 12C). Moreover, TLB also reduced the amount of LDH release in a concentration-dependent manner. However, 25 μM Tr1 did not alter the LDH level (Fig. 12D). In addition, OGD/R led to neuron shrink and reduction in cell numbers, even death. Whereas TLB reversed these changes after OGD/R, as evidenced by observation of light converted microscopy. However, 25 μM Tr1 did not alter these changes (Fig. 12E).

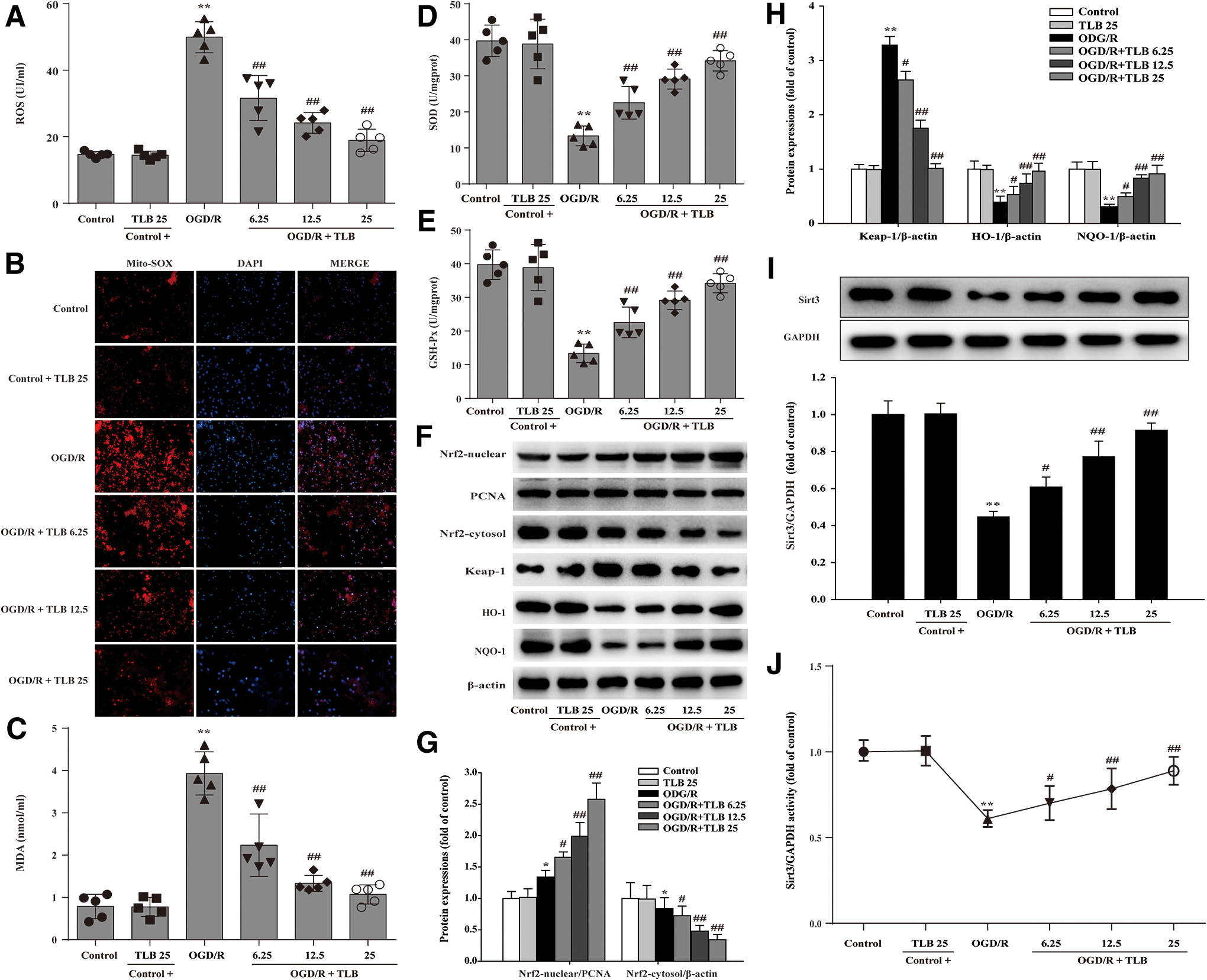
TLB attenuated oxidative injury and activated Nrf2/Sirt3 signaling pathway after OGD/R insult in primary cortical neurons
MDA level, SOD, GSH-Px activities, intracellular ROS, and mitochondrial superoxide anion (O2 •−) generation were determined using according kits and MitoSOX staining, respectively, and NRF2, Keap1, HO-1, and NQO1 expressions were detected by Western blot. The red fluorescence was augmented after OGD/R than that of control, which suggested that OGD/R accelerated O2 •− accumulation in mitochondria. Moreover, the generation of intracellular ROS and MDA was also increased, and SOD and GSH-Px activities were decreased after OGD/R; however, these changes were reversed by TLB (Fig. 13A–E). Furthermore, nuclear Nrf2 level was increased and cytosol Nrf2 level was decreased after OGD/R than that of control group; However, TLB upregulated Nrf2 expression of the nucleus and downregulated it of the cytoplasm (Fig. 13F, G). Notably, TLB not only downregulated the Keap-1 expression and upregulated NQO-1 and HO-1 expressions than those of OGD/R group (Fig. 13F, H), but also increased Sirt3 expression and its activity (Fig. 13I, J). Whereas 25 μM Tr1 did not alter Sirt3 expression and its activity (Fig. 11D–F).
