
Abstract
Aims:
A high-salt diet can aggravate oxidative stress, and renal fibrosis via the brain and renal renin-angiotensin system (RAS) axis in chronic kidney disease (CKD) rats. (Pro)renin receptor (PRR) plays a role in regulating RAS and oxidative stress locally. However, whether central PRR regulates salt-induced renal injury in CKD remains undefined. Here, we hypothesized that the reduction of central PRR expression could ameliorate central lesions and thereby ameliorate renal injury in high-salt-load CKD rats.
Results:
We investigated RAS, sympathetic nerve activity, oxidative stress, inflammation, and tissue injury in subfornical organs and kidneys in high-salt-load 5/6 nephrectomy CKD rats after the silencing of central PRR expression by intracerebroventricular lentivirus-RNAi. We found that the sympathetic nerve activity was reduced, and the levels of inflammation and oxidative stress were decreased in both brain and kidney. Renal injury and fibrosis were ameliorated. To explore the mechanism by which central inhibition of PRR expression ameliorates kidney damage, we blocked central MAPK/ERK1/2 and PI3K/Akt signaling pathways as well as angiotensin converting enzyme 1–angiotensin II–angiotensin type 1 receptors (ACE1-Ang II-AT1R) axis. Salt-induced overexpression of renal RAS, inflammation, oxidative stress, and fibrosis in CKD rats were prevented by central blockade of the pathways.
Innovation:
This study provides new insights into the mechanisms underlying salt-induced kidney damage. Targeting central PRR or PRR-mediated signaling pathway may be a novel strategy for the treatment of CKD.
Conclusions:
These results suggested that the silencing of central PRR expression ameliorates salt-induced renal injury in CKD through Ang II-dependent and -independent pathways.
Introduction
It has been recognized that the renin-angiotensin system (RAS) plays a pivotal role in the development of chronic kidney disease (CKD) (25, 47, 58, 60). During the past decade, (pro)renin receptor (PRR), as a newly discovered component of RAS, has been known to play important roles during many physiological and pathophysiological states in a wide variety of tissues, including the brain, kidney, and heart (39, 35, 63). Amilolide-sensitive epithelial sodium channels (ENaC) are important transporters that play a key role in maintaining Na+ homeostasis by enhancing their sodium ion absorption. During CKD, PRR is likely to affect sodium transport and further regulate blood pressure through ENaC (43, 44). Moreover, PRR can promote kidney injury and fibrosis by amplifying Wnt/β-catenin signaling (30). PRR also regulates vacuolar acidification in intercalated A cells of the renal collecting ducts and cardiomyocytes (1, 20).
Moreover, the main components of RAS can be found in the brain, and we have recently demonstrated that the blockade of RAS in the brain can downregulate the expression of local RAS in the kidney (4, 5). The “reno-cerebral RAS axis” also plays an extremely important role in the progression of CKD (4, 5).
In addition, studies have shown that a high-salt diet can aggravate kidney damage of CKD, including inflammation, oxidative stress, and fibrosis (4, 5). Mizuguchi et al. have shown that a high-sodium diet and PRR can synergistically activate mineralocorticoid receptors, thereby increasing arterial pressure (32). Therefore, it seems reasonable to assume that silencing or reduction of central PRR could inhibit local RAS in the kidney, consequently ameliorating renal injury in a high-salt-load CKD rat model.
To our best knowledge, this study is the first to investigate the effect of central PRR on the renal damage in the high-salt-load CKD. Simultaneously, we try to explore the mechanism by which central PRR ameliorates renal injury.
Since PRR is mainly expressed in neuronal cells (17), it has been shown that PRR regulates the expression of Nox2 and Nox4 by activating the MAPK/ERK1/2 and PI3K/Akt signal pathways, thereby mediating the production of reactive oxygen species (ROS) (40). Further, NADPH oxidase catalyzes the transfer of electrons from NADPH to molecular oxygen, through the Nox catalytic subunit, to produce ROS (46). Further, Huber et al. also found that increasing the level of PRR in paraventricular nucleus stimulated the production of brain ROS, thereby enhancing sympathetic activity and promoting the development of hypertension in anesthetized rats (17). These studies suggest that central PRR may affect kidney damage through oxidative stress and sympathetic activity.
Innovation
A high-salt diet can aggravate renal injury via the brain and renal renin-angiotensin system (RAS) axis in chronic kidney disease (CKD). (Pro)renin receptor (PRR) is a major component in the brain and renal RAS axis. However, whether central PRR regulates salt-induced renal injury in CKD remains undefined. This study indicates that the silencing of central PRR ameliorates salt-induced renal injury in CKD through angiotensin II-dependent and -independent pathways. To our best knowledge, this is the first study demonstrating that central PRR is implicated in the pathogenesis of salt-induced renal injury. These findings may provide new insights into the mechanisms underlying salt-induced kidney damage.
In this study, we used a 5/6 nephrectomy (5/6 Nx) rat model of CKD to investigate whether central PRR promotes salt-induced oxidative stress, inflammation, renal lesions, and fibrosis in CKD and the underlying mechanism.
We found that a high-salt diet contributes to progression of these renal lesions in CKD, and they were ameliorated by the silencing of central PRR. This provides new insights into the regulatory mechanism underlying salt-induced renal damage and potential new strategies for the prevention of CKD progress by central blockade of PRR. To explore the mechanism by which the central PRR ameliorates renal damage, we blocked the MAPK/ERK1/2 and PI3K/Akt signaling pathways as well as angiotensin converting enzyme 1–angiotensin II–angiotensin type 1 receptors (ACE1-Ang II-AT1R) axis in the brain. Thereafter, the experimental results were analyzed to explore the possible pathways for central PRR to ameliorate salt-induced renal injury in CKD.
Results
Lentivirus screening in adult neural stem cells and validation of lentivirus intervention of PRR expression in the brain
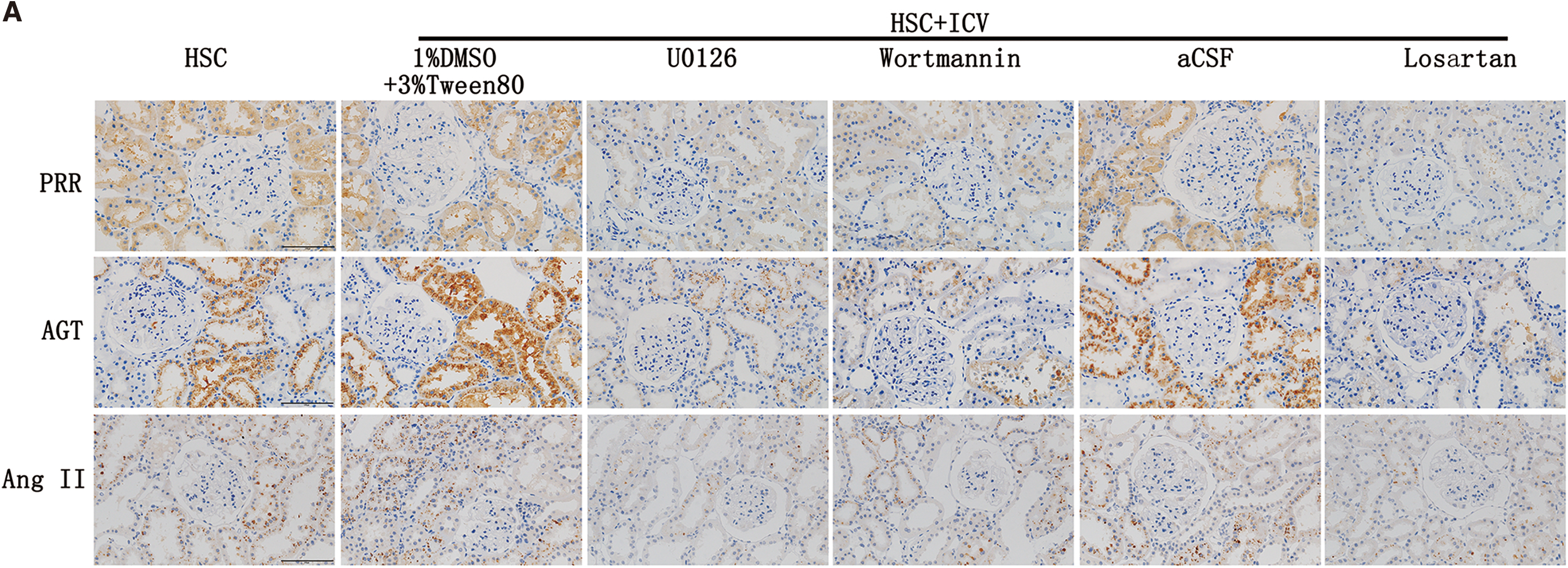
Three lentivirus RNAi were developed and used to intervent PRR expression in adult neural stem cells (ANSCs) of rat (Fig. 1A). Seventy-two hours after lentivirus transfection, the neural stem cells grew well, and more than 80% of the cells were transfected (Fig. 1A). Compared with the negative control, intervention by the lentivirus RNAi 19634-1, but not the lentivirus RNAi 19635-1 or lentivirus RNAi 19636-1, significantly decreased the messenger RNA (mRNA) level of PRR (p < 0.05) (Fig. 1C). Thus, lentivirus RNAi 19634-1 was chosen for intervention in this study.
