
Abstract
Aims:
Due to their significant biological activity, thiosemicarbazones (TSCs) are promising candidates for anticancer therapy. In part, the efficacy of TSCs is linked to their ability to chelate essential metal ions such as copper and iron. Triapine, the best-studied anticancer TSC, has been tested clinically with promising results in hematological diseases. During the past few years, a novel subclass of TSCs with improved anticancer activity was found to induce paraptosis, a recently characterized form of cell death. The aim of this study was to identify structural and chemical properties associated with anticancer activity and paraptosis induction of TSCs.
Results:
When testing a panel of structurally related TSCs, compounds with nanomolar anticancer activity and paraptosis-inducing properties showed higher copper(II) complex solution stability and a slower reduction rate, which resulted in reduced redox activity. In contrast, TSCs with lower anticancer activity induced higher levels of superoxide that rapidly stimulated superoxide dismutase expression in treated cells, effectively protecting the cells from drug-induced redox stress.
Innovation:
Consequently, we hypothesize that in the case of close Triapine derivatives, intracellular reduction leads to rapid dissociation of intracellularly formed copper complexes. In contrast, TSCs characterized by highly stable, slowly reducible copper(II) complexes are able to reach new intracellular targets such as the endoplasmic reticulum-resident protein disulfide isomerase.
Conclusion:
The additional modes of actions observed with highly active TSC derivatives are based on intracellular formation of stable copper complexes, offering a new approach to combat (drug-resistant) cancer cells.
Innovation
In conclusion, the work presented here, on the one hand, confirms that interaction with copper ions plays an important role in the anticancer activity of nanomolar-active thiosemicarbazones (TSCs). On the other hand, it also raises strong doubts on the dogma of “activation by reduction”-induced redox stress via superoxide production as the main executer of (metal-free applied) drug effectivity. In contrast, we propose that due to their high solution stability, copper(II) complexes of nanomolar-active TSCs are able to reach additional intracellular protein targets such as the endoplasmic reticulum-resident protein disulfide isomerase, resulting in paraptotic cell death induction and increased anticancer activity.
Introduction
Thiosemicarbazones (TSCs) possess significant biological activity, which resulted in their development as pharmaceuticals against several diseases, including cancer (24). In part, the efficacy of TSCs is linked to their ability to chelate essential metal ions such as copper and iron. Cancer cells, in particular, require higher amounts of these metal ions due to their increased rate of replication (3, 60). As a result, the ability of α-N-heterocyclic TSCs to form stable metal complexes is an important property for their development as anticancer agents (26).
Initially, the mechanism of action of α-N-heterocyclic TSCs was believed to primarily rely on the depletion of iron and the consequential inhibition of the iron-containing enzyme ribonucleotide reductase (RR) (57). However, the role of other metals (especially copper), metalloenzymes, and metal-interacting proteins is gaining more attention (16, 22). For example, the ability of copper(II)-TSC complexes to undergo redox cycling in the presence of reducing agents, with production of reactive oxygen species (ROS) [a process also called “activation by reduction” (22)], and the disruption of the cellular thiol redox homeostasis are increasingly discussed as relevant contributions to TSC activity (11, 23, 27, 44). Furthermore, the interaction with copper ions has been recently suggested to be involved in collateral sensitivity of P-glycoprotein (P-gp, ABCB1)-overexpressing multidrug-resistant (MDR) cancer cells to the nanomolar-active TSC di-2-pyridylketone 4,4-dimethyl-3-thiosemicarbazone (Dp44mT) (21). Interestingly, recently performed studies not only confirmed the enhanced sensitivity of certain P-gp-overexpressing cells (e.g., MES-SA/Dx5) to several metal chelators (including Dp44mT) but also suggested that the collateral sensitivity of these MDR cells may rely on other (more complex) mechanisms independent of P-gp transport function (9, 40).
With regard to the clinical situation, the best studied anticancer TSC, Triapine, has been already tested in several phase I and II trials with promising results, especially against hematological diseases (12, 25, 54, 56, 59). Currently, this drug is being investigated as a chemo- and radiosensitizer in an ongoing clinical phase III study assessing Triapine in combination with cisplatin and radiation therapy (NCT02466971) (30). In addition, two other α-N-heterocyclic TSCs, di-2-pyridylketone 4-cyclohexyl-4-methyl-3-thiosemicarbazone (DpC) and 4-(pyridine-2-yl)-N-{[(8E)-5,6,7,8-tetrahydroquinolin-8-ylidene]amino})piperazine-1-carbothioamide (Coti-2) recently entered clinical phase I trials.
Noteworthy, these two compounds as well as Dp44mT (the predecessor of DpC) and our dimethylated Triapine derivative Me2NNMe2 represent a subclass of TSCs characterized by a ∼500-fold higher anticancer activity compared to Triapine in cell culture (20, 37). Recent studies suggested that this efficiency could be based on an additional mode of action, associated with the formation of intracellular copper complexes (16, 19, 34). In particular, our group discovered that these compounds are able to induce paraptosis, a novel form of programmed cell death (13). Paraptosis is a caspase-independent cell death discernable by the appearance of cytoplasmic vesicles originating from the endoplasmic reticulum (ER) (33, 50). TSC-induced paraptosis appears to be associated with the (copper-dependent) inhibition of the ER-resident protein disulfide isomerase (PDI) (13).
In light of these recent data, the aim of this study was to elucidate the role of copper complex formation of various TSCs in anticancer activity, paraptosis induction, and collateral sensitivity. For this purpose, we investigated solution stability and redox properties of a selected panel of structurally related α-N-heterocyclic TSCs and their respective copper(II) complexes. The measured physicochemical properties and biological parameters (anticancer activity, resistance ratio of MDR cancer cells, paraptosis induction, and PDI inhibition) were then correlated with each other.
Thus, we show that highly active TSCs (with IC50 values in the nanomolar range) form copper(II) complexes characterized by high stability and slow reduction kinetics. Our data suggest that these stability and redox properties protect the copper(II) complexes of this TSC subclass from premature reduction-induced ligand liberation, allowing the interaction of the copper(II) complexes with intracellular targets such as the ER-resident PDI. In contrast, the copper complexes of TSCs such as Triapine are sensitive to rapid reduction and thus ligand liberation inside the cancer cells, which are efficiently protected from the generated superoxide by the upregulated enzyme superoxide dismutase (SOD) in cell culture as well as in a tumor model in vivo.
Results
Anticancer activity and paraptosis-inducing potential of the TSC panel
As a first step, the in vitro anticancer activity of the selected α-N-pyridyl TSCs (Table 1 with their chemical formulae) and their preformed (i.e., in situ generated) copper(II) complexes (in a 1:1 metal-to-ligand ratio) was measured in human colon adenocarcinoma SW480 and uterine sarcoma MES-SA cells as well as the MDR subline MES-SA/Dx5. SW480 cells were investigated by conventional MTT assay, whereas the fluorescently labeled MES-SA and MES-SA/Dx5 cells were tested in co-culture by using an automated system (Fig. 1A, B and Tables 1, 2 as well as Supplementary Tables S1–S4, Supplementary Fig. S1) (55).

Structure and IC50 (72 h) Values of Tested Compounds in SW480 Cells
IC50 (72 h) Values of Tested Compounds in MES-SA and MES-SA/Dx5 Cells
FTSC, 2-formylpyridine thiosemicarbazone; PTSC, pyridine-2-carbaldehyde thiosemicarbazone.
In accordance to previous reports (27 –29, 47, 52), after a 72 or 144 h incubation, the three terminally dimethylated compounds pyridine-2-carbaldehyde thiosemicarbazone (PTSC), Me2NNMe2, Dp44mT and the disubstituted derivative DpC showed distinctly higher anticancer activities than the other compounds (2-formylpyridine thiosemicarbazone [FTSC], Triapine, H2NNHMe, H2NNMe2, MeHNNMe2, Me2NNH2, Me2NNHMe). This was especially pronounced in the MES-SA/Dx5 cell model, reflecting the collateral sensitivity of the P-gp-overexpressing cells to this subtype of TSCs (Tables 1, 2 and Fig. 1B).