The effect of TLB in Sirt3-knockout (Sirt3-KO) rats and Sirt3-KO astrocytes or neurons
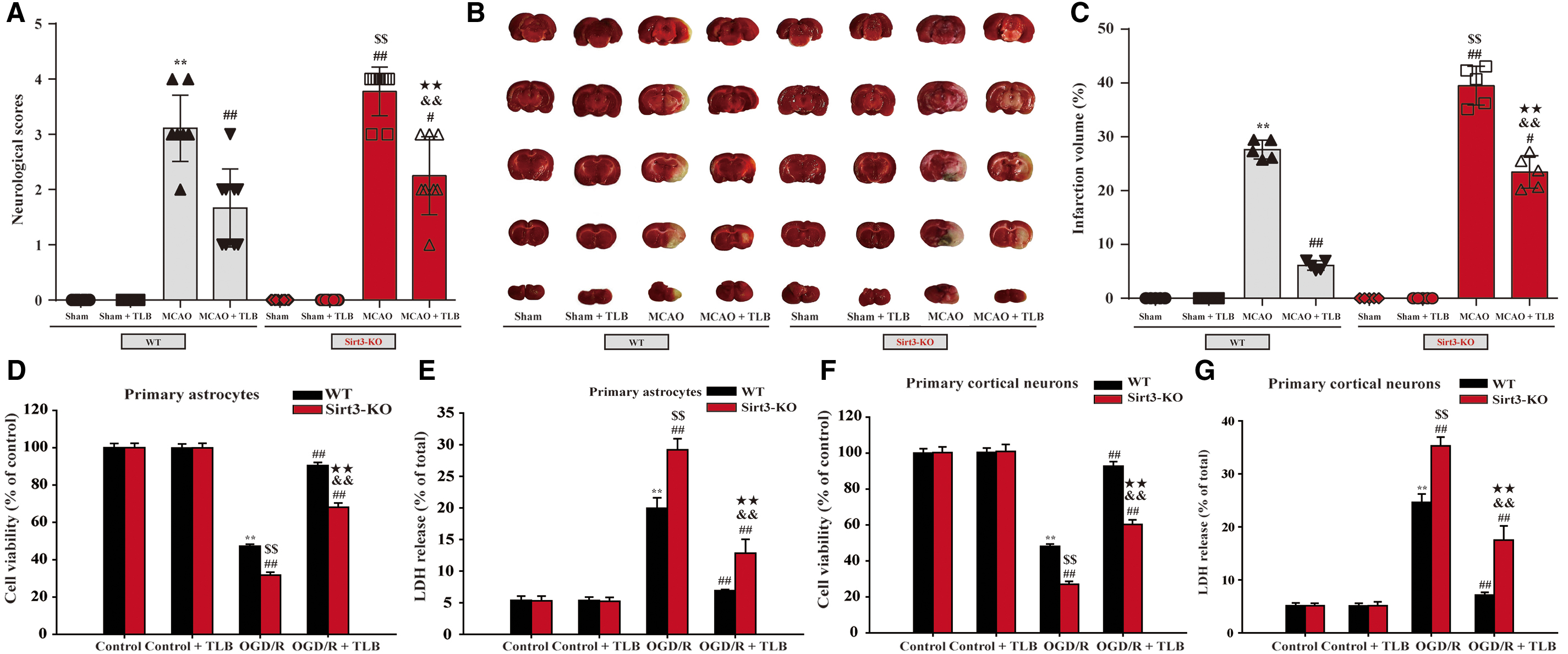
Sirt3-KO rats and Sirt3-KO astrocytes or neurons were generated using the CRISPR/Cas9 system resulting in >80% loss in Sirt3 mRNA levels relative to sham group (Supplementary Fig. S1A) or control cells (Supplementary Fig. S1B). To further confirm that TLB works via binding to and activating Sirt3, we conditionally deleted Sirt3 both in rats and in neurons and astrocytes using CRISPR-Cas9 Sirt3 lentivirus. The results showed that Sirt3 KO rats showed more severe injury after MCAO insult than that of wild-type (WT) rats, which was keeping with the previous report (39). Whereas TLB treatment partly lost its ability in the reduction of MCAO-induced injury in Sirt3-KO rats than those of WT rats (Fig. 14A–C). Interestingly, consistent with the results in vivo, Sirt3-KO neurons or astrocytes also showed more severe injury after OGD/R insult than that of WT neurons or astrocytes. However, the protective effects of TLB on OGD/R-induced injury were partially occluded in Sirt3-KO astrocytes (Fig. 14D, E) and Sirt3-KO neurons (Fig. 14F, G) than those of WT astrocytes or neurons. These findings highlighted that TLB-mediated protection is, at least partly, dependent on the presence of Sirt3, and Sirt3 might be a potential target of TLB, which was consistent with the findings of molecular docking mentioned above.
Reciprocity between TLR4 and Nrf2 was involved in the beneficial effects of TLB on OGD/R-induced injury
Furthermore, small interfering RNA (siRNA) was applied to verify whether TLB regulated the TLR4/Nrf2 signaling pathway to attenuate OGD/R-induced injury in astrocytes. First, the levels of TLR4 and Nrf2 in TLR4 and Nrf2 siRNA-treated groups drastically decreased than those of the scrambled siRNA-transfected group (Supplementary Fig. S2). Without any OGD/R stimulus, silencing of TLR4 or Nrf2 exerted no effect on cell viability and cytotoxicity. Whereas upon OGD/R stimulus, the cell viability and cytotoxicity of TLR4 siRNA-transfected cells were significantly increased and decreased, respectively, than those of scrambled siRNA-transfected cells; However, the beneficial effect of TLB was markedly promoted by the TLR4 siRNA. Meanwhile, the cell viability and cytotoxicity of Nrf2 siRNA-transfected cells were significantly reduced and augmented, respectively, than those of scrambled siRNA-transfected cells; however, the beneficial effect of TLB was significantly abolished by the Nrf2 siRNA (Supplementary Fig. S3). Next, the effect of TLB on inflammatory cytokine in primary rat astrocytes was also further examined by measuring the levels of IL-1β, IL-6, TNF-α, and iNOS in the serum of rats with siTLR4 and siNrf2 by ELISA. The results indicated that the inhibitory effects of TLB in inflammatory cytokines were enhanced by TLR4 siRNA and partially abolished by Nrf2 siRNA (Supplementary Fig. S4A–D). Moreover, the inhibitory effects of TLB in ROS and MDA levels were increased by TLR4 siRNA and almost abolished by Nrf2 siRNA; while the elevation effects of TLB in antioxidant enzymes, including SOD and GSH-Px, were elevated by TLR4 siRNA and almost abolished by Nrf2 siRNA, respectively (Supplementary Fig. S4E–H).
Of note, the results further demonstrated that knockdown of Nrf2 enhanced TLR4 expression in response to OGD/R, while the attenuative effects of TLB on OGD/R-induced TLR4 increase were almost abolished by Nrf2 siRNA (Fig. 15A, B). Moreover, knockdown of TLR4 significantly increased Nrf2 expression in the nucleus and decreased it in the cytoplasm after OGD/R, while the promotion effects of TLB were also elevated by TLR4 siRNA (Fig. 15C, D). Furthermore, the docking of Nrf2 and TLR4 was carried out by the ZDOCK and RDOCK method, as in a previous study. The results showed that there were strong interactions between Nrf2 and TLR4 as evidenced by the output values of score of E_RDOCK (Table 1). Of note, the results further demonstrated that the hydrogen bond and charge interactions were generated through the amino acid residues, including HIS3, ASN409, HIS5, PHE263, PHE429, GLN430, SER9, and HIS7; and Pi interactions with the residues such as TRP332 and PHE330, TRP332 and PHE313, TRP332 and PHE330, TRP332 and PHE313, PHE408 and HIS431 (Fig. 15E–G). Notably, we further verified the direct interaction between Nrf2 and TLR4 by surface plasmon resonance (SPR). The results demonstrated that Nrf2 directly bound to TLR4 in a concentration-dependent manner with a KD value of 2.139e−9 M (Fig. 15H). These findings suggested that there existed potent affinity between Nrf2 and TLR4.