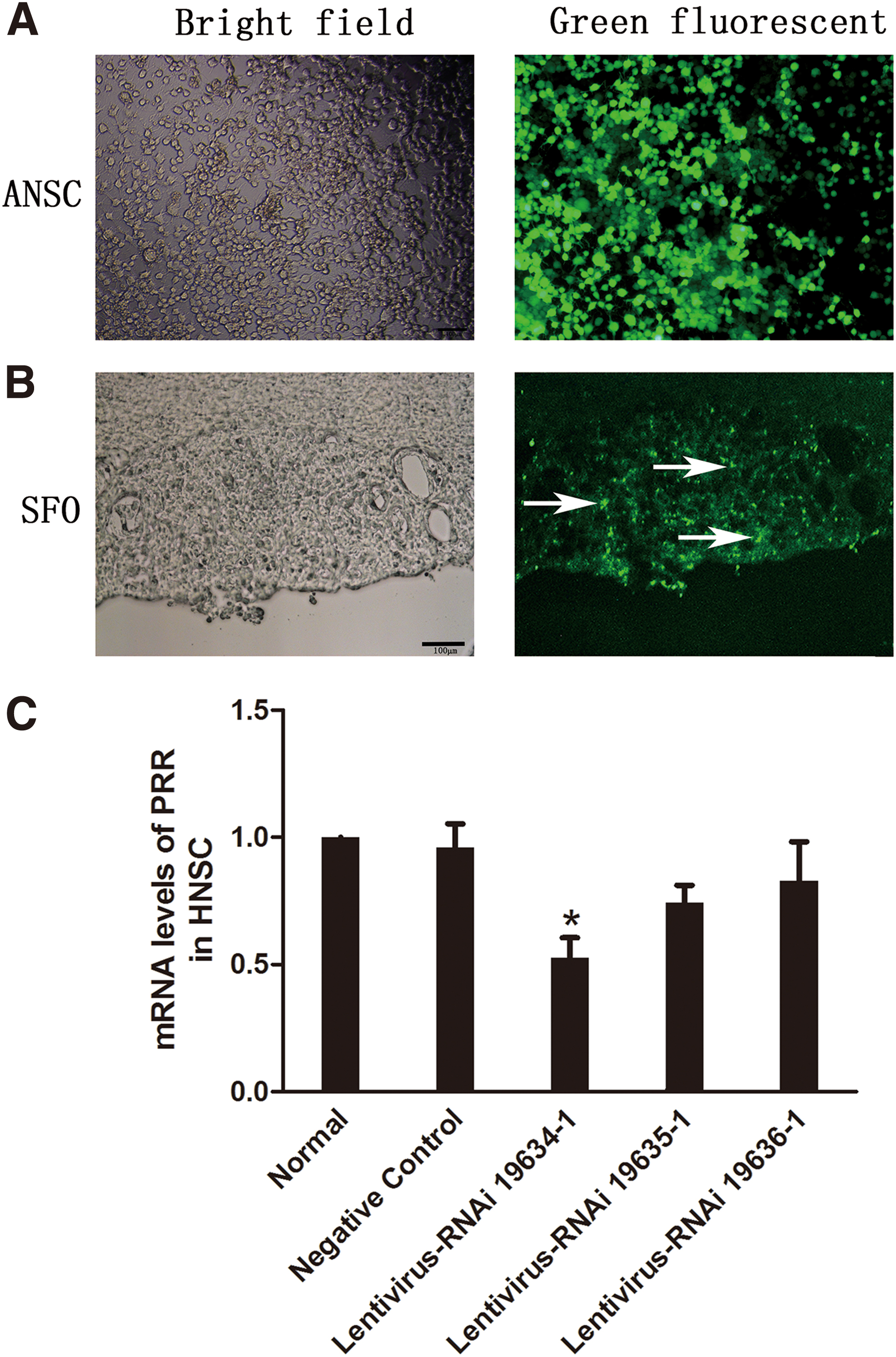
Four weeks after lateral ventricle injection of lentivirus in CKD rats, the cells in the subfornical organ (SFO) exhibited significant green fluorescence, suggesting that lentivirus transfection in SFO was successful (Fig. 1B).
Physiological parameters
We examined body weight, serum creatinine, and systolic blood pressure (SBP) in 5/6 Nx and sham rats to ensure the successful establishment of the CKD model. Based on protocol 2, high-salt load induced a higher kidney weight/body weight ratio and SBP in CKD rats relative to a normal salt diet, whereas these parameters were decreased after intracerebroventricular (ICV) Lv-RNAi compared with ICV artificial cerebrospinal fluid (aCSF), but there was no significant difference in body weight and serum creatinine (Supplementary Table S1). Based on protocol 3, as compared with the HSC, ICV U0126, ICV Wortmannin, and ICV Losartan exhibited a significant decrease in SBP and kidney weight/body weight ratio, but there was no significant difference in serum creatinine and body weight (Supplementary Table S2).
Silencing of central PRR expression ameliorates RAS, inflammation, and oxidative stress in SFO
Compared with the HSC+ICV aCSF, the expression of PRR was decreased after ICV Lv-RNAi (p < 0.05) (Fig. 2A, C, E). High salt intake in CKD rats evoked an upregulation of the expression of RAS components (PRR, angiotensinogen [AGT], Ang II, ACE1), inflammation markers (glial fibrillary acidic protein [GFAP], intercellular cell adhesion molecule-1 [ICAM1], monocyte chemotactic protein 1 [MCP1]), NADPH oxidase (Noxs) (NADPH oxidase 2 [Nox2], NADPH oxidase 4 [Nox4]), and oxidative stress parameters (3-nitrotyrosine [3NT], 4-hydroxynonenal [4HNE]), and decreased the expression of antioxidant enzymes (catalase, glutathione peroxidase 1 [GPx1], superoxide dismutase 1 [SOD1]) in SFO, as demonstrated by immunohistochemistry (Fig. 2A–D and Supplementary Fig. S1A, C) and Western blotting analysis (Fig. 2E–H).