To gain more insight into the time dependency of these effects, the activity of the compounds was also assessed after 3 and 24 h. None of the metal-free ligands exhibited relevant anticancer activity after 3 h, resulting in IC50 values above the highest tested concentration of 25 μM (Supplementary Tables S1–S4). With the exception of PTSC, Dp44mT, and DpC, the compounds did not decrease viability after a 24 h-long incubation with the tested cancer cells. Interestingly, in contrast to the later time points (Fig. 1B), collateral sensitivity of MES-SA/Dx5 cells was not observed at 24 h (Supplementary Tables S2 and S3), indicating that the collateral sensitivity might be based on enhanced cell cycle arrest rather than enhanced apoptosis.
The anticancer activity of copper(II) complexes was slightly different (Tables 1 and 2 as well as Supplementary Tables S1–S4). Several complexes were already active after a relatively short incubation time (24 h) in SW480 cells (Supplementary Tables S1–S3), showing the same activity pattern as observed in the 72 h experiment (Table 1). However, after 72 h, IC50 values of all tested copper complexes were in a similar range as the respective metal-free ligands (Fig. 1A and Table 1), with the exception of Me2NNHMe, which was significantly more active as a copper(II) complex. Thus, the long-term anticancer activity of the copper complexes of the nanomolar-active TSCs was similar to that of the respective metal-free ligand.
To investigate the paraptosis-inducing potential of the compounds, perinuclear vesicle formation was investigated by microscopy after drug treatment (Fig. 1C). In agreement with previous results (13), Me2NNMe2, Dp44mT, and DpC induced high levels of vacuolization in SW480 cells already at a 0.1 μM drug concentrations (Fig. 1C, D). In addition, incubation with the terminally dimethylated PTSC, H2NNMe2, and MeHNNMe2 likewise induced paraptosis (however, vacuolization at 0.1 μM was observed only in the case of PTSC, whereas for the other two derivatives, the effect was visible only at 1 μM, reflecting their higher IC50 values). Similar results were obtained in MES-SA cells (Supplementary Fig. S2).
Proton dissociation processes and pK a values of the investigated TSCs
As there is a distinct structure-activity relationship in our TSCs panel, subsequently, we characterized whether these effects are reflected by differences in their chemical properties. For Triapine and some derivatives, several reports on proton dissociation processes are available (5 –7); whereas for most of the compounds in this study, this parameter has not been characterized so far (especially not in pure aqueous solution). First, pK a values were determined by UV-visible (UV-vis) spectrophotometric titrations in pure water at compound concentrations of 10–50 μM. In the case of DpC, addition of dimethyl sulfoxide (DMSO) was necessary and the pK a values of DpC in pure water were estimated by extrapolation (Table 3; Supplementary Fig. S3A).
pK a Values of the TSC Ligands Determined by Spectrophotometric Titrations and Overall Stability Constants for the Copper(II) TSC Complexes (logβ) a Calculated from the Conditional Stability Constants (logβ’5.9 of [CuL]+) and pK a Values of the Copper(II) Complexes
pCu = −log [Cu(II)] values calculated at pH 7.4 cCu(II) = cTSC = 1 μM and fraction of [CuL(OH)].
pK a taken from Ref. (20).
Estimated values from the pK a values of DpC ligand measured in 5% and 30% (w/w) DMSO/H2O mixtures.
Data taken from Ref. (5).
TSC, thiosemicarbazone.
For all compounds, the measured UV-vis spectra revealed characteristic changes on increasing pH, as depicted exemplarily for Me2NNMe2 in Figure 2A. The pH dependence of the absorbance spectra always showed two well-separated deprotonation steps: one at pH 2.5-6.0 and one at 8.0-11.5. Therefore, two pK a values could be determined (Table 3). The λ max and ɛ values of the ligand species in the different protonation states are collected in Supplementary Table S5. The calculated individual spectra (e.g., for Me2NNMe2 in Supplementary Fig. S3B) represent significant differences between the molar absorptivities of the various ligand species (Supplementary Table S5).
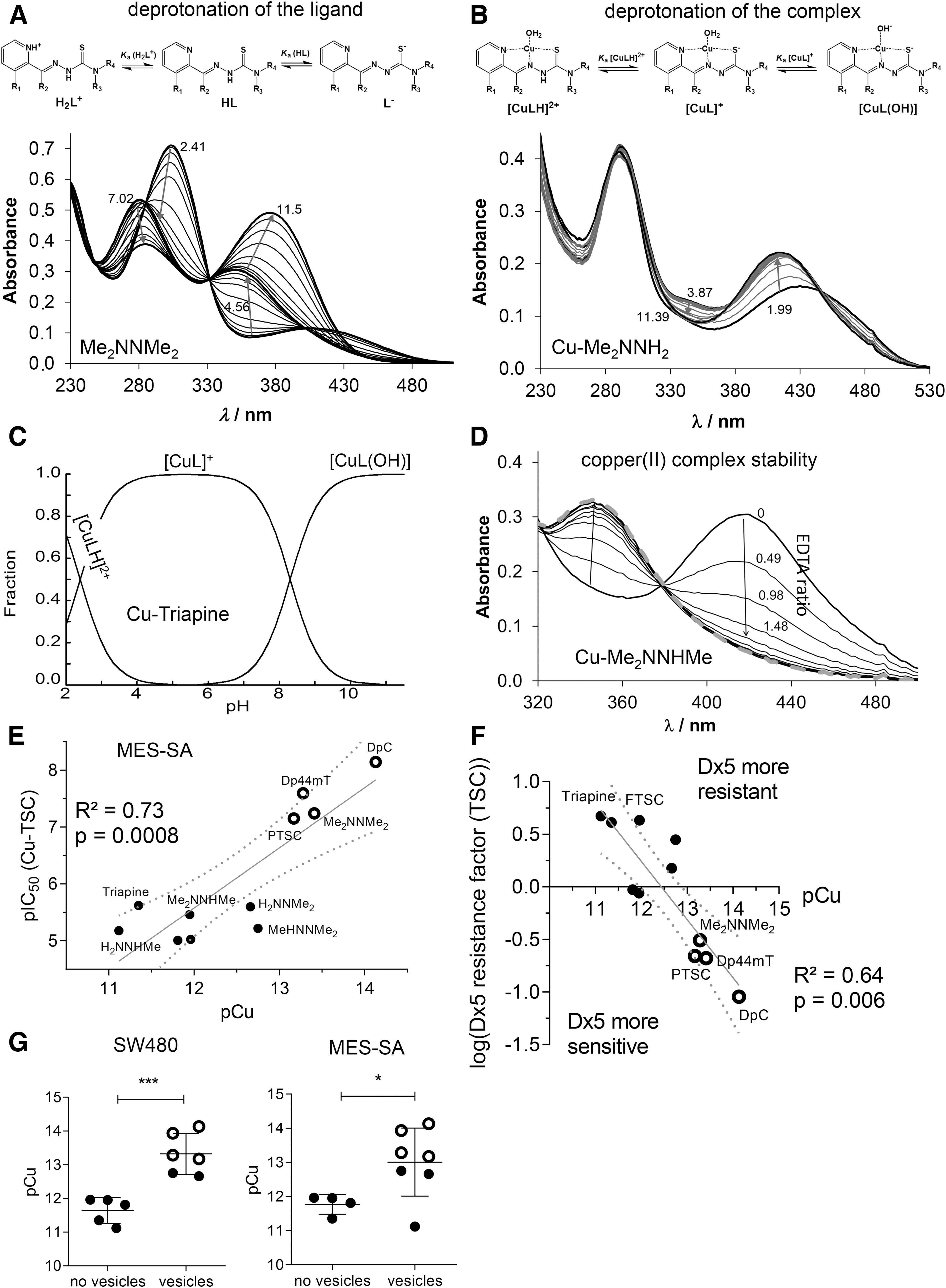
Notably, the pK a of the second pyridyl moiety in Dp44mT and DpC could not be determined under the applied conditions due to its fairly acidic nature. Analysis of the pK a values revealed that N-terminal dimethylation results in slightly higher pK a (H2L+) and lower pK a (HL) (Triapine vs. H2NNMe2 or FTSC vs. PTSC etc.), whereas dimethylation of the pyridine amine slightly increases both pK a values (Triapine vs. Me2NNH2). At the same time, monomethylations have only a minor influence. The dipyridyl derivatives Dp44mT and DpC as well as FTSC and PTSC possess decreased pK a (H2L+) by almost one order of magnitude, but no clear trend was evident for pK a (HL).