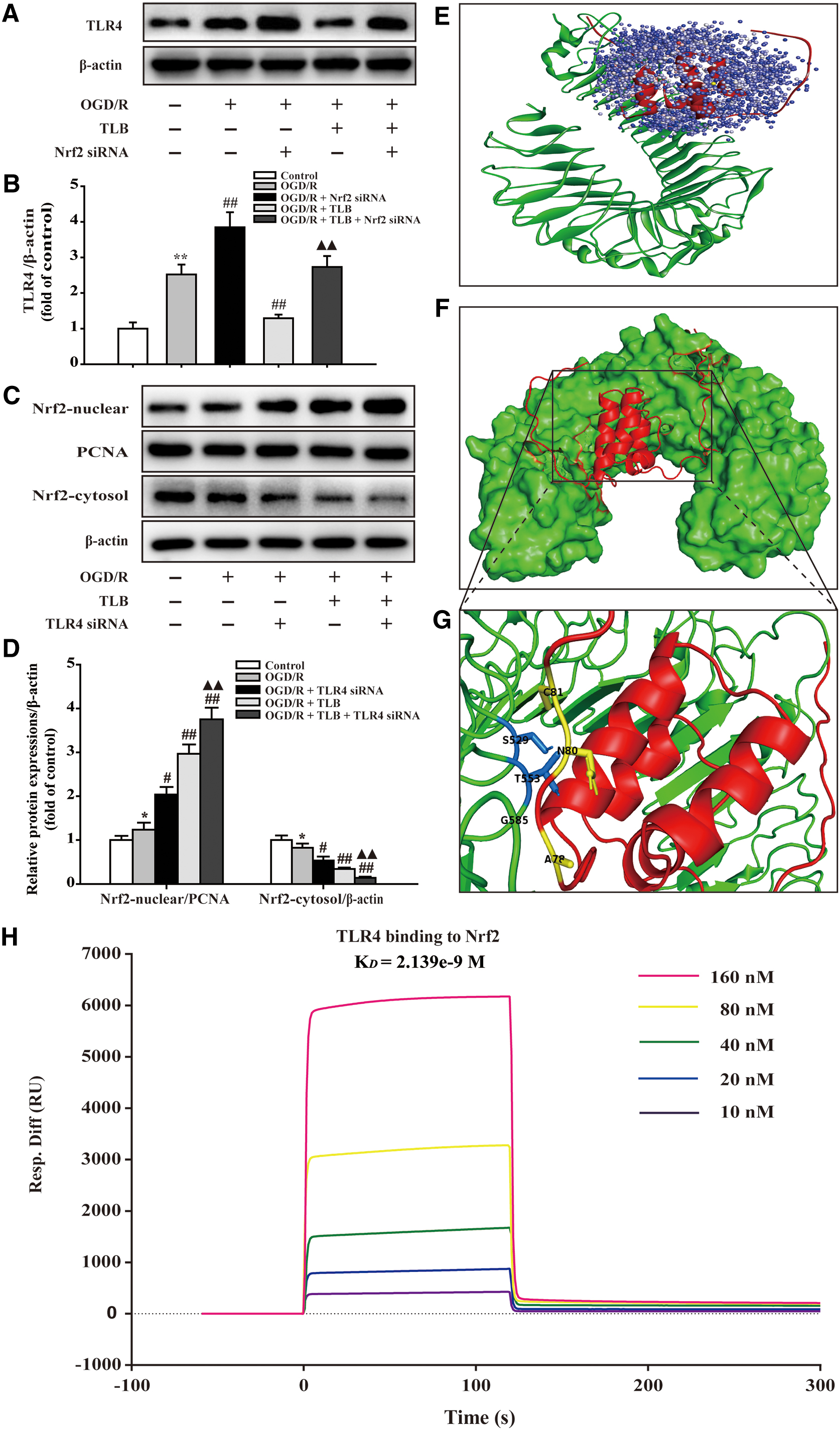
ZDOCK and RDOCK Score of Nrf2 and TLR4
Lower values of ZRANK score and E_RDOCK and higher ZDOCK score indicate top docking of Nrf2 and TLR4.
Clash “0” indicates no stearic clash between the proteins after refined using RDOCK.
Nrf2, nuclear factor erythroid 2-related factor 2; TLR4, Toll-like receptor 4.
Discussion
The present study, for the first time, discovered that (i) TLB, a naturally occurring Sirt3 agonist derived from herbal L. polystachyus Rehd., exerted neuroprotection against MCAO-induced injury in rats and OGD/R-induced injury in primary cultured astrocytes or neurons in vitro. (ii) The inhibitory effects of TLB were due to inhibition of neuroinflammation through suppressing TLR4 signaling pathway, as well as reduction of oxidative stress injury via activating Nrf2 signaling pathway. (iii) Reciprocity between TLR4 and Nrf2 was involved in the neuroprotection of TLB against cerebral I/R injury (Fig. 16).
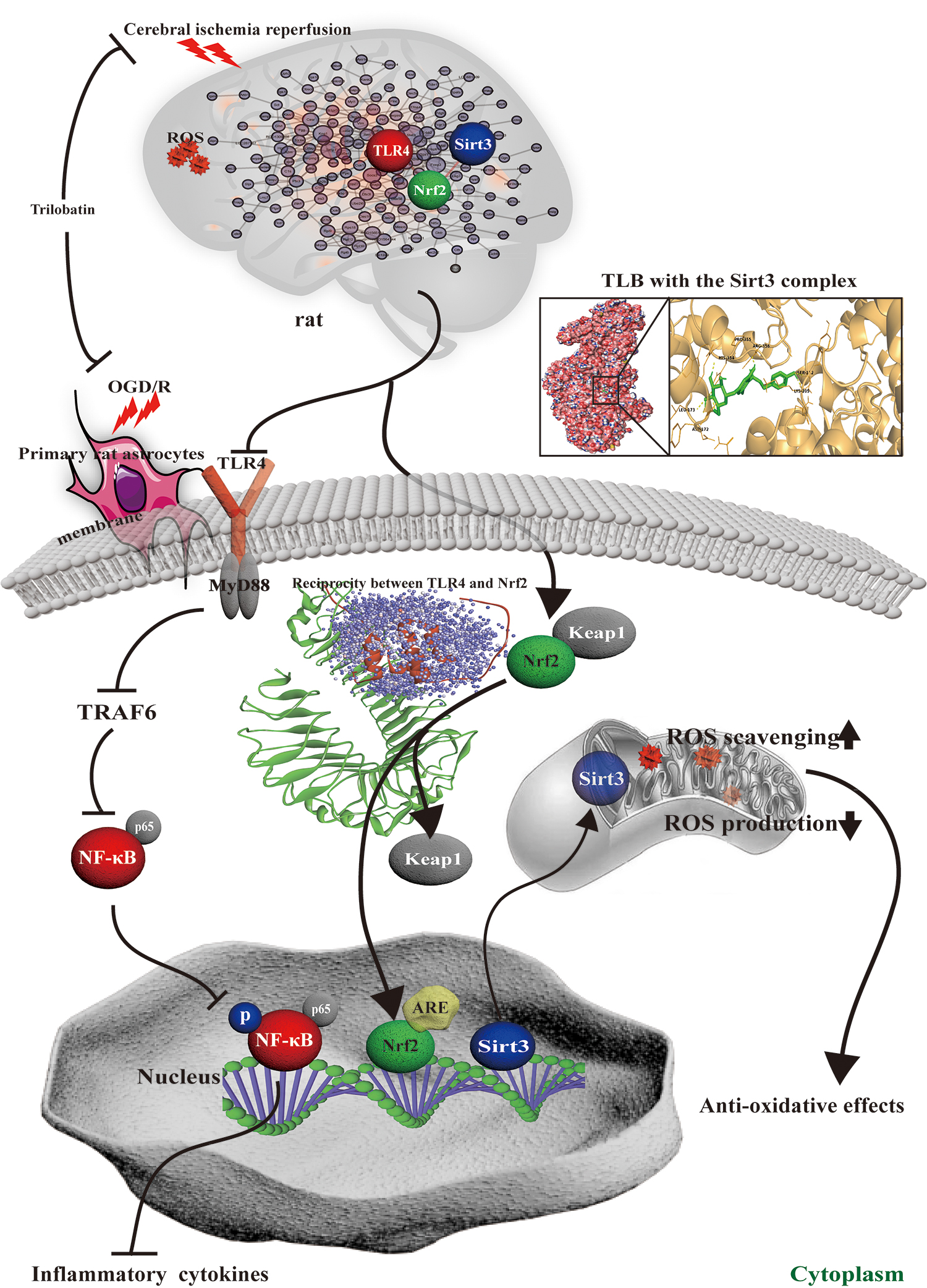
On account of lacking effective prophylaxis and treatment strategies for ischemic stroke, a mass of pharmacological neuroprotectants have been researched and developed; unfortunately, with limitation of clinical success or failure of preclinical due to a complicated pathological process with manifold mechanisms during cerebral I/R injury (19, 26). Thus, exploring an effective neuroprotectant is an extreme clinical demand. In the current study, we explored the neuroprotection of TLB, a natural small molecule monomer, against cerebral I/R injury and endeavored to elucidate its underlying mechanisms. First, our findings demonstrated that TLB effectively protected against outcomes of cerebral I/R injury as evidenced by reduced MCAO-induced neurological deficits, cerebral edema, and infarction volume in vivo. Next, of note, TLR4, Nrf2, and Sirt3 were identified as DEGs as proved by Microarray Data Analyses (Venn diagram, hierarchical clustering analysis, GeneMANIA analysis, and GO terms) and TLB also reduced TLR4 mRNA level and increased Nrf2 and Sirt3 mRNA levels in line with the array data as affirmed by qRT-PCR. Therefore, it is reasonable to speculate that TLR4-mediating neuroinflammation and Nrf2/Sirt3-mediating oxidative stress were involved in TLB-induced neuroprotection against cerebral I/R injury. As noted, neuroinflammation is a crucial step and a secondary damage mechanism in the cerebral I/R injury (34). When neuronal cells are challenged by cerebral I/R, neurogliocytes (astrocytes and microglia) are activated and release proinflammatory factors, including IL-1β, IL-6, TNF-α, and iNOS, thereby exacerbating brain injury (28). As we expected, MCAO induced activation of astrocyte and microglia, and also promoted release of inflammatory cytokines (IL-1β, IL-6, TNF-α, and iNOS), which are the premier triggers of activating astrocytes in cerebral I/R injury; whereas TLB reversed these change after MCAO, suggesting that TLB-mediated neuroprotection against cerebral I/R injury, at least partially, through inactivation of neurogliocyte and inhibition of proinflammatory factor release. Furthermore, since TLR4 is expressed by neurogliocyte and contributes to cerebral I/R-induced inflammatory injuries (13), the role of TLR4 signaling pathway during the beneficial effects of TLB on cerebral I/R injury was determined. The results revealed that the expressions of TLR4, MyD88, and TRAF6 significantly increased after MCAO, which suggested that the TLR4-regulated signaling pathway was activated after cerebral I/R and heightened inflammatory responses and further aggravated brain injury, in keeping with the previous study. However, TLB inhibited MCAO-elicited changes. Moreover, TLR4 signaling was stimulated by cerebral I/R injury, resulting in activating the transcription factors NF-κB involved in activation of proinflammatory genes and cytokines (30). Our findings also demonstrated that the phosphorylation level of NF-κBp65 was upregulated by MCAO, while TLB obviously repressed the phosphorylation level of NF-κBp65 after MCAO. These findings suggested that the attenuative neuroinflammation effects of TLB on cerebral I/R injury were mainly due to inactivation of the TLR4/MyD88/TRAF6/NF-κB pathway.