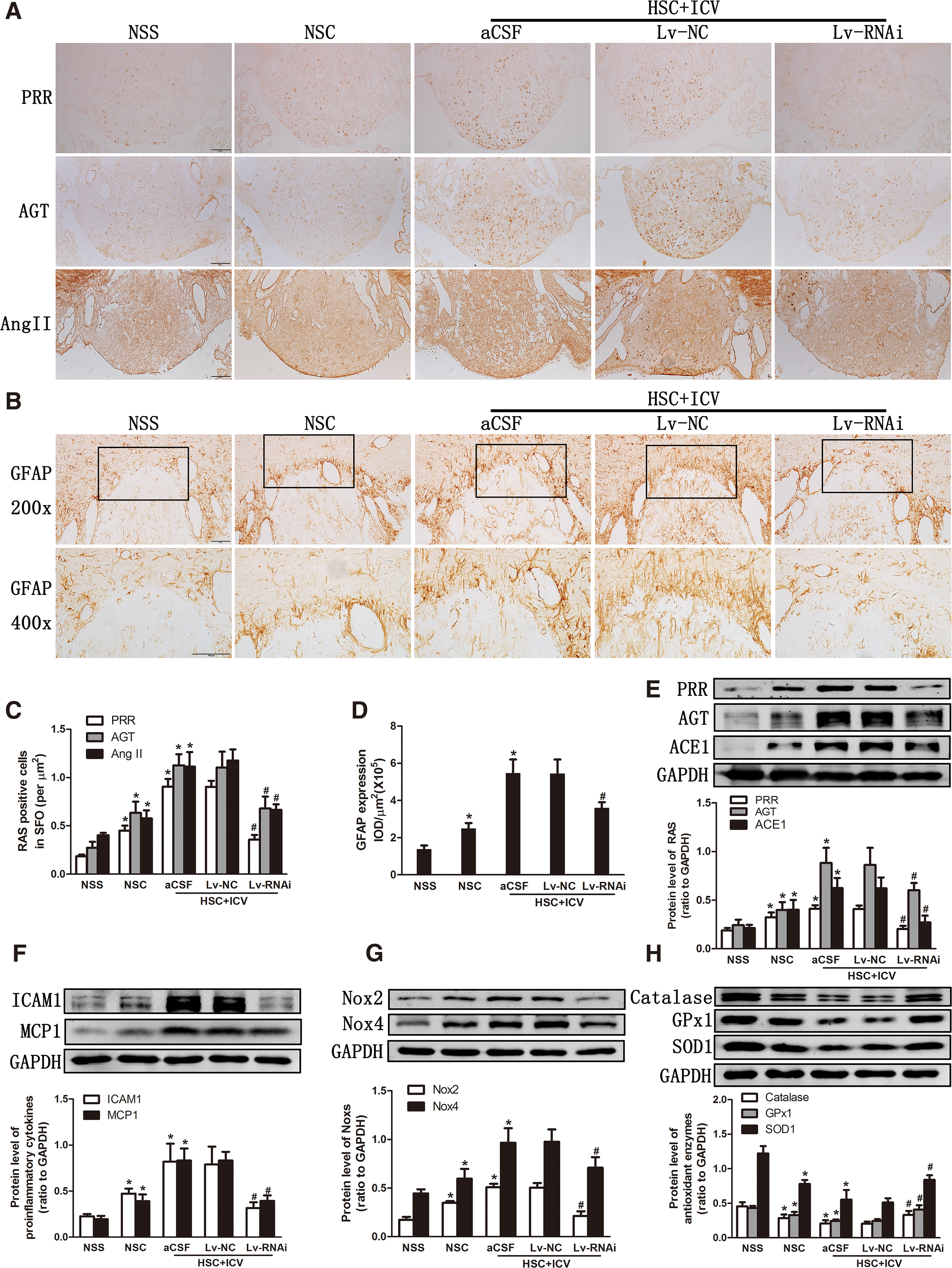
The silencing of central PRR expression could inhibit the overexpression of RAS, inflammation markers, Noxs, and oxidative stress parameters, and it increased the expression of the antioxidant enzymes; however, HSC+ICV Lv-NC did not affect these changes (Fig. 2).
Kidney injury and fibrosis were attenuated by silencing of central PRR expression
High salt intake induced a significant increase in renal tissue injury score, glomerulosclerosis index, and tubulointerstitial fibrosis in CKD rats. The silencing of central PRR expression significantly decreased high-salt-induced renal tissue injury score (Fig. 3A, B) and the glomerulosclerosis index (Fig. 3A, C). The silencing of central PRR also alleviated the tubulointerstitial fibrosis, as demonstrated by Masson staining (Fig. 3A, D), and a decrease of fibronectin (FN), Collagen I, and α-smooth muscle actin (α-SMA) expression based on Western blotting analysis (Fig. 3E) and immunohistochemistry (Fig. 3F) in CKD rats. However, ICV Lv-NC was ineffective.

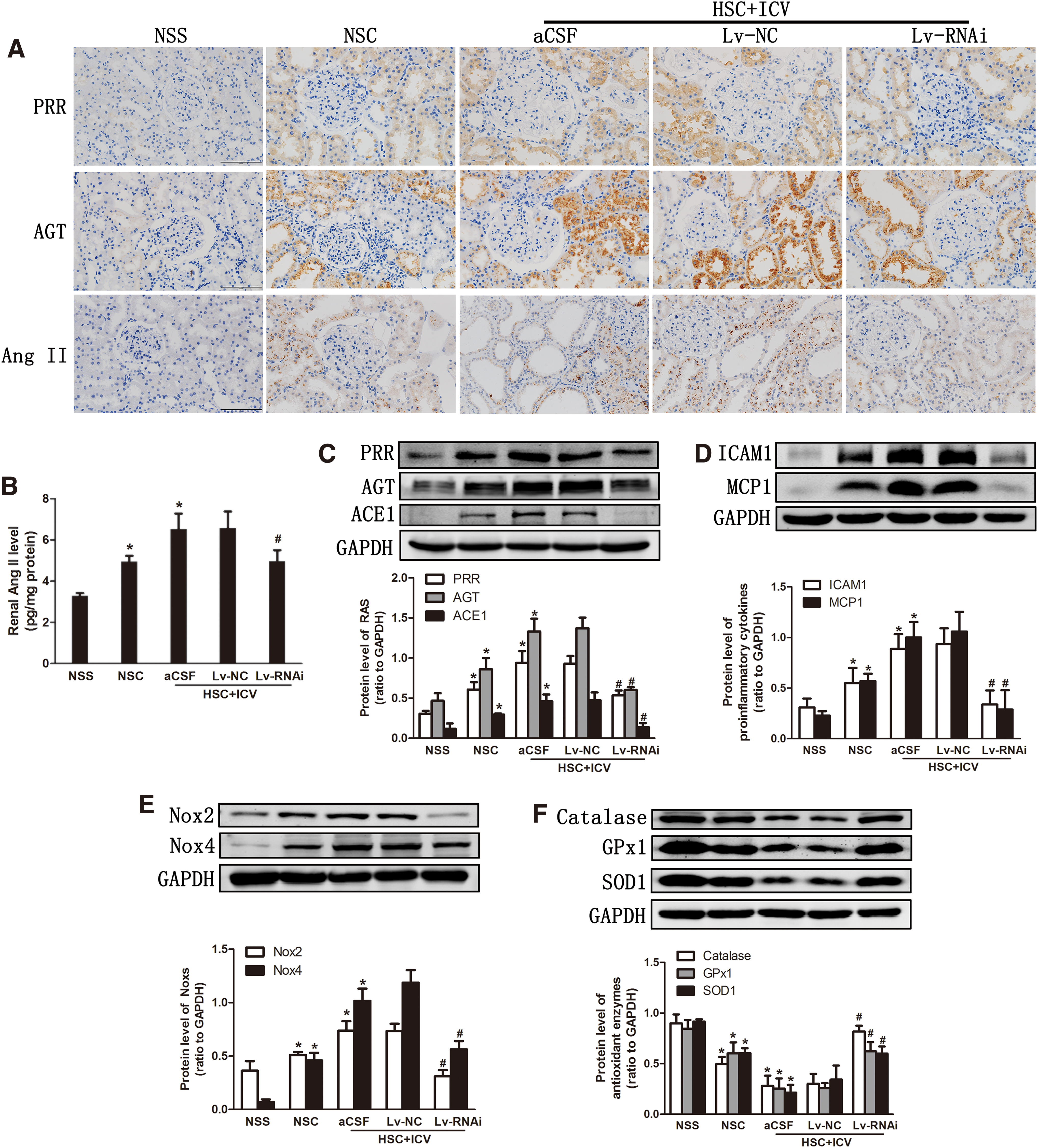
Silencing of central PRR expression ameliorates renal RAS, inflammation, and oxidative stress
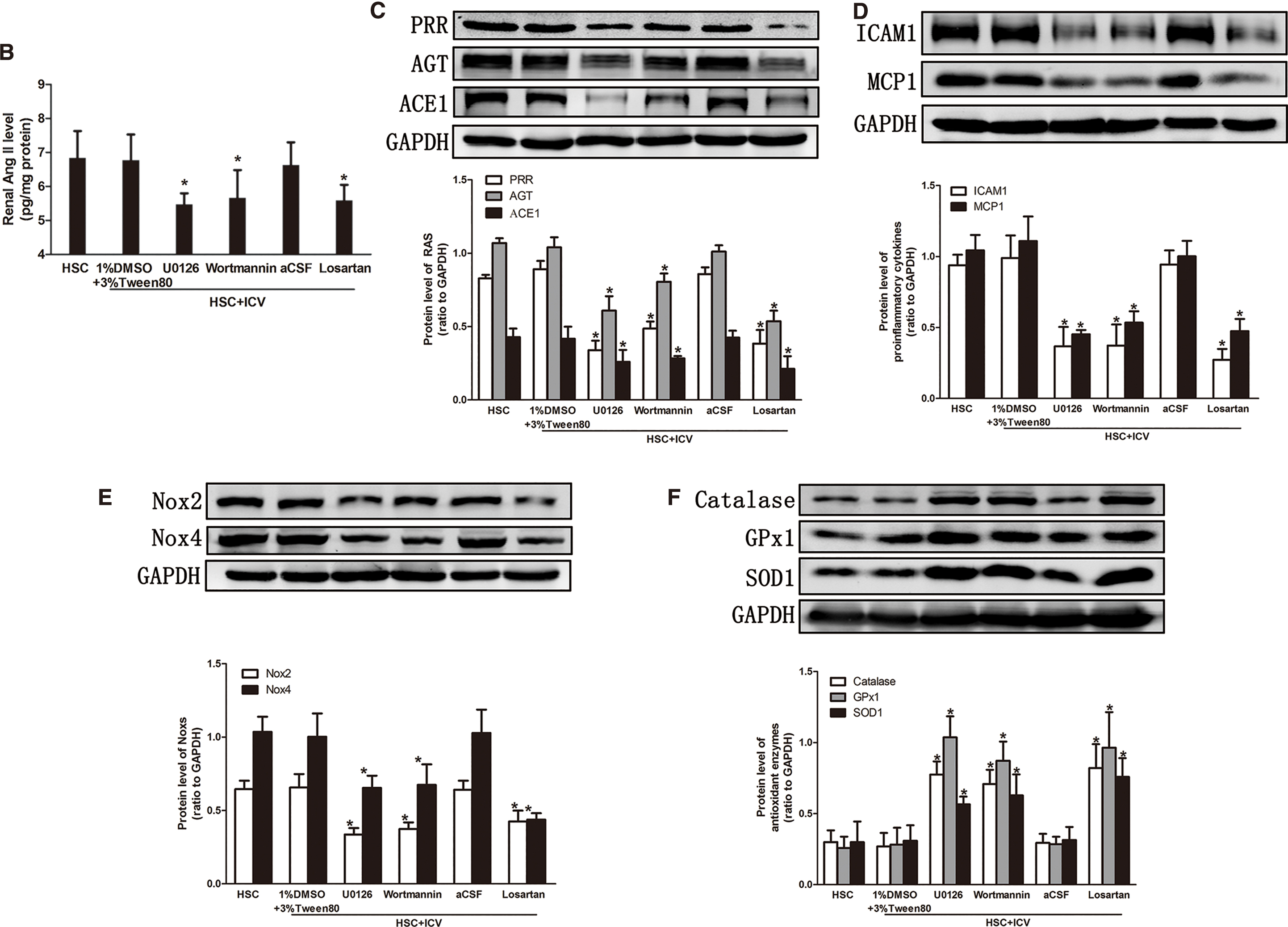
High salt intake in CKD rats upregulated the expression of RAS, inflammation, and Noxs, and it decreased antioxidant enzymes in the kidney (Fig. 4). The silencing of central PRR expression inhibited the overexpression