In general, not only the electron-donating/withdrawing properties of the substituents should be considered to explain differences in pK a values, but also the ability of the derivatives to be stabilized by mesomeric/resonance effects (e.g., the thione/thiol equilibrium). As apparent from the determined pK a values, all TSCs investigated in this study are charge neutral (in HL form) at physiological pH.
High copper(II) complex solution stability is an important parameter for anticancer activity
Having characterized the (de)protonation behavior of our TSCs, we evaluated the stability of the respective copper complexes in aqueous solution. Based on our previous findings (5, 6), we assumed that some of the studied α-N-pyridyl TSCs (Triapine, FTSC, PTSC, H2NNMe2) form very stable [CuL]+ complexes in a wide pH range. At lower pH, the balance shifts to [CuLH]2+ complexes, containing the protonated ligand. In contrast, a mixed hydroxido complex [CuL(OH)] is prevalent in the basic pH range, whereas at ligand excess further species (e.g., [CuL2], [Cu2L3]+) can be found (6).
Equimolar aqueous solutions of the metal ion and the respective ligands (10 or 25 μM concentration) were titrated, and the deprotonation processes of the complexes were followed spectrophotometrically (Fig. 2B for Cu-Me2NNH2). Deconvolution of the spectra resulted in the pK a values of the copper(II) complexes and their individual absorbance spectra (Supplementary Table S6 and Supplementary Fig. S3C). All pK a values were in the range of 2.1–2.6 (for [CuLH]2+) and 8.1–8.8 (for [CuL]+) and were, thus, well comparable to the reported data of FTSC (with 1% DMSO) (1). In general, the pK a value of [CuLH]2+ was by 7.2–8.6 orders of magnitude lower compared with that of the metal-free HL form, revealing that displacement of the dissociable proton in the complex is mediated by the metal ion coordination. As the formation of the complex [CuL]+ is predominant in a wide pH range (4.0-6.5) (Fig. 2C for Cu-Triapine) and assumed to be quantitative due to the high solution stability, apparent (conditional) formation constants (β′) for this type of complexes were subsequently determined by competition experiments with ethylenediaminetetraacetic acid (EDTA) (24) (Fig. 2D for 1:1 Cu-Me2NNHMe and Supplementary Fig. S3D). Since the displacement was found to be relatively slow, 1–2 h equilibration time was applied for this reaction (the reverse experiment using Cu-EDTA + FTSC resulted in the same endpoint; see Supplementary Fig. S4).
Increasing amounts of EDTA resulted in decreasing absorbance at the wavelength characteristic for the S→Cu charge transfer band (e.g., 422 nm in case of Me2NNHMe in Fig. 2D). Taking into account the conditional stability constants determined (Supplementary Table S6) as well as the proton dissociation constants of the ligands (Table 3), the overall stability constants (β) of the complexes [CuL]+ were calculated (Table 3). Furthermore, also the β values of the other two types of complexes [CuLH]2+ and [CuL(OH)] were computed (Table 3) by using the pK a of [CuLH]2+ and [CuL]+ (Supplementary Table S6). Based on these data, it can be concluded that at physiological pH, [CuL]+ is the most predominant species, accompanied by a smaller fraction (3.9–16.3%) of [CuL(OH)] (Table 3 and Fig. 2C).
Since a direct comparison of the logβ [CuL]+ constants is not adequate due to the different basicity of the ligands, pCu (−log [Cu(II)]) values were calculated to compare the copper(II)-binding ability of the studied TSCs at pH 7.4 (a higher pCu value indicates a stronger metal ion-binding ability of the ligand) (Table 3). The calculated pCu values revealed that dimethylation at both the terminal and the pyridine amino group increased complex stability. On the contrary, the effect of monomethylation on the copper(II)-binding ability was minor at both positions. Undoubtedly, the four compounds with activity in the nanomolar range form copper(II) complexes with the highest stability, followed by the two trimethylated compounds MeHNNMe2 and Me2NNHMe and all other derivatives (Table 3).
To evaluate the effect of copper complex stability on the biological activity, the pCu and 72 h pIC50 values of either the ligands or their in situ generated copper complexes were correlated. These analyses revealed that the ligands with high pIC50 values possess a higher copper(II)-binding ability (indicated by higher pCu values) and the correlation is even more pronounced for the pIC50 values of the copper(II) complexes (Fig. 2E and Supplementary Fig. S5). Accordingly, the higher copper complex stability was also associated with higher activity in the MDR cells after long-term incubations (Fig. 2F) and paraptosis induction (Fig. 2G), indicating an important role of the copper(II) complex in these TSC-induced effects.
Reduced reduction rate of the copper(II) complex has a strong impact on TSC activity
In addition to the stability of the formed copper(II) complexes, also their redox properties may have an impact on their biological activities. To investigate whether there are differences in the reduction rates of our TSC panel, the redox reactions of the in situ generated copper(II) complexes with two physiological reducing agents, namely ascorbic acid (AA) and L-glutathione (GSH), were studied. Reduction of the copper(II) complexes was followed spectrophotometrically in aqueous solution at pH 7.4 under anaerobic conditions.
In good agreement with the literature (11), no time-dependent spectral changes were observed in the case of AA, which suggests that these metal complexes cannot be reduced by AA under the applied conditions (Supplementary Fig. S6). This could be explained by the relatively weak reducing power of ascorbate (formal potential at pH 7.0: +0.06 V for dehydro-L-ascorbate/AA) (15). In contrast, the stronger reducing agent GSH (formal potential at pH 7.4: −0.26 V for GSSG/GSH) (45) reduced all studied copper(II) TSC complexes, although with different rates (Fig. 3A, B).