In addition, mounting evidence suggests that oxidative stress, termed as an imbalance between the antioxidase system and the generation of ROS, is known as an early event during cerebral I/R injury for that the brain is very suggestible to oxidative stress (22). Moreover, accumulation of ROS is primary in activating TLR4-regulated signaling pathways and induced proinflammatory factor release in cerebral I/R injury (23). Our results manifested that MCAO induced an increase in ROS generation and level of MDA, derived from lipid peroxidation and considered a biomarker of oxidative stress, consistent with the theory that cerebral I/R injury can attract a remarkable amount of MDA generation in the ischemic brain hemisphere (25); also, MCAO induced a decrease in SOD and GSH-Px activities, which were the antioxidant enzymes to maintain redox homeostasis and affect the inflammatory response (9). Conversely, TLB reversed these change by MCAO insult, inferring that TLB-mediated neuroprotection against cerebral I/R injury, partially, was via suppressing oxidative lesions. Thereafter, our results further clarified that MCAO promoted Nrf2 translocation from the cytoplasm into the nucleus; however, TLB significantly facilitated nuclear translocation of Nrf2. Whereas the change of Nrf2 protein expression after MCAO was not consistent with the results of the mRNA level of Nrf2, which did not change. The reason might be due to that there is a more intrinsic and complex dependence between mRNA and protein, such as translation and transcription of gene expression existing in time/space span, and there would be post-transcriptional processing, degradation of transcriptional products, translation, post-translational processing, and modification after transcription. Furthermore, MCAO also decreased ARE, including HO-1 and NQO1 expressions, while TLB increased HO-1 and NQO1 expressions after MCAO, indicating that TLB prompted Nrf2 dissociated from Keap1 and then translocated into the nucleus, thereby activating ARE to protect against oxidative stress in cerebral I/R injury. Thus, we believed herein that TLB alleviated neuroinflammatory responses and oxidative stress during cerebral I/R injury. Nevertheless, what is the potential target of TLB and whether reciprocity between TLR4 and Nrf2 was involved in the TLB-mediated neuroprotection against cerebral I/R injury were still unclear. Therefore, we further explore the uncovered problem in neurons and astrocytes in vitro. Notably, astrocytes are the most plentiful non-neuronal cell type in the central nervous system and have been indicated to have intensive resistance to cerebral I/R injury compared with neurons (10, 45); while susceptibility of neurons to cerebral I/R will be augmented once astrocytes dysfunction (14). Most importantly, astrocytes not only expressed TLR4 but also enriched transcription factor Nrf2 (21). Moreover, since in ischemic stroke, a dramatical decrease in rCBF elicits deprivation of oxygen and glucose and eventually leads to cerebral injury. Thus, we used OGD/R-induced astrocyte and neuron injury to mimic cerebral I/R injury in vitro to further investigate the mechanism of TLB-mediated neuroprotection. The results showed that TLB effectively increased cell viability and decreased cytotoxicity after OGD/R insult both in neurons and astrocytes, respectively. Furthermore, TLB also restrained proinflammatory factor release as well as inactivated the TLR4/MyD88/TRAF6/NF-κB pathway after OGD/R insult in astrocytes. Meanwhile, some oxygen free radicals and their derivatives are accumulated after cerebral I/R injury, including O2 •−, which generated in mitochondria. The results indicated that both intracellular and mitochondrial ROS generation exhibited a significant decrease by TLB after OGD/R, as well as activated Nrf2 signaling pathway accompanied with elevated ARE genes and enzymes both in neurons and astrocytes. Our findings further verified that TLB effectively attenuated cerebral I/R injury through inactivation of TLR4-mediated pathway and activation of Nrf2-mediatied pathway to suppress neuroinflammation and oxidative stress, in keeping with the findings in vivo.