of RAS (Fig. 4A–C), pro-inflammatory chemokines (Fig. 4D), Noxs (Fig. 4E), and oxidative stress parameters (Supplementary Fig. S1B, D) and it increased the expression of the antioxidant enzymes (Fig. 4F). However, ICV Lv-NC did not affect these changes.
Activation of sympathetic nerve system was prevented by silencing of central PRR expression
Our previous study has shown that a high-salt diet increases production of tyrosine hydroxylase (TH) in the neurons of CKD rats, which is the rate-limiting enzyme in cerebral norepinephrine synthesis, indicating increased central sympathetic drive and, consequently, increased renal efferent sympathetic activity (5). In this study, ICV Lv-RNAi inhibited the overexpression of TH in activated neurons (c-fos-positive) in the rostral ventrolateral medulla (RVLM), which is a gateway for activation of the central sympathetic nervous system (Fig. 5A, B), and attenuated both serum and renal cortex norepinephrine levels (Fig. 5C, D).
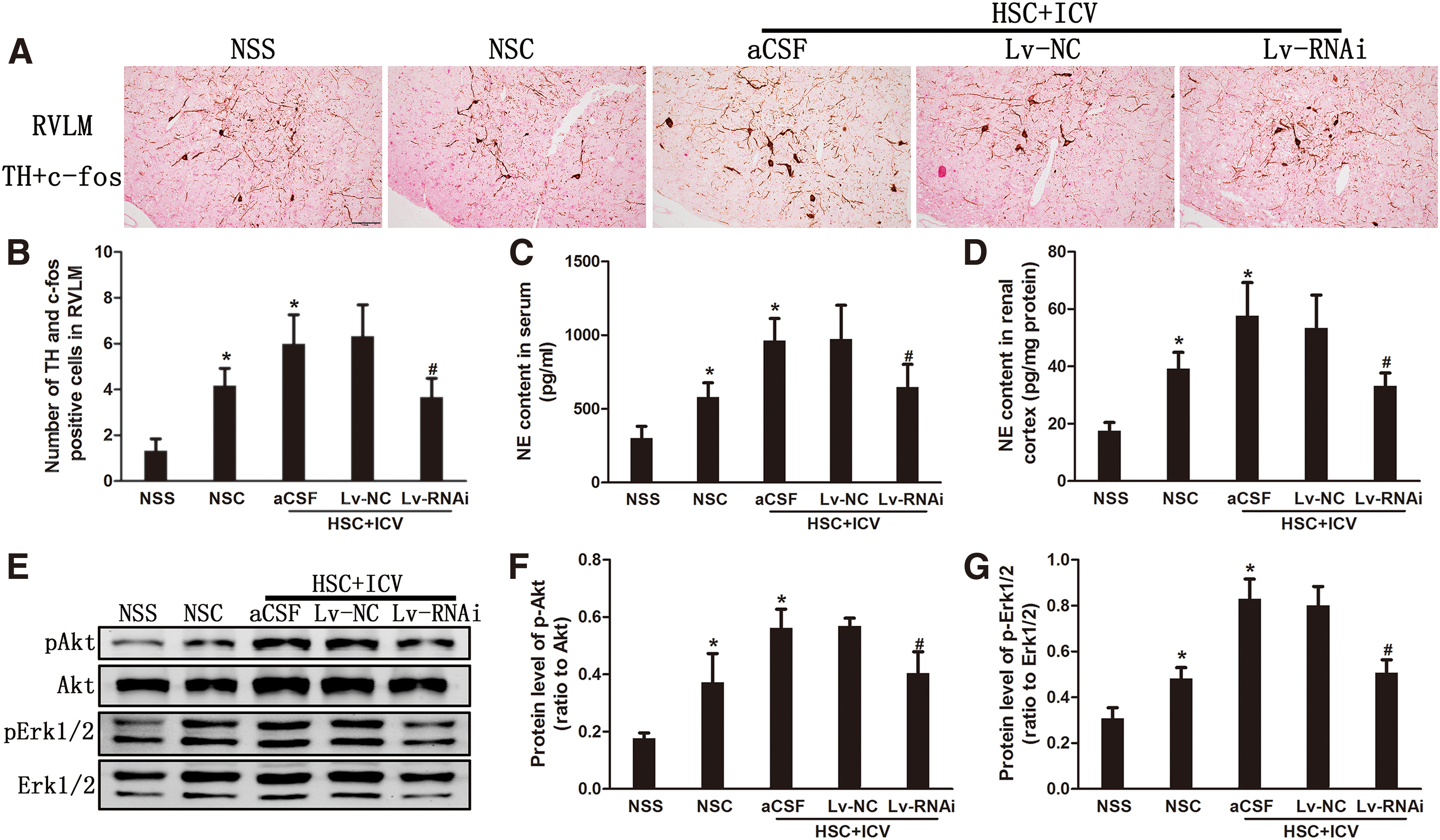
This suggested that activation of the sympathetic nerve system in high-salt-load CKD rats was prevented by the silencing of central PRR expression. Further, we also found that a high-salt diet upregulated Akt and extracellular-signal-regulated kinase 1 and 2 (Erk1/2) phosphorylation levels in brain. However, the upregulation of Akt and Erk1/2 phosphorylation levels was inhibited after ICV Lv-RNAi, but not ICV Lv-NC (Fig. 5E–G).