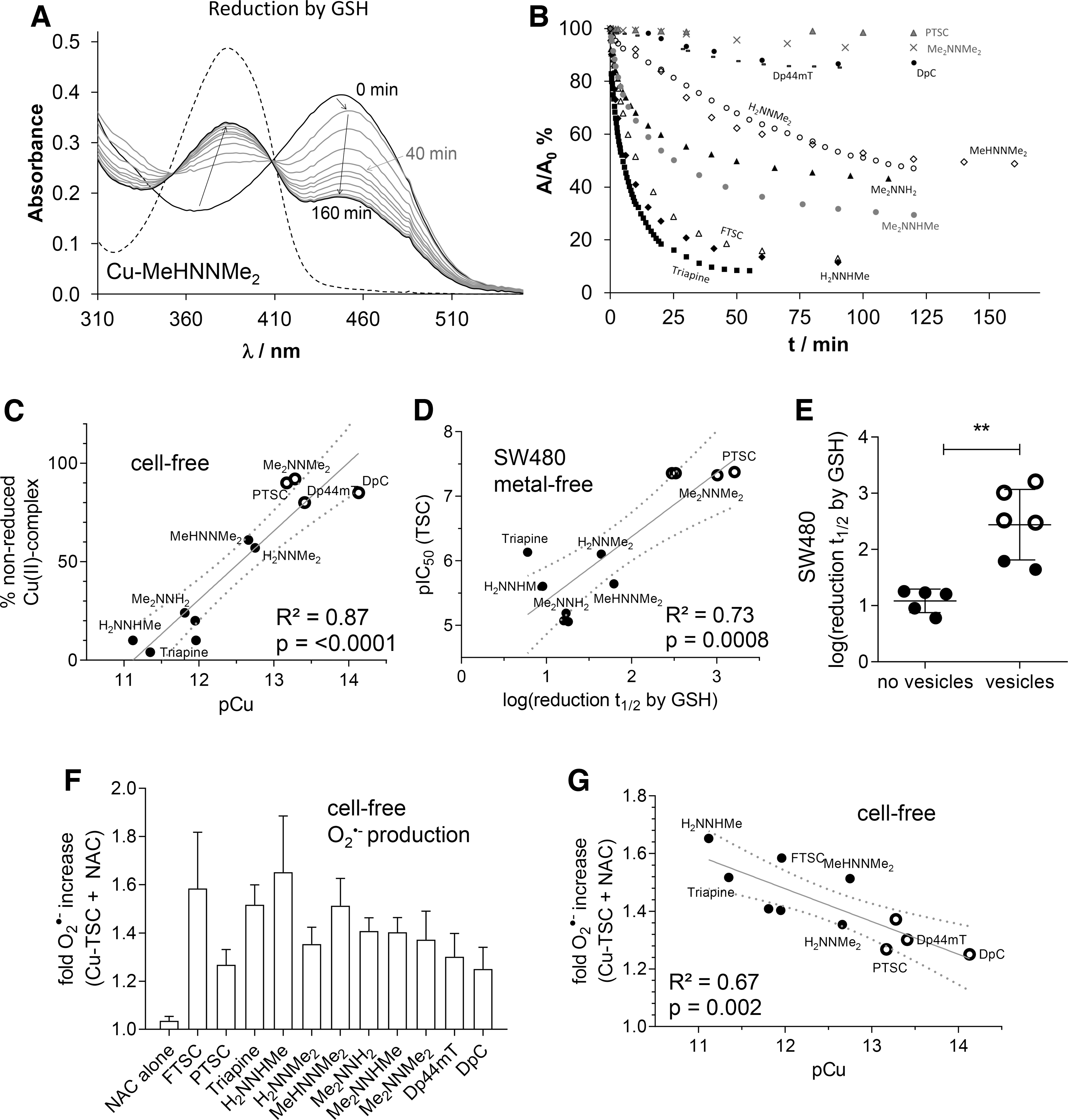
The first recorded spectrum after mixing the reactants showed a small shift of the λ max value (e.g., 448 → 452 nm, as shown for Cu-MeHNNMe2 in Fig. 3A), most probably due to the formation of a mixed ligand complex with GSH, as is reported for various TSC complexes (22, 44). This shift was followed by a significant decrease of the absorbance at this λ max, whereas the absorbance value at the λ max of the free ligand (∼382 nm) increased, probably as a result of the decomposition of the generated unstable copper(I) complex. Noteworthy, oxygenizing (bubbling O2 into) the solution regenerated the original copper(II) complexes, confirming the reversibility of the redox process (data not shown). The other studied TSCs behaved similarly. However, significant differences were observed regarding the reduction rates of the respective complexes (Fig. 3B).
To obtain comparable data, the recorded absorbance/time curves were further analyzed at the λ max of the complexes. The calculated k obs, half-lives (t 1/2) and percentage of nonreduced complex after 1 h in the presence of 50 equiv. (1.25 mM) of GSH are collected in Table 4. For selected complexes (Triapine, PTSC), kinetic runs were additionally performed at other equivalents of GSH, and k obs were found to be very sensitive to the concentration of the reducing agent, namely slower reaction rates with decreasing excess of GSH were observed (data not shown).
Observed Rate Constants (k obs) and Half-Lives (t 1/ 2) in the Copper(II)–TSC–GSH (1:1:50) Systems from the Spectral Changes at the λ max of the Copper(II) Complex and Absorbance Values Measured at 1 h Compared with That of the Ligand
k obs = 1.03 × 10-3 min-1, t 1/2 = 675 min in the presence of 100 eq. GSH (20).
k obs = 6.30 × 10-2 min-1, t 1/2 = 11 min in the presence of 20 eq. GSH; k obs = 5.90 × 10-2 min-1, t 1/2 = 12 min in the presence of 10 eq. GSH.
In accordance to the reports from Santoro et al. (44) under the applied conditions, the reduction of the copper(II) complexes was incomplete, especially with nanomolar-active compounds (see plateau in Fig. 3B and % nonreduced copper(II) complex after 1 h in Table 4). Interestingly, the remaining fraction of nonreduced copper(II) complex after 1 h (Table 4) showed a strong correlation with the pCu value and, therefore, with the solution stability of the copper(II) complexes (Fig. 3C). These data demonstrate that the copper(II) complexes of α-N-pyridyl TSC ligands bearing higher solution stability can only be reduced by GSH in a slower and much less effective way.
When these reduction rates were subsequently correlated with the anticancer activity (after 72 h), it became apparent that a slower reduction rate is associated with higher anticancer activity (Fig. 3D and Supplementary Fig. S7A) and paraptosis-inducing potential (Fig. 3E and Supplementary Fig. S7B) of the metal-free ligands as well as the copper complexes.
Overall, these results are surprising, as so far it has been assumed that the anticancer activity of copper TSC complexes is mainly based on intracellular redox activity, either by the copper(II) TSC itself or by the copper release from the complex in the cell (22, 44).
Copper(II) complex stability influences redox behavior of TSCs under cell-free conditions
It is widely accepted in the literature (22, 27, 38, 44) that the reduction of copper(II) complexes to copper(I) and their subsequent re-oxidation under aerobic conditions results in the generation of superoxide radicals and thus redox stress in treated cells. To compare the obtained reduction rates of the copper(II) complexes by GSH with their superoxide production potential, formation of cell-free superoxide was measured spectrophotometrically by using the nitroblue tetrazolium (NBT) assay.
In line with the activation by reduction theory for this compound class (16), without the addition of a reducing agent, none of the tested TSCs (neither as metal-free ligand nor as copper(II) complex) induced any positive signal (data not shown). In contrast, as expected when the GSH precursor and reducing agent N-acetyl cysteine (NAC) was added to the copper(II) complexes, superoxide generation (up to 1.6-fold compared with the control) was detected (Fig. 3F). In agreement with the results depicted earlier, distinct differences between the individual ligands were observed. Thus, indeed copper(II) complexes with a higher stability and slower reduction rate also produced less cell-free superoxide (Fig. 3G and Supplementary Fig. S8). In contrast, copper ions, which under certain conditions have been reported to induce ROS on their own (10, 41), did not result in positive measurable NBT signals (at 5 μM) neither alone nor in combination with NAC (data not shown).
Consequently, these data indicate that despite the widely accepted hypothesis that the anticancer activity of copper(II) complexes is based on (intracellular) reduction-induced ROS (superoxide) formation, in our hands especially the TSC complexes showing reduced redox activity are characterized by distinctly enhanced cytotoxicity.
Stimulation of antioxidant enzymes and stress-response genes in TSC-treated cells
To investigate whether we can also detect superoxide production on drug treatment in living tumor cells, the dihydroethidium (DHE) assay was used at conditions similar to the cell-free assay. Comparable to the results of the cell-free experiments, neither the ligands nor copper ions (5 μM) alone did significantly increase the fluorescence signals (data not shown). Unexpectedly, for the copper(II) complexes a different picture emerged compared with the cell-free experiments, as no significant increase in superoxide levels could be detected neither in the presence nor in the absence of NAC (Fig. 4A). Hypothesizing that cancer cells efficiently protect themselves from TSC-induced redox stress by upregulation of antioxidant response signaling, we used our previously published (13) whole-genome gene expression data of Triapine- and Me2NNMe2-treated SW480 cells. In these experiments, cells were treated with Triapine (1 μM), Me2NNMe2 (0.1 and 1 μM), or solvent for 15 h and mRNA levels were analyzed for drug-induced changes in gene transcription. When looking for altered gene sets associated with response to oxidative stress (e.g., “regulation of response to oxidative stress”) by gene set enrichment analysis (GSEA), no significant gene set enrichment was detected (lowest false discovery rate [FDR] values: 0.10 for Me2NNMe2 and 0.35 for Triapine [Supplementary Fig. S9]). However, when looking at individual genes, we found upregulation of the superoxide scavenger enzymes SOD2 and SOD3, but not SOD1 on Triapine treatment (Fig. 4B). In contrast, in Me2NNMe2-treated cells, no comparable upregulation was observed for SOD2 (at IC50 concentrations) and SOD3 (Fig. 4B).