Although multiple studies demonstrated that the TLR4 signaling pathway and Nrf2 signaling are all activated during neuroinflammation (11), little is known about whether reciprocity between TLR4 and Nrf2 was involved in cerebral I/R injury. Interestingly, our results revealed that knockdown of TLR4 with siRNA evidently partly strengthened the protective roles of TLB in promoting astrocyte survival, decreasing proinflammatory cytokine production and ROS generation, as well as facilitating Nrf 2 nuclear translocation in astrocytes after OGD/R insult. Moreover, knockdown of Nrf 2 with siRNA apparently partly abolished the protective roles of TLB in facilitating astrocyte survival, decreasing inflammatory cytokine and ROS generation, as well as reducing TLR4 protein expression in astrocytes under OGD/R condition. Thus, it is speculated that there might be a direct reciprocity between TLR4 and Nrf2. Of particular interest was that TLR4 directly bound to Nrf2 as confirmed by ZDOCK and RDOCK as well as SPR. These findings disclosed the effect of TLB against cerebral I/R injury via reciprocity between the Nrf2 and TLR4 signaling pathways. Of note, Sirt3 is a mitochondrial nicotinamide adenine dinucleotide-dependent protein, which localized to the mitochondrial matrix (38). Recently, Sirt3 has been deemed to not only mediate oxidative stress and mtROS homeostasis, but also orchestrate suppression of inflammation (15, 29). Noteworthily, the results in this study demonstrated that Sirt3 was apparently decreased after MCAO and OGD/R, in line with the theory that Sirt3 deficiency impairs neurovascular recovery in ischemic stroke (4); whereas TLB increased Sirt3 expression both in vivo and in vitro consistent with our previous study (8). Moreover, TLB also significantly enhanced Sirt3 activity after OGD/R insult. Intriguingly, in fact, a direct interaction between TLB and Sirt3 was evidenced by molecular docking. Furthermore, the beneficial effects of TLB on cerebral I/R injury were partially abolished in Sirt3 deficiency rats although Sirt3-KO rats showed normal under common conditions, which indicated that Sirt3 plays a vital role under stress conditions and further verified that TLB might be a naturally occurring Sirt3 agonist. Thus, it is plausible to suppose that Sirt3 was likely to be a promising therapeutic target of TLB against cerebral I/R injury. Emerging evidence demonstrates that a mass of neuroprotective agents exhibited valid effects in in vivo experiments, but did not display significant effects in a clinical trial due to a narrow therapeutic window. Therefore, it is necessary to explore the therapeutic window of neuroprotective agents in in vivo experiments. Our findings indicated that TLB exerted significantly therapeutic effects on cerebral I/R injury in rats within 4 h, and although its beneficial effects were partially lost after 6 h, it still effectively attenuated the injury after MCAO in rats. Thus, whether TLB can induce an extended window of ischemic tolerance in the rat brain will be investigated in our next study. It should be noted that the present study has evaluated the neuroprotection of TLB against cerebral I/R injury and its preliminary mechanisms. Whether TLB can cross the blood/brain barrier and mitigate cerebral I/R-induced astrocyte injury to further reinforce the resistance of neurons to neuroinflammation and oxidative injury still needs to be in-depth explored. In fact, these problems could be solved by means of pharmacokinetics, BBB-3D model, and cocultured astrocytes and neurons in transwell in our next study.
Collectively, our findings afford new insight into that TLB inhibited cerebral I/R-induced neuroinflammation and oxidative injury via the TLR4/Nrf2/Sirt3 signaling pathway. These findings highlight the feasibility that TLB might be a promising Sirt3 agonist against ischemic stroke.
Materials and Methods
Chemicals and reagents
TLB was from Guangdong Kedi Medical Technology Corporation (purity ≥98%). The synthesis of Tr1 was implemented as described in Figure 9A. The nuclear magnetic resonance spectra and mass spectrum data of the TLB and Tr1 are shown in Supplementary Data and Supplementary Figures S5–7. 2,3,5-Triphenyltetrazolium chloride (TTC) and MTT were purchased from Sigma-Aldrich, Neurobasal™-A medium, B-27™ Supplement (50 × ), and MitoSOX red were obtained from Invitrogen (Eugene, OR). The LDH Cytotoxicity Assay Kit, IL-1β, IL-6, TNF-α, ROS, MDA, SOD, and GSH-Px assay kits were obtained from Shanghai Renji Bioengineering Institute (Shanghai, China). The primary antibodies used in present study, including GFAP, Iba-1, iNOS, TLR4, MyD88, TRAF, NF-κBp65, Nrf2, Keap1, HO-1, NQO1, Sirt3, and Sirt3 activity assay kit (Fluorometric), were purchased from Abcam (Cambridge, United Kingdom). TLR4 siRNA (r) and Nrf2 siRNA (r) were purchased from Santa Cruz Biotech (Santa Cruz, CA). Lipofectamine™ RNAiMAX transfection reagent and scrambled siRNA were obtained from Invitrogen.
Animals
Male adult Sprague-Dawley rats (8–10 weeks old, 250–280 g) were purchased from the Experimental Animal Center of Daping Hospital (Certificate No. SCXK 2014-0011). All rats were housed in five to six per cage with 12-h light/dark cycles and provided free access to standard rodent diet and tap water and kept under a temperature (23°C ± 1°C)- and humidity (55% ± 5%)-controlled environment. Randomization was used to allocate animals to various experimental groups, and the data analyses were performed by a blinded investigator. All animal experimental protocols in the present study were operated according to the Guide for the Care and Use of Laboratory Animals published by the U.S. National Institutes of Health (National Institutes of Health Publication 85–23, revised 1996), and were approved by the Experimental Animal Ethics Committee of the Zunyi Medical University (Guizhou, China).