Effect of blockade central signaling pathways on central RAS, oxidative stress, and inflammation
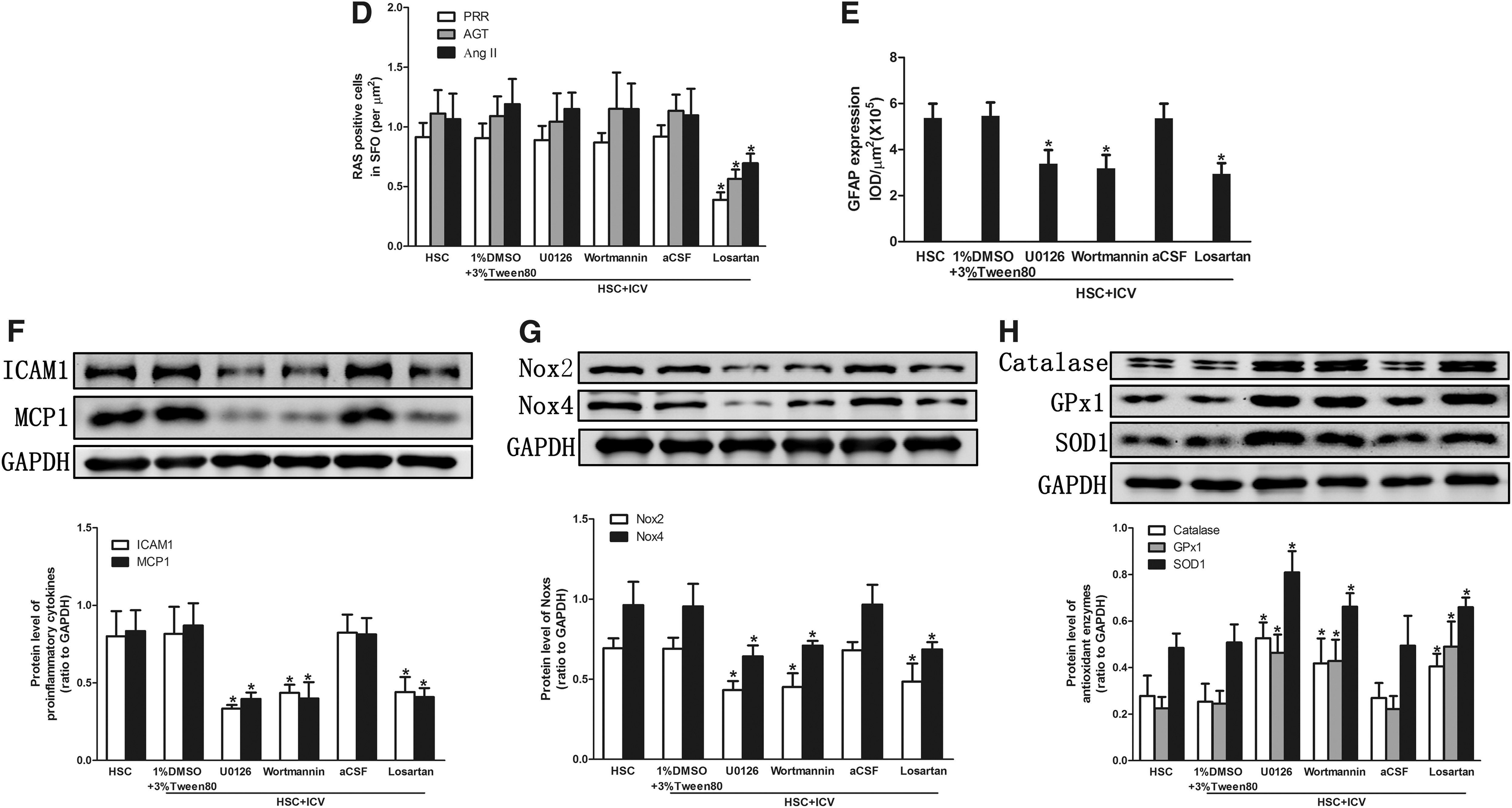
To investigate the potential pathways to mediate central oxidative stress and inflammation, we inhibited the activity of MAPK kinase by U0126, PI3K/Akt signaling by Wortmannin, and AT1R by Losartan. Inhibition of MAPK or inhibition of PI3K signaling attenuated the inflammation (Fig. 6C, E, F) and oxidative stress (Fig. 6G, H and Supplementary Fig. S2A, C), whereas these processes did not affect the activity of the RAS system (Fig. 6A, B, D). Blockading of AT1R suppressed inflammation and oxidative stress (Fig. 6).

Kidney injury and fibrosis were attenuated by blockade of central signaling pathways
Blockading the central MAPK/ERK1/2 or PI3K/Akt signaling pathway or AT1R decreased high-salt-induced renal tissue injury score (Fig. 7A, B), attenuated the glomerulosclerosis (Fig. 7A, C), and alleviated the tubulointerstitial fibrosis as demonstrated by Masson staining (Fig. 7A, D) and a decrease of FN, Collagen I, and α-SMA expression (Fig. 7E, F).


High-salt-induced overexpression of renal RAS, inflammation, oxidative stress, and fibrosis were prevented by blockade of central signaling pathways
Blockading of central MAPK/ERK1/2, PI3K/Akt signaling pathways or ACE1-Ang II-AT1R axis suppressed the overexpression of renal RAS (Fig. 8A–C), pro-inflammatory chemokines (Fig. 8D), Noxs (Fig. 8E), oxidative stress parameters (Supplementary Fig. S2B, D), and increased the expression of the antioxidant enzymes (Fig. 8F). These data demonstrated that central PRR may contribute to kidney damage through ACE1-Ang II-AT1R axis as well as MAPK/ERK1/2 and PI3K/Akt signaling pathways.
The change of sympathetic nerve activity after blockading the central signaling pathways
ICV U0126 reduced Erk1/2 phosphorylation level but not Akt phosphorylation level (Fig. 9E–G). However, both Erk1/2 and Akt phosphorylation levels decreased after ICV Wortmannin and Losartan (Fig. 9E–G).