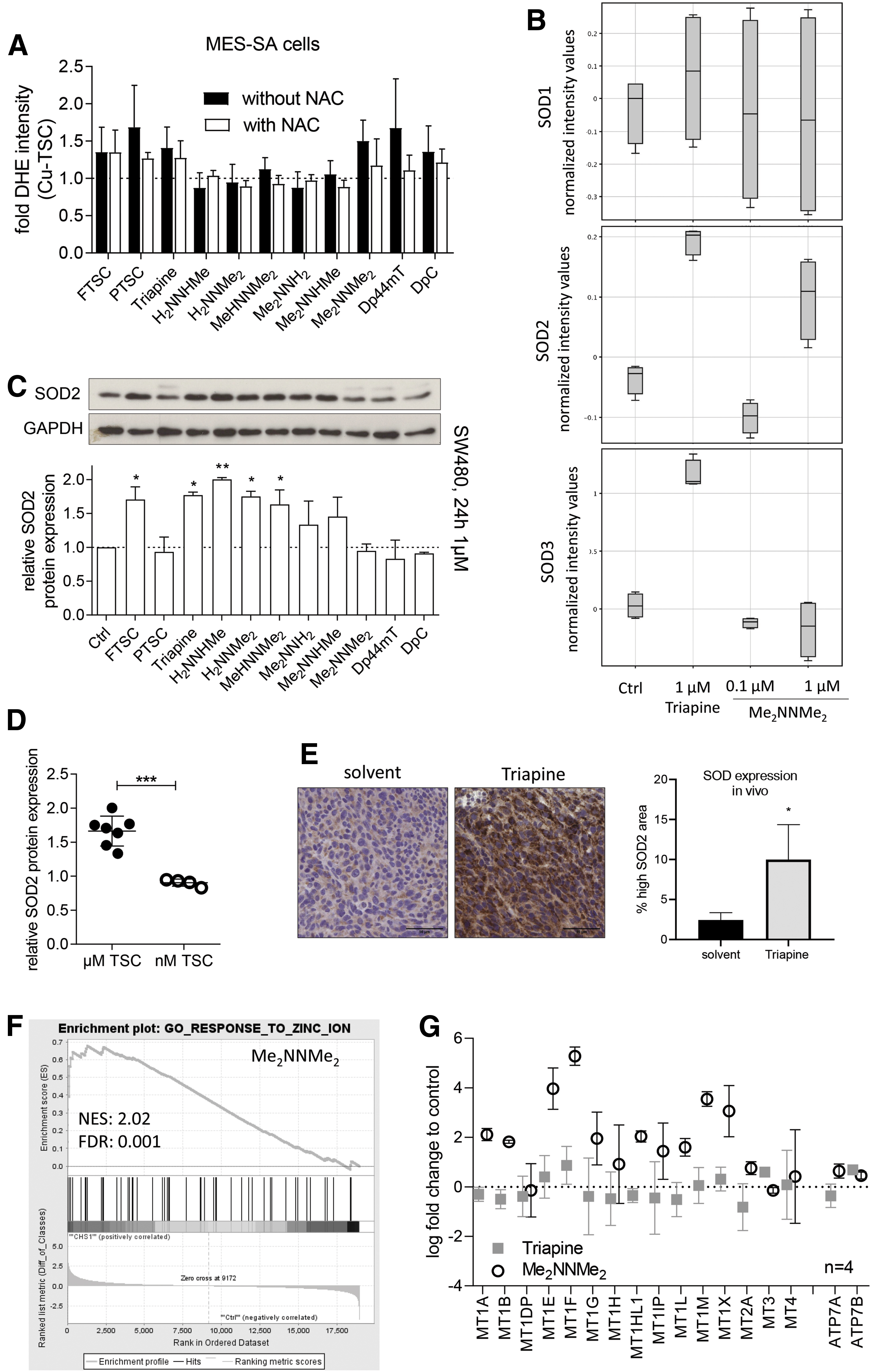
For confirmation at the protein level, Western blot analysis of SOD2 with lysates from cells treated with our TSC panel was performed. Indeed, SOD2 was upregulated after treatment with Triapine and all other micromolar-active TSCs, whereas none of the compounds with activity in the nanomolar concentration range induced SOD2 expression (Fig. 4C, D).
To test whether SOD2 was also upregulated by Triapine treatment in tumor cells in vivo, CT-26 colon carcinoma-bearing mice were treated orally with either solvent or 10 mg/kg Triapine. As shown in Supplementary Figure S10, Triapine had significant anticancer activity and was well tolerated in this setting. On the last day of treatment (day 15), the tumors were collected and immunohistochemically stained for SOD2. In accordance with the array data as well as the Western blot analysis, also tumors from Triapine-treated animals showed strong stimulation of SOD2 expression compared with the solvent control (Fig. 4E).
Overall, the significant upregulation of SOD is in line with the hypothesis of efficient degradation of TSC-induced ROS and might explain the discrepancy of superoxide production between the cell-free and cell culture experiments observed earlier. Despite its much higher cytotoxicity, the SOD stimulation was weaker in the case of Me2NNMe2 and the nanomolar-active TSCs, indicating that (in contrast to Triapine) at drug doses in the IC50 range no significant generation of superoxide occurs in the treated cells with these compounds. Consequently, it can be hypothesized that due to the higher resistance of the copper(II) complexes toward reduction, the compounds are less efficient in ROS production inside of cells.
Based on the assumed importance of metal chelation in the mode of action of TSCs, we also investigated changes in pathways regarding metal homeostasis in our arrays. Interestingly, we found only one significantly changed gene set, namely upregulation of genes involved in “response to zinc ions” after treatment with Me2NNMe2 (Fig. 4F). This is of interest, as a closer look on this gene set revealed that it mainly contains metallothioneins (which are cysteine-rich, low-molecular-weight proteins being responsible also for copper homeostasis) as well as the copper transporters ATP7A and ATP7B (Fig. 4G).
High copper complex stability is important for PDI inhibition potential
In a previous study, we showed that Me2NNMe2 induces paraptosis by inhibition of the ER-resident PDI and consequent disruption of the ER thiol redox homeostasis (13). Therefore, the copper-TSC complexes studied here were tested for their potential to inhibit this enzyme.
In line with our previous report (13), copper in form of a simple salt was already able to inhibit the PDI enzymes to some extent. This inhibition distinctly increased when copper was complexed by Me2NNMe2 or other TSCs with terminal dimethylation/di-substitution (Fig. 5A). Interestingly, the inhibition followed an “all-or-nothing” pattern, with complexes showing either the same weak inhibition as copper ions alone or strong PDI-disrupting ability comparable to the Me2NNMe2 complex. Dividing the compounds in these two categories, it can be clearly seen that PDI inhibition is associated with higher complex stability, higher anticancer activity, and enhanced vesicle formation (Fig. 5B and Supplementary Fig. S11).

Thus, although inhibition of PDI is probably not the only target of the nanomolar-active TSCs (other targets may include the RR inhibition) (29, 58), this indicates that there is a link between redox properties and copper complex stability with PDI inhibition and paraptosis induction, which needs to be further investigated in future studies.
Discussion
TSCs have long been known for their anticancer activity and their metal-chelating abilities (16, 23). In case of the currently in phase III clinically investigated derivative Triapine, especially an interaction with the iron homeostasis was suggested based on its strong inhibition of the iron-containing RR and the occurrence of methemoglobinemia as the main adverse effect in patients (16).
With the aim of developing TSCs with improved efficiency, derivatives with activity in the nanomolar IC50 range (such as DpC, Dp44mT, and Me2NNMe2) came into the focus of interest during the past decade, of which DpC also recently entered clinical trials (2, 20, 28, 29). An increasing body of evidence indicates that these drugs have additional modes of action that are responsible for their increased cytotoxicity. The particularities of nanomolar-active TSC are indicated by the induction of (i) a specific form of ER stress associated with disruption of the ER thiol redox homeostasis and inhibition of the ER protein PDI (13, 14, 39, 53), (ii) a just recently discovered form of programmed cell death called paraptosis (13), and (iii) increased activity in some MDR cancer cells (9, 40, 46).