Induction of focal cerebral ischemia and drug treatments
Focal cerebral ischemia was induced by MCAO as described in the previous study (43). In brief, the rats were anesthetized with 1% sodium pentobarbital (45 mg/kg, i.p.) and the right common carotid artery, external carotid artery, and internal carotid artery were separated carefully. Then, a piece of 5/0 monofilament nylon suture with diameter 0.36 mm was inserted into the internal carotid artery through the external carotid artery stump to occlude the origin of middle cerebral artery. After 2 h, the filament was withdrawn to establish reperfusion and the rat body temperature was kept at 37°C during surgery. Laser Doppler flowmetry (Moor Instruments, United Kingdom) was applied to monitor MCAO severity rCBF during the surgical procedure and at the reperfusion as described previously. A successful MCAO model was accepted when rCBF lowered to below 20% and recovered to higher than 80% of baseline. In the first study of TLB treatment on cerebral I/R, the rats were randomly divided into six groups: sham group, sham + TLB (20 mg/kg) group, MCAO group, MCAO + TLB (5 mg/kg) group, MCAO + TLB (10 mg/kg) group, and MCAO + TLB (20 mg/kg) group. Sham group underwent the same operation as mentioned above except MCAO. Animals were treated with TLB by gavage at doses of 5, 10, and 20 mg/kg at the onset of reperfusion twice a day for 3 days, and the rats of sham and model groups were given volume-matched saline, instead. The second group was designed to evaluate the time window of TLB for the treatment of cerebral I/R, and TLB was administered at the doses of 20 mg/kg at 1, 2, 3, 4, and 6 h after MCAO. The sham group and MCAO group of rats received volume-matched saline. In the third group, to discover the effect of TLB on functional recovery after MCAO, TLB was administered at the dosage of 5, 10, and 20 mg/kg at the onset of reperfusion twice daily for 28 days after MCAO. The sham group and MCAO group of rats were administered volume-matched saline. In the final group, to determine the effects of TLB on the MCAO model, rats were divided into the following four groups 3 weeks after CRISPR-Cas9 Sirt3 lentivirus microinjection: WT + sham, WT + sham + TLB (20 mg/kg), WT + MCAO, WT + MCAO + TLB (20 mg/kg), Sirt3-knockout (Sirt3-KO) + sham, Sirt3-KO + sham + TLB (20 mg/kg), Sirt3-KO + MCAO, Sirt3-KO + MCAO + TLB (20 mg/kg).
Determination of neurological deficit scores
Neurological injury after MCAO was determined by a neurobehavioral test that was scored on a five-point scale: grade 0, observable deficit; grade 1, failure to fully extend left forepaw; grade 2, circling to the left; falling 3, falling to the left; 4, unable to walk spontaneously accompanied with a depressed level of consciousness. A neurobehavioral test was carried out 3 days after MACO in a double-blind manner as described previously (16).
Assessment for long-term functional recovery after cerebral I/R injury in rats
Neurological deficit scores were determined after day 28 as mentioned above. In addition, rats were trained in the sensorimotor tests for successive 3 days before MCAO. Rotarod and adhesive tape removal tests were performed as described in previous reports (31, 48). The rotarod was applied to determine general fitness and motor co-ordination. In brief, rats were placed on an accelerating rotating beam (acceleration from 4 to 40 rpm within 5 min) and a stop-clock was started. The latency for each rat to fall off the rotarod onto the sensing platform below was registered. Each rat performed three trials. Moreover, the sensorimotor impairment induced by MCAO was detected by adhesive tape removal tests. In brief, a piece of adhesive tape (3 × 1 cm) was applied as bilateral tactile stimuli occupying the distal-radial area on the wrist of each forelimb in rats. Then, the rats were placed back to their cages. The mean latency to remove adhesive tapes from the forepaws was registered for the lesioned forepaws. The times to detect and remove the tapes were recorded with a maximum limitation of 120 s. Furthermore, spatial memory and learning were detected using Y-Maze test, as in the previous study (44). In brief, three same arms of the maze apparatus (30 cm × 10 cm × 20 cm) were randomly assigned as the beginning arm, novel arm, or other arm. First, rats were subjected to investigate the beginning arm and the other arm for 8 min. Thereafter, rats were returned back to the maze in the same beginning arm and allowed to investigate for 5 min with access to all three arms at liberty. The number of entries and percentage of time spent in the novel arm that rats detected were recorded. Spontaneous alternation (%) = [(number of actual alternations)/(number of total arm entries-2)] × 100 was applied as an index to evaluate spatial memory. Moreover, NOR tasks were use to determine learning and memory after MCAO as described in the previous report (42). In brief, rats were accustomed to an open-field box (50 × 50 × 50 cm) at 28 days after MCAO. During the first phase (10 min), two of the same objects (A1 and A2, green cubes) were placed symmetrically from the wall. During familiarization phase (10 min), two dissimilar objects (object A2 was replaced with a novel object B1, a brown box) were placed in the same box for 1 h after the first trial. The total time that a rat spent detecting each object during two phases was recorded. The objects' DI was calculated using a DI as described in the previous report (42).
Measurement of infarct volume
After the neurological test, the rats were anesthetized using 1% sodium pentobarbital and decapitated under anesthesia. Thereafter, the rat brains were promptly removed and frozen at −20°C for 15 min. Then, five coronal brain sections of 2 mm thickness were stained with TTC at 37°C for 15 min in darkness, and fixed with 4% formaldehyde for 24 h in the dark. The red color area and the pale gray color area of the slices were evaluated by ImageJ software in a double-blind manner, as described previously (14).
Microarray processing and data analysis
Total RNA was extracted from the brain tissues of rats in sham, MCAO + MCAO, and MCAO + TLB (20 mg/kg) groups using TRIzol buffer on the basis of experimental protocol, and then quantified using an Agilent 2100 (Agilent Technologies Co. Ltd., Palo Alto, CA) analysis meter. Qualified RNA transcriptome was sequenced on the platform of BGISEQ-500RS RNA-Seq supported by the Beijing Genomics Institute (Shenzhen, China). The gene expression was calculated using the FPKM value of each sample, and gene expression with an FC greater than 1.5 and p-value less than 0.05 were identified as DEGs in this study. OmicShare tools was used to plot volcano plot and advanced bubble diagram, and Venny 2.1.03 was applied to draw Venn diagram. PPI network was constructed by Cytoscape 3.6.0 software. DAVID web-based tool was used to carry out the functional enrichment analysis, the enrichment of pathways and functional processing were formed based on KEGG and annotations of GO, respectively. Furthermore, STRING web-based tool was adopted to analyze PPIs.
IHC staining
IHC was used to determine the activation of astrocyte and microglia using GFAP staining and Iba-1 staining as described previously (3). In brief, the brain sections, paraffin embedded (4 μm), were rinsed with 3% H2O2 for 10 min to block endogenous peroxide activity and incubated with 10% goat serum albumin for 30 min to block nonspecific binding. Then, the sections were incubated with the primary antibodies anti-GFAP (1:100) and anti-Iba1 (1:200) overnight at 4°C; after incubation, sections were incubated with a corresponding secondary antibody (anti-goat IgG-HRP, 1:200). Immunoreactions were visualized using 3,30-diaminobenzidine tetrahydrochloride. Counterstaining was performed using hematoxylin. Negative control sections were stained only with a secondary antibody to control for possible specific staining. For the semiquantitative analysis of the IHC results, three sections from each brain, with each section containing three microscopic fields from the ischemic boundary zone (penumbra) in the cerebral cortex, were digitized under a 40 × objective. The immunoreactivity of the target proteins was quantified based on the integrated optical density of immunostaining per field, using Image Pro Plus 6.0 software.