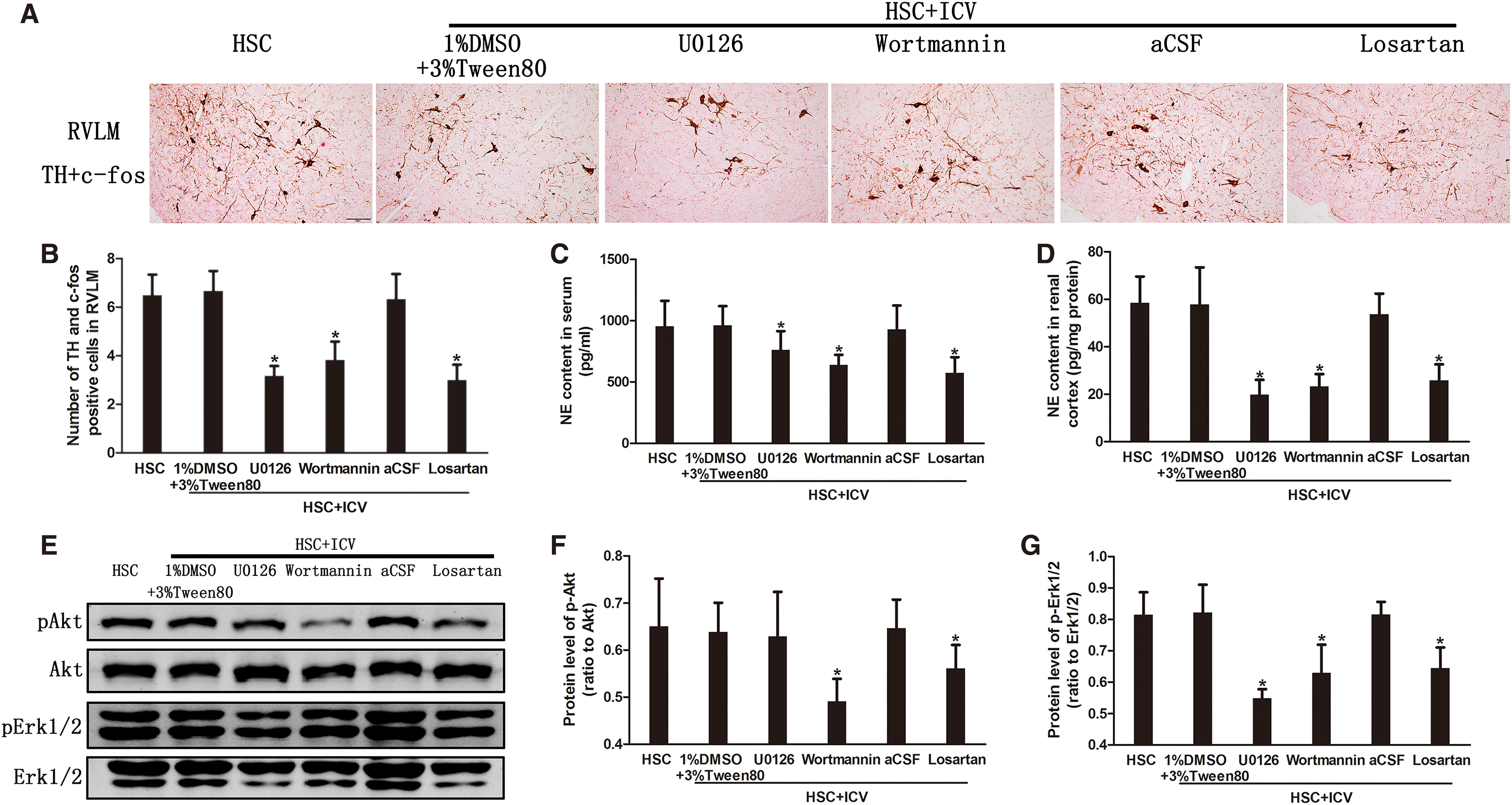
Blockading the central MAPK/ERK1/2, PI3K/Akt signaling pathway or ACE1-Ang II-AT1R axis inhibited the expression of TH in the activated neurons of RVLM (Fig. 9A, B), and attenuated both serum and renal cortex norepinephrine levels (Fig. 9C, D). This suggests that activation of the sympathetic nervous system in high-salt-load CKD rats was inhibited by central blockading of these pathways. Thus, in addition to the ACE1-Ang II-AT1R axis, MAPK/ERK1/2 and PI3K/Akt signaling pathways were also involved in the progression of renal disease through the sympathetic nervous system.
Discussion
This study has uncovered two major findings: (i) Silencing of central PRR ameliorated salt-induced kidney damage in CKD rats, and (ii) central PRR mediated the progression of CKD with a high salt load through Ang II-dependent and -independent pathways. Figure 10 showed the co-activation of oxidative stress, inflammation, and RAS by high salt intake in both damaged kidney and brain and its cross-linking in the positive feedback mode by afferent and efferent renal sympathetic nerves. The potential mechanism was proposed by which central PRR affects the peripheral pathology through Ang II-dependent and -independent pathways.

PRR can be combined with renin and prorenin, and it can play a pivotal modulatory role in local tissue RAS activity (2, 19, 36 –38). On the one hand, after binding to renin in the brain, PRR can increase renin catalytic activity to convert AGT to Angiotensin I and mediate local Ang II formation. On the other hand, after PRR binds to prorenin in the brain, it can also mediate the formation of Ang II in the brain through nonproteolytic activation (29).
What is more, when the “handle region” of prorenin is bound to PRR, it changes its conformation to become enzymatically active, and triggers intracellular signaling cascades of PRR, resulting in cell proliferation, neurogenic hypertension, oxidative stress, fibrosis, and so on (9, 30, 36, 52). Because the content of renin in the brain is extremely low and PRR, which is highly expressed in the brain (38), binds to prorenin with a higher affinity than renin (33, 41), the binding of prorenin to PRR is more likely to play a pivotal role in the brain. Further, activation of brain PRR can increase the production of Ang II, oxidative stress, and pro-inflammatory chemokines and enhance sympathetic activity (17, 40, 48, 66).
However, there is still some controversy as to whether prorenin exists in the brain. Van Thiel et al. challenged the possibility of prorenin interaction with PRR in the brain (57), although some studies showed the presence of prorenin in the brain (24, 28). Shan et al. suggested that neuronal PRR can increase Ang II production by binding renin or prorenin. Solid-phase enzyme catalysis can provide thousands of times the rate of angiotensin formation, compared with the liquid phase that occurs in plasma. Thus, even a small amount of renin or prorenin can produce physiologically relevant concentrations of Ang II locally in the brain (49).
It is possible that brain renin levels may reflect trapped plasma renin since circulating renin can reach the brain through circumventricular organs that lack the blood–brain barrier and interact with PRR in these regions, such as SFO and organum vasculosum laminae terminalis. However, renin-like activity and immunoreactivity have been confirmed in the hypothalamic nucleus (10, 13), suggesting local renin production.
In this study, we have no evidence of prorenin or renin presence in the brain, although other studies have shown that PRR exists in the brain and plays an important pathophysiological role (6, 17, 51). Here, we just propose possible mechanisms for PRR to play a role in renal disease. Whether prorenin, renin, or other ligands (such as Par3, PDHB, LRP6) interact with PRR in brain needs further study (18).
Consistent with a previous study (5, 23, 34, 59), we demonstrated that high salt intake upregulated RAS, promotes inflammation and oxidative stress in the brain and kidney. Simultaneously, a high-salt diet also facilitated kidney injury and development of fibrosis. Similar observations have been made in cardiac damage (12).
Interestingly, the renin expression of the kidney was reduced and confined to the juxtaglomerular apparatus (JGA) in a high salt state, although renal AGT, ACE1, and Ang II increased (Supplementary Fig. S3A, C). Combined with our previous research, we believe that the salt-induced changes in the intrarenal RAS were fully dissociated from JGA renin (5).
However, Matsusaka et al. have demonstrated that liver-derived AGT is the primary source of renal AGT protein and Ang II (31), but they did not verify this result under pathological conditions, such as hypertension and CKD. Studies have shown that there is an abnormal increase in renal RAS under high salt, which is consistent with our previous study (5, 54). It is also shown that the salt-induced increase in renal Ang II may largely originate from the increase of AGT and ACE in the proximal tubules in the kidneys of CKD rats (22). What is more, in addition to the classic RAS pathway, prorenin receptors and chymase also participate in the generation of Ang II in the kidney (21).
Therefore, it is believed that in CKD rats with a high salt load, there is a separation of local RAS in the kidney and systemic RAS. The reason for elevated Ang II level in the kidney may be that local RAS activation in the kidney and other factors, such as oxidative stress, under the pathological state lead to an increase in the production of Ang II in the kidney. What is more, to further testify the effect of silencing PRR, we used lentivirus to knock down central AGT and the results are shown in Supplementary Fig. S4. The results showed that in the AGT knockdown group, the expression of Ang II decreased accompanied by AGT knockdown, which suggests that the brain AGT is an important source of Ang II in the brain.