Significantly, for all of these effects, the interactions with (intracellular) copper pools leading to formation of redox-active copper complexes were suggested to be crucial (16, 19, 34). Thus, it was proposed that copper complexes induce ROS (superoxide) by a redox reaction with intracellular reductants, a mechanism also referred to as “activation by reduction” (11, 34, 36, 44, 49). Accordingly, a strong synergism between the metal-free ligand and free copper salts has been repeatedly reported for nanomolar-active TSCs (19, 28, 49).
However, the experiments supporting the ROS model were often performed either with preformed copper(II) complexes (11, 34) or by preincubation of the ligands with high levels of extracellular copper (28, 36, 49). In contrast, in many cases, incubation with the metal-free ligands alone did not induce global ROS detectable, for example by the DCF-DA stain (13, 28, 34). Thus, it might be hypothesized that although there are indications that copper plays an important role in the mode of action of nanomolar-active TSCs, under physiological conditions, this might not be due to reductant-induced global redox stress production. However, localized and slower non-ROS-producing redox reactions (e.g., formation of disulfide bridges) might still be involved in their activity.
The aim of this study was to further investigate the role of copper in the activity of nanomolar-active TSCs in comparison to micromolar-active derivatives such as Triapine. To this end, a selected panel of structurally related α-N-heterocyclic TSCs and their respective copper(II) complexes was investigated with regard to solution stability, cell-free as well as intracellular redox properties, toxicity against sensitive and MDR cancer cell lines, and paraptosis-inducting potential. We show that the activity of the compounds has a strong correlation with these properties, confirming the relevance of their interaction with copper ions (Fig. 5C).
To analyze the interaction of the compounds with copper in more detail, we compared the anticancer activity of the preformed copper complexes with that of the metal-free ligands, together with their potential to induce collateral sensitivity in a P-gp-overexpressing cell model. Therefore, crucial differences were observed between short- and long-term drug incubation.
Although short incubation times (3 and 24 h) resulted in higher anticancer activity for especially nanomolar-active TSC copper(II) complexes compared with metal-free ligands, after longer incubation times (72 and 144 h), mostly no enhanced efficiency of the copper complexes was observed. The increased activity is in line with the literature showing (mainly for the nanomolar-active compounds) an enhanced activity of the metal-free ligands when co-applied with an excess of simple copper salts (19, 28, 49) or of preformed 1:1 or 1:2 copper(II):Dp44mT complexes compared with the metal-free ligand (20). An exception to the increased activity were copper(II) complexes of Triapine, H2NNHMe, and H2NNMe2, which showed no activity after a short time and even lower activity compared with the metal-free ligand after long-term incubation.
This may be explained by a less efficient absorption and cellular uptake of the charged [CuL]+ species compared with the neutral HL ligand, which are the predominating forms in solution at physiological pH based on our solution speciation studies in all cases. After longer incubation times, the differences in activity between the copper(II) complexes and the respective metal-free ligands disappeared, which could be explained by the hypothesis of different kinetics of the underlying cell killing mechanisms as described by Ishiguro et al. (19).
Thus, although the rapid cell killing mechanism is conducted by extracellular TSC copper complexes and characterized by redox reactions (27), the slow activity is redox independent and depends more on the ligand and/or intracellular copper complexation. In this regard, MES-SA/Dx5 cells were found to be more resistant against the rapid cell killing mechanism of both the metal-free ligands and preformed complexes compared with the parental MES-SA cells (seen in the 24 h IC50 values), whereas they were (slightly) more sensitive to the long-term activity of most copper complexes compared with the respective ligands.
The exceptions were complexes of TSCs such as Triapine, H2NNMeH, and H2NNMe2, with lower activity compared with metal-free ligands, that also did not exhibit increased activity in MES-SA/Dx5 cells as either ligand or complex. In fact, collateral sensitivity of MES-SA/Dx5 cells correlated with higher copper(II) complex stability, which further points to the importance of copper chelation for nanomolar TSC activity. This is in good agreement with previous studies, as also P-gp-overexpressing and Triapine-resistant SW480 or colchicine-resistant KB-3-1 cells showed no cross-resistance against terminally or pyridine amino di-substituted (especially nanomolar) TSCs after long-term incubation (28, 52).
It is important to note that the copper(II) complexes of the studied TSC ligands have the same composition ([CuL]+) with the same coordination mode (Npyridyl,N,S−)(H2O) at pH 7.4 in aqueous solution. However, significant differences were seen regarding their stability. Namely, the nanomolar-active TSCs form complexes of significantly higher stability with this metal ion compared with the micromolar compounds. On the other hand, the copper(II) complexes of the nanomolar TSCs could be reduced by GSH in a much slower redox reaction. When we investigated the role of redox activity, opposed to the current theory of activation by reduction (16, 20, 27), nanomolar-active TSCs were characterized by copper(II) complexes with higher stability and less efficient reduction by reducing agents.
This observation is in line with other studies such as from Garcia-Tojal et al. and from Santoro et al., both of whom found slower reduction by GSH in the case of the copper(II) complex of Dp44mT, as compared with the copper(II) complexes of Triapine and FTSC (11, 44). Moreover, for both Triapine as well as FTSC, reduction of the copper(II) complex resulted in efficient binding of the copper(I) ion to GSH, leaving a metal-free ligand able to interact with other metal ions such as iron or zinc (44). In agreement with a slower reduction, also cell-free superoxide production (which results from re-oxidation of the copper(I) complex) was lower with these complexes. Interestingly, the confirmation of these data in living cells turned out to be difficult. Thus, at physiologically relevant conditions, none of the TSC complexes increased DHE fluorescence (indicating intracellular superoxide) neither in the presence of a thiol-containing reducing agent (NAC) nor in its absence. Consequently, we hypothesized that cells rapidly adapt to redox stress and efficiently degrade the drug-generated superoxide.
In line with a higher, cell-free superoxide production of Triapine and its closest (micromolar-active) derivatives, upregulation of the superoxide-degrading enzymes SOD2/3 on mRNA and protein levels was identified as a possible protection mechanism. This is of interest, as comparable upregulation and increased activity of SOD enzymes was also reported and shown to protect against treatment with TSC copper complexes by other groups (11, 48, 51). This stimulation of SODs only in case of micromolar-active TSCs resulted in the generation of the theory that in case of Triapine and its close derivatives, the rather fast (intracellular) reduction leads to rapid dissociation of the copper complex and thus liberation of the metal-free ligands [which is also in good agreement with the very recently published data of Santoro et al. (44)]. Consequently, metal-free (or iron-/zinc-bound) Triapine could be the main species occurring inside the cell, playing a primary role in anticancer activity.
In contrast, as copper(II) complexes of nanomolar-active TSCs are much slower in their reduction and thus more stable, the copper(II) complex is able to reach intracellular targets such as the ER-resident protein PDI (Fig. 5D). In line with this hypothesis, paraptosis induction, measured by vacuole formation, was most pronounced with the nanomolar-active TSCs. This increase in paraptosis induction also correlated with the solution stability of their copper complexes as well as a slower reduction rate. In addition, also PDI inhibition by the TSC complexes highly correlated with copper complex stability and vacuole formation.
In line with a more pronounced role of copper in the anticancer activity of nanomolar-active TSCs, our array analysis revealed that Me2NNMe2 induces upregulation of metallothioneins, which strongly bind diverse metal ions including copper(I). Noteworthy, metallothioneins also contain multiple thiol groups, and thus take part in the cellular thiol redox homeostasis (43), which seems to be disrupted by the nanomolar-active TSC copper complexes (13). Interestingly, it has been suggested by Santoro et al. that metallothioneins play a crucial role in the removal of the reduced copper(I) from Triapine and FTSC after reduction, whereas in the case of Dp44mT the interaction of zinc-loaded metallothioneins with the copper(I)-TSC-GSH complex resulted in zinc transmetalation (44). In accordance, also our data indicate that there might be an interaction with metallothioneins in the cytosol (but not in the ER) of living cells after treatment with the metal-free TSC ligand. The exact nature of this interaction definitely warrants further investigations.