Primary rat astrocyte and cortical neuron culture and drug treatment
Primary rat astrocyte culture was obtained as described in the previous study (3). In brief, the astrocytes were collected and identified using the anti-GFAP antibody. The astrocytes cultured on 96-well plates or 6-well plates were washed with HBSS, the culture medium was shifted with a glucose-free Dulbecco's modified Eagle's medium (DMEM)/F12, and then the astrocytes were cultured for 1.5 h in oxygen-free N2/CO2 (95%/5%) gas. The 1.5-h time point was chosen because more than 50% cell death occurred. Then, the OGD/R group medium was replaced with standard culture medium, or treated with various concentrations of TLB (12.5, 25, 50 μM) for another 48 h.
Primary rat cortical neurons were obtained from new born Sprague-Dawley rats as mentioned in the previous study (47). In brief, the primary cortical neurons were cultured in 96-well plates or 6-well plates with poly L-lysine coating and resuspended in Neurobasal medium containing 10% FBS and 2% B27 supplement; then, the cells were cultured in a humidified incubator with 5% CO2 at 37°C for a successive 10 days. Cultures contain >95% neurons were identified by neuron-specific enolase. Thereafter, neurons were insulted by OGD for 2 h to mimic ischemic damage in vitro as mentioned in the previous report (32). Brief, neurons were incubated for 2 h in oxygen-free N2/CO2 (95%/5%) gas, and the neurons of control group were cultured in EBSS with 10 mM glucose. Then, the OGD/R group medium was replaced with standard culture medium, or treated with various concentrations of TLB (6.25, 12.5, 25, 50 μM) for another 24 h.
Determination of cell viability and neurotoxicity
The primary rat astrocytes or cortical neurons were treated as mentioned above. In brief, at the endpoint of the treatment, each well was cultured with MTT (5 mg/mL) for another 4 h. Then, the medium was discarded and dimethyl sulfoxide (150 mL) was added to dissolve the formazan, after which the absorbance of formazan formation was evaluated by a microplate reader at 490 nm wavelength. In addition, in parallel, neurotoxicity was also detected by an LDH kit as described previously (7). In brief, at the endpoint of the treatment as described above, the supernatants were obtained, centrifuged for 5 min at 400 g. The amount of LDH released from astrocytes was lysed in 1% Triton X-100 following the manufacturer's introduction. Moreover, cellular morphologic changes were observed by a phase-contrast microscope.
Determination of inflammatory factors
The astrocytes were treated as mentioned above. Briefly, the tissues or cells were collected and homogenized using 0.1 M phosphate-buffered saline (PBS; pH 7.4). Thereafter, the tissue homogenates were centrifuged at 3000 g for 20 min at 4°C. Then, levels of inflammatory factors, including IL-1β, IL-6, and TNF-α, were detected using ELISA kits according to the manufacturer's protocol.
Determination of ROS level
The astrocytes or cortical neurons were treated as mentioned above. In brief, the tissues or cells were collected and homogenized using 0.1 M PBS (pH 7.4). Thereafter, the tissue homogenates were centrifuged at 3000 g for 20 min at 4°C. Then, levels of ROS were determined using related ELISA kits according to the manufacturer's protocol. In addition, mitochondrial O2 •− generation was measured by MitoSOX red staining, a peculiar fluorescent probe for detecting mitochondrial O2 •− generation (7). In brief, astrocytes or cortical neurons were treated as mentioned above and then were washed with balanced salt solution and stained with 5 μM MitoSOX Red in the dark at 37°C for 20 min. Then, the astrocytes were observed using a fluorescence microscope (Olympus IX73; Olympus, Tokyo, Japan) with excitation/emission (510/580 nm) filters. ROS level was quantified by the Image Pro Plus software.
Measurement of SOD and GSH-Px activities
The astrocytes or cortical neurons were treated as mentioned above. Briefly, the tissues or cells were collected and homogenized using 0.1 M PBS (pH 7.4). Thereafter, the tissue homogenates were centrifuged at 3000 g for 20 min at 4°C. Then, activities of SOD and GSH-Px were determined using related ELISA kits according to the manufacturer's protocol.
Quantitative real-time PCR
Total RNA was obtained with the TRIzol reagent, which was reverse transcribed to complementary DNA (cDNA) with the PrimeScript™ RT Reagent Kit. The CFX96 real-time PCR detection system (Bio-Rad Laboratories Ltd, Hertfordshire, United Kingdom) was used to perform qRT-PCR. The specific primers and according sequences are displayed as follows: GAPDH, forward 5′-AACGACCCCTTCATTGACCT-3′ and reverse 5′-CCCCATTTGATGTTAGCGGG-3′; Nrf2, forward 5′-GTTCAGTCGGTGCTTTGACA-3′ and reverse 5′-CTCTGATGTGCGTCTCTCCA-3′; TLR4, forward 5′-CTGGGTGAGAAAGCTGGTAA-3′ and reverse 5′-AGCCTTCCTGGATGATGTTGG-3′; and Sirt3, forward 5′-TACTTCCTTCGGCTGCTTCA-3′ and reverse 5′-AAGGCGAAATCAGCCACA -3′. In the reaction, 1 μL cDNA of each sample was mixed with SYBR®GREEN PCR Master Mix according to the manufacturer's protocol. The PCR conditions are listed as follows: 30 s at 95°C, then 40 cycles at 95°C for 5 s, followed by 56°C for 30 s. Results were normalized to GAPDH mRNA level and presented as the FC (2–ΔΔCt).
Western blot analysis
In vivo, at 3 days after reperfusion, the rats were sacrificed with 1% sodium pentobarbital after treatment with TLB, and then, the ischemic penumbra was collected as described previously (24). In vitro, primary rat astrocytes or cortical neurons were washed three times with ice-cold PBS and then for following protein extraction after treatment with TLB as abovementioned. Protein extracts from nuclear and cytosolic fractions were extracted by a nuclear extraction kit according to the manufacturer's introduction, and the protein concentration was measured using bicinchoninic acid assay. The brain tissues and primary rat astrocytes or cortical neurons were homogenized with radio-immunoprecipitation assay buffer. Thereafter, the lysates were normalized to equal amounts of protein, and 10 μg protein from tissue lysates or 20 μg protein of cell lysates was divided in sodium dodecyl sulfate/polyacrylamide gel electrophoresis (10%) and transferred to a nitrocellulose membrane, blocked with 5% nonfat milk in Tris Buffered Saline Tween for 1 h at room temperature. Then, the membranes were incubated with primary antibodies, including iNOS (1:1000), TLR4 (1:1000), MyD88 (1:1000), TRAF6 (1:1000), p-NF-κBp65 (1:1000), NF-κBp65 (1:1000), Nrf2 (1:1000), Keap1 (1:1000), HO-1 (1:1000), NQO1 (1:1000) and Sirt3 (1:1000) overnight at 4°C; after which the proteins were determined with the according species-specific HRP-conjugated secondary antibodies for 2 h at room temperature. Representative bands then were visualized by ECL Western blot detection reagents, and ImageJ software was applied to quantify the band optical intensity.