Aliskiren, a renin inhibitor, can effectively inhibit the activity of RAS from the upstream of RAS. It remains unclear as to whether Aliskiren plays a role in our model. Here, we found that central administration of Aliskiren can reduce the expression of GFAP in SFO, the number of TH+c-fos-positive cells in RVLM, and the level of NE in the kidney; ameliorate kidney damage and fibrosis; reduce the expression of AGT and MCP1; and increase the expression of SOD1 (Supplementary Fig. S5). However, compared with Aliskiren, the ICV Lv-RNAi group has a better effect on the brain and kidney. The possible reason is that Aliskiren can only inhibit the RAS axis, but it cannot inhibit the downstream ERK 1/2 phosphorylation or kinase activity (8, 45). Therefore, in this model, silencing of PRR is more effective than Aliskiren administration.
What is more, we found that the SFO and kidney damage were significantly alleviated through silencing of central PRR expression. To our best knowledge, although it has been reported that inhibition of central PRR expression can reduce blood pressure and ameliorate heart injury (49), this is the first study demonstrating that central PRR is implicated in the pathogenesis of salt-induced renal injury.
In this study, we found that after the silencing of central PRR expression, salt-load CKD rats exhibited a decreased generation of cerebral TH, in c-fos-positive neurons in RVLM, thereby indicating a decrease in central sympathetic drive. Moreover, the content of norepinephrine in serum and kidney cortex in the Lv-RNAi group significantly declined compared with ICV aCSF and Lv-NC groups. These results were consistent with the finding that central RAS, including PRR, activated the sympathetic nervous system (5, 17, 61). This is consistent with our previous report that central RAS activation is upstream of central sympathetic outflow (5).
Together, these prove that the silencing of PRR decreased renal afferent nerve activity, which, in turn, ameliorated kidney damage. One of the possible causes of decreased sympathetic activity is that the level of Ang II was decreased after silencing PRR, resulting in a decrease in subsequent central sympathetic output, which is consistent with our previous studies (4, 5). In addition, we also found that high salt activated central MAPK/ERK1/2 and PI3K/Akt signaling pathways by increasing phosphorylation levels of ERK1/2 and Akt; nevertheless, silencing PRR normalized this increased phosphorylation. Apparently, MAPK/ERK1/2 and PI3K/Akt signaling pathways are intracellular signaling pathways of PRR, which is an Ang II-independent signaling pathway (40).
Taken together, we conclude that central PRR regulates renal lesions in high salt load-CKD rats through Ang II-dependent and -independent signaling pathways.
Interestingly, renal function and fibrosis improved after silencing central PRR, but the improvement in blood pressure did not decrease significantly in value (Supplementary Table S1). Why did this happen? First of all, our previous study showed that salt-induced activation of the central RAS and SNS, and the progressive renal fibrosis are not mediated by hypertension, but they are susceptible to blockade of RAS or the SNS (5). Therefore, in this model, kidney damage, fibrosis, and other lesions are related to the activation of RAS and SNS independent of BP. Indeed, BP decreases after PRR knockdown in the cardiovascular-related nucleus. For example, knockdown of PRR in PVN can ameliorate elevated blood pressure (51).
In our study, the elevated blood pressure after silencing PRR was also ameliorated, and the results were statistically significant, although the drop in blood pressure is not as dramatic as expected. The possible reason is that the measurement time has a certain effect.
To investigate the potential pathway to regulate salt-induced renal injury by PRR, we blocked the MAPK/ERK1/2 pathway by U0126, PI3K/Akt pathways by Wortmannin, or the ACE1-Ang II-AT1R axis by Losartan in the brain of salt-loaded CKD rats. After blockading any of the pathways, pro-inflammatory chemokines and NADPH oxidase subunits were downregulated, whereas antioxidant enzymes were upregulated in the brain. Blockade of MAPK/ERK1/2 or PI3K/Akt did not significantly affect RAS components. These data demonstrated that PRR activated the Ang II-dependent and -independent pathways, leading to brain pathophysiology.
Simultaneously, ICV administration of either Wortmannin or Losartan could inhibit the activation of MAPK/ERK1/2 and PI3K/Akt, whereas ICV administration of U0126 could only suppress the activation of MAPK/ERK1/2. This suggests that PI3K/Akt is upstream of the MAPK/ERK1/2. Previous research has shown that Ang II stimulates Noxs to increase the production of ROS (11), and then ROS mediates both p38 MAPK and Akt activation (55, 56). Therefore, in this study, after administration of Losartan, it is possible to reduce the activation of MAPK/ERK1/2 and PI3K/Akt by reducing ROS production. These findings are in agreement with previous findings (7, 40, 55, 56).
Blockading all three signaling pathways can mitigate kidney damage, inflammation, oxidative stress, and fibrosis. Interestingly, blockading MAPK/ERK1/2 and PI3K/Akt had an inhibitory effect on renal RAS. However, it has no effect on renin of JGA (Supplementary Fig. S3B, D). Central sympathetic drive and the content of norepinephrine in the serum and kidney were decreased after administration of Losartan as well as U0126 and Wortmannin. This suggested that blockading MAPK/ERK1/2 and PI3K/Akt could attenuate the increase in sympathetic activity, which may be attributed to the fact that PRR activation promoted sympathetic activity by MAPK/ERK1/2 or PI3K/Akt or ACE1-Ang II-AT1R axis.
In addition to the sympathetic pathway, we cannot rule out that other pathways are involved in the effect of brain PRR on kidney injury, such as the humoral pathway. However, further studies are required to elucidate the mechanism between brain PRR and kidney injury.
Materials and Methods
Animals
The CKD model was induced in male Sprague–Dawley rats (Southern Medical University Animal Experiment Center, Guangzhou, China) at 5 weeks of age with a body weight of 150 to 180 g. The rats were maintained in a pathogen-free facility under controlled temperature (24°C ± 2°C) and humidity (55% ± 5%), with a 12-h light/dark cycle. All the surgeries were performed under anesthesia with 3% sodium pentobarbital. All animal experiments were approved by the Animal Ethics Committee of Nanfang Hospital.
Study design
Protocol 1
Lentivirus screening: The lentivirus coding for RNA interfering PRR (lentivirus RNAi) was developed in Genechem Technology Co, Ltd (Shanghai, China). ANSCs were purchased from Millipore (Temecula, CA).
Laminin was first coated overnight and then poly-ornithine, and ANSC was resuscitated for culture. Cells were digested with Accutase (Innovative Cell Technologies, San Diego, CA), and at the second passage, the cells were cultured overnight and divided into five groups: (i) Normal, (ii) Negative Control, (iii) Lentivirus-RNAi 19634-1 (GCGTCATCTCTTACCCTTT), (iv) Lentivirus-RNAi 19635-1 (CCCTTTGGAGAATGCAGTT), and (v) Lentivirus-RNAi 19636-1 (GCTCCGTAATCGCCTGTTT). The next day, the lentivirus with green fluorescence (10 μL, 5 × 108TU/mL) was added as well as polybrene to enhance the transfection efficiency. The transfection efficiency was observed under an inverted microscope 72 h later. When the transfection efficiency was higher than 80%, the cells were collected for real-time quantitative polymerase chain reaction (PCR).
Real-Time PCR detection of PRR expression was performed as previously described (64). The experiment was repeated six times, and a relative quantity value is used for statistics. We chose a group with a significant decrease in PRR expression as an intervention lentivirus.
Protocol 2
After a week of adaptive feeding, 5/6 Nx or sham operation of Sprague–Dawley rats were performed at 6 weeks of age as previously described (26).
After operation, all rats received a normal salt diet for 8 weeks, and then SBP was measured by using a tail cuff with a sphygmomanometer (BP-98A; Softron, Japan) according to the manufacturer's protocol; venous blood was collected for measuring serum creatinine. Then, all rats were randomly assigned to five groups (n = 6 per group), and they received the following treatments for 4 weeks: (i) NSS: normal salt diet (0.4% NaCl) + sham operation, (ii) NSC: normal salt diet +5/6 Nx, (iii) HSC+ICV aCSF: high-salt diet (4% NaCl) +5/6 Nx + ICV aCSF, (iv) HSC+ICV Lv-NC: high-salt diet +5/6 Nx + ICV lentivirus negative control, or (v) HSC + ICV Lv-RNAi: high-salt diet +5/6 Nx + ICV lentivirus RNAi. HSC+ICV aCSF, HSC+Lv-NC and HSC +Lv-RNAi were injected with aCSF(10 μL), lentivirus (10 μL, 5 × 108 TU/mL), and lentivirus RNAi (10 μL, 5 × 108TU/mL) in the lateral ventricle, respectively (14, 15).