Materials and Methods
Chemicals
FTSC, PTSC, Triapine, H2NNHMe, H2NNMe2, MeHNNMe2, Me2NNH2, Me2NNHMe, Me2NNMe2, FTSC, MeHNNH2, MeHNNHMe, Dp44mT, and DpC were prepared as previously described (28, 29, 35). EDTA, KCl, KOH, and HCl were obtained from Reanal (Hungary); 2-(N-morpholino)ethanesulfonic acid (MES) and 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) were purchased from Sigma-Aldrich and used without further purification. Copper(II) stock solution was prepared by the dissolution of CuCl2 in water and its concentration was determined by complexometry with EDTA. For cell culture experiments, TSCs were first diluted in DMSO (10 mM), after which further dilutions were performed in aqueous solutions (double distilled (dd) H2O, buffer, or cell culture media depending on the assay) or with which copper complexes were formed in combination with CuCl2 (10 mM in ddH2O). Therefore, concentrations of DMSO in cell culture did not reach toxic doses. For spectrophotometric measurements, TSCs were directly diluted in ddH2O at low concentrations (255 μM) and dissolution was improved by ultrasound bath and adding a low concentration of HCl.
Spectrophotometric titrations
A Hewlett Packard 8452A diode array spectrophotometer was used to record the UV-Vis spectra in the interval 200–800 nm. The path length was 1 cm. Proton dissociation constants (pK a) of the TSC ligands, the copper(II) mono complexes, and the individual spectra of the species in the various protonation states were calculated by the computer program PSEQUAD (31). Spectrophotometric titrations were performed on samples containing the ligands at 10–50 μM concentration by a KOH solution in the presence of 0.1 M KCl at 25.0°C ± 0.1°C in the pH range from 2 to 11.9. An Orion 710A pH-meter equipped with a Metrohm combined electrode (type 6.0234.100) and a Metrohm 665 Dosimat burette were used for the pH-metric titrations. The electrode system was calibrated to the pH = −log[H+] scale by means of blank titrations (HCl vs. KOH) according to the method suggested by Irving et al. (18). The average water ionization constant (pKw) is 13.76 ± 0.05 in water. Argon was also passed over the solutions during the titrations.
Due to the limited water solubility of DpC, pK a values of the ligand were determined in 5% and 30% (w/w) DMSO/H2O solvent mixture, whereas pK a values of the copper(II) complexes were determined in 30% (w/w) DMSO/H2O. The pKa values obtained at various DMSO content were plotted against the 1/ɛr values of the solvent medium, where ɛr is the relative permittivity (or dielectric constant) of the solvent medium and values for the pure aqueous solution were obtained by extrapolation. The ɛr values are interpolated data taken from Covington and Dickinson (4). Namely, the pKa values for DpC in pure water were extrapolated from the values obtained in the DMSO/H2O mixtures with the slopes of the linear curves of DpC (5%, 30%), Triapine (0%, 30%), and H2NNMe2 (0%, 30%).
The conditional stability constants (β’) of the copper(II) complexes were calculated at pH 5.90 based on the spectral changes via the displacement reaction with EDTA in the presence of 50 mM MES and 0.1 M KCl (using a 1–2 h incubation). Data for pK a of EDTA and its Cu(II) complex taken from (8) and logβ’5.90 = 13.89 were calculated for [Cu(EDTA)]2−. In the competition experiments, the samples contained 25 μM copper(II), 25 μM ligand and the concentration of EDTA was varied in the range from 0 to 400 μM. It should be noted that EDTA and its copper(II) complex have negligible contribution to the measured absorbance values in the monitored wavelength range (320–550 nm), and only [CuL]+ and HL absorb light. In the case of DpC and Triapine, the completion reaction was performed in 30% (w/w) DMSO/H2O solvent mixture. The conditional stability constants of the metal complexes (β' (CuL)) and the individual spectra of the species were calculated by the computer program PSEQUAD (31). The overall stability constants of the [CuL]+ complexes (β) were calculated from the conditional stability constants: β [CuL]+ = β' [CuL]+ × αH , where αH = 1 + [H+]/Ka (HL) + [H+]2/(Ka (HL) × Ka (H2L+)); [H+] = 10-5.90 M. The overall stability constants of the protonated [CuLH]2+ and the mixed hydroxido [CuL(OH)] complexes were calculated as follows: log β [CuLH]2+ = log β [CuL]+ + pK a [CuLH]2+. Log β [CuL(OH)] = log β [CuL]+-pK a [CuL]+. pCu = −log [Cu(II)] values were calculated at pH 7.4 by using the determined stability constants.
Spectrophotometric kinetic measurements
The redox reaction of the copper(II) complexes with GSH and AA was studied at 25.0°C ± 0.1°C on a Hewlett Packard 8452A diode array spectrophotometer by using a special, tightly closed tandem cuvette (Hellma Tandem Cell, 238-QS). The reactants were separated until the reaction was triggered. Both isolated pockets of the cuvette were completely deoxygenated by bubbling a stream of argon for 10 min before mixing the reactants. Spectra were recorded before and then immediately after the mixing, and changes were followed till no further absorbance change was observed. One of the isolated pockets contained the reducing agent (GSH or AA) and its concentration was in the range of 250–2500 μM and the other contained the copper(II) complex, which was prepared in situ by using 25 μM of the metal ion and the ligand respectively. The pH of all the solutions was adjusted to 7.40 by 50 mM HEPES buffer, and an ionic strength of 0.1 M (KCl) was applied. The stock solutions of the reducing agents and the complexes were freshly prepared every day.
During the calculations, the absorbance (A), time (t) curves were fitted and analyzed at the λ max of the complex. (A 0-A final) × e(-a×t) + A final equation was used, where A 0, A final, and a parameters were refined and accepted at the minimal value of the weighted sum of squared residuals (difference between the measured and calculated absorbance values) at the given wavelength. Then observed rate constants (k obs) of the redox reaction were obtained from the data points of the simulated absorbance-time curves as the slope of the ln(A/A 0) versus t plots.
Cell lines and culture conditions
Human uterine sarcoma MES-SA and the doxorubicin selected MES-SA/Dx5 cells expressing mCherry and eGFP proteins, respectively, were engineered from MES-SA and MES-SA/Dx5 (ATCC; MES-SA: No. CRL-1976™, MES-SA/Dx5: No. CRL-1977™) by using a lentiviral system (55). The phenotype of the resistant cells was verified by using cytotoxicity assays (not shown). Before the experiments, MES-SA/Dx5 cells were cultured in 500 nM doxorubicin, to ensure Pgp expression. Cells were cultured in Dulbecco's modified Eagle's medium (DMEM; SigmaAldrich, Hungary) supplemented with 10% fetal bovine serum, 5 mM glutamine, and 50 U/mL penicillin and streptomycin (Life Technologies). Human colorectal adenocarcinoma SW480 cells (obtained from ATCC; No. CCL-228™) were cultured in MEME supplemented with 10% fetal calf serum (PAA, Austria). CT-26 murine colon carcinoma cells (CRL-2638, purchased from ATCC) were cultured in DMEM/F12 medium (1:1 from Sigma; #D6421) supplemented with 10% heat-inactivated fetal calf serum. All cell lines were cultivated at 37°C, 5% CO2.
Cell viability assay
In the co-culture system, after trypsinization, suspensions of MES-SA mCherry and MES-SA/Dx5 eGFP cells were mixed and seeded on 384-well plates at a 2500 cells/well density (1250 cells/well per cell model) in 20 μL of medium, 1 day before drug addition. Cells were then treated with a serial dilution of the drugs, so that the final volume was 60 μL. Liquid handling was fully automated by a Hamilton StarLet robotic pipetting workstation (Hamilton, Switzerland). Plates were incubated and measured twice: after 72 and 144 h of drug addition. Growth inhibition of the cells was assessed based on the detection of the respective fluorescent intensities scanned from the wells by an EnSpire plate reader (eGFP: 485ex/510em; mCherry: 585ex/610em, Perkin Elmer, United Kingdom). Raw measurement files were exported, and automated data evaluation was performed by our custom program, which was written by Judit Sessler in C#. Data were normalized to the negative (live cells, maximal fluorescence) and positive (dead cells, minimal fluorescence) controls; then, growth inhibition data points (plotted against the respective concentrations) were connected with a line, and IC50 was considered as the point at which the connecting line reached the 50% inhibition according to the y axis.