Transient silencing by siRNAs
Transfection was implemented when the primary rat astrocytes achieved 70%–80% confluence in 96-well or six-well plates. The Nrf2-targeted siRNA or TLR4-targeted siRNA was diluted with Opti-MEM and balanced for 15 min at room temperature. Then astrocytes were transfected with Nrf2 siRNA, TLR4 siRNA, or scrambled siRNA by the Lipofectamine™ RNAiMAX transfection reagent following the manufacturer's protocol. The knockdown of endogenous Nrf2 or TLR4 with siRNA was verified using qRT-PCR and Western blot, respectively. After being transfected for 24 h, the transfected astrocytes were exposed to OGD/R and treated with TLB as described above. Thereafter, cell viability, LDH release, inflammatory factors, ROS, MDA, antioxidant enzymes, expression of TLR4, and levels of cytoplasmic Nrf2 and nuclear Nrf2 were also detected.
Molecular docking analysis
Molecular docking analysis between TLB and Sirt3 was performed using AutoDock 4.2 and AutoDock Tools (ADT). The human X-ray crystal structure of Sirt3 (PDB ID: 3GLS) was obtained from the Protein Data Bank (PDB) archives and used as target for molecular docking. The molecular docking results were written as a pose viewer file, and the protein/ligand complex interactions were studied using the PyMOL molecular graphics system. The interactions between TLR4 (PDB ID: 2Z63) and Nrf2 (PDB ID: 2LZL) were detected using ZDOCK and RDOCK, which was a widespread admissive method to implement detailed prediction of protein/protein docking. Subsequently, the poses of predicted protein were scored from ZDOCK, and then RDOCK was used to refine and rerank the scores. Thereafter, the binding affinity of TLR4 and Nrf2 was predicted by E_RDOCK, which was used as the default scoring function. At last, the 18 poses of docked conformations that had lower E_RDOCK were chosen for subsequent analysis.
Surface plasmon resonance
Biacore X100 instrument (GE Healthcare, Uppsala, Sweden) with Biacore X100 and sensor chip CM5 (BR-1003-99; GE Healthcare) was performed to further confirm the interaction between TLR4 and Nrf2 (36). Briefly, TLR4 (ab233665; Abcam) was dissolved in 10 mM sodium acetate (PH 5.0) at a concentration of 20 μg/mL, and absorbed as immobilization. Nrf2 (ab132356; Abcam) was diluted at 10, 20, 40, 80, and 160 μM in HBS-EP buffer. The protein interaction time was set to 120 and 300 s for dissociation. Glycine-HCl (PH 2.0, , BR-1003-55; GE Healthcare) was applied for regeneration. The Biacore evaluation 3.1 analysis software (GE Healthcare) was used to analyze the data for evaluating binding affinity between TLR4 and Nrf2. Equilibrium dissociation constants (K D ) were determined by global fitting of the kinetic data from various concentrations of Nrf2 with a 1:1 Langmuir binding model.
Measurement of Sirt3 activity
The SIRT3 activity was determined using the Sirt3 activity assay kit. Briefly, the astrocytes or neurons were treated with TLB or Tr1 as described above. Then the samples were added to double-distilled water, Sirt3 assay buffer, flour-substrate peptide, and NAD, then developed to each well of the microtiter plate, mixed well, and initiated reactions by adding 5 μL of enzyme sample or buffer of enzyme sample or recombinant Sirt3 to each well and mixing thoroughly at room temperature according to the manufacturer's introduction. Thereafter, Sirt3 activity was determined by kinetic measurements at 2-min intervals by a microtiter plate fluorometer with excitation at 340–360 nm and emission at 440–460 nm for 30 min.
Generation of Sirt3-KO rats and Sirt3-KO primary rat astrocytes or cortical neurons
Sirt3-KO rats and Sirt3-KO primary rat astrocytes or cortical neurons were generated by a CRISPR-Cas9 system. The lentivirus-based CRISPR/Cas9 KO plasmid, pHBLV-U6-gRNA-EF1-CAS9-PURO, with the Sirt3 gRNA sequences 5′-TGGTAGTCATGCGTGTTGGG-3′ (forward) and 5′-CTCAGAACCCAGAAGGTGTG-3′ (reverse) primers. Promoter U6 drived gRNA expression, and CMV drived Cas9 expression. In brief, the lateral ventricles of 6-week-old Sprague-Dawley rats were microinjected with a lentiviral-packed CRISPR-Cas9 Sirt3 (5 μL, multiplicity of infection (MOI) of 100; Hanbio Biotechnology Co., Ltd., Shanghai, China) at a constant rate (1 μL/min) using a syringe pump system at the subsequent microinjection coordinates: 1.0 mm behind the bregma and 1.0 mm lateral from the sagittal midline, at a depth of 3.5 mm from the skull surface. Three weeks after lentivirus microinjection, the rats were treated with or without TLB for 3 days after MCAO. Thereafter, the neurological function and infarct volume were determined using a five-point scale and TTC staining, respectively. Moreover, primary rat astrocytes or cortical neurons were transfected with lentiviral-packed CRISPR-Cas9 Sirt3 at an MOI of 100 for 48 h. Thereafter, the knockout of endogenous Sirt3 by CRISPR-Cas9 Sirt3 lentivirus was confirmed by RT-PCR and Western blot, respectively. Then, the transfected cells were cultured and treated with or without TLB after OGD/R insults for further analysis.
Statistical analysis
Data are expressed as mean ± SD, and “n” represents the number of independent experiments and not replicates. GraphPad Prism 5 (GraphPad Software, Inc., San Diego, CA) was used to analyze all data. Some data were normalized to control for unwanted sources of variation as follows. First, the data were normalized to bring all of the variations into proportion with one another after deleting any outliers in various groups. Then, the coefficients associated with each variable will scale properly to adjust for the disparity in the variable sizes. The cell viability of control group was deemed to be 100%, and the cell viability of TLB-treated group was displayed as a percentage of the control group. The iNOS, TLR4, MyD88, TRAF6, Keap1, HO-1, and NQO1expression was normalized to β-actin, and Sirt3 expression was normalized to GAPDH. In addition, the cytoplasmic Nrf2 level was normalized to the cytoplasmic β-actin, and nuclear Nrf2 level was normalized to nuclear PCNA. The protein expressions or levels of TLB-treated group were presented as FC of the sham or control group, the expression of which was set to 1. Two or multiple groups were compared using Student's unpaired t-test or one-way analysis of variance followed by Bonferroni post hoc test with F at p < 0.05 and no significant variance inhomogeneity. p < 0.05 was considered statistically significant.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by the Natural Science Foundation of China (Grant No. 81760727), National key R & D plan for Research on modernization of Traditional Chinese Medicine (Grant No. 2017YFC1702005), Postsubsidy project of State key R & D plan in social development field (Grant No. SQ2017YFC170204-05), the “hundred” level of high-level innovative talents in Guizhou Province (Grant No. QKHRCPT 20165684), Program for Changjiang Scholars and Innovative Research Team in University, China (Grant No. IRT_17R113), and Program for Outstanding Youth of Zunyi Medical University (Grant No. 15zy-002).
Supplementary Material
Supplementary Data
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Figure S7
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.