The accuracy of the ICV injection was confirmed by using the tracer Evans blue. After 4 weeks of salt-diet feeding and RNA intervention, the SBP was measured. The tissue collection was performed as previously described (27, 65). The scheme of Protocol 2 is provided in Supplementary Figure S6A.
Protocol 3
As mentioned in protocol 2, the CKD rat model was prepared, SBP was measured, and venous blood was collected for measuring serum creatinine. The 5/6Nx rats were fed with a high-salt diet and randomly assigned to six groups (n = 6 in each group).
They received the following treatments for 4 weeks: (i) HSC: high-salt diet +5/6 Nx; (ii) HSC+ICV (1% DMSO +3% Tween80): DMSO and Tween80 were dissolved in aCSF to a final concentration of 1% DMSO +3% Tween 80, as a solvent control of group 3 and 4; (iii) HSC+ICV U0126: The dosage was 2.5ug per day; (iv) HSC+ICV Wortmannin: The dosage was 2.5ug per day; (v) HSC+ICV aCSF: This group was used as the solvent control of group 6; (vi) HSC+ICV Losartan: The dosage was 1 mg/kg per day. U0126, Wortmannin and Losartan were purchased from Selleck Chemicals (Houston). All groups were administered by an Alzet osmotic minipump (Durect Corp.). After 4 weeks of treatment, the blood pressure was measured. The tissue collection was performed as previously described (27, 65). The scheme of Protocol 3 is provided in Supplementary Figure S6B.
Measurement of renal function and mean arterial pressure
Serum creatinine concentration was measured as a marker of renal function by using an automatic biochemical analyzer (AU480; Beckman Coulter). The blood pressure was obtained with a tail cuff method as mentioned earlier.
Renal histologic analyses
The kidney tissues were sliced into 2 μm sections. The paraffin-embedded sections were processed for hematoxylin-eosin staining, periodic acid-Schiff staining, and Masson's trichrome staining, respectively, as previously described (3, 42, 53).
Evaluation of sympathetic activity
Concentrations of norepinephrine in serum and kidney cortex were assessed with an ELISA kit (DEE5200; Demeditec Diagnostics, Germany) according to the manufacturer's protocol.
The number of c-fos-positive and TH-expressing neurons in the RVLM was determined by immunohistochemistry (4, 62). Five-micrometer-thick brain stem sections were double stained with antibodies against TH (BM1609; Boster, Wuhan, China) and c-fos (PC05; Millipore, Temecula, CA), and the number of labeled TH and c-fos immunopositive neurons located within the defined borders of the RVLM was assessed as delineated in the atlas of Paxinos and Watson.
Western blotting analyses
Renal cortex and SFO samples were homogenized in a lysis buffer, and Western blot was performed as previously described (53). Briefly, proteins were extracted from kidney and SFO samples, and they were separated by using sodium dodecyl sulfate–polyacrylamide gel electrophoresis.
Primary antibodies used included the following: anti-PRR (HPA003156, 1:500, rabbit; Sigma-Aldrich, St. Louis), anti-AGT (IBL, 405, 1:400, rabbit; Fujioka-Shi, Japan), anti-ACE1 (ab216436, 1:500, rabbit; Abcam, Cambridge, United Kingdom), anti-Nox2 (ab129068, 1:5000, rabbit; Abcam), anti-Nox4 antibody (sc30141, 1:400, rabbit; Santa Cruz Biotechnology, TX), anti-Catalase (ab16731, 1:1000, rabbit; Abcam), anti-GPx1 (ab22604, 1:1000, rabbit; Abcam), anti-SOD1 antibody (PB0453, 1:1000, rabbit; Boster, Wuhan, China), anti-ICAM1 (PB9018, 1:1000, rabbit; Boster), anti-MCP1 (BM1255, 1:200, mouse; Boster), anti-FN (F3648, 1:20,000, rabbit; Sigma-Aldrich), anti-Collagen I (BA0325, 1:200, rabbit; Boster), anti-α-SMA (A5228, 1:2000, mouse; Sigma-Aldrich), anti- phospho-extracellular-signal-regulated kinase 1 and 2 (p-Erk 1/2, 9101, 1:1000, rabbit; Cell Signaling Technology, Boston), anti-Erk1/2 (9102, 1:1000, rabbit; Cell Signaling Technology), anti-phospho-Akt (Ser473, p-Akt, 4060, 1:500, rabbit; Cell Signaling Technology), anti-Akt (4691, 1:1000, rabbit; Cell Signaling Technology), and GAPDH(5174, 1:5000, rabbit; Cell Signaling Technology).
These primary antibodies were incubated overnight, and they were then incubated with secondary antibodies for 1 h. The protein bands were detected with an Odyssey color infrared laser scan-imaging instrument (Li-Cor, Lincoln).
The protein levels were determined as previously described (27). All protein content was normalized to GAPDH amounts. p-Erk1/2 and p-Akt were normalized to total Erk1/2 and Akt, respectively. The entire Western blots were shown in Supplementary Figure S7.
Immunohistochemistry analyses
Paraffin-embedded SFO (5 μm) and kidney (2 μm) sections were processed by using anti-PRR (1:500, rabbit; Sigma-Aldrich), anti-AGT (1:250, rabbit; IBL), anti-Ang II (T-4007, 1:200, rabbit; Peninsula Laboratories, San Carlos, CA), anti-GFAP (MAB360, 1:5000, mouse; Millipore), anti-FN (1:6000, rabbit; Sigma-Aldrich), anti-Collagen I (1:500, rabbit; Boster), anti-α-SMA (1:3000, mouse; Sigma-Aldrich), anti-Renin (14291-1-AP, 1:300, rabbit; Proteintech, Rosemont), anti-3NT (ab61392, 1:400, mouse; Abcam), and anti-4HNE (ab46545, 1:400, rabbit; Abcam) primary antibodies overnight at 4°C, and secondary antibodies for 1 h. The expression was semiquantitated as previously described (5, 42).
Real-time PCR
Total RNA from ANSC was extracted with TRIzol reagent (162901; Invitrogen, Carlsbad) according to the manufacturer's instructions. Specific primers for rat PRR, and rat GAPDH were purchased from Invitrogen (PRR, Forward: 5′-GCATTGTCCATGGGCTTCTC-3′ Reverse: 5′-TAGCCCGAGGACGATGGAAT-3′; GAPDH, Forward: 5′-TGCCAAGTATGATGACATCAAGAA-3′ Reverse: 5′-AGCCCAGGATGCCCTTTAGT-3′). The expression levels of targeted mRNAs were normalized based on the expression levels of the GAPDH, which was used as an internal control mRNA.
Radioimmunoassay
Ang II concentrations in homogenates of renal cortex were determined with radioimmunoassay kits according to the manufacturer's instructions (Beijing North Institute of Biological Technology, Beijing, China). Renal cortex was homogenized on ice with 0.045 M HCl in ethanol containing 0.9 mM p-hydroxylmercuribenzoate, 131.5 mM 1,10-phenanthroline, 0.9 mM phenylmethylsulfonyl fluoride, 1.75 mM pepstatin A, 1.1 mM ethylene diamine tetraacetic acid, and 0.0043% protease-free bovine serum albumin (16).
Statistical analyses
All data were expressed as mean ± standard deviation of at least three independent experiments. Continuous variables among groups were compared by using one-way analysis of variance, followed by the least-significant difference test. Statistical analyses were conducted with SPSS 22.0 for Windows (SPSS, Inc., Chicago). A value of p < 0.05 was considered statistically significant.
Footnotes
Author Disclosure Statement
The authors declare that there is no conflict of interests regarding the publication of this article.
Funding Information
This study was supported by grants from the National Nature and Science Grants (81270825 and 81770727), GDUPS (2017), Science and Technology Planning Project of Guangdong Province (2017A010103041), and Key Project of Guangzhou Science Technology and Innovation Commission (201804020054).
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Figure S7
Supplementary Table S1
Supplementary Table S2
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.