For single-cell viability assay, SW480 cells were plated (2 × 103 cells/well) in 96-well plates and allowed to recover for 24 h. Then, cells were treated with increasing concentrations of TSCs for 72 h. Cell viability was measured by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)-based vitality assay (EZ4U; Biomedica, Vienna, Austria) as published (17). GraphPad Prism software was used to calculate cell viability expressed as IC50 values calculated from full dose-response curves. For further analysis and comparison, pIC50 values were used instead of IC50 values (Supplementary Fig. S1). pIC50 values were calculated as the –log10 from IC50 values in molar.
Microscopy
Cells were seeded into a 24-well plate with 2 × 104 cells/well and left to recover for 24 h. Then, cells were treated with indicated concentrations of TSC ligands. After 24 or 48 h, microscopic phase-contrast images were taken with a Zeiss primo vert microscope with a Zeiss axio cam ERc5s camera. Percentage of vacuolated cells was counted in at least three different parts of a well.
Quantification of superoxide radicals
To examine the cell-free production of superoxide radicals, the reduction of NBT was analyzed as previously reported (27). Briefly, 0.6 mM NBT was incubated with 5 μM copper(II) complexes with or without 2 mM NAC. The experiments were performed in PBS (pH 7.4). The extent of NBT reduction was determined spectrophotometrically by measuring the absorbance at 560 nm after 45 min of incubation. No superoxide radicals were observed without NAC (data not shown).
Intracellular superoxide determination using DHE
DHE (#D7008; Sigma-Aldrich, MO) was used to detect the production of intracellular superoxide. Briefly, 5 × 105 MES-SA cells per sample in 500 μL of PBS (78.1 mM Na2PO4 × 2 H2O, 14.7 mM KH2PO4, 26.8 mM KCl, 1.37 M NaCl) were incubated with or without 2 mM NAC for 15 min at 37°C. Then, 5 μM of indicated TSC complexes were added for further 60 min. Subsequently, DHE (10 μM) was added 30 min before measurement. After incubation, the mean fluorescence intensity was measured by flow cytometry using an FACSCalibur instrument (Becton Dickinson, Palo Alto, CA). Antimycin A (AMA, 10 μM) was used as positive control.
PDI reduction activity measurement
PDI reduction activity was measured by using PROTEOSTAT PDI assay kit (#ENZ-51024, Enzo Life Sciences, Switzerland). Experiments were performed according to the manufacturer's instructions. Briefly, drugs alone or preincubated with CuCl2 (1:1) were added to a prepared insulin PDI solution. Then, dithiothreitol (DTT) (1 mM) was added to start PDI reduction activity. After 30 min, the reaction was stopped by the Stop reagent and the insulin precipitate was fluorescently labeled with Proteostat PDI detection reagent for 15 min. Fluorescence intensity was measured at 500 nm excitation and 603 nm emission by using the spectrophotometer Tecan infinite 200Pro (Tecan Group, Männedorf, Switzerland).
Protein expression
After drug treatment, total protein lysates were prepared, 20 μg per sample, separated by SDS-PAGE, and transferred onto a polyvinylidene difluoride membrane for Western blotting as previously described (17). The following antibodies were used: Cell Signaling Technology: SOD2 (#13141), GAPDH (#5174). Primary antibodies were used: 1:1000. Secondary anti-rabbit (#7074) horseradish peroxidase-labeled antibodies from Cell Signaling Technologies were used in working dilutions of 1:10 000.
Total-RNA isolation and whole-genome gene expression array
Total RNA from SW480 cells (either untreated or treated [0.1 or 1 μM for 15 h]) was isolated by using RNeasy Mini kit (#74106, Quiagen, Germany) following the manufacturer's instruction. Transcriptional profiles of cells were determined by performing a 4 × 44K whole-genome oligonucleotide gene expression array (Agilent, CA) as previously described (32). Normalization was performed in R by using the Bioconductor (version 3.7) package “limma” if not otherwise indicated (42). Whole-genome gene expression array and GSEA were performed as previously described (13).
Animal experiments
Six- to eight-week-old BALB/c mice were purchased from Janvier (France). The animals were kept in a pathogen-free environment, and every procedure was carried out in a laminar airflow cabinet. Experiments were done according to the regulations of the Ethics Committee for the Care and Use of Laboratory Animals at the Medical University Vienna (proposal number BMWF-66.009/0081-WF/V/3b/2015), the U.S. Public Health Service Policy on Human Care and Use of Laboratory Animals, as well as the United Kingdom Coordinating Committee on Cancer Prevention Research's Guidelines for the Welfare of Animals in Experimental Neoplasia. To ensure animal welfare throughout the experiment, the body weight of the mice was assessed once a day. At weight loss exceeding 10% (in less than 2 days) or occurrence of ascites, animals were sacrificed by cervical dislocation.
In vivo analysis of SOD2 expression
CT-26 cells (5 × 105 cells in 50 μL) were injected subcutaneously into the right flank of female Balb/c mice. Starting on day 4, Triapine (10 mg/kg in 10% DMSO) or solvent treatment was given orally for 5 consecutive days a week for 2 weeks. Animals were sacrificed on day 14 per cervical dislocation, and tumor tissue was isolated and fixed in 4% paraformaldehyde (Carl Roth, #P087.3) for 24 h. Tumor tissue was paraffin-embedded with the KOS machine (Milestone) and sliced in 4 μm-thick sections. For SOD2 staining, sections were incubated with a SOD2-specific antibody (1:1000; Cell Signaling, #13141) in a humid chamber for 1 h at room temperature after antigen retrieval by boiling for 30 min in 10 mM citrate buffer (pH 6.0, DAKO; #S1699),. Antibody binding was detected by using the UltraVision LP detection system according to the manufacturer's instructions (Thermo Fisher Scientific, Inc.; #TL-125-HL). Color was developed by using 3,3′-diaminobenzidine (Dako; #K3468), followed by a nuclear counterstain with hematoxylin. Stained tissue slides were scanned and analyzed by using Definiens Software.
Correlation analysis
Correlations were performed in the GraphPad Prism 8 software. R2 from linear or one-phase decay regression is given in correlation diagrams. Regressions lines are shown in correlation plots with 95% confidence interval. p-values were calculated by using Pearson correlation coefficient, which is a measure of linear correlation. For the correlation matrix (Fig. 5D), p-values of correlations were corrected for multiple comparisons with a two-stage linear step-up procedure of Benjamini, Krieger, and Yekutieli with an FDR of 1%. The thereby generated q-values are shown with a grayscale code.
Footnotes
Author Disclosure Statement
The authors declare no conflict of interest.
Funding Information
This work was supported by the National Research, Development and Innovation Office-NKFI through project FK 124240 and FIKP program TUDFO/47138-1/2019-ITM. Furthermore, this work was in part funded by the Austrian Science Fund (FWF) grant number P31923 (to C.R. Kowol and P. Heffeter) as well as by the bilateral program Scientific and Technological Cooperation of the OeAD. Part of the data was generated in course of the research exchange program Aktion “Österreich-Ungarn” as well as in a short-term fellowship of the European Molecular Biology Organization (EMBO, grant ASTF335-2016) both to P. Heffeter. S. Hager is a recipient of a DOC Fellowship of the Austrian Academy of Sciences. The funding sources had no involvement in the collection, analysis, and interpretation of data as well as in the decision to submit the article for publication.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Figure S7
Supplementary Figure S8
Supplementary Figure S9
Supplementary Figure S10
Supplementary Figure S11
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
Supplementary Table S5
Supplementary Table S6